 ,1, 张丽雯,2, 贺彩红2,3, 阮梅花2, 季勇,4, 于建荣,2,3, 周红文,1
,1, 张丽雯,2, 贺彩红2,3, 阮梅花2, 季勇,4, 于建荣,2,3, 周红文,1Progress on familial hypercholesterolemia
Wanzi Jiang,1, Liwen Zhang,2, Caihong He2,3, Meihua Ruan2, Yong Ji,4, Jianrong Yu,2,3, Hongwen Zhou,1通讯作者: 季勇,博士,教授,研究方向:心血管疾病分子机制及药物防治。E-mail:yongji@njmu.edu.cn;于建荣,硕士,研究员,研究方向:科技情报。E-mail:yujianrong@sibs.ac.cn;周红文,博士,教授,主任医师,研究方向:肥胖、脂代谢和罕见代谢病。E-mail:drhongwenzhou@njmu.edu.cn
编委: 孟卓贤
收稿日期:2021-06-22修回日期:2021-08-12
| 基金资助: |
Received:2021-06-22Revised:2021-08-12
| Fund supported: |
作者简介 About authors
蒋琬姿,在读博士研究生,专业方向:肥胖、脂代谢和罕见代谢病。E-mail:
张丽雯,硕士,馆员,研究方向:生物情报学。E-mail:

摘要
家族性高胆固醇血症(familial hypercholesterolemia, FH)是以肌腱黄瘤、低密度脂蛋白胆固醇(low density lipoprotein cholesterol, LDL-C)显著升高和早发冠心病(premature coronary artery disease, PCAD)为特征的一种常染色体显性或隐性遗传病。本文分析了FH的国内外研究现状,总结了目前中国已报道的FH人群相关的基因突变位点及治疗现状,同时统计了FH相关专利及药物研发情况。在论文发表方面,FH致病机制与治疗、未成年FH患者研究等成为研究热点;在专利方面,再生元制药、阿斯利康、默克等大型药企在FH检测、诊断、治疗等方面积极探索;在药物研发方面,已有12种药物在美国、日本、欧洲等国家/地区上市,为FH患者带来希望。
关键词:
Abstract
Familial hypercholesterolemia (FH) is an autosomal inherited disease characterized by a significant increase in low density lipoprotein cholesterol (LDL-C), tendon xanthoma and premature coronary artery disease (PCAD). In this paper, we analyze the current research status of FH, summarize the reported mutation gene loci in Chinese FH patients and treatment for them, and elaborate the current status of patents and drug researches. The results show that scientific outcomes of FH are increasing with a good developmental trend and the most popular topics of FH study are pathogenesis, treatment of FH, and research on juvenile FH patients. In terms of patents, large pharmaceutical companies, such as Regeneron Pharmaceuticals Inc, AstraZeneca Plc, Merck & Co Inc, are actively engaged in FH detection, diagnosis and treatment. In addition, 12 drugs have been launched in the United States, Japan, Europe and other countries or regions, bringing hope to FH patients.
Keywords:
PDF (1246KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
蒋琬姿, 张丽雯, 贺彩红, 阮梅花, 季勇, 于建荣, 周红文. 家族性高胆固醇血症研究进展. 遗传[J], 2021, 43(11): 1011-1022 doi:10.16288/j.yczz.21-218
Wanzi Jiang.
家族性高胆固醇血症(familial hypercholesterolemia, FH)是以肌腱黄瘤、低密度脂蛋白胆固醇(low density lipoprotein cholesterol, LDL-C)显著升高和早发冠心病(premature coronary artery disease, PCAD)为特征的一种常染色体遗传性疾病[1]。临床上分为杂合子FH(HeFH)和纯合子FH(HoFH),杂合子及纯合子患者血浆LDL-C水平分别为正常人的2~3倍和6~8倍[2]。流行病学研究表明,全球FH的总体患病率约为1/500,FH纯合子患病率约为1/100万[3];近年来也有研究表明,FH的总体发病率为1/200~1/500,FH纯合子预估患病率为1/30万~1/60万[4,5,6]。但是世界上绝大部分国家和地区FH患者诊断率仍然小于1%[7],且治疗状况差,大部分患者未达到指南推荐的LDL-C目标水平[8]。中国作为世界上人口最多的国家,FH患者约占全球所有FH患者的8%[9]。协和医院张抒扬教授依据中国总人口预估的纯合子FH患病人数约为2205~4609例,杂合子患病人数约为276万~691万[9]。
FH主要为常染色体显性遗传,部分为常染色体隐性遗传。已鉴定出的致FH突变基因中,显性遗传基因主要包括低密度脂蛋白受体(low density lipoprotein receptor, LDLR)、前蛋白转化酶枯草溶菌素9 (proprotein convertase subtilisin/kexin type 9, PCSK9)及载脂蛋白B (apolipoprotein B, Apo B)基因[10];隐性遗传基因则主要为LDLR衔接因子蛋白1 (low-density lipoprotein receptor adapter protein 1, LDLRAP1)基因[11]。近年来也有文献报道FH致病新基因,如环氧化物水解酶2 (epoxide hydrolase 2, EPHX2)、生长激素受体(growth hormone receptor, GHR)、载脂蛋白E (apolipoprotein E, Apo E)基因等[12]。
《家族性高胆固醇血症筛查与诊治中国专家共识》[13]指出,成人符合下列标准中的2项即可诊断为FH:(1)未经调脂药物治疗的患者血清LDL-C水平≥4.7 mmol/L (180 mg/dL);(2)有皮肤/腱黄色瘤或<45岁的人存在脂性角膜弓;(3)一级亲属中有FH或早发动脉粥样硬化性心血管疾病,特别是冠心病患者。儿童FH的诊断标准:未治疗的血LDL-C水平≥3.6 mmol/L (140 mg/dL)且一级亲属中有FH患者或早发冠心病患者。检测出LDLR、Apo B、PCSK9和LDLRAP1基因致病性突变也可诊断为FH。本文从FH基因突变位点、治疗现状、专利、药物研发等方面分析FH的总体研究现状,为我国FH的研究与治疗提供参考。
1 FH主要突变基因
LDLR、PCSK9、Apo B、LDLRAP1、EPHX2、GHR及Apo E基因的突变大多数是由外显子区域碱基替换及小片段缺失导致,本文将进一步介绍LDLR、PCSK9、Apo B、LDLRAP1、EPHX2、GHR及Apo E基因的突变位点研究现状(截至2020年中国FH患者主要突变基因相关情况详见附表1~4)。1.1 LDLR
LDLR基因位于染色体19p13.1-13.3,包含18个外显子和17个非编码区,大约89% FH患者为LDLR基因突变[14]。LDLR主要位于肝细胞膜表面,该基因突变可导致LDLR的功能和活性发生变化,无法清除外周血中的LDL-C,从而导致血LDL-C浓度升高[5]。据LOVD数据库(
FH人群中LDLR主要突变形式有:c.2177C> T、c.1775G>A、c.2054C>T、c.1048C>T、c.682G>A、c.829G>A、c.1285G>A、c.798T>A、c.(940+1_941-1)_ (1186+1_1187-1)del、c.1567G>A、c.530C>T、c.1222G>A、c.1432G>A等。
据统计[15],1950~2019年,中国大陆、中国香港、中国澳门地区共报道177个LDLR突变,主要包括:c.1747C>T、c.1448G>A、c.1879G>A、c.313+1G> A、c.2054C>T、c.986G>A、c.1765G>A、c.1879G> A、c.1432G>A等,其中E10上c.1448G>A (第1448位碱基G被A取代)为最常见突变,其氨基酸改变为p.Trp483X (第483位色氨酸被终止密码子取代);中国台湾地区共报道81个LDLR突变,主要包括:c.1747C>T、c.986G>A、c.268G>A、c.1322T>C、c.1432G>A、c.1246C>T等,其中E12上c.1747C>T (第1747位碱基C被T取代)为最常见突变,其氨基酸改变为p.His583Tyr (第583位组氨酸被酪氨酸取代)。
1.2 Apo B
Apo B基因位于染色体2p24.1,该基因主要编码LDL中的载脂蛋白B-100 (ApoB-100),是与LDLR结合的主要配体,大约9% FH患者为该基因突变[14]。Apo B基因突变可导致ApoB-100的LDLR结合域发生改变,导致载有脂质的LDL无法与肝细胞膜表面的 LDLR正确结合,从而导致血中LDL清除减少,血LDL-C浓度升高[16]。截至2020年9月,LOVD数据库共记录有全球1148个Apo B基因突变的相关文献报道,涉及958个Apo B突变位点,且有759种突变仅见一次报道。在Apo B突变中,约90%为碱基置换突变,8%为缺失突变,2%为插入及复制突变。
