全文HTML
--> --> -->材料中的缺陷及缺陷演化对材料性质有重要影响. 如何有效地反映缺陷行为对高熵合金性能的影响, 是构建理论计算模型时首先要考虑的问题. 由于高熵合金为无序固溶体合金, 晶格中原子排布的无序性对其晶格结构的搭建构成极大挑战. 目前在无序固溶体合金计算模拟中, “有效近似方法”是一种常用的构建无序固溶体结构的方法, 如虚拟晶格近似(virtual crystal approximation, VCA)和相干势近似方法(coherent popential approximation, CPA)[8-10], 该方法通过有效介质近似, 将每个原子, 平均地视作一种“有效原子”, 从而避免了需要点对点确定各类原子在晶格点位上排布的问题. 但与此同时, 这种力求平均的效果, 忽略了晶格中原子局域环境的变化, 无法反映原子畸变、原子对间的配位差异性等. 而高熵合金由于原子成分的复杂性, 缺陷的形成和演化必定与不同原子周围的局域环境有关, 因此, 此类方法在高熵合金缺陷行为的研究中并不适用. 在目前的诸多报导中, 研究者多采用超胞近似的方法来开展对高熵合金缺陷的研究. 其中, 较为常用的是准随机近似方法(special quasirandom structures, SQS), 该方法寻求无序固溶体理想的无序状态[11-13], 构建得到的结构和原子排布的随机性接近理想随机状态. 这种随机结构可以很好地反映晶格中原子周围局域环境的变化, 被广泛用于对高熵合金的点缺陷、层错缺陷等的研究. 不过该方法构建过程中, 只考虑了数学上的随机性, 虽然保证了原子点对点排列的随机性, 但对于原子尺寸、原子电负性等物理性质并未考虑. 构建得到的结构在高温条件下可能是合理的, 但在低温条件下合理性上仍有待验证; 并且在有限晶胞尺寸下, 晶格中原子的理想随机排布状态不只一种, 通常在研究同一问题时, 需要同时构建多个结构进行模拟, 并统计结果的平均值. 这在实际研究中, 带来了结果的不确定性和计算成本的上升.
至今, 高熵合金距离最早被文献报导已有15年, 并伴随着越来越多的高熵合金体系被开发和研究, 研究者对高熵合金的认识和理解也越发成熟. 最初, 研究者认为由于高熵合金原子种类的复杂性带来的高熵效应, 将使该合金能保持稳定的单相无序固溶体状态, 典型的如FeCuCrMnMo合金(也被称为Cantor合金)[1,2], 显微结构分析结果表明该合金中的原子排布达到了近似理想的随机混合状态. 但随着研究的深入, 目前学界认为, 高熵合金中原子的排布并非是理想的无序状态, 短程序或者多相共存现象是普遍存在的. Pickering等[14]在FeCrCoNiMn合金热处理后的晶界处, 发现了Cr原子的偏析现象. Santodonata等[15]在Al1.3CoCrCuFeNi合金中发现了多处富Cu相、富Al-Ni相和富Cr-Fe相的存在. Singh等[16]在AlCoCrCuFeNi合金中发现了富Cu区的存在. 包括本研究中的FeCuCrMnMo合金, 退火处理后组织结构中有明显的Cu偏析现象. 而对于固溶体合金短程有序(short range order, SRO)结构的构建, 将是高熵合金理论计算的基础和难点.
在此基础上, 本研究组提出了用蒙特卡洛结合密度泛函理论(Monte Carlo/density functional theory, MC/DFT)构建高熵合金的平衡态结构的计算方法. 该方法充分考虑了原子本身的物理性质, 以及原子在不同配位环境下排布的合理性, 最终得到的平衡态结构可以有效反映高熵合金中的SRO现象, 并与实验结果相一致. 同时, 分析了短程序形成的影响因素及短程序的出现对高熵合金性质的影响.
2.1.第一性原理计算
计算采用蒙特卡洛(Monte Carlo, MC)方法与基于密度泛函的第一性原理(density functional theory, DFT)计算方法. 体系能量计算采用的DFT方法通过VASP(Vienna ab-initio simulation package)软件包[17]实现, 其中交换关联泛函采用广义梯度近似(generalized gradient approximation, GGA)下的PBE(Perdew-Burke-Ernzerhof)修正, PBE方法被认为是目前为止最有效的计算铁磁性材料性质的方法. 由于MC/DFT方法在交换原子过程中, 涉及到上千个结构能量的计算, 基于计算成本上的考量, 本工作采用两套不同精度的计算条件设置: 1)中等精度. 在MC/DFT方法构建结构的计算中, 平面波基组的截断能设为330 eV, K网格点仅设置为Γ点, 能量收敛标准设为0.1 meV, 力的收敛标准为20 meV/?, 原子弛豫为全弛豫. 在交换原子过程中的平衡态阶段, 新旧结构间的能量差为5 meV量级, 该精度设置可以有效保证MC抽样过程中平衡态的收敛. 2)高精度. 