1.College of Science, Guangxi University of Science and Technology, Liuzhou 545006, China 2.Materials Science and Engineering Research Center, Guangxi University of Science and Technology, Liuzhou 545006, China
Fund Project:Project supported by the National Natural Science Foundation of China (Grant Nos. 11464003,11864005), the Natural Science Foundation of Guangxi, China (Grant No. 2017GXNSFAA198315), the Key Science and Technology Program of Liuzhou, China (Grant No. 2016B040202), and the Science-Technology Foundation for Middle-aged and Young College Teacher of Guangxi, China (Grant No. 2018KY0324).
Received Date:24 December 2018
Accepted Date:10 April 2019
Available Online:01 May 2019
Published Online:20 May 2019
Abstract:Exploring new type of optoelectronic materials has fundamental scientific and practical significance in the development of society and economy. Recently, intense research has focused on the use of the wide band-gap bipolarity semiconductor material CuInO2 which will allow to the fabrication of that total transparent optoelectronic materials. However, the conductivity of CuInO2 is significantly lower than other n-type conductivity of other TCOs. As a result, one of the key question is how to improve the electric properties of CuInO2 by doping method. Motivated by this observation, in this paper, using the first-principles methods, the formation energetics properties of dopant (Be, Mg, Mn) in transparent conducting oxides CuInO2 were studied within the local-density approximation. Substituting dopant (Be, Mg, Mn) for In, substituting dopant (Be, Mg, Mn) for Cu and dopant as interstitial in their relevant charge state are considered. By systematically calculating formation energies and transition energy level of defect, the calculated results show that, substituting Mg for In does not induce the large structural relaxation. in CuInO2. One can expect that substituting the Mg and Mn for In introduces acceptor because the relative lower formation energies, furthermore, Be atoms would be substitute for In atoms when the Ef move to CBM. In addition, the donor-type extrinsic defects(such as substituting dopant for Cu and dopant as interstitial) have difficulty in inducing n-conductivity in CuInO2 because of their deep transition energy level or the higher formation energies. Considering the transition energy level position, BeIn, MgIn, and MnIn have transition energy levels at 0.06, 0.05, and 0.40 eV above the VBM, respectively. Thus, for all the acceptor-type extrinsic defects, substituting Mg for In is the most prominent doping acceptor with relative shallow transition energy levels in CuInO2 under O-rich condition. Based on our calculated results and discussion mentioned above, in order to increase p-type conductivity in CuInO2, we could substitute Mg atoms for In atoms by the sit-selective doping method through atomic layer epitaxy growth or controlling the oxygen partial pressure in the molecular beam epitaxy or metal-organic chemical vapor deposition crystal growth process. The calculation results will not only provide the guide for design of new type In-based optoelectronic materials, but will also further understand the potential properties in CuInO2. Keywords:CuInO2/ doping/ formation energy
2.计算方法和计算模型CuInO2的晶体结构属于R3m空间群, 晶胞中具有3个特征结构单元: 1)平行C轴分布的O-Cu-O哑铃结构; 2)垂直C轴的六角Cu层; 3) InO6 八面体结构, 其中In位于八面体内(见图1). 总能计算中原子赝势则采用 投影缀加波(PAW)赝势[22,23], 交换关联势采用局域密度近似(LDA)[24,25], 采用软件包VASP进行计算[26,27]. 能带结构计算采用包含四个原子的原胞, 在计算的超晶胞中则包括108个原子, 计算过程中所有的原子都被弛豫到H-F受力最小. 截断能设置为400 eV. 布里渊区的K点设置为1 × 1 × 1 Monkhorst–Pack网格[28,29]. 对于带电荷的缺陷, 计算中引入一个均匀电荷背景以保持整个周期性超晶胞的电中性, 而且所有超晶胞计算都采用同样的K点设置. 单质的化学势也是在LDA和PAW势的总能计算中获得. CuInO2材料中所考虑的价电子为Cu的3d和4p电子, O的2s和2p电子, In的5d和6s电子; 对于掺杂元素Be, Mg, Mn, 价电子包括Be的2s电子, Mg的3s和3p电子, Mn的3d和4s电子. 图 1 CuInO2晶体结构图, 图中红色原子为O原子, 灰色原子为In原子, 棕色原子为Cu原子 (a)黄色掺杂原子替代Cu原子的情况; (b)绿色掺杂原子替代In原子的情况 Figure1. The crystal structure of the CuInO2, the red atoms are O atoms, the brown atoms are Cu atoms, the purple atoms are In atoms: (a) Substituting yellow dopant atom for Cu atom; (b) substituting green dopant atom for In atom.