FH人群中Apo B主要突变形式有:c.10579C> T、c.10580G>A、c.10913G>A、c.7696G>A、c.5741A> G、c.13444A>G、c.4838G>C、c.12382G>A、c.10294C> G、c.3383G>A、c.293C>T等。
亚洲人群中Apo B突变的主要形式为p. Arg3527Trp。1950~2019年,中国大陆、中国香港与中国澳门地区FH人群共报道47个Apo B突变,其突变形式有:c.10579C>T、c.1594C>T、c.6110T> C、c.7223C>T、c.8267G>T、c.8462C>T、c.889C>T、c.9164A>G、c.2870T>C、c.10748A>T、c.581C>T、c.11052A>T、c.11585T>C,其中以c.10579C>T及c.11585T>C突变较为常见[12,17~21];中国台湾地区共报道58个Apo B突变,较常见的突变形式有c.10707C>T、c.10579C>T、c.10828 C>T[22,23,24]。
1.3 PCSK9
PCSK9基因位于染色体1p32.3,有12个外显子,属于前蛋白转换酶家族[10],大约2% FH患者为该基因突变[16]。PCSK9可与肝细胞表面LDLR的表皮生长因子A (epidermal growth factor-A, EGF-A)结构域结合,通过增加LDLR内吞和溶酶体降解作用来减少肝脏LDL-C的吸收,进而导致血LDL-C升高[25]。截至2020年9月,LOVD数据库共记录有全球286个PCSK9基因突变的相关文献报道,涉及258个PCSK9突变位点,且有233种突变仅见一次报道。在PCSK9突变中,约87%为碱基置换突变,8%为缺失突变,其余为插入突变、复制突变。
根据突变对PCSK9功能的影响,可分为功能获得型和功能缺失型突变两种,前者导致胆固醇水平升高,FH人群中PCSK9主要突变形式有:c.1487G> A、c.1120G>T、c.10G>A、c.94G>A、c.1486C>T、c.1405C>T、c.658G>A、c.277C>T、c.7454A>G等[26]。
亚洲人群中最常见的突变为c.94G>A;中国FH人群中PCSK9主要的突变形式有:c.277C> T、c.1792G>A、c.10G>A、c.644G>A、c.626C>T、c.63-65insCTG、c.287G>T、c.313C>T、c.918G>T等,研究表明c.287G>T及c.313C>T错义突变在中国FH人群中最为常见[19]。
1.4 LDLRAP1
LDLRAP1基因位于染色体1p36.11,由9个外显子组成。该基因编码308个氨基酸,LDLRAP1与LDLR可结合成LDL-LDLR复合物进行内吞并输运到胞内[27]。LDLRAP1突变导致LDLRAP1蛋白的分子缺陷,严重减少LDL-C摄取,从而降低LDL-C的代谢。截至2020年9月,据LOVD数据库,全球共有88个LDLRAP1基因突变的相关文献报道,涉及86个LDLRAP1突变位点,且有84种突变仅见一次报道。在LDLRAP1突变中,约91%为碱基置换突变,6%为缺失突变,3%为复制突变。
LDLRAP1主要突变形式有:c.396C>T、c.654A> G、c.65G>A、c.71dup、c.71G>T、c.223G>A、c.284G> A、c.397G>A、c.406C>T、c.413A>G、c.423C>T等。
1.5 EPHX2
EPHX2基因位于染色体8p21.2-p21.1,包含19个外显子,编码555个氨基酸,是α/β-水解酶家族成员[28]。EPHX2广泛存在于哺乳动物组织中,在自然界中其N端磷酸酶结构域及C端水解酶结构域以二聚体形式稳定存在[29]。其中N端具有去磷酸化作用可调节胆固醇水平和信号传递,而C端可催化花生四烯酸类环氧化合物(epoxyeicosatrienoic acids, EETs)水解产生二醇类物质[30]。通过EETs和其他脂质介质的代谢,可溶性环氧化物水解酶在高血压、心脏肥大、动脉硬化、脑和心脏缺血/再灌注损伤等多种疾病中发挥作用。EPHX2突变降低细胞LDLR结合及内吞能力,从而使得血LDL-C浓度升高[28]。截至2020年9月,根据LOVD数据库,全球共有6个EPHX2基因突变的相关文献报道,涉及6个EPHX2突变位点,均仅见一次报道。在EPHX2突变中,约66.7%为碱基置换突变,33.3%为缺失突变。
EPHX2主要突变形式有:c.-86G>C、c.-5G> A、c.230G>A、c.313A>G、c.502-19A>G、c.577-3C>T。
1.6 GHR
GHR基因位于染色体5p13.1-p12,包含10个外显子,编码620个氨基酸,是属于细胞因子受体超家族的单一跨膜糖蛋白[31]。生长激素与跨膜受体GHR结合,激活的GHR通过细胞内信号转导途径介导细胞反应,导致胰岛素样生长因子的合成和分泌。GHR突变可导致莱伦氏综合征,临床表现有生长迟缓、骨龄延迟、马鞍鼻、声音音调高亢、男性生殖器较小、骨质疏松、肌肉发育不良、运动能力发展迟缓等。代谢特点是空腹低血糖,至少50%的婴儿和儿童有过明显的低血糖症状,另可伴有血LDL-C水平升高。有研究表明,GHR突变导致GH上调肝脏LDLR mRNA表达、加速LDL-C分解代谢和降低组织的脂质含量刺激LDL-C清除等作用减弱[32]。截至2020年9月,据LOVD数据库,全球共有140个GHR基因突变的相关文献报道,涉及78个GHR突变位点,且有39种突变仅见一次报道。在GHR突变中,约86%为碱基置换突变,9%为缺失突变,其余为插入突变、复制突变。
GHR主要突变形式有:c.181C>T、c.558A> G、c.703C>T、c.1483C>A、c.1735C>A等。
1.7 Apo E
Apo E基因位于染色体19q13.32,编码317个氨基酸的Apo E前体,18个氨基酸的信号肽裂解和糖基化后,成熟的Apo E以299个氨基酸的蛋白质分泌[33]。Apo E作为脂蛋白的结构蛋白,构成脂蛋白的外层,主要分布于极低密度脂蛋白、高密度脂蛋白、乳糜微粒中,作为LDLR家族的配体,调节脂质代谢等多种生物学反应。截至2020年9月,据LOVD数据库,全球共有240个Apo E基因突变的相关文献报道,涉及88个Apo E突变位点,且有72种突变仅见一次报道。在Apo E突变中,约86%为碱基置换突变,9%为缺失突变,其余为插入突变、复制突变。
Apo E主要突变形式有:c.388T>C、c.526C> T、c.942C>T、c.90C>G、c.149G>A等。
2 研究趋势
2.1 致病机制与疾病治疗为FH研究热点
在Web of Science核心合集数据库检索1( 检索时间:2021-03-16,数据库更新时间:2021-03-15,文献类型:Article+Review,在主题中检索,检索式为:TS = ("familial hypercholesterolemia" or "Familial hypercholesterolaemia" or "familial hyperbetalipoproteinaemia" or "familial hypercholesteremia" or "Fredrickson type IIa hyperlipoproteinemia" or "Fredrickson type IIa lipidaemia" or "Hyperlipoproteinemia type IIA" or "Type II Hyperlipidemia"))FH相关论文,得到论文8676篇(截至2020年)。选取2016~2020年的2159篇论文对研究热点、主要国家/地区进行分析。Web of Science核心合集数据库收录的最早的FH相关论文始于1948年。1974年,Brown等[34]在Science杂志上首次发文报道了LDLR基因突变可导致FH,推动了FH相关发病机制的研究(图1),Brown也因此获得了1985年的诺贝尔生理学及医学奖。2003年,Abifadel等[10]通过对PCSK9基因的12个外显子进行测序,首先报道了PCSK9新的基因突变位点,揭示了一种新的致FH机制,促进了FH领域研究的发展。
图1
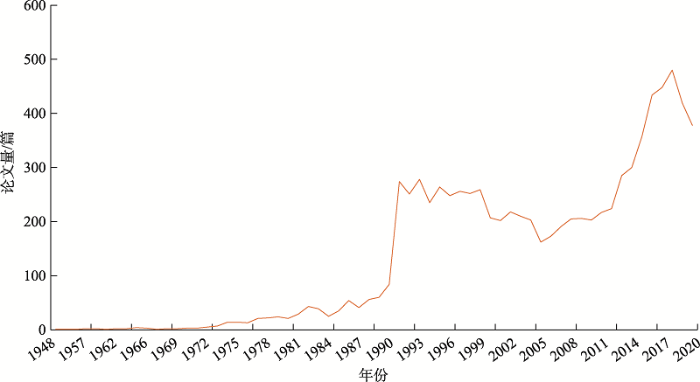 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1FH论文年度趋势
Fig. 1Trend of publications on FH
在论文发表方面,2016~2020年,FH领域发文量排名前10位国家/地区分别为:美国、英国、意大利、加拿大、中国、荷兰、澳大利亚、德国、法国、日本。从年度趋势来看,2016~2019年中国论文发表量不断增长,随着发文量上升,于2019年超过英国、意大利等居第二位,而2020年发文量较2019年出现一定程度下降。使用VOSviewer软件进行关键词聚类分析,可以看出,FH的研究重点主要围绕基因表达与疾病发生、流行病学研究、临床试验与治疗、未成年人家族性高胆固醇血症疾病发生等四方面展开(图2)。
(1)基因表达与疾病发生(红色):包括LDLR、PCSK9、Apo B、LDLRAP1等致病基因的表达、变异与疾病的关系及相关诊断方式、技术等。
(2)流行病学研究(绿色):主要对FH等心血管疾病的发病率、危险因素、临床症状、预防与控制等展开研究。
(3)临床试验与治疗(蓝色):主要包括FH的LDLR、PCSK9、Apo B、LDLRAP1等主要致病基因抑制剂的临床试验及相关抗体、药物的制备等。
(4)未成年人家族性高胆固醇血症疾病发生(黄色):主要对青少年、儿童等未成年人FH等心血管疾病发生、治疗及防控手段进行研究。
图2
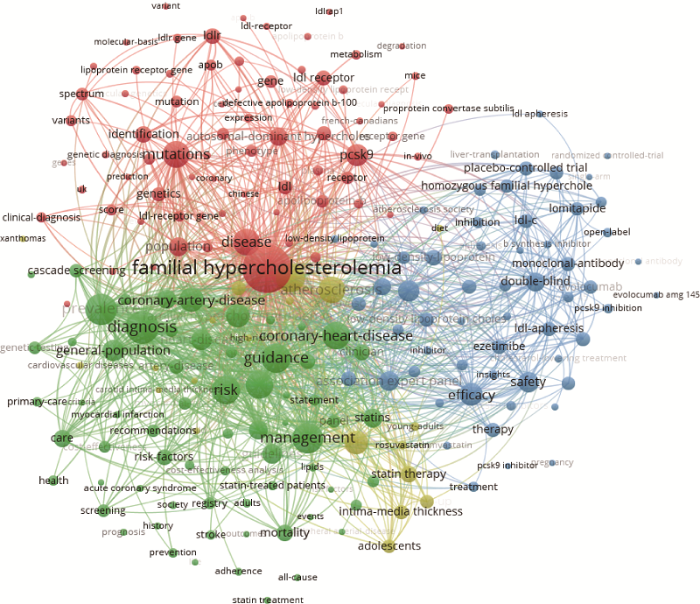 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图22016~2020年FH领域研究热点分析
Fig. 2Research hotspots of FH, 2016-2020
2.2 诊断治疗成为FH专利技术研发重点
在Incopat数据中检索FH相关专利,共得到相关专利541件,并对这些专利进行深入分析。FH领域公开专利最早可追溯到2002年,2002~2004年,专利申请数有较大程度增长,到2014年,申请与公开专利数量均呈波动下降趋势。2014~ 2018年,FH相关专利申请与公开数量上升;2018年后专利公开数量呈波动上升趋势(图3)。
图3
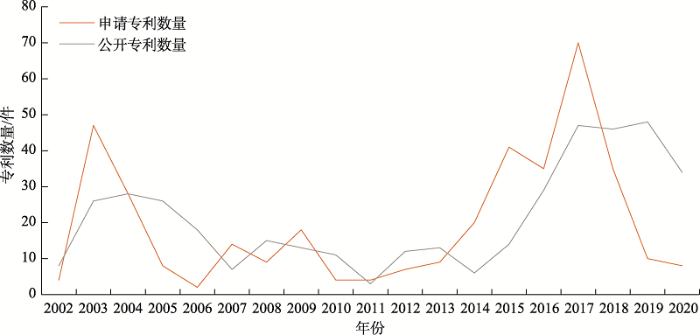 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3FH领域专利申请与专利公开年度趋势