在获得平衡态结构后, 对该结构进行全弛豫优化, 及对电子性质、磁性、力学性质的计算, 此时将计算精度调高, 截断能设为350 eV, K网格点设为4 × 4 × 2, 能量收敛标准设为0.01 meV, 力收敛标准设为10 meV/?, 计算中均考虑电子自旋极化作用. 在MC/DFT计算中, 分别测试了尺寸为2 × 2 × 5(40个原子), 2 × 3 × 5(60个原子)和3 × 3 × 5(90个原子)的超胞结构, 以及较大尺寸的5 × 5 × 5(125个原子)BCC原胞结构, 结果表明四种结构的平衡态结构中均有富Cu原子短程序产生, 基于尺寸效应及计算成本的考量, 本工作选取包含90个原子的3 × 3 × 5进行MC/DFT中各温度下的计算, 以及后续较高精度的电子、力学性质等的计算. 为了比较短程序结构对材料性质的影响, 采用ATAT(alloy theoretic automated toolkit)软件中的SQS代码, 同时构建了6个准随机结构, 该结构保证了最近邻原子间的理想随机性, 并对其进行结构全弛豫优化, 及电子、磁性和力学性质的计算. 最后, 比较了FeCuCrMnMo合金短程序结构在铁磁性状态、顺磁性状态的结构优化总能, 以得到该合金的最稳定结构状态.2
2.2.MC方法
MC理论方法通过自己编译程序实现. 将MC与DFT方法的计算技术相结合, 在很多材料体系的结构预测中得到应用[18-20]. MC方法的优势在于, 在沿Markov链搜索稳定态的过程中, 可以避免体系陷入局域能量极小的亚稳态中, 最终得到全局体系能量最小的稳态状态. 在本工作中, 通过自编代码与VASP软件结合实现. 将MC方法和DFT理论计算相结合, 初始晶胞选采用SQS方法构建的准随机结构, 采用等温等压(NPT)系综, 每步能量为全弛豫条件下计算得到. 其具体计算流程如下: 1)采用DFT方法计算初始晶胞结构能量; 2)随机交换晶胞中的两个原子, 得到一个新结构, 每交换一次原子, 视为进行一步MC步; 3)用DFT方法计算每个MC步得到的新结构的能量; 4)根据Metropolis算法, 新结构被接受的概率为

2
2.3.序参数定义
根据Warren-Cowley[21]规则, 有序参数定义为


3.1.FeCuCrMnMo合金平衡态晶格结构
同时计算了800, 1000, 1200和1500 K四个温度下的平衡态结构, 并跟踪结构中原子对有序参数的变化, 其中有序参数由公式(2)计算得到(图1). 如图1(a)所示, 在800 K下, 当MC程序运行至近3000步时, 能量曲线趋于平缓, 此时体系结构达到平衡态. 通过观察有序参数变化(图1(b)), 此时有明显的Cu-Cu和Mo-Mn原子对富集现象, 同时Mo-Mo和Fe-Mn原子对相对贫乏. 比较不同温度下的结果可以发现, 随着温度的上升, 能量曲线和序参数曲线振荡加剧, 说明温度的上升加剧了结构的无序性, 但即使在高温条件下, 如1200 K, 仍可以观察到Cu-Cu短程序的产生. 选取800 K下得到的平衡态结构作为后续计算使用的结构, 其优化后的结果如图2所示, 从晶格结构中可以观察到明显的Cu原子SRO现象, 该结构中Cu-Cu有序参数的值为–0.53. 图 1 温度分别在800, 1000, 1200和1500 K下, FeCuCrMnMo合金结构总能和有序参数随MC步数的演化 (a), (c), (e)和(g)为MC步数-结构总能关系, (b), (d), (f)和(h)为MC步数-有序参数关系图
图 1 温度分别在800, 1000, 1200和1500 K下, FeCuCrMnMo合金结构总能和有序参数随MC步数的演化 (a), (c), (e)和(g)为MC步数-结构总能关系, (b), (d), (f)和(h)为MC步数-有序参数关系图Figure1. System evolution vs MC steps at 800, 1000, 1200 and 1500 K. (a), (c), (e) and (g) System energy; (b), (d), (f), and (h) SRO parameters for atomic pairs.
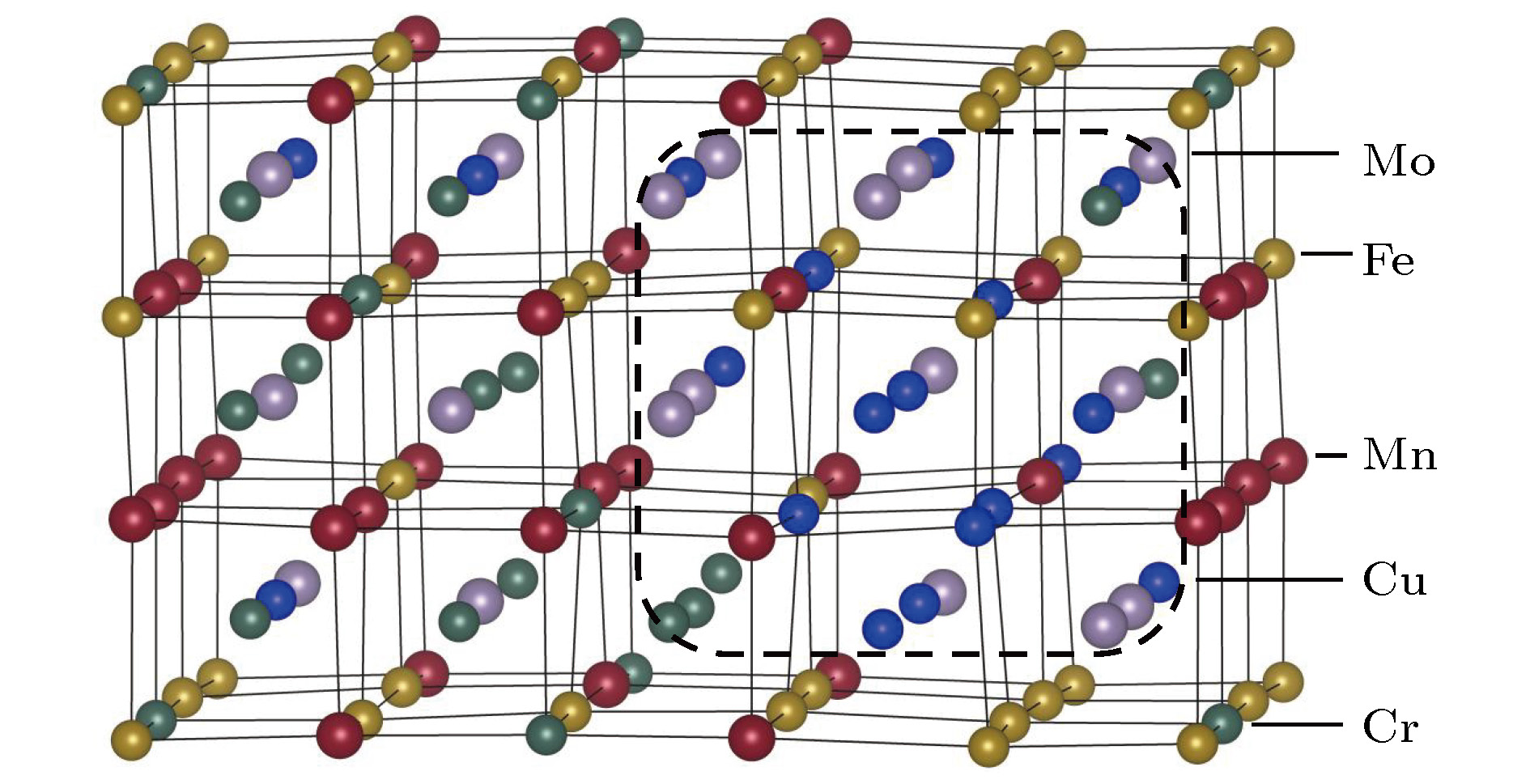 图 2 FeCuCrMnMo合金平衡态晶格结构, 其中Cu原子(蓝色原子球)的SRO现象用黑色虚线框标出
图 2 FeCuCrMnMo合金平衡态晶格结构, 其中Cu原子(蓝色原子球)的SRO现象用黑色虚线框标出Figure2. The lattice structure of FeCuCrMnMo alloy, in which Cu-rich short range order (SRO) is framed by a dotted box. Blue spheres represent Cu atoms.