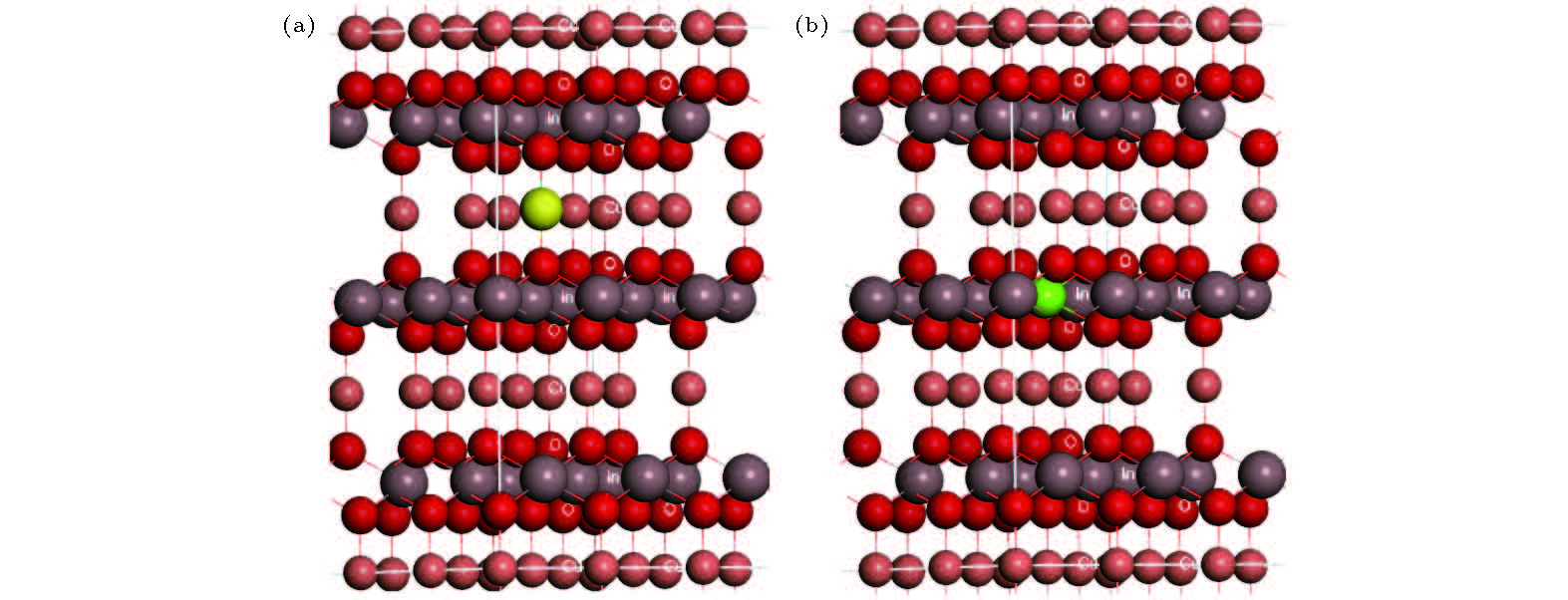 图 1 CuInO2晶体结构图, 图中红色原子为O原子, 灰色原子为In原子, 棕色原子为Cu原子 (a)黄色掺杂原子替代Cu原子的情况; (b)绿色掺杂原子替代In原子的情况
图 1 CuInO2晶体结构图, 图中红色原子为O原子, 灰色原子为In原子, 棕色原子为Cu原子 (a)黄色掺杂原子替代Cu原子的情况; (b)绿色掺杂原子替代In原子的情况


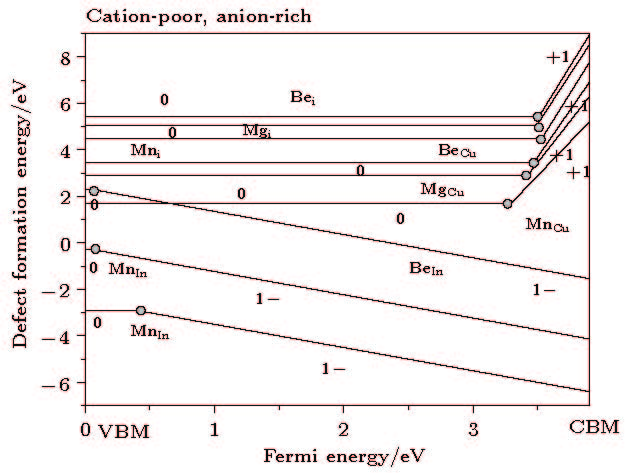 图 2 掺杂形成能在cation-poor, anion-rich的变化图
图 2 掺杂形成能在cation-poor, anion-rich的变化图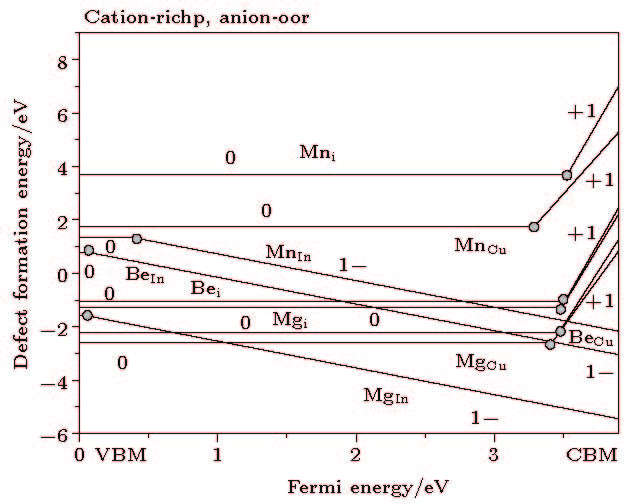 图 3 掺杂形成能在cation-rich, anion-poor的变化图
图 3 掺杂形成能在cation-rich, anion-poor的变化图 图 4 掺杂元素在CuInO2的缺陷跃迁能级
图 4 掺杂元素在CuInO2的缺陷跃迁能级