由于专利申请后评审需要18个月后才能公开,因此2019和2020年的专利量不全,仅供参考。
Fig. 3Trend of patent application and disclosure on FH
对FH专利主题词进行聚类分析,主要可分以下四类。
(1) FH基因疗法及他汀外药物降脂活性剂研究:涉及ANGPTL8抑制剂、ANGPTL3抑制剂、内皮脂肪酶抑制剂、PCSK9抑制剂等药物研发及FH的基因疗法研究。
(2) FH他汀类药物研发与应用:主要包括瑞舒伐他汀、阿托伐他汀、辛伐他汀等HMG-CoA还原酶抑制剂类药物的研究。
(3) FH检测/诊断/预后方法研究:主要涉及LDLR、Apo B、PCSK9基因突变检测方法与试剂盒开发及FH易感性相关SNP位点的引物、检测方法研究、FH的体外诊断方法、饮食辅助疗法预后等研究。
(4) FH片剂/胶囊剂/粉剂药物制备方法与优化:主要为辛伐他汀胶囊剂、匹伐他汀钙片剂、考来替兰分散片、奥美沙坦酯/瑞舒伐他汀复方制剂、阿托伐他汀钙化合物等药物制备及优化(如添加碱性金属增强稳定性)方法研究。
对FH领域全球Top5专利权人(美国再生元制药、美国宾夕法尼亚大学、英国/瑞典阿斯利康、美国默克、美国德克萨斯大学)的专利布局进行分析,前5专利权人申请的专利集中在FH检测、诊断及治疗方面,又各有侧重。再生元制药申请的专利主要围绕PCSK9抑制剂、ANGPTL8抑制剂、ANGPTL3抑制剂等药物的研发;宾夕法尼亚大学重点围绕FH诊断方法及LDLR、Apo B、PCSK9基因突变检测方法进行布局;阿斯利康与默克专利布局主要集中在瑞舒伐他汀等他汀类药物研发应用方面;德克萨斯大学则主要围绕FH的检测方法进行布局。
3 治疗进展
由于FH患者出生后即处于高LDL-C水平暴露状态,其罹患动脉粥样硬化性心血管疾病(arteriosclerotic cardiovascular disease, ASCVD)风险明显增高。因此尽早开展级联筛查,早期诊断和早期治疗是改善FH患者临床预后的重要措施。我国FH患者的治疗目标如下:合并与不合并ASCVD的成人FH患者血LDL-C的目标值分别为<1.8 mmol/L (70 mg/dL)和<2.6 mmol/L (100 mg/dL);儿童FH患者血LDL-C的目标值<3.4 mmol/L (130 mg/dL)。若难以达到上述目标值,建议至少将血清LDL-C水平降低50%[13]。目前针对FH患者的降脂治疗的主要方式包括改善生活方式、药物干预治疗、脂蛋白血浆置换及肝脏移植等。
3.1 改善生活方式
健康科学的生活方式是FH治疗的基础措施,鼓励患者戒烟,坚持低饱和脂肪酸、低胆固醇饮食。控制体重,建议患者积极参加体育锻炼(在体育活动开始之前仔细评估心血管风险)[13]。3.2 药物干预治疗
FH诊断后应立即启动降胆固醇药物治疗[13]。3.2.1 全球已有12个FH药物上市
在Cortellis数据库中检索FH相关药物,共获得有效药物23种(包括已上市、已注册、预注册、临床3期、临床2期、临床1期、临床阶段、发现阶段等药物)。
其中,已上市药物12种,分别为:米泊美生(mipomersen)、依折麦布-瑞舒伐他汀钙片(ezetimibe- rosuvastatin calcium)、依折麦布-阿托伐他汀(ezetimibe-atorvastatin)、依折麦布-辛伐他汀(ezetimibe- simvastatin)、依洛尤单抗(evolocumab)、匹伐他汀(pitavastatin)、依折麦布(ezetimibe)、氟伐他汀(口服,缓释型) (fluvastatin, oral, extended-release)、阿托伐他汀(atorvastatin)、阿利珠单抗(alirocumab)、瑞舒伐他汀(rosuvastatin)、洛美他派(lomitapide);处于临床3期的药物有2种,分别为LIB-003、resmetirom。
3.2.2 HMG-CoA还原酶和PCSK9等为FH药物主要靶标
目前,治疗FH的药物主要是他汀类,可联合胆固醇吸收抑制剂或胆汁酸螯合剂,降低LDL-C水平。多种新型降胆固醇药物已被批准用于FH的治疗,包括HMG-CoA还原酶抑制剂(7个)、PCSK9抑制剂(5个)、白细胞介素-1β受体抑制剂(2个)、尼曼-匹克C1型类似蛋白1抑制剂(2个)、微粒体甘油三酯转运蛋白(microsomal triglyceride transfer protein, MTTP)抑制剂(1个)、Apo B合成抑制剂等(1个)、血管生成素样3 (angiopioetin-like protein 3, ANGPTL3)抑制剂(1个)等,其主要作用见表1。
Table 1
表1
表1不同类别药物在降LDL-C或抗炎方面的作用
Table 1
| 药物类别 | 名称 | 作用机制 | 参考文献 |
|---|---|---|---|
| HMG-CoA 还原酶抑制剂 | 瑞舒伐他汀、匹伐 他汀、氟伐他汀等 | 通过增强胆固醇调节元件结合蛋白2激活,上调 LDLR表达,促进对血液中胆固醇的吸收,降低血液中的LDL-C 水平 | [35] |
| 尼曼-匹克C1型 类似蛋白1抑制剂 | 依折麦布 | 作用于小肠绒毛刷状缘,通过抑制胆固醇转运蛋白尼曼-匹克C1型类似蛋白1,选择性地抑制肠道对胆固醇的吸收,减少肝胆固醇储备,?上调肝细胞表面LDLR,从而增加对血LDL-C的清除,且不影响肠道对其他脂质的吸收 | [36] |
| 胆汁酸螯合剂 | 考来烯胺、考来替泊 | 胆酸螯合剂为碱性阴离子交换树脂,可阻断肠道内胆汁酸中胆固醇的重吸收,促进肝细胞内胆固醇向胆汁酸转化并上调LDLR的表达,从而降低血LDL-C水平 | [16] |
| PCSK9抑制剂 | 依洛尤单抗 | 直接阻止循环中PCSK9与LDLR相结合,减少PCSK9 介导的LDLR的分解,加强其对LDL-C的清除能力 | [37] |
| MTTP抑制剂 | 洛美他派 | 直接抑制MTTP活性,抑制甘油三酯向Apo B的转运,减少极低密度脂蛋白的合成,从而降低血LDL-C的水平 | [38] |
| Apo B合成抑制剂 | 米泊美生 | 可与Apo B mRNA结合并抑制其翻译从而减少载脂蛋白Apo B合成的反义寡核苷酸,降低血LDL-C的水平 | [38] |
| ANGPTL3抑制剂 | 依维苏单抗 | 可独立于LDLR降低LDL-C水平,并可降低发生心血管事件的风险 | [39] |
| 白细胞介素-1β 配体抑制剂 | 卡那津单抗 | 抗炎作用,抑制白细胞介素6信号通路,可降低发生心血管事件的风险 | [40] |
新窗口打开|下载CSV
他汀类药物是治疗FH的一线药物。LDLR具有50%以上功能的HeFH患者对他汀类药物反应良好,但HoFH患者对他汀反应较差。治疗效果可能与种族有关,中国患者接受常规剂量他汀类药物治疗反应不佳,对大剂量他汀类药物的耐受性普遍差于西方人[41]。
FH患者除他汀类药物治疗外,还应给予降脂药物,包括依折麦布、胆酸螯合剂、烟酸、洛美他派、米泊美生、PCSK9抑制剂等。但目前洛美他派及米泊美生在中国暂未上市,国内获批上市的药物只有他汀类、依折麦布、PCSK9抑制剂和烟酸。其他药物缺乏针对中国人群的安全性和有效性数据,在上市前仍处于研发或II期和III期临床试验阶段[9]。此外,当烟酸和他汀类药物一起服用时,肌肉毒性的风险会增加。因此,大多数中国FH患者使用他汀类药物联合依折麦布降低血脂水平[42]。研究表明,近年来开发的新型治疗药物PCSK9抑制剂与他汀类药物联合依折麦布可以更好地降低LDL-C水平[43]。2018年8月,依洛尤单抗成为国内首个用于治疗HoFH成人或12岁以上青少年的PCSK9抑制剂,从而推动了中国FH患者治疗新方法的开发,为患者带来希望[9]。
3.2.3 默克、拜耳、诺华等大型制药企业引领FH药物研发
FH领域在研药物数量最多的为美国默克,有3个,分别为依折麦布-瑞舒伐他汀钙片、依折麦布-辛伐他汀、依折麦布,均已上市,主要为HMG-CoA还原酶抑制剂及尼曼-匹克C1型类似蛋白1抑制剂类药物;其次为德国拜耳2个(依折麦布-阿伐他汀、依折麦布-瑞舒伐他汀钙片,均已上市)、美国诺华制药2个(其中,氟伐他汀(口服,缓释型)已上市)、美国再生元制药2个(其中,阿利珠单抗已上市)、日本安斯泰来制药1个(依洛尤单抗,已上市)等。
3.3 脂蛋白血浆置换
?若患者药物联合治疗效果欠佳,可考虑脂蛋白血浆置换。血浆置换为有创治疗且费用昂贵,需每1~2周进行1次维持治疗[44],主要适用于HoFH患者,对伴有冠心病的高危HeFH患者或对他汀类药物不耐受或药物治疗下血LDL-C水平仍较高的HeFH患者。有研究表明,脂蛋白血浆置换后,LDL-C和Lp(a)水平可显著降低50%~70%[45]。脂蛋白血浆置换与降脂药物联合使用,可能会进一步改善血脂水平,降低心血管风险;最常见的不良反应是轻度至重度低血压和恶心[45]。3.4 肝脏移植
肝脏作为清除血胆固醇的主要器官,约80%的LDLR位于肝细胞中,肝移植成为可选治疗方案[46]。通过肝移植纠正肝细胞上LDLR、PCSK9、Apo B等基因的分子缺陷,虽然可以降低LDL-C水平,但由于移植后手术并发症和死亡率高以及供体匮乏等因素,难以作为主要的FH治疗手段[13]。4 结语与展望
近年来,FH的发病率逐年上升,且有年轻化的趋势,FH的就医率及FH的社会认知度处于较低水平。中国FH患者人数预计将达到276万~691万,但诊治率不足1%,从而导致高血脂患者早期心血管事件的发生极高。但对于FH并发冠心病患者,未治疗率也高达20.6%,即便经低、中、高强度降脂治疗(治疗率分别为6.0%、68.3%、5.0%),患者的LDL-C水平也均未达到≤2.6 mmol/L[47]。在FH患者治疗方面,国内外指南推荐使用他汀类药物,而他汀可能存在不耐受以及肌痛、痉挛、肌酸激酶升高等药物相关肌肉症状,临床迫切需要一种安全性好且能有效降低LDL-C水平的新药物及新疗法。随着基因检测与基因编辑技术、精准医学和个性化医疗的发展,大数据与人工智能在医疗中的广泛应用,FH领域将进一步快速发展。随着对LDLR、Apo B、PCSK9、LDLRAP1等主要致FH基因及EPHX2、GHR、Apo E等新发现致病基因的研究,相应突变基因靶向药将拥有巨大市场。如大量研究证明,PCSK9单克隆抗体不仅能够降低FH患者LDL-C水平、耐受性良好,且具有额外的心血管获益,可作为我国FH患者治疗新选择,助力我国FH的防治。
值得注意的是,由于FH治疗方案的信息大多来自国外研究,迫切需要国内FH患者的临床研究数据,以便在国家层面进一步开展治疗。此外,中国FH儿童长期服用降脂药物的安全性和耐受性仍需进一步关注。