FeCuCrMnMo合金中富Cu相的产生, 在实验中也同样被检测到[7]. 图3为温度773 K下退火1 h后FeCuCrMnMo合金采用扫描透射电子显微术(scanning transmission electron microscopy, STEM)连同能量弥散X射线谱分析(energy-dispersive X-ray spectroscopy, EDS)技术对该合金的显微结构的检测分析图片. 从TEM图中可以观察到明显的元素偏析现象, 进一步分析表明, 合金中的偏析区域主要是有富Cu相产生, 说明理论计算结果与实验现象是一致的.
 图 3 FeCuCrMnMo合金773 K下退火1 h后的STEM-EDS微观组织图片[7]
图 3 FeCuCrMnMo合金773 K下退火1 h后的STEM-EDS微观组织图片[7]Figure3. STEM-EDS maps of the FeCuCrMnMo alloys at 773 K for 1 h[7].
2
3.2.FeCuCrMnMo合金晶格结构分析
采用SQS方法同时构建了FeCuCrMnMo合金的理想随机结构. 由于五元合金的理想随机结构不只一种, 总共构建了6个SQS结构作为统计. 如图4所示, 这些结构中各原子对的有序参数值均在 ± 0.05以内, 展示出很高的随机性. 但由于原子排布的不同, 各结构优化后的总能出现微小变化(见图4(b)). 将其统计平均值列于表1中.| 金属 | 磁性 | 晶格 | 内聚能/eV | 体积/?3 | 晶格常数 | |
| a (理论值) | a0 (实验值) | |||||
| HEA-SRO | FM | BCC | –8.13 | 12.15 | 2.896 | 2.878[7] |
| NM | BCC | –8.12 | 12.05 | 2.889 | ||
| FM | FCC | –8.08 | 12.31 | 3.666 | ||
| HEA-SQS | FM | BCC | –8.09 | 12.53 | 2.926 | |
| Fe | FM | BCC | –8.32 | 11.78 | 2.832 | 2.834[22] |
| Cu | NM | FCC | –3.72 | 11.81 | 3.636 | 3.615[23] |
| Cr | AFM | BCC | –9.51 | 11.97 | 2.835 | 2.882[23] |
| Mn | FM | BCC | –8.29 | 14.38 | 3.080 | 3.080[23] |
| Mo | NM | BCC | –10.95 | 15.58 | 3.148 | 3.147[23] |
表1FeCuCrMnMo合金的SRO结构、SQS结构及相关纯金属的平均单个原子能、平均单个原子体积和晶格常数值. 其中FM, NM和AFM分别代表铁磁性、顺磁性和反铁磁性
Table1.Cohesive energy per atom, structural volume per atom, and lattice parameters for SRO and SQS structures, and Fe, Cu, Cr, Mn, Mo, and W metals. FM, AFM, and NM denote ferromagnetism, antiferromagnetism, and nonmagnetic, respectively.
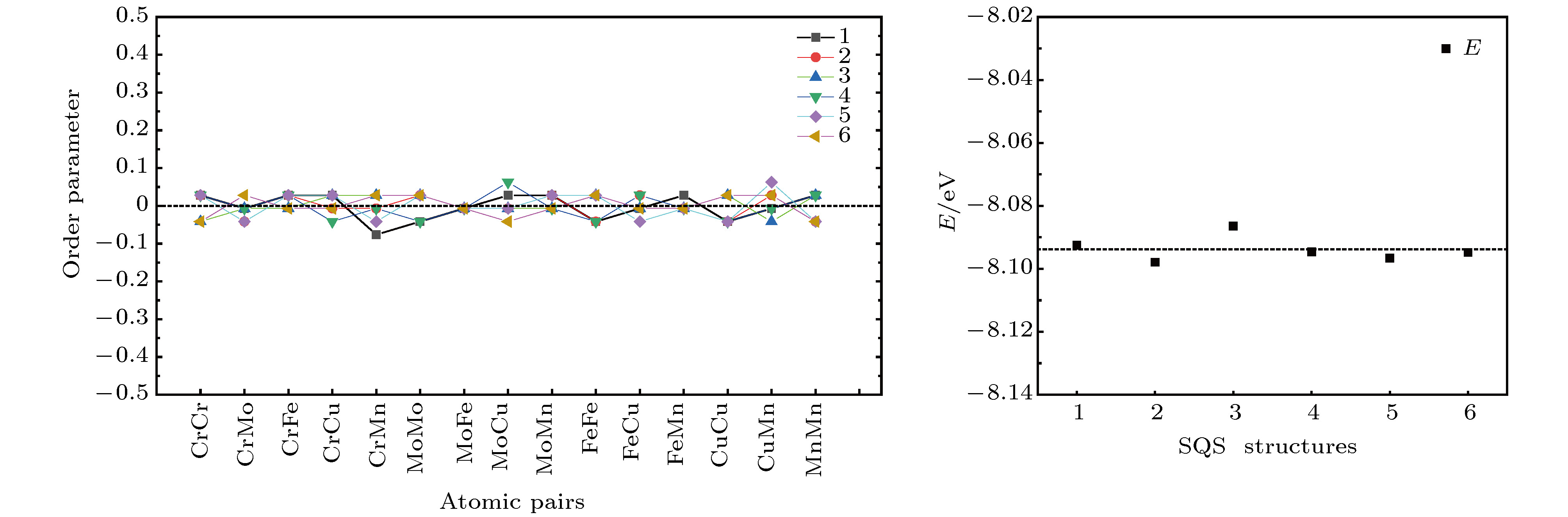 图 4 FeCuCrMnMo合金的6个SQS结构 (a)有序参数; (b)结构总能(单个原子)
图 4 FeCuCrMnMo合金的6个SQS结构 (a)有序参数; (b)结构总能(单个原子)Figure4. Six SQS structures of FeCuCrMnMo alloy: (a) Order parameters of each atomic pairs; (b) total energy per atom.
表1为FeCuCrMnMo合金的SRO结构和SQS结构及合金对应的纯金属的结构优化结果. 从表1可以看出, SRO结构能量比随机结构低0.04 eV/atom, 对应于总能量(90个原子), 则总能量降低3.6 eV, 而且体积更加紧凑. 说明短程序结构中原子排布更符合低能组态, 换言之, SRO的出现使结构更稳定. 同时, 相比于SRO结构的FCC状态和顺磁性(paramagnetism, NM)状态, 具有BCC结构的铁磁性(ferromagnetism, FM)状态最为稳定. 在本工作后续研究中, 均采用800 K下的SRO结构(BCC), 并考虑电子自旋作用.
为了表征高熵合金结构优化后原子对间的畸变状态, 分别对SRO结构和理想随机结构进行径向分布函数分析. 径向分布函数可以统计不同原子对在不同距离下的分布状态, 其计算公式为








图5为结构优化前的BCC结构(无畸变)、优化后的高熵合金的SRO结构和SQS结构的径向分布函数图. 结果表明, 优化后的SRO结构和SQS结构仍表现出明显的BCC结构的特征, 但峰值变低, 峰宽增大, 说明结构中存在明显的晶格畸变. SRO结构峰的位置的相对左移与该结构的晶格常数较小有关, 见表1.
