Fund Project:Project supported by the National Natural Science Foundation of China (Grant Nos. 61874070, 61674099, 61274067) and the R&D Foundation of SHU-SOEN’s PV Joint Lab (Grant No. SS-E0700601).
Received Date:13 January 2019
Accepted Date:15 March 2019
Available Online:01 May 2019
Published Online:20 May 2019
Abstract:An amorphous mixing layer (3.5–4.0 nm in thickness) containing silicon (Si), oxygen (O), molybdenum (Mo) atoms, named α-SiOx(Mo), is usually formed by evaporating molybdenum trioxide (MoO3) powder on an n-type Si substrate. In order to investigate the process of adsorption, diffusion and nucleation of MoO3 in the evaporation process and ascertain the formation mechanism of α-SiOx(Mo) on a atomic scale, the first principle calculation is used and all the results are obtained by using the Vienna ab initio simulation package. The possible adsorption model of MoO3 on the Si (100) and the defect formation energy for substitutional defects and vacancy defects in α-SiO2 and α-MoO3 are calculated by the density functional theory. The results show that an amorphous layer is formed between MoO3 film and Si (100) substrate according to ab initio molecular dynamics at 1500 K, which are in good agreement with experimental observations. The O and Mo atoms diffuse into Si substrate and form the bonds of Si—O or Si—O—Mo, and finally, form an α-SiOx(Mo) layer. The adsorption site of MoO3 on the reconstructed Si (100) surface, where the two oxygen atoms of MoO3 bond with two silicon atoms of Si (100) surface, is the most stable and the adsorption energy is -5.36 eV, accompanied by the electrons transport from Si to O. After the adsorption of MoO3 on the Si substrate, the structure of MoO3 is changed. Two Mo—O bond lengths of MoO3 are 1.95 ? and 1.94 ?, respectively, elongated by 0.22 ? and 0.21 ? compared with the those before the adsorption of MoO3 on Si substrate, while the last bond length of MoO3 is little changed. The defect formation energy value of neutral oxygen vacancy in α-SiO2 is 5.11 eV and the defect formation energy values of neutral oxygen vacancy in α-MoO3 are 0.96 eV, 1.96 eV and 3.19 eV, respectively. So it is easier to form oxygen vacancy in MoO3 than in SiO2, which implies that the oxygen atoms will migrate from MoO3 to SiO2 and forms a 