同时,优化FH的诊疗及随访体系,未来可建立FH患者国家电子档案数据库,与各临床医疗中心的电子病历系统对接,对早期筛查、整合与分析患者信息、指导治疗方案、评估治疗效果、实现长期监管、进一步制定适合中国FH患者的诊断标准及治疗方案、改善FH患者远期预后具有重要意义,甚至可为国内外专家的沟通与交流提供支持。同时,在中国人群中开展FH新致病基因的筛选,也具有重要临床与科研价值。
通过健康教育、饮食控制、改善生活方式以及有规律的运动,可有效调节机体脂质以及脂蛋白代谢,实现对FH及相关心脑血管疾病的早期防治。通过基因诊断,可实现FH的早发现、早诊断,未来可结合基因编辑个体化医疗,有望治愈越来越多的罕见病,大幅度提高患者的生活质量。
责任编委: 孟卓贤
附录:
附加材料详见文章电子版附加材料:
Supplementary Table 1附表1
附表1中国FH患者LDLR基因突变的致病位点总结
Supplementary Table 1
| 一般情况 | 治疗前血脂 | 临床表现 | 基因诊断 | 发表 时间 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 序号 | 性别 (女/男) | 年龄 (岁) | 省/市 | TC (mmol/L) | LDL-C (mmol/L) | 冠心 病 | 黄色 瘤 | 角膜 弓 | 纯合子/ 复合杂合 | cDNA 突变 | 氨基酸突变 | |
| 1 | 男 | 6 | 天津 | 6.88 | 5.25 | 纯合子 | c. 611G>A | p.Cys204Tyr | 2020 [1] | |||
| 2 | 男 | 7 | 陕西 | 21.24 | 15.51 | + | 纯合子 | c.418G>A | p.Glu140Lys | 2020 [2] | ||
| 3 | 198/ 110 | 48.3±11.2 | 11.06?±?1.58 | 8.41?±?1.39 | c.12G>A | p.Trp4* | 2020[3] | |||||
| c.17G>A | p.Trp6* | |||||||||||
| c.81C>A | p.Cys27* | |||||||||||
| c.97C>T | p.Gln33* | |||||||||||
| c.138C>A | p.Cys46* | |||||||||||
| c.224G>A | p.Cys75Tyr | |||||||||||
| c.268G>A | p.Asp90Asn | |||||||||||
| c.285C>A | p.Cys95* | |||||||||||
| c.301G>A | p.Glu101Lys | |||||||||||
| c.327C>A | p.Cys109* | |||||||||||
| c.400T>C | p.Cys134Arg | |||||||||||
| c.418G>T | p.Glu140* | |||||||||||
| c.510delC | p.Asp170fs | |||||||||||
| c.622G>A | p.Glu208Lys | |||||||||||
| c.682G>T | p.Glu228* | |||||||||||
| c.718G>A | p.Glu240Lys | |||||||||||
| c.769C>T | p.Arg257Trp | |||||||||||
| c.974G>A | p.Cys325Tyr | |||||||||||
| c.1135T>C | p.Cys379Arg | |||||||||||
| c.1206delC | p.Phe402fs | |||||||||||
| c.1222G>A | p.Glu408Lys | |||||||||||
| c.1285G>A | p.Val429Met | |||||||||||
| c.1448G>A | p.Trp483* | |||||||||||
| c.1474G>A | p.Asp492Asn | |||||||||||
| c.1538delG | p.Arg513fs | |||||||||||
| c.1567G>A | p.Val523Met | |||||||||||
| c.1599G>A | p.Trp533* | |||||||||||
| c.1618G>A | p.Ala540Thr | |||||||||||
| c.1633G>C | p.Gly545Arg | |||||||||||
| c.1747C>T | p.His583Tyr | |||||||||||
| 一般情况 | 治疗前血脂 | 临床表现 | 基因诊断 | 发表 时间 | ||||||||
| 序号 | 性别 (女/男) | 年龄 (岁) | 省/市 | TC (mmol/L) | LDL-C (mmol/L) | 冠心 病 | 黄色 瘤 | 角膜 弓 | 纯合子/ 复合杂合 | cDNA 突变 | 氨基酸突变 | |
| c.1765G>A | p.Asp589Asn | |||||||||||
| c.1864G>T | p.Asp622Tyr | |||||||||||
| c.1879G>A | p.Ala627Thr | |||||||||||
| c.1948delG | p.Glu650fs | |||||||||||
| c.2054C>T | p.Pro685Leu | |||||||||||
| c.2389G>A | p.Val797Met | |||||||||||
| c.2439G>A | p.Trp813* | |||||||||||
| c.10579C>T | p.Arg3527Trp | |||||||||||
| c.1336C>T | p.Arg446* | |||||||||||
| c.461G>A | p.Arg154His | |||||||||||
| 4 | 男 | 30 | 22.31 | 12.25 | + | 纯合子 | c.1470G>A | p.Trp469* | 2019[4] | |||
| 5 | 男 | 35 | 海南 | 9.96 | 7.44 | c.986G>A | p.Cys329Tyr | 2019[5] | ||||
| 6 | 37/188 | 46.64±7.21 | 4.86 | 225 | c.241C?>?T | p.Arg81Cys | 2019[6] | |||||
| 3.89 | c.292G?>?A | p.Gly98Ser | ||||||||||
| 8.00 | c.1525A?>?G | p.Lys509Glu | ||||||||||
| 7.25 | c.1691A?>?G | p.Asn564Ser | ||||||||||
| 5.72 | c.1867A?>?T | p.Ile623Phe | ||||||||||
| 7.74 | c.2054C?>?T | p.Pro685Leu | ||||||||||
| 7 | 1/9 | 44.80± 6.36 | 5.04 (4.03-5.91) | 3.39 (2.58-4.08) | + | 杂合子 | c.129G>C | p.Lys43Asn | 2019[7] | |||
| + | 杂合子 | c.241C>T | p.Arg81Cys | |||||||||
| + | 杂合子 | c.292G>A | p.Gly98Ser | |||||||||
| + | 杂合子 | c.1525A>G | p.Ile509Glu | |||||||||
| + | 杂合子 | c.1691A>G (2人) | p.Ala564Ser | |||||||||
| + | 杂合子 | c.1867A>T | p.Ile623Phe | |||||||||
| + | 杂合子 | c.2054C>T (2人) | p.Pro685Leu | |||||||||
| 8 | 女 | 4 | 河北 | 16.4 | 15.06 | + | + | 复合杂合 | c.727T>A, c.1003G>T | p.Cys243Ser, p.Gly335Cys | 2019[8] | |
| 9 | 女 | 52 | 8.10 | 5.02 | 杂合 | c.1731G>A | p.Trp577* | 2018[9] | ||||
| 男 | 22 | 8.20 | 4.86 | + | 杂合 | c.1731G>A | p.Trp577* | |||||
| 女 | 45 | 9.40 | 4.93 | 杂合 | c.2054C>T | p.Pro685Leu | ||||||
| 男 | 27 | 18.30 | 11.60 | + | + | 杂合 | c.1731G>A,c.2054C>T | p.Trp577*,p.Pro685Leu | ||||
| 10 | 男 | 10 | 20.31 | 14.87 | + | 杂合子 | c.724C>T,c.1747C>T | p.Gln242*,p.His583Tyr | 2018[10] | |||
| 11 | 52/41 | M: 50.2±11.9 F: 53.2±10.3 | 中国香港 | M: 9.3±1.9 F: 9.7±2.3 | M: 7.6±1.9 F: 7.8±2.2 | M: 18 F: 20 | M: 9 F: 8 | 纯合子 | c.1474G>A | p.Asp492Asn | 2018[11] | |
| 一般情况 | 治疗前血脂 | 临床表现 | 基因诊断 | 发表 时间 | ||||||||
| 序号 | 性别 (女/男) | 年龄 (岁) | 省/市 | TC (mmol/L) | LDL-C (mmol/L) | 冠心 病 | 黄色 瘤 | 角膜 弓 | 纯合子/ 复合杂合 | cDNA 突变 | 氨基酸突变 | |
| 12 | 女 | 16 | 20.15 | 13.16 | + | 复合杂合 | c.28893T>G, deletions in exon 18 | p.Cys377Gly,NA | 2018[12] | |||
| 13 | 女 | 13 | 14.73 | 13.44 | + | 纯合子 | c.655_c.660 delGGCCCC | p.219Ala_220Prodel | 2018[13] | |||
| 14 | 男 | 12 | 北京 | 23.51 | 14.17 | + | + | 复合杂合 | c.1129G>T, c.1268T>C | p.Cys365Gly, p.Ile402Thr | 2018[14] | |
| 15 | 女 | 8 | 7.6 | 5.37 | + | 杂合子 | c.357delG | p.Gly119fs*86 | 2018[15] | |||