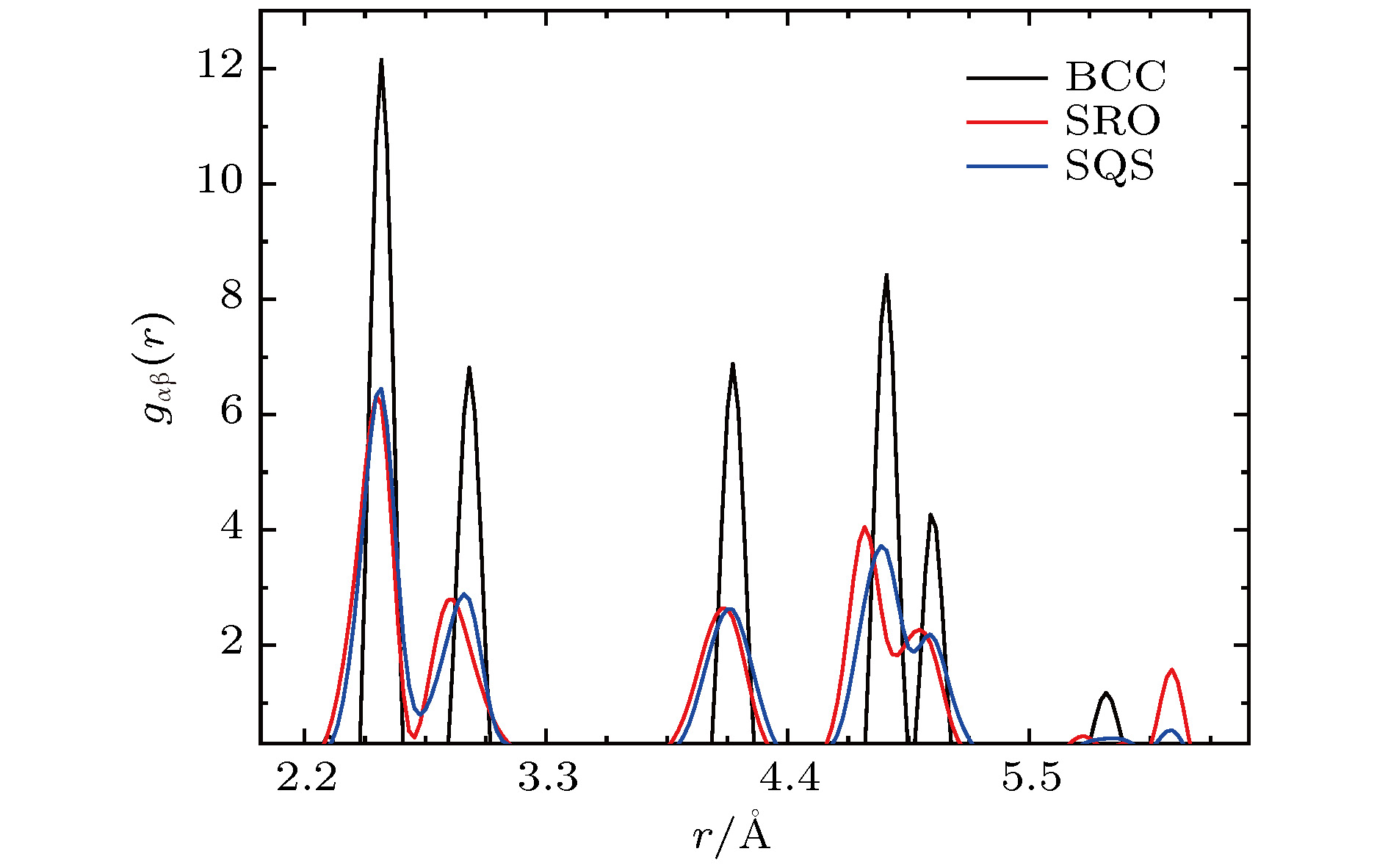 图 5 高熵合金理想BBC结构, 结构优化后的SRO结构和SQS结构的径向分布函数计算结果
图 5 高熵合金理想BBC结构, 结构优化后的SRO结构和SQS结构的径向分布函数计算结果Figure5. Average radial distribution functions for BCC structure (unrelaxed), SRO structure (relaxed) and SQS structure (relaxed) of FeCuCrMnMo alloy.
在此基础上, 进一步分析不同原子对间的径向分布结果. 如图6(a)和图6(b)所示, SQS结构中的异类原子概率分布相对均匀, 不同原子对在第一近邻分布的概率相对一致, 但峰宽较宽, 说明原子对间距离的畸变较大, 不同原子对距离的分布峰距离第一近邻距离偏移较大, 原子对距离分布范围在2.16—2.84 ?区间. 而SRO结构中不同原子对的概率分布极不均匀, Cu-Cu峰高且尖锐(图6(c)), 说明Cu-Cu出现在第一近邻的概率极大, 这与结构中出现的Cu-Cu短程序现象相吻合, 且峰宽较窄; 而Mo-Mo和Mn-Mn等原子对多出现在第二近邻, 类似于简单立方短程有序排布; 同时从图6(d)可以看出, Fe-Mo, Cr-Mo和Mn-Mo等原子对出现在第一近邻的概率较高, Cu-Mo和Fe-Mn等概率较小, 这与图1结果相吻合; 且不同原子对距离峰的分布相对集中, 峰宽较窄, 第一近邻的原子对的距离分布范围在2.25—2.7 ?区间.