3.5–4.0-nm-thick α-SiOx(Mo) layer. As for the substitutional defects in MoO3 and SiO2, Mo substitutional defects are most likely to form in SiO2 in a large range of Mo chemical potential. So based on our obtained results, the forming process of the amorphous mixing layer may be as follows: the O atoms from MoO3 bond with Si atoms first and form the SiOx. Then, part of Mo atoms are likely to replace Si atoms in SiOx. Finally, the ultra-thin buffer layer containing Si, O, Mo atoms is formed at the interface of MoO3/Si. This work simulates the reaction of MoO3/Si interface and makes clear the interfacial geometry. It is good for us to further understand the process of adsorption and diffusion of atoms during evaporating, and it also provides a theoretical explanation for the experimental phenomenon and conduces to obtaining better interface passivation and high conversion efficiency of solar cell. Keywords:first principle/ MoO3/Si interface reaction/ molybdenum-doped amorphous silica/ defect formation energy
为了模拟MoO3/Si界面反应及钼掺杂非晶氧化硅层的形貌特征, 构造了如图1(a)所示的周期性结构模型, 该模型包含152个原子(80个Si, 18个Mo, 54个O), 结构尺寸为10.84 × 11.20 × 28.58 ?, Si和MoO3分别选择(100)面和(010)面. 为了表现出硅的块体性质, 将模型中的最上和最下两层固定, 其余原子在模拟中可自由移动. 从头算分子动力学模拟采用正则系综[13], 考虑到MoO3的沸点为1155 ℃, 因此温度控制在1500 K, 步长为2 fs, 模拟时间为6 ps, 布里渊区K点选取为4 × 4 × 1. 图 1 MoO3(010)/Si(100)分子动力学模型 (a)扩散反应前; (b)扩散反应后; 灰色球、蓝色球、红色球分别代表钼原子、硅原子和氧原子 Figure1. The structure model of MoO3(010)/Si(100) interface: (a) Before the ab initio molecular dynamics; (b) after the ab initio molecular dynamics. The grey, blue and red balls stand for Mo atoms, Si atoms, and O atoms, respectively
MoO3薄膜的形成过程是历经了气化后的原子、分子的吸附、扩散、成团形岛并取向生长, 最终形成薄膜, 而了解沉积初始时的吸附模型及化学反应, 有助于深入理解缓冲层的形成过程. 由于MoO3粉末在蒸发过程中存在多种团簇成分[14], 这里仅以MoO3分子为例. 结构优化后的MoO3分子结构在图2(a)中显示, 其中三个Mo—O键长均为1.73 ?, 三个O—Mo—O键角分别为107.73°, 107.74°和107.77°, 形成四面体结构, 与Oliveira等[15]的计算结果符合得很好. 另外, 洁净的硅表面由于存在悬挂键, 为了保持稳定, 通常会发生重构现象. 我们选取Si(100)表面, 真空层厚度设置为15 ?, 采用分子动力学模拟了Si(100)表面的重构, 获得经过表面重构后的Si(100)结构, 如图2(a)所示. 由该图可知, 在硅表面发生了重构现象, 表面的硅原子两两成键, 形成二聚体, 成键的两个原子高低不同, 且前后形成高低交替的皱折结构[16]. 在蒸发过程中, MoO3分子可能吸附在Si(100)面的不同位置, 因此用吸附能来衡量不同位置的稳定性. 在Si(100)表面考虑了七个不同的位置作为初始的吸附位置[17], 图2(a)中用不同颜色来表示不同的初始位点, 其中包括位点1(黄)、2(橙)、3(绿)、4(蓝)、5(粉红)、6(红)、7(紫). 我们采用了吸附能的定义[18]: 图 2 MoO3在Si(100)不同吸附位点的结构示意图 (a) MoO3分子结构及重构后Si(100)表面形貌; (b)?