| 16 | 112/173 | ?49 ± 12 | 7.03±2.53 | 5.22±2.12 | 233 | 28 | c.285C > G | p.Cys95Trp | 2018[16] | |||
| c.302A > T | p.Glu101Val | |||||||||||
| c.393delC | p.Asp131fs | |||||||||||
| c.524A > G | p.Asp175Gly | |||||||||||
| c.728G > A | p.Cys243Tyr | |||||||||||
| c.1206delC | p.Phe402fs | |||||||||||
| c.1934A > T | p.Asn645Ile | |||||||||||
| c.1448G > A | p.Trp483* | |||||||||||
| c.1879G > A | p.Ala627Thr | |||||||||||
| c.769C > T | p.Arg257Trp | |||||||||||
| c.1765G > A | p.Asp589Asn | |||||||||||
| c.1187-10G > A | NA | |||||||||||
| c.1747C > T | p.His583Tyr | |||||||||||
| c.1864G > T | p.Asp622Tyr | |||||||||||
| 17 | 28 | 中国台湾 | 9.03-12.05 | 6.47-9.67 | 杂合子 | c.1186+2T>G(6人) | NA | 2018[17] | ||||
| 中国台湾 | 15.00 | 13.78 | 复合杂合 | c.986G>A, c.1291G>A | p.Cys329Tyr p.Ala431Thr | |||||||
| 中国台湾 | 8.30 | 6.05 | 杂合子 | c.12G>A | p.Trp4* | |||||||
| 中国台湾 | 9.03 | 7.08 | 杂合子 | c.298G>A | p.Asp100Asn | |||||||
| 中国台湾 | 12.77 | 9.44 | 杂合子 | c.809G>A | p.Cys270Tyr | |||||||
| 中国台湾 | 8.69-11.45 | 5.95-8.10 | 杂合子 | c.986G>A(2人) | p.Cys329Tyr | |||||||
| 中国台湾 | 9.72 | 7.50 | 杂合子 | c.1176C>A | p.Cys392* | |||||||
| 中国台湾 | 11.43 | 9.31 | 杂合子 | c.1216C>A | p.Arg406= | |||||||
| 中国台湾 | 15.36 | 8.69 | 杂合子 | c.1246C>T | p.Arg416Trp | |||||||
| 中国台湾 | 11.53 | 9.30 | 杂合子 | c.1322 T>C | p.Ile441Thr | |||||||
| 中国台湾 | 9.67 | 6.75 | 杂合子 | c.1432G>A | p.Gly478Arg | |||||||
| 中国台湾 | 10.34 | 8.40 | 杂合子 | c.1592 T>A | p.Met531Lys | |||||||
| 中国台湾 | 8.87 | 6.80 | 杂合子 | c.1952_1953del | p.Asp651fs | |||||||
| 中国台湾 | 9.28 | 5.97 | 杂合子 | c.2054C>T | p.Pro685Leu | |||||||
| 中国台湾 | 9.18 | 5.95 | 杂合子 | c.2205_2206insTT | p.Ala735fs | |||||||
| 中国台湾 | 20.43 | 9.96 | 杂合子 | c.2237delT | p.Val746fs | |||||||
新窗口打开|下载CSV
Supplementary Table 1
附表1
附表1续
Supplementary Table 1
| 一般情况 | 治疗前血脂 | 临床表现 | 基因诊断 | 发表 时间 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 序号 | 性别 (女/男) | 年龄 (岁) | 省/市 | TC (mmol/L) | LDL-C (mmol/L) | 冠心 病 | 黄色 瘤 | 角膜 弓 | 纯合子/ 复合杂合 | cDNA 突变 | 氨基酸突变 | |
| 18 | 女 | 23 | 北京 | 17.66 | 15.78 | + | 复合杂合 | c.2390-2AbG, c.1687C>T | p.Pro563Ser | 2017[18] | ||
| 女 | 29 | 北京 | 13.38 | 11.16 | + | + | 复合杂合 | c.292G>A, c.1864G>A | p.Gly98Ser, p.Asp622Asn | |||
| 19 | 男 | 52 | 山东 | 9.34 | 7.88 | + | + | c.1885_1889delinsGATCATCAACC | p.Phe629_ser630delinsAspHisGlnPro | 2017[19] | ||
| 女 | 68 | 山东 | 9.79 | 7.59 | + | |||||||
| 女 | 48 | 山东 | 9.52 | 6.88 | + | |||||||
| 男 | 13 | 山东 | 10.45 | 8.46 | + | |||||||
| 20 | 男 | 7 | 河南 | 19.79 | 13.93 | 复合杂合 | c. 97 C > T, c. 890 A > C, c. 892delA | p. Gln12*, p. Asn276Thr, p. Met277Trpfs*72 | 2017[20] | |||
| 男 | 1 | 河南 | 24.83 | 17.80 | + | 复合杂合 | c. 292 G > A, c. 971delG, c. 1864 G > A | p. Gly77Ser, p. Gly303Alafs*46, p. Asp601Asn | ||||
| 21 | 女 | 23 | 河南 | 19.0 | 16.7 | + | 纯合子 | c.97C>T | p.Gln33* | 2017[21] | ||
| 22 | 119/162 | 50.3±15.6 | 9.12±3.35 | 6.5±2.46 | 178 | 15 | c.280G>A | p.Asp94Asn | 2017[22] | |||
| c.327C>A | p.Cys109* | |||||||||||
| c.470G>A | p.Cys157Tyr | |||||||||||
| c.724G>A | p.Val242Met | |||||||||||
| c.1066T>G | p.Trp356Gly | |||||||||||
| c.1343T>C | p.Leu448Pro | |||||||||||
| c.1393G>A | p.Gly465Arg | |||||||||||
| c.1562C>A | p.Ser521Tyr | |||||||||||
| c.1522G>C | p.Gly508Arg | |||||||||||
| c.1810A>T | p.Lys604* | |||||||||||
| 23 | 115/104 | ?31.15 ± 15.21 | ?11.15 ± 3.96 | ?8.32 ± 3.83 | ?46 | ?25 | ?c.97C > T | ?p.Gln33* | 2017[23] | |||
| ?c.268G > A | ?p.Asp90Asn | |||||||||||
| ?c.284G > C | ?p.Cys95Ser | |||||||||||
| ?c.485C > T | ?p.Pro162Leu | |||||||||||
| ?c.494G > A | ?p.Trp165* | |||||||||||
| ?c.530C > T | ?p.Ser177Leu | |||||||||||
| ?c.605T > C | ?p.Phe202Ser | |||||||||||
| ?c.682G > T | ?p.Glu228* | |||||||||||
| ?c.718G > A | ?p.Glu240Lys | |||||||||||
| ?c.760C > T | ?p.Gln254* | |||||||||||
| ?c.763T > C | ?p.Cys255Arg | |||||||||||
| ?c.796G > C | ?p.Asp266His | |||||||||||
| ?c.986G > A | ?p.Cys329Tyr | |||||||||||
| ?c.1048C > T | ?p.Arg350* | |||||||||||
| ?c.1132C > T | ?p.Gln378* | |||||||||||
| 一般情况 | 治疗前血脂 | 临床表现 | 基因诊断 | 发表 时间 | ||||||||
| 序号 | 性别 (女/男) | 年龄 (岁) | 省/市 | TC (mmol/L) | LDL-C (mmol/L) | 冠心 病 | 黄色 瘤 | 角膜 弓 | 纯合子/ 复合杂合 | cDNA 突变 | 氨基酸突变 | |
| ?c.1253A > G | ?p.Glu418Gly | |||||||||||
| ?c.1258C > A | ?p.Trp419* | |||||||||||
| ?c.1448G > A | ?p.Trp483* | |||||||||||
| ?c.1747C > T | ?p.His583Tyr | |||||||||||
| ?c.1860G > A | ?p.Trp620* | |||||||||||
| ?c.1879G > A | ?p.Ala627Thr | |||||||||||
| ?c.1897C > T | ?p.Arg633Cys | |||||||||||
| ?c.1954delAT | ?p.Met652Glyfs*16 | |||||||||||
| ?c.2030G > A | ?p.Cys677Tyr | |||||||||||
| ?c.2000delG | ?p.Cys667Leufs*6 | |||||||||||