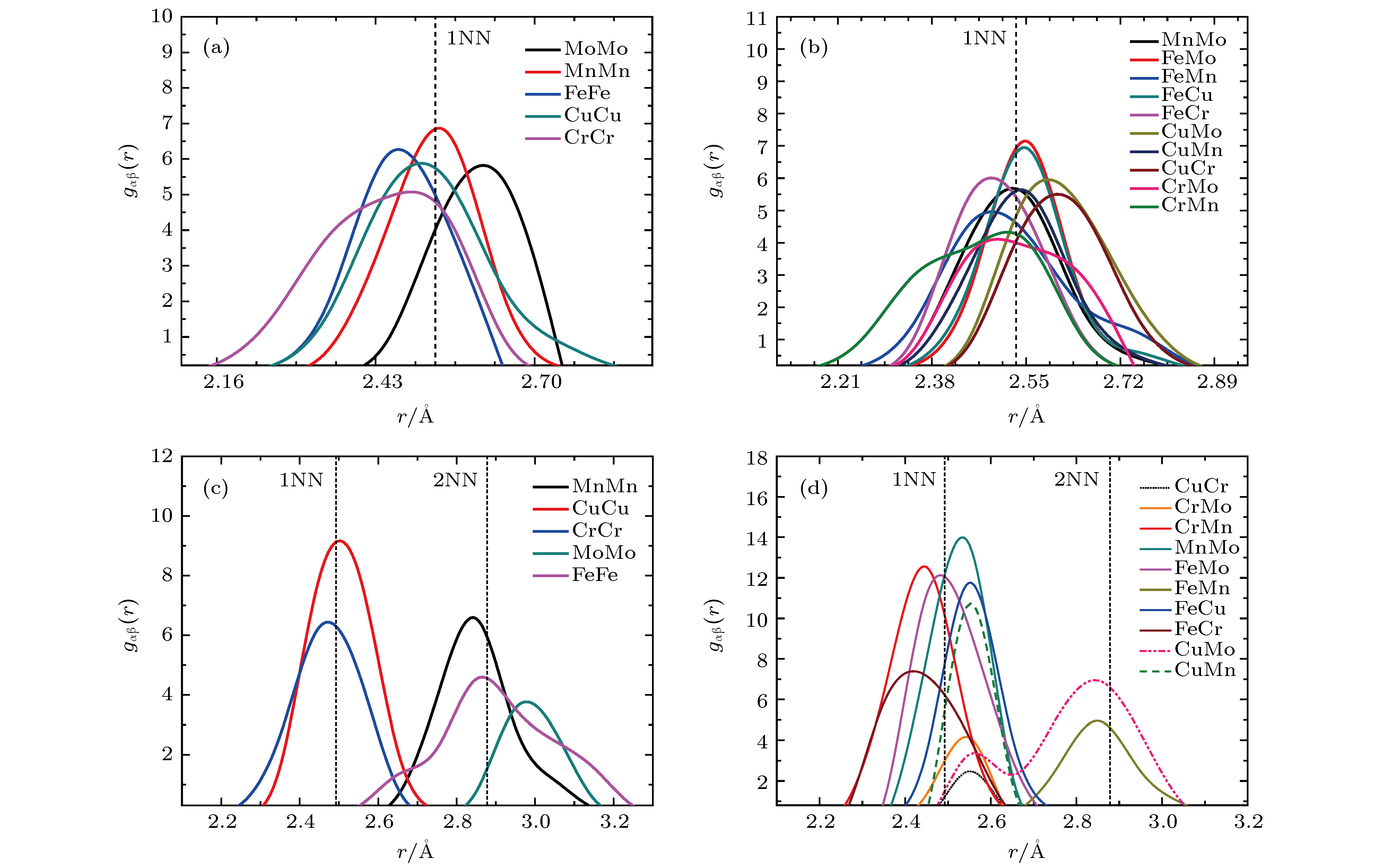 图 6 高熵合金结构优化后原子对间径向分布函数图 (a), (b) SQS结构; (c), (d) SRO结构. 虚线标明无畸变晶格中第一近邻(1NN)和第二近邻(2NN)的距离值
图 6 高熵合金结构优化后原子对间径向分布函数图 (a), (b) SQS结构; (c), (d) SRO结构. 虚线标明无畸变晶格中第一近邻(1NN)和第二近邻(2NN)的距离值Figure6. Partial pair distribution functions of FeCuCrMnMo alloy: (a), (b) SQS structure; (c), (d) SRO structure. Dotted lines show the distances of first nearest neighbor (1NN) and second nearest neighbor in unrelaxed lattice.
径向分布函数分析说明, 结构优化后, 不同原子对距离均出现畸变, 相比于理想无畸变晶格, 原子间距离被拉近或被拉远. SQS结构的原子对间的距离分布更大, 说明其晶格畸变也更大, 而SRO结构晶格畸变相对较小. 这与之前的结论相一致, SRO结构中原子在空间排列更密集.
2
3.3.SRO现象物理分析
高熵合金中原子短程序的出现与不同原子的物理性质密切相关, 而这在SQS方法中是被忽略的. 下面将通过原子尺寸、原子混合焓和原子间相互作用等分析短程序特别是Cu-Cu富集现象出现的物理原因.1)原子尺寸
根据对合金中不同原子对应的纯金属结构的优化, 得到相应原子的单位体积尺寸为: Fe(11.78) < Cu(11.81) < Cr(11.97) < Mn(14.38) < Mo(15.58), 单位为?3/atom. 根据晶胞密堆积原理, 晶体中原子排布趋向于相互配位数高, 堆积密度大, 能充分利用空间, 因而体系稳定的结构. 可以推测, 尺寸较大的原子更倾向于被尺寸较小的原子所包围, 以减小原子畸变带来的局部应力. 这可能是Mo-Mo和Mn-Mn等原子对较少在第一近邻距离下出现的原因, 而Cr-Mo和Fe-Mo等更容易在第一近邻出现.
2)原子间混合焓
对于金属元素, 合金化后对应的混合焓为:



不同原子间混合焓为以上两种因素综合作用的结果, 其值可以从文献[24]中查到(表2). 混合焓小于0, 表示原子间更容易混合, 该原子对的结合降低了体系内能, 混合焓大于0则表示原子间更难混合, 需要消耗能量使该原子对结合. 根据混合焓的数据, 一个很明显的特征是Cu原子与其它四种原子的混合焓均为正值, 其中值最大的为Cu-Mo原子对, 这与Cu-Mo原子对在结构中第一近邻距离下出现概率较小相吻合(图6(d)). 同时, Fe-Mo, Fe-Cr和Cr-Mo等更容易混合, 这也与之前的结果相一致.
| 混合焓 | Cr | Mn | Mo | Fe |
| Cu | 12 | 4 | 19 | 13 |
| Cr | 2 | 0 | –1 | |
| Mn | 5 | 0 | ||
| Mo | –2 |
表2FeCuCrMnMo合金中不同原子对间的混合焓[24] (单位: KJ/mol)
Table2.Enthalpy of mixing of binary systems containing the elements in FeCuCrMnMo alloy(Unit: KJ/mol).
3)原子间相互作用
由于高熵合金中原子排布的无序性, 所以不同的原子配位环境不同. 为尽可能分析原子间相互作用, 在晶格中找到以Mo原子为中心, 周围最近邻原子均包含另外4类原子(Cu, Cr, Fe, Mn)的典型情况. 计算其电子分波态密度(PDOS), 结果如图7所示. 从图7可以看出, 过渡族金属的DOS主要来自3d电子的贡献, Cu原子的电子分布相对局域化, 主要集中在–4—–2 eV, 这与Cu原子d电子轨道恰好为全满状态有关. 根据洪特规则: 在同一轨道, 当电子分布为全满、半满或全空时体系最为稳定, 而Cu原子恰好为3d轨道电子全满, 4s轨道半满的状态, 所以其外层电子最为不活泼. 同时, 在Mo原子和Mn原子的–2.2 eV附近观察到微小的共振峰, 说明Mn-Mo会有相对较强的相互作用.