(h) MoO3在吸附位点1?7时优化后的吸附模型; (i)最佳吸附位点7的差分电荷密度(黄色和绿色表示得失电子) Figure2. Adsorption configurations of MoO3 on Si (100) surface: (a) The optimized geometries of MoO3 molecule and reconstructed Si (100); (b)?(h) the adsorption configurations of MoO3 adsorbed on the different adsorption sites of Si (100) surface; (i) the difference charge density of MoO3 on the best adsorption site 7 of Si (100)
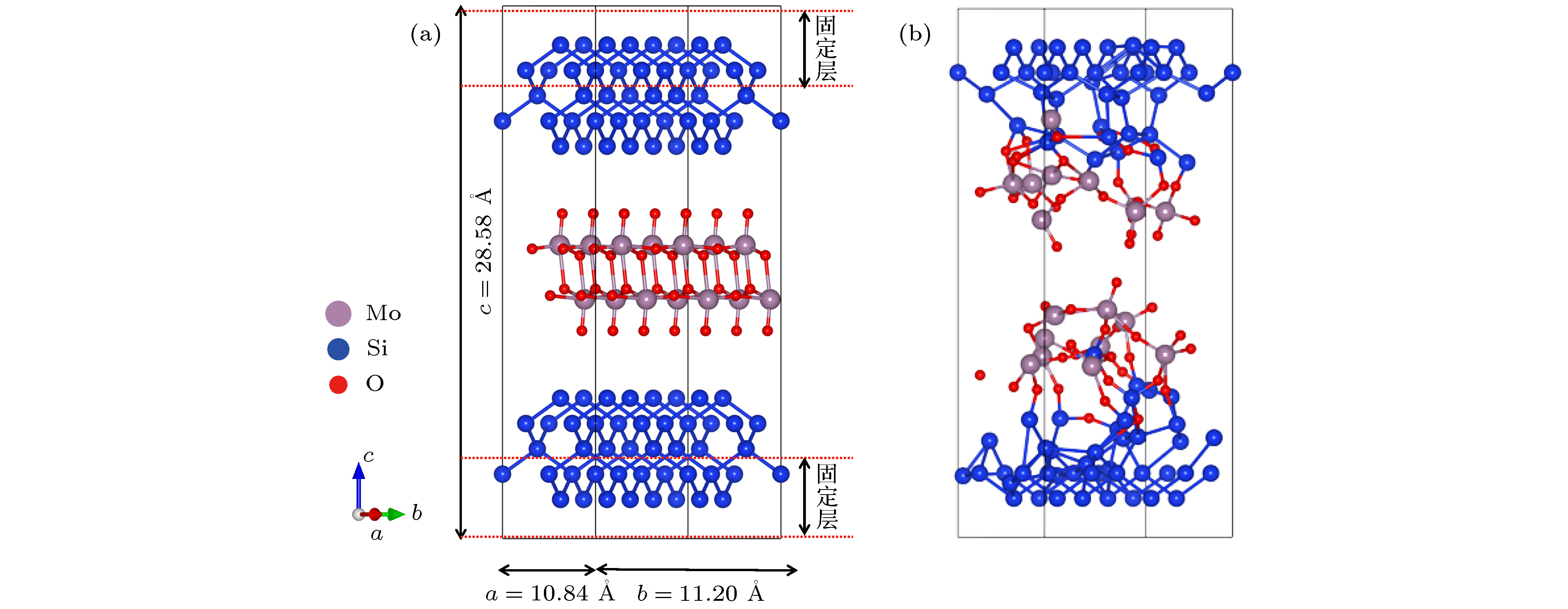 图 1 MoO3(010)/Si(100)分子动力学模型 (a)扩散反应前; (b)扩散反应后; 灰色球、蓝色球、红色球分别代表钼原子、硅原子和氧原子
图 1 MoO3(010)/Si(100)分子动力学模型 (a)扩散反应前; (b)扩散反应后; 灰色球、蓝色球、红色球分别代表钼原子、硅原子和氧原子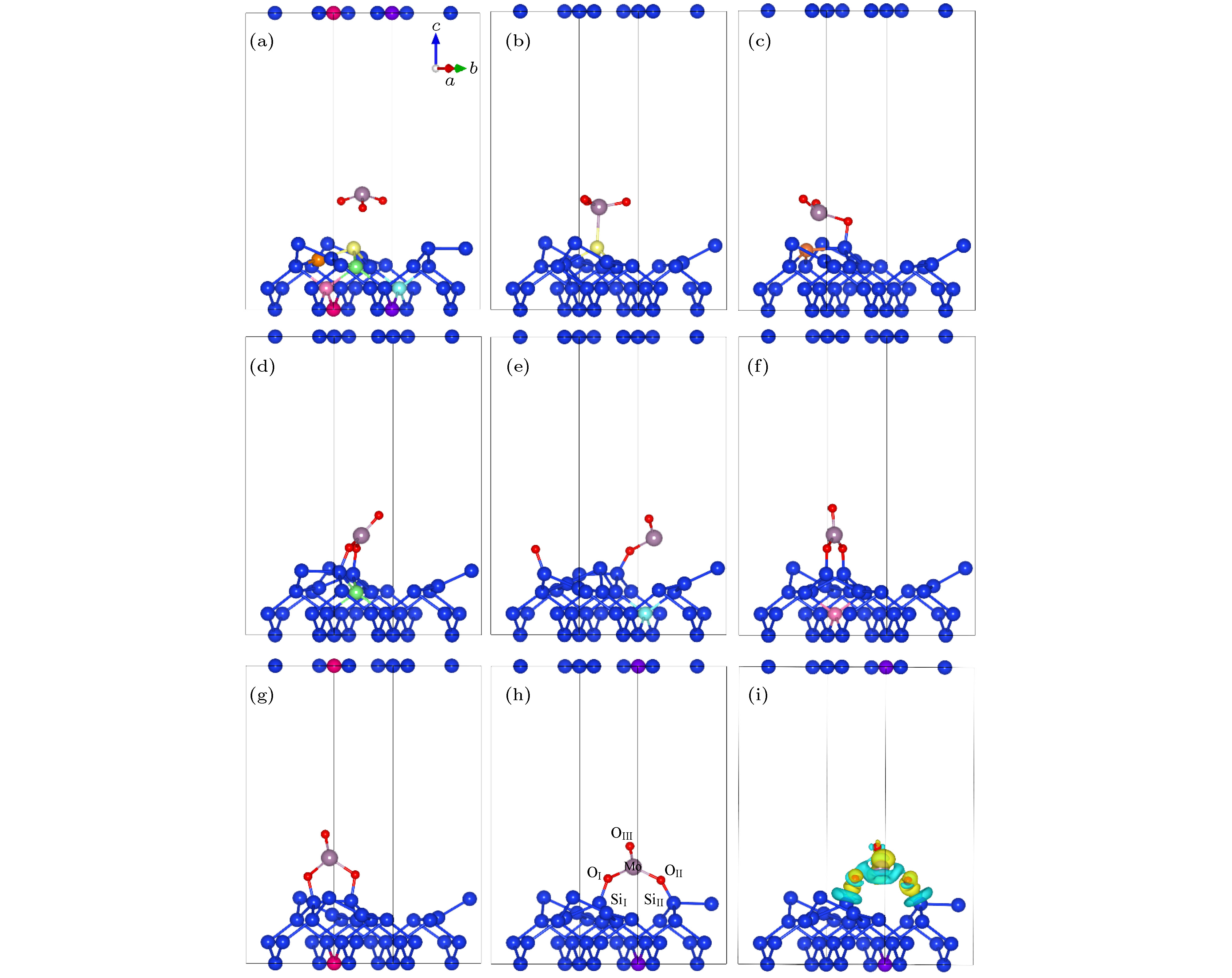 图 2 MoO3在Si(100)不同吸附位点的结构示意图 (a) MoO3分子结构及重构后Si(100)表面形貌; (b)?(h) MoO3在吸附位点1?7时优化后的吸附模型; (i)最佳吸附位点7的差分电荷密度(黄色和绿色表示得失电子)
图 2 MoO3在Si(100)不同吸附位点的结构示意图 (a) MoO3分子结构及重构后Si(100)表面形貌; (b)?(h) MoO3在吸附位点1?7时优化后的吸附模型; (i)最佳吸附位点7的差分电荷密度(黄色和绿色表示得失电子)









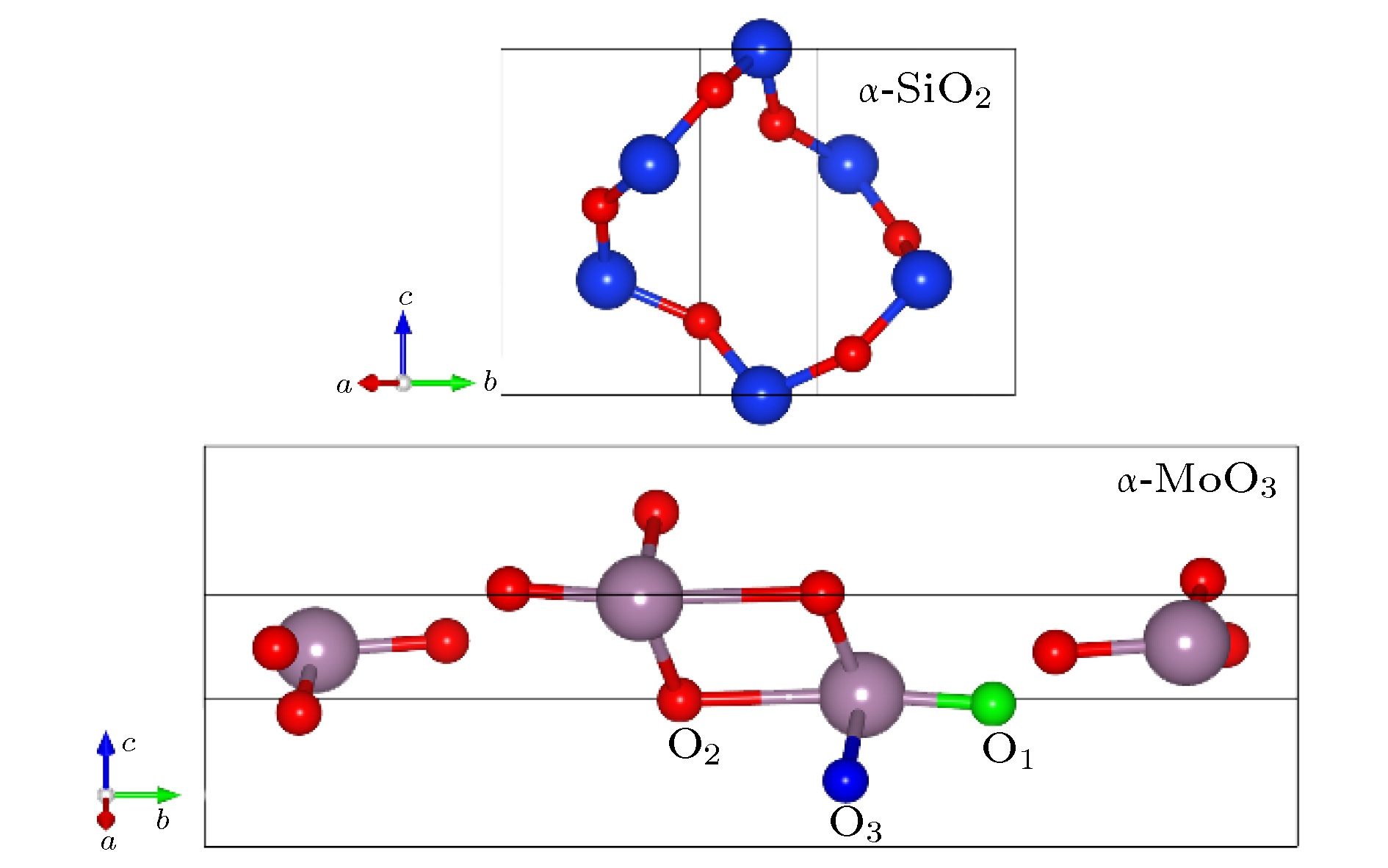 图 3 α-SiO2和α-MoO3晶胞结构
图 3 α-SiO2和α-MoO3晶胞结构


























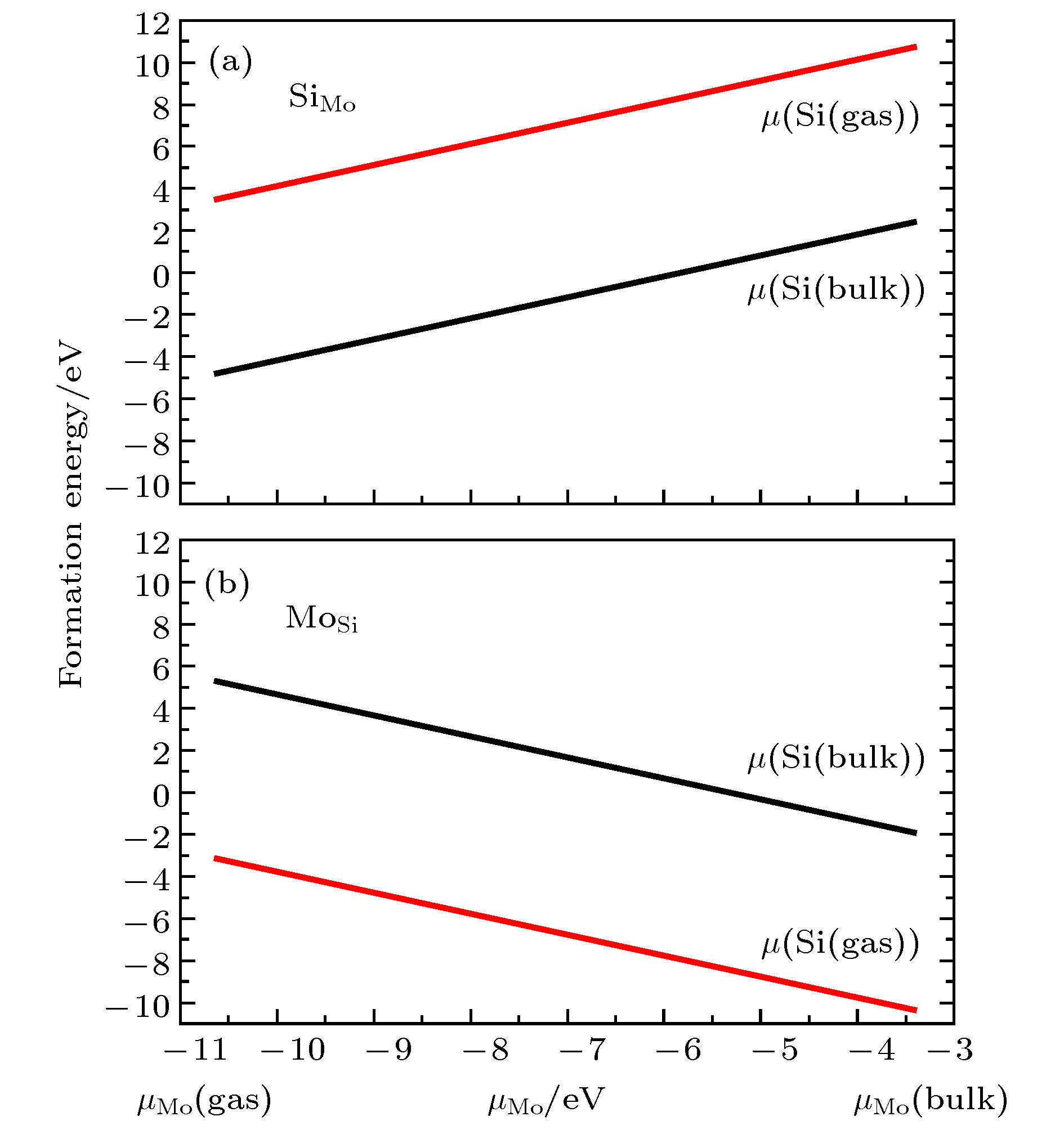 图 4 不同生长条件下替位杂质形成能 (a) Si替位Mo; (b) Mo替位Si; 黑线表示晶体硅的化学势, 红线表示富氧条件下硅的化学势
图 4 不同生长条件下替位杂质形成能 (a) Si替位Mo; (b) Mo替位Si; 黑线表示晶体硅的化学势, 红线表示富氧条件下硅的化学势