| ?c.2416insG | ?p.Val806Glyfs*11 | |||||||||||
| ?c.1984dupA | ?p.Arg662Lysfs*7 | |||||||||||
| ?c.2381_2382delTC | ?p.Leu794Profs*22 | |||||||||||
| ?c.218T > G | ?p.Phe73Cys | |||||||||||
| ?c.516C > A | ?p.Asp172Glu | |||||||||||
| ?c.1279C > T | ?p.Arg427Trp | |||||||||||
| ?c.1601C > T | ?p.Thr534Ile | |||||||||||
| ?c.1720C > A | ?p.Arg574Ser | |||||||||||
| 24 | 10/10 | 14.8±8.8 | 16.1±2.9 | 13.2±2.4 | 3 | 20 | 17 | 复合杂合 | c.1439C>T,c.1729T>G | p.Ala480Val,p.Trp577Gly | 2016[24] | |
| c.1864G>T | p.Asp622Tyr | |||||||||||
| c.1879G>A | p.Ala627Thr | |||||||||||
| 复合杂合 | c.611G>A,c.673delA | p.Cys204Tyr,p.Lys225Asnfs*40 | ||||||||||
| c.691T>C | p.Cys231Arg | |||||||||||
| c.2054C>T | p.Pro685Leu | |||||||||||
| 复合杂合 | c.1448G>A,c.1363delC | p.Trp483*,p.Gln455Serfs*52 | ||||||||||
| 复合杂合 | c.517T>C,c.1757C>A | p.Cys173Arg,p.Ser586* | ||||||||||
| 复合杂合 | c.428G>A,c.1744C>T | p.Cys143Tyr,p.Leu582Phe | ||||||||||
| 复合杂合 | c.1448G>A,c.1879G>A | p.Trp483*,p.Ala627Thr | ||||||||||
| 复合杂合 | c.1129T>G,c.1268T>C | p.Cys377Gly,p.Ile423Thr | ||||||||||
| 复合杂合 | c.818-2A>G,c.1448G>A | NA,p.Trp483* | ||||||||||
| 复合杂合 | c.1448G>A,c.2475C>G | p.Trp483*,p.Asn825Lys | ||||||||||
| 复合杂合 | c.1448G>A,c.1216C>A | p.Trp483*,p.Arg406Arg | ||||||||||
| 复合杂合 | c.514G>C,c.1723C>T | p.Asp172His,p.Leu575Phe | ||||||||||
| 复合杂合 | c.1246C>T,c.1747C>T | p.Arg416Trp,p.His583Tyr | ||||||||||
| 一般情况 | 治疗前血脂 | 临床表现 | 基因诊断 | 发表 时间 | ||||||||
| 序号 | 性别 (女/男) | 年龄 (岁) | 省/市 | TC (mmol/L) | LDL-C (mmol/L) | 冠心 病 | 黄色 瘤 | 角膜 弓 | 纯合子/ 复合杂合 | cDNA 突变 | 氨基酸突变 | |
| 复合杂合 | c.1448G>A,c.2132G>A | p.Trp483*,p.Cys711Tyr | ||||||||||
| 复合杂合 | c.665G>T,c.2054C>T | p.Cys222Phe,p.Pro685Leu | ||||||||||
| 25 | 女 | 26 | 天津 | 12 | 9.8 | 纯合子 | c.1864G>T | p.Asp601Tyr | 2016[25] | |||
| 26 | 女 | 14 | 湖南 | 17.1 | 16.6 | 纯合子 | c.516C>A | p.Asp172Glu | 2016[26] | |||
| 女 | 25 | 湖南 | 20.2 | 18.2 | + | 复合杂合 | c.760C>T, c.1216C>A | p.Gln254*, p.Arg406= | ||||
| 女 | 31 | 湖南 | 18.5 | 16.5 | + | 复合杂合 | c.1132C>T, c.1448G>A | p.Gln378*, p.Trp483* | ||||
| 女 | 16 | 安徽 | 20.3 | 18.4 | + | + | 纯合子 | c.1448G>A | p.Trp483* | |||
| 27 | 男 | 8 | 安徽 | 15.9 | 13.9 | + | + | + | 复合杂合 | c.1448G>A, c.1879G>A | p.Trp483*, p.Ala627Thr | 2016[27] |
| 男 | 26 | 安徽 | 14.8 | 11.3 | + | + | + | 复合杂合 | c.1448G>A, c.1363delC | p.Trp483*,p.Gln455Serfs | ||
| 女 | 6 | 辽宁 | 17.3 | 12.8 | + | + | + | 复合杂合 | c.1448G>A, c.2132G>T | p.Trp483*, p.Cys711Tyr | ||
| 女 | 21 | 湖南 | 15.7 | 14.7 | + | + | 复合杂合 | c.1448G>A, c.2475C>G | p.Trp483*, p.Asn825Lys | |||
| 男 | 12 | 安徽 | 17.0 | 11.6 | + | + | + | 纯合子 | c.1448G>A | p.Trp483* | ||
| 女 | 13 | 江苏 | 16.3 | 12.8 | + | + | + | 复合杂合 | c.1448G>A, c.818-2A>G | p.Trp483*, NA | ||
| 女 | 22 | 四川 | 15.7 | 14.4 | + | + | 复合杂合 | c.1448G>A, c.418G.A | p.Trp483*, p.Glu140Lys | |||
| 男 | 9 | 广西 | 16.1 | 12.6 | + | + | 复合杂合 | c.1448G>A, c.1729T>G | p.Trp483*, p.Trp577Gly | |||
| 男 | 2.5 | 安徽 | 20.0 | 18.4 | + | 复合杂合 | c.1448G>A, c.760C>T | p.Trp483*, p.Gln254* | ||||
| 男 | 20 | 河北 | 16.0 | 15.4 | + | + | + | 复合杂合 | c.1448G>A, c.1216C>A | p.Trp483*, p.Arg406= | ||
| 28 | 女 | 22 | 江苏 | 22.6 | 18.9 | + | 纯合子 | c.632_634delACT | p.His211_Ser212delinsPro | 2016[28] | ||
| 29 | 女 | 3 | 湖南 | 16.2 | + | 复合杂合 | c.796G>C, c.1048C>T | p.Asp266His, p.Arg350* | 2016[29] | |||
| 男 | 8 | 湖南 | 15.0 | + | 复合杂合 | c.1448G>A, c.1720C>A | p.Trp483*, p.Arg574Ser | |||||
| 男 | 3 | 湖南 | 19.2 | + | 纯合子 | c.605T>C | p.Phe202Ser | |||||
| 女 | 3 | 湖南 | 19.8 | + | 复合杂合 | c.2030G>A, c.1257C>A | p.Cys677Tyr, p.Tyr419* | |||||
| 30 | 男 | 25 | 湖北 | 10.7# | 8.7# | + | + | 复合杂合 | c.1879G>A, c.482T> C | p.Ala627Thr, p.Ile161Thr | 2015[30] | |
| 31 | 男 | 5 | 河南 | 21.2 | 19.8 | + | + | 纯合子 | c.1187-10G> A | NA | 2015[31] | |
| 32 | 男 | 15 | 湖南 | 11.6# | 10.3# | + | + | 纯合子 | c.298G>A | p.Asp100Asn | 2015[32] | |
| 33 | 女 | 22 | 陕西 | 11# | 9.2# | + | 复合杂合 | c.400T>C, c.1246C> T | p.Cys134Arg, p.Arg416Trp | 2015[33] | ||
| 34 | 男 | 32 | 湖南 | 9.9 | 9.0 | + | 纯合子 | c.605T>C | p.Phe202Ser | 2015[34] | ||
| 男 | 12 | 湖南 | 19.0 | 16.4 | + | 纯合子 | c.2030G>A | p.Cys677Tyr | ||||
| 男 | 14 | 湖南 | 21.5 | 18.6 | 纯合子 | c.605T>C | p.Phe202Ser | |||||
| 35 | 男 | 7 | 河南 | 19.8 | 13.9 | + | + | 复合杂合 | c.97C>T, c.892delA, c.890A>C | p.Gln33*, p.Met298Cysfs, p.Asn297Thr | 2015[35] | |
| 36 | 男 | 23 | 中国台湾 | 14.95 mg/dl | 12.69 mg/dl | + | 复合杂合 | c.1449G>A, c1879G>A | p.Trp483*, p.Ala627Thr | 2015[36] | ||
| 37 | 6 | 中国台湾 | 595 mg/dl | 438 mg/dl | 复合杂合 | p.Cys308Tyr, p.Ala410Thr | 2015[37] | |||||
| 21 | 中国台湾 | 517 mg/dl | 404 mg/dl | 复合杂合 | p.Cys308Tyr, p.His562Tyr | |||||||
| 18 | 中国台湾 | 460 mg/dl | NA mg/dl | 复合杂合 | p.Cys308Tyr, p.Asp69Asn | |||||||