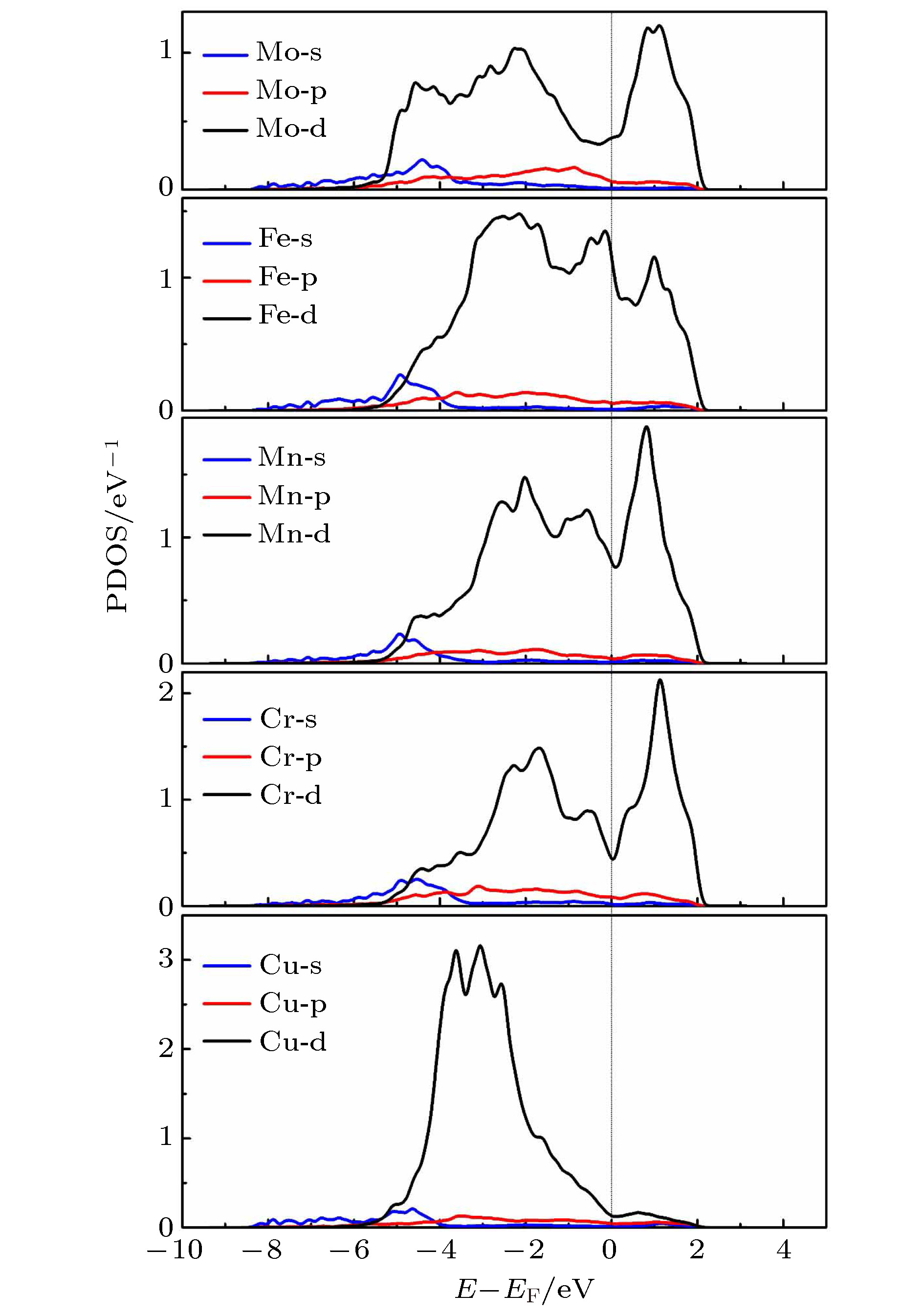 图 7 FeCuCrMnMo合金中原子的电子(自旋向上)分波态密度, 虚线处代表费米能级
图 7 FeCuCrMnMo合金中原子的电子(自旋向上)分波态密度, 虚线处代表费米能级Figure7. Partial density of states of FeCuCrMnMo alloy (spin-up). Dotted line shows the Fermi level.
为进一步量化评估原子间的相互作用, 将FeCuCrMnMo合金中的各原子对进行晶体轨道重叠布局(crystal orbital Hamilton population, COHP)分析. COHP值通过将不同电子轨道间相互作用乘以相应的态密度得到[25], 可以量化描述原子间键强及成键态(值为负时)或反键态(值为正时)的相互作用. 将COHP曲线对能量积分, 可以得到量化的(integrated COHP)ICOHP的值(表3).
| 原子对 | Cu-Fe | Cu-Mn | Cu-Mo | Cu-Cr | Cu-Cu |
| ICOHP | –0.54 | –0.59 | –0.88 | –0.66 | –0.39 |
| 原子对 | Fe-Cr | Fe-Mo | Mn-Mo | Cr-Mo | Cr-Mn |
| ICOHP | –1.45 | –1.68 | –1.72 | –1.78 | –1.61 |
表3FeCuCrMnMo合金中不同原子对的ICOHP平均值
Table3.Mean values of ICOHPs for each atomic pair in FeCuCrMnMo alloy.
这里, 仍以一个Mo原子为中心, 计算其周围最近邻的另外4种原子与该原子的成键状态(图8). 原子对CrMo在约0.5 eV以下为成键态, 0.5 eV以上为反键态, 并在1.2 eV附近达到一个强烈的峰值. Fe-Mo和Mn-Mo原子对分别在–1.2和–0.5 eV以下为成键态, 且其反键态峰较弱. 这三种原子对在费米能级以下均有两个成键态峰. 但对于Cu-Mo原子对来说, 成键态在–2.5 eV处即转变为反键态, 在此能量下只有成键态峰. 因此, Cr-Mo, Fe-Mo和Mn-Mo原子对具有相似的成键强度, 并且都强于Cu-Mo间成键强度. 其它原子对间的成键强度量化结果平均值见表3.
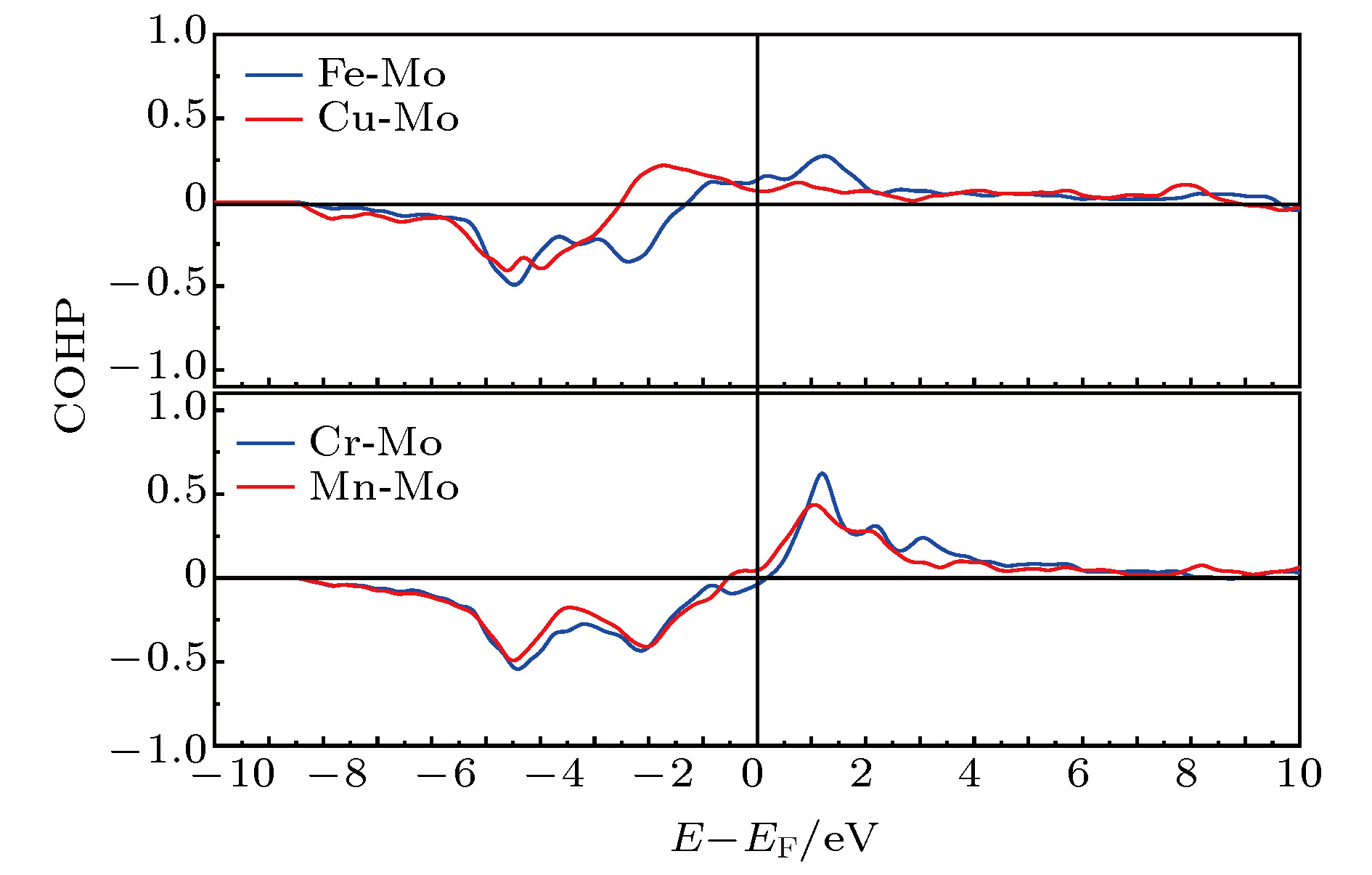 图 8 FeCuCrMnMo合金中Fe-Mo, Cu-Mo, Cr-Mo和Mn-Mo原子对间的COHP分析
图 8 FeCuCrMnMo合金中Fe-Mo, Cu-Mo, Cr-Mo和Mn-Mo原子对间的COHP分析Figure8. COHP for FeCuCrMnMo alloy describing Fe-Mo, Cu-Mo, Cr-Mo and Mn-Mo interactions.
对于多组元合金体系, 由于原子尺寸不同, 各原子的配位环境不同, 导致结构优化后原子间距离产生变化, 即使同一原子对, 由于所处位置不同, 原子间相互作用强度也会不同. 所以, 需要对ICOHP求平均值进行分析.
从表3可以看出, 含Cu原子对间的成键强度均小于其它原子对, 而Fe-Mo和Mn-Mo成键强度较强, 这可能也是Fe-Mo和Mn-Mo原子对在结构优化后距离被拉近的原因, 如图6(d)所示, 结构优化后, Fe-Mo和Mn-Mo原子间距离均在第一近邻距离左侧, 而Cu-Mn和Cu-Fe等原子对间距离则被拉远.
2
3.4.SRO对结构性质的影响
1)对电子分布的影响分别对FeCuCrMnMo合金的SRO结构和SQS结构的电子态密度进行计算(图9). SRO并没有对电子态密度曲线产生太大影响, 最大的差异在费米能附近, 这可能会对电子的传输有影响. 同时, 分别比较了SRO和SQS结构的电荷密度(图10). 由于Cu的核外电子排布为3d轨道全满, 4s轨道半满状态, 其价电子最为不活泼, 所以Cu原子周围电荷密度较低. Cu原子短程序的出现使得Cu原子聚集, 将进一步降低Cu富集区域的电荷密度, 形成“电荷密度阱”区域, 如图10(a)所示, 可能对富Cu区域间隙原子的溶解、空位缺陷的形成等产生重要影响, 这在本小组之后的研究中会有所涉及. 与纯金属的电荷密度图相比(图10(c)), 不论是SRO结构还是SQS结构, 电荷密度分布的周期重复对称性均被破坏, 在结构中不同的间隙位置, 如四面体间隙和八面体间隙, 电荷密度的大小也不再有明显的区分, 这必定会对高熵合金的杂质类缺陷, 如H/He等杂质原子的溶解[26], 包括扩散行为等产生影响.
 图 9 FeCuCrMnMo合金SRO结构和SQS结构电子态密度
图 9 FeCuCrMnMo合金SRO结构和SQS结构电子态密度Figure9. Density of state of FeCuCrMnMo alloy in SRO lattice and SQS lattice.