| 一般情况 | 治疗前血脂 | 临床表现 | 基因诊断 | 发表 时间 | ||||||||
| 序号 | 性别 (女/男) | 年龄 (岁) | 省/市 | TC (mmol/L) | LDL-C (mmol/L) | 冠心 病 | 黄色 瘤 | 角膜 弓 | 纯合子/ 复合杂合 | cDNA 突变 | 氨基酸突变 | |
| 17 | 中国台湾 | 576 mg/dl | 474 mg/dl | 复合杂合 | p.Cys308Tyr, p.Gly457Arg | |||||||
| 4 | 中国台湾 | 384 mg/dl | 334 mg/dl | 复合杂合 | p.His562Tyr, p.Ala410Thr | |||||||
| 11 | 中国台湾 | 598 mg/dl | 415 mg/dl | 复合杂合 | p.Trp462*, p.Asp679Gly | |||||||
| 8 | 中国台湾 | 672 mg/dl | 528 mg/dl | 复合杂合 | p.Cys308Tyr, p.Ala410Thr | |||||||
| 11 | 中国台湾 | 848 mg/dl | NA mg/dl | 复合杂合 | p.Arg395Trp, exon 3-5 duplication | |||||||
| 4 | 中国台湾 | 1200 mg/dl | 1125 mg/dl | 复合杂合 | c.64del G, c.1953-1954 delTA | |||||||
| 21 | 中国台湾 | >800 mg/dl | NA mg/dl | 复合杂合 | p.Asp47Asn, NA | |||||||
| 38 | 男 | 12 | 上海 | 18.7 | 18.3 | + | + | + | 纯合子 | c.2389G>A | p.Val797Met | 2014[38] |
| 男 | 3 | 上海 | 27.6 | 22.9 | + | + | 杂合子 | c.513delC, c.1959T>C | p.Pro171fs, p.Val653= | |||
| 39 | 女 | 5 | 北京 | 11.98 | 9.22 | + | + | 杂合子 | c.1257C>A | p.Tyr398* | 2014 [39] | |
| 40 | 女 | 17 | 上海 | 21.5 | 19.4 | + | + | 纯合子 | c.541C>T | p.Pro181Ser | 2014[40] | |
| 41 | 男 | 2 | 北京 | 24.7 | 14.8 | + | + | 复合杂合 | c.1268T>C, c.1129T>G | p.Ile425Thr, p.Cys377Gly | 2014[41] | |
| 42 | 女 | 40 | 北京 | 16.9# | 13.9# | + | + | 杂合子 | c.1907G>T | p.Gly615Val | 2014[42] | |
| 43 | 男 | 12 | 北京 | 19.5 | 17.1 | + | + | + | 杂合子 | c.97C>T | p.Gln33* | 2013[43] |
| 44 | 女 | 17 | 安徽 | 18.9 | 10.9 | + | + | 杂合子 | c.1448G>A | p.Trp462* | 2012[44] | |
| 45 | 男 | 9 | 上海 | 14.0 | 11.3 | + | 复合杂合 | c.1747C>T, c.2054C>T | p.His583Tyr, p.Pro685Leu | 2012[45] | ||
| 46 | 3/1 | 20(12-45) | 中国台湾 | 477(410-694) mg/dl | 411(321-570) mg/dl | 2 | 3 | 复合杂合 | c.986G>A, c.268G>A, c.1291G>A, 1432G>A, c.769C>T/1765G>A | p.Cys329Tyr, p.Asp90Asn, p.Ala431Thr, p.Gly478Arg, p.Arg257Trp, p.Asp589Asn | 2012[46] | |
| 7/6 | 50(11-69) | 中国台湾 | 343(314-445) mg/dl | 263(204-370) mg/dl | 5 | 4 | 杂合子 | c.986G>A | p.Cys329Tyr | |||
| 8/6 | 45(6-69) | 中国台湾 | 332(282-453) mg/dl | 233(204-354) mg/dl | 4 | 3 | 杂合子 | c.1747C>T | p.His583Tyr | |||
| 47 | 男 | 19 | 山东 | 12.6# | 11.1 | + | + | + | 纯合子 | c. 1581G>A | NA | 2011[47] |
| 男 | 17 | 山东 | 15.5 | 13.0 | + | + | + | 纯合子 | c. 1581G>A | NA | ||
| 女 | 16 | 山东 | 14.5 | 12.0 | + | + | 纯合子 | c. 1581G>A | NA | |||
| 48 | 组1:26/15 组2:54/31 | 40±20 43±16 | 中国台湾 | 408±83 mg/dl 338±49 mg/dl | 311±78 mg/dl 250±45 mg/dl | c.268G>A | p.Asp90Asn | 2011[48] | ||||
| c.310T>C | p.Cys104Arg | |||||||||||
| c.344G>A | p.Arg115His | |||||||||||
| c.516C>G | p.Asp172Glu | |||||||||||
| c.590G>A | p.Cys197Tyr | |||||||||||
| c.599T>G | p.Phe200Cys | |||||||||||
| c.682G>T | p.Glu228* | |||||||||||
| c.769C>T | p.Arg257Trp | |||||||||||
| c.828C>A | p.Cys276* | |||||||||||
新窗口打开|下载CSV
Supplementary Table 1
附表1
附表1续
Supplementary Table 1
| 一般情况 | 治疗前血脂 | 临床表现 | 基因诊断 | 发表 时间 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 序号 | 性别 (女/男) | 年龄 (岁) | 省/市 | TC (mmol/L) | LDL-C (mmol/L) | 冠心 病 | 黄色 瘤 | 角膜 弓 | 纯合子/ 复合杂合 | cDNA 突变 | 氨基酸突变 | |
| c.947A>G | p.Asn316Ser | |||||||||||
| c.986G>A | p.Cys329Tyr | |||||||||||
| c.1016T>C | p.Leu339Pro | |||||||||||
| c.1048C>T | p.Arg350* | |||||||||||
| c.1054T>A | p.Cys352Ser | |||||||||||
| c.1216C>T | p.Arg406Trp | |||||||||||
| c.1246C>T | p.Arg416Trp | |||||||||||
| c.1247G>T | p.Arg416Leu | |||||||||||
| c.1291G>A | p.Ala431Thr | |||||||||||
| c.1322T>C | p.Ile441Thr | |||||||||||
| c.1329G>A | p.Trp443* | |||||||||||
| c.1384G>A | p.Val462Ile | |||||||||||
| c.1432G>A | p.Gly478Arg | |||||||||||
| c.1474G>A | p.Asp492Asn | |||||||||||
| c.1586+1G>T | NA | |||||||||||
| c.1618G>A | p.Ala540Thr | |||||||||||
| c.1691A>G | p.Asn564Ser | |||||||||||
| c.1747C>T | p.His583Tyr | |||||||||||
| c.1765G>A | p.Asp589Asn | |||||||||||
| c.1807A>T | p.Lys603* | |||||||||||
| c.1783C>T | p.Arg595Trp | |||||||||||
| c.1851_1862del | p.Val618_Thr621del | |||||||||||
| c.1879G>A | p.Ala627Thr | |||||||||||
| c.1897C>T | p.Arg633Cys | |||||||||||
| c.1953_1954del | p.Met652GlyfsX16 | |||||||||||
| c.2215C>T | p.Gln739* | |||||||||||
| c.2389G>A | p.Val797Met | |||||||||||
| c.2446A>T | p.Lys816* | |||||||||||
| c.510delC | p.Asp171Profs*35 | |||||||||||
| c.562delT | p.Tyr188Thrfs*18 | |||||||||||
| c.656 661del | p.Gly219Asp221del | |||||||||||
| 49 | 女 | 25 | 北京 | 13 | 9.8 | + | + | 杂合子 | c.1448G>A | p.Trp462* | 2010[49] | |
| 50 | 女 | 23 | 17.66 | 15.78 | + | 复合杂合 | c.2390-2A>G, c.1687C>T | p.Pro563Ser | 2010[50] | |||
| 51 | 男 | 10 | 陕西 | 17.1 | 14.0 | + | + | 纯合子 | c.1864G>T | p.Asp601Tyr | 2010[51] | |
| 52 | 女 | 5 | 北京 | 16.8 | 13.1 | + | + | 杂合子 | c.1879G>A | p.Ala627Thr | 2010[52] | |
新窗口打开|下载CSV