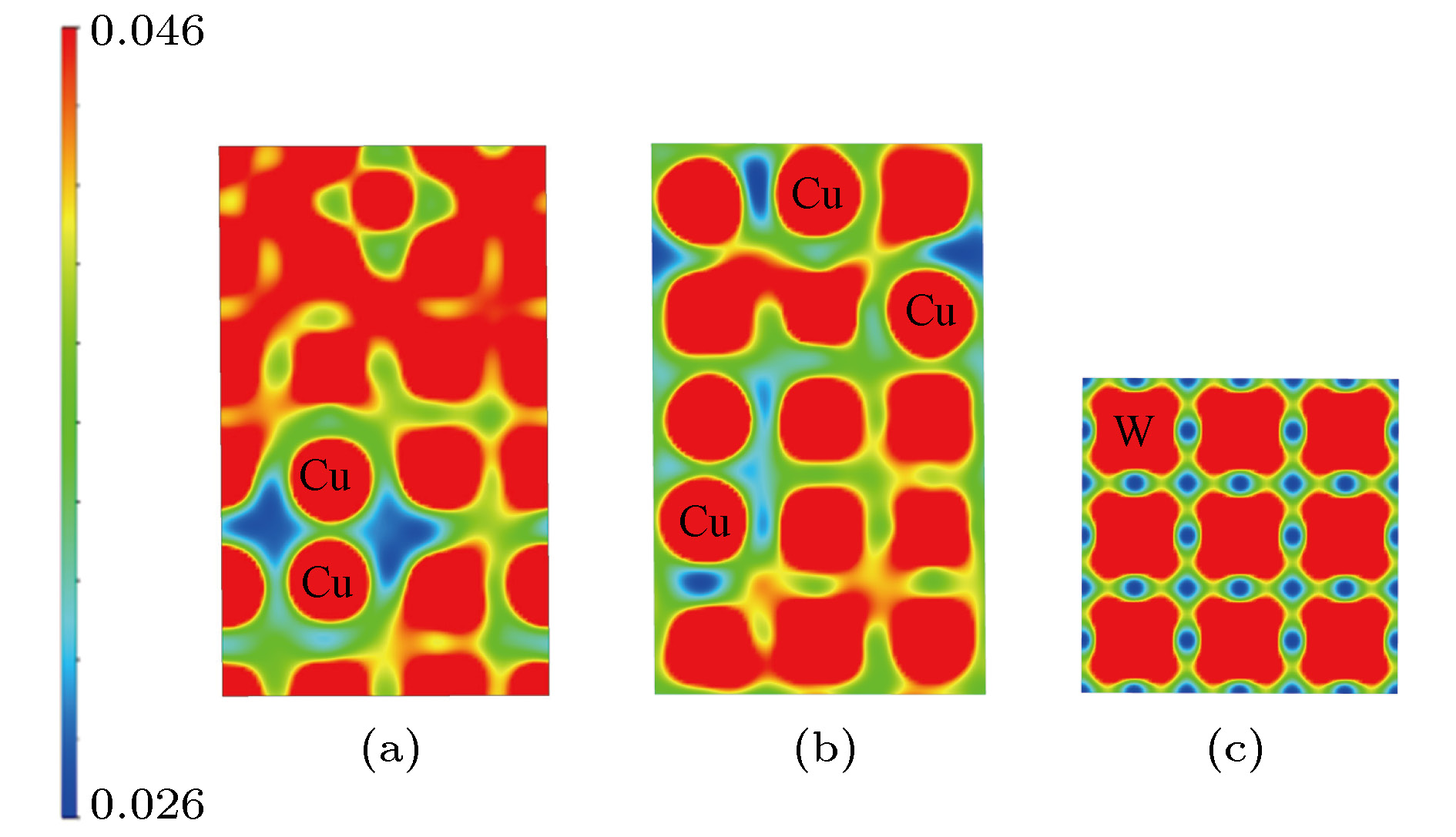 图 10 FeCuCrMnMo合金和纯金属W的{100}方向电荷密度图 (a) SRO结构; (b)SQS结构, 其中蓝色区域附近的“圆形状原子”为Cu原子; (c)纯金属W
图 10 FeCuCrMnMo合金和纯金属W的{100}方向电荷密度图 (a) SRO结构; (b)SQS结构, 其中蓝色区域附近的“圆形状原子”为Cu原子; (c)纯金属WFigure10. Electron density of {100} atomic plane: (a) SRO structure; (b) SQS structure; (c) W lattice.
2)对磁性的影响
材料的磁性由电子磁矩决定, 原子磁矩主要来自未填满的电子壳层的贡献, 其值为自旋向上态和自旋向下态的电子占据数之差. 对于3d过渡族金属, 原子磁矩主要来自3d电子自旋磁矩的贡献. 由于不同金属原子化学势不同, 合金化后, 近邻原子间的电子转移将改变原子自旋向上/下电子占据状态, 带来原子磁矩的变化. 原子磁矩的变化对原子周围的配位环境变化非常敏感. 在针对FeM(M = Fe, Co, Ni, Al, Si)二元合金的磁性计算中[18], BCC结构Fe的平均磁矩为2.18 μB, 但是当Fe处于不同合金中时, 由于其原子配位环境改变磁性也发生改变. 通常Co和Ni元素会提升合金中Fe的平均磁矩, 而Al和Si则降低Fe的平均磁矩. 在FeSi合金中, Fe原子的平均磁矩甚至接近于0.
在FeCuCrMnMo合金中, SRO的出现改变了理想随机晶格中的原子配位关系. 计算结果表明, 相对于理想随机结构(平均磁矩为0.55 μB), SRO降低了结构的平均磁矩(平均磁矩为0.13 μB). 图11为合金中每个原子的磁矩统计结果, 相比于SQS结构, Cu原子的磁矩变化不大, 个别Fe原子以及Mn原子的磁矩下降导致其平均磁矩减小, 因此降低了晶体中的平均磁矩. 这可能与这些原子第一近邻下配对的概率降低有关.
 图 11 FeCuCrMnMo合金SRO和SQS结构中各原子的磁矩
图 11 FeCuCrMnMo合金SRO和SQS结构中各原子的磁矩Figure11. Magnetic moments of individual atoms in SRO structure and SQS structure of FeCuCrMnMo alloy.
3)对力学性质的影响
根据经典的应力应变关系:









| 结构 | C11 | C12 | C44 | B | BEOS | G | E | ν | G/B | AZ |
| SRO | 263.4 | 187.5 | 100.5 | 212.8 | 188.7 | 88.2 | 232.5 | 0.318 | 2.415 | 2.65 |
| SQS | 156.1 | 139.3 | 85.1 | 144.9 | 154.4 | 43.4 | 118.4 | 0.364 | 3.323 | 10.1 |
表4FeCuCrMnMo合金SRO结构和SQS结构的力学性质, 其中弹性模量的单位为GPa
Table4.Calculated mechanical properties for SRO structure and SQS structure of FeCuCrMnMo alloy. The unit for the elastic moduli is GPa.
根据Pugh规则[31], Pugh比(B/G)结合泊松比ν可以用来评估材料的延展性, 当B/G > 1.75, ν > 0.31时, 材料表现为韧性, 反之则为脆性. SRO结构和SQS结构均表现出韧性特征, 但SRO结构具有较低的Pugh比及泊松比, 意味着短程序的出现降低了材料的韧性. 为了分析结构的各向异性性质, 采用Zener比[32], 即AZ值来评估, 其计算公式为