Supplementary Table 1
附表1
附表1续
Supplementary Table 1
| 一般情况 | 治疗前血脂 | 临床表现 | 基因诊断 | 发表 时间 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 序号 | 性别 (女/男) | 年龄 (岁) | 省/市 | TC (mmol/L) | LDL-C (mmol/L) | 冠心 病 | 黄色 瘤 | 角膜 弓 | 纯合子/ 复合杂合 | cDNA 突变 | 氨基酸突变 | |
| 53 | 男 | 16 | 北京 | 15.5 | 13.8 | + | + | + | 纯合子 | c.1448G>A | p.Trp462* | 2009[53] |
| 男 | 13 | 北京 | 28.8 | 27.7 | + | + | + | 纯合子 | c.313+1G>A | p.Leu64_Pro105delinsSer | ||
| 女 | 13 | 北京 | 14.7 | 13.3 | + | + | 复合杂合 | c.428G>A, c.1211T>C | p.Cys122Tyr, p.Thr383Ile | |||
| 男 | 34 | 北京 | 17.1 | 14.5 | + | 纯合子 | c.691T>C | p.Cys231Arg | ||||
| 54 | 女 | 34 | 湖北 | 16.2 | 13.8 | + | 纯合子 | c.1879G>A | p.Ala627Thr | 2009[54] | ||
| 男 | 23 | 8.36 | 6.47 | 杂合子 | c. 1511G>A | p.Trp483* | ||||||
| 55 | 男 | 6 | 安徽 | 16.4 | 14.7 | + | + | 复合杂合 | c.1448G>A, c.1879G>A | p.Trp462*, p.Ala606Thr | 2009[55] | |
| 56 | 女 | 4 | 河南 | 23.3 | 19.9 | + | + | + | 复合杂合 | c.1879A>G, c.1864G>T | p.Ala606Thr, p. Asp601Tyr | 2009[56] |
| 57 | 男 | 6 | 安徽 | 19.5 | 18.0 | + | 纯合子 | c.1075C>T | p.Gln359* | 2009[57] | ||
| 58 | 男 | 8 | 14.91 | 12.85 | 纯合子 | c.1439C>T | p.Ala459Val | 2008[58] | ||||
| 59 | 男 | 23 | 北京 | 11.81 | 8.33 | + | 杂合子 | c.358G>T | p.Asp108Tyr | 2008[59] | ||
| 60 | 男 | 9 | 北京 | 19.5 | 15.3 | 复合杂合 | c.685del A, c.386A>G | NA, p.Asp129Gly | 2007[60] | |||
| 61 | 男 | 12 | 20.34 | 16.78 | 杂合子 | c.558G>A | p.Trp165* | 2007[61] | ||||
| 男 | 9 | 北京 | 20.3 | 16.8 | + | + | 杂合子/ 纯合子 | c.495G>A/IVS5-1G>A | p.Trp165*/NA | |||
| 62 | 男 | 9 | 19.49 | 15.25 | + | 复合杂合 | c.80T>C,c.1413G> A,c.685delA,c.386A>G | p.Trp6=,p.Gly450=, p.685del 1,p.Asp129Gly | 2007[60] | |||
| 男 | 41 | 10.64 | 8.75 | 复合杂合 | c.80T>C,c.1413G> A,c.685delA | p.Trp6=,p.Gly450=, p.685del 1 | ||||||
| 女 | 40 | 8.11 | 6.33 | 杂合子 | c.80T>C,c.386A>G | p.Trp6=,p.Asp129Gly | ||||||
| 63 | 29/22 | 55 (48-64) | 中国台湾 | 311 (281-343) mg/L | 208 (189-245) mg/L | + | + | c.1747C>T | p.His562Tyr | 2006[62] | ||
| + | + | + | c.1953delTA | NA | ||||||||
| c.1592T>A | p.Met510Lys | |||||||||||
| + | c.1597T>C | P.Trp512Arg | ||||||||||
| c.1816G>A | p.Ala606Thr | |||||||||||
| c.986G>A | p.Cys329Tyr | |||||||||||
| 复合杂合 | c.769C>T,c.1765G>A | p.Arg236Trp,p.Asp568Asn | ||||||||||
| c.1246C>T | p.Arg395Trp | |||||||||||
| c.1322T>C | p.Ile420Thr | |||||||||||
| c.268G>A | p.Asp90Asn | |||||||||||
| c.681C>A | p.D206E | |||||||||||
| c.682G>T | p.E207X | |||||||||||
| 64 | 女 | 29 | 13.38 | 11.16 | + | 复合杂合 | ?c.292G>A, ?c.1864G>A | ?p.Gly98Ser, ?p.Asp622Asn | 2005[63] | |||
| 65 | 男 | 36 | 中国台湾 | 8.33 | 6.67 | 杂合子 | c.2043C>G | p.Cys660Trp | 2005[64] | |||
| 66 | 男 | 26 | 浙江 | 19.1 | 17.1 | + | + | 纯合子 | c.2048A>C | p.Glu683Ala | 2004[65] | |
| 67 | 女 | 4 | 17.81 | 15.22 | + | 纯合子 | c.1304A>G | p.Glu414Gly | 2004[66] | |||
| 一般情况 | 治疗前血脂 | 临床表现 | 基因诊断 | 发表 时间 | ||||||||
| 序号 | 性别 (女/男) | 年龄 (岁) | 省/市 | TC (mmol/L) | LDL-C (mmol/L) | 冠心 病 | 黄色 瘤 | 角膜 弓 | 纯合子/ 复合杂合 | cDNA 突变 | 氨基酸突变 | |
| 女 | 33 | 14.93 | 10.44 | + | + | 纯合子 | c.1329delG | p.422fs | ||||
| 男 | 12 | 12.09 | 10.44 | + | 纯合子 | c.1544A>G | p.Asn494Ser | |||||
| 男 | 7 | 21.3 | 19.2 | + | + | 纯合子 | c.2400insC | p.779fs | ||||
| 女 | 19 | 15.66 | 14.68 | + | 复合杂合 | c.1849 A>G, c.1877C>T | p.Lys596Glu, p.Glu605Val | |||||
| 女 | 6 | 22.3 | 18.29 | + | 纯合子 | c. 2021A>G | p.Asn653Ser | |||||
| 男 | 15 | 15.48 | + | 纯合子 | c. 1100T>A | p.Leu346His | ||||||
| 女 | 19 | 15.66 | 14.68 | + | 纯合子 | c. 826T>G | p.Cys255Arg | |||||
| 68 | 男 | 10 | 北京 | 14.7 | 12.5 | + | 杂合子 | c. 444T>A | p.Cys127Trp | 2004[67] | ||
| 69 | 女 | 10 | 北京 | 14.7 | 13.4 | + | + | 复合杂合 | c. 428G>A, c.1211C>T | p.Cys122Tyr, p.Thr383Ile | 2003[68] | |
| 70 | 男 | 11 | 江苏 | 15.7 | 15.1 | + | 纯合子 | c.890delA | p.Asn297Thrfs | 2003[69] | ||
| 男 | 2 | 江苏 | 17.4 | 16.4 | + | 纯合子 | c.890delA | p.Asn297Thrfs | ||||
| 71 | 男 | 浙江 | 纯合子 | c.683A>C | p.Gln207Ala | 2002[70] | ||||||
| 72 | 女 | 14 | 北京 | 15.4 | 11 | + | + | 纯合子 | c.1816G>A | p.Ala606Thr | 2001[71] | |
| 女 | 12 | 北京 | 18.6 | 13.4 | + | + | 纯合子 | c.1816G>A | p.Ala606Thr | |||
| 男 | 10 | 北京 | 23.9 | 19.8 | + | + | 纯合子 | c.850T>C | p.Cys263Arg | |||
| 73 | 男 | 3 | 北京 | 23.9 | 16.2 | + | 纯合子 | c.850T>C | p.Cys263Arg | 2001[72] | ||
| 74 | 男 | 11 | 山东 | 21.3 | 19.6 | + | + | 复合杂合 | c.811G>A, c.814C>T, c.830G>A | p.Val271Ile, NA | 2001[73] | |
| 75 | 男 | 62 | 8.3 | 6.7 | + | 纯合子 | c. ?-152 G>T | NA | 2000[74] | |||
| 女 | 53 | 9.9 | 8.1 | 纯合子 | c. ?2108insTGCTGGC | p.682fs | ||||||
| 女 | 71 | 7.1 | 5 | + | 纯合子 | c.2048G>A | p.Ala663Thr | |||||
| 男 | 44 | 9.6 | 6.7 | + | 纯合子 | c.2087G>A | p.Cys675Tyr | |||||
| 男 | 44 | - | 11 | 9.3 | + | 纯合子 | c.757C>T | p.Arg232Trp | ||||
| 女 | 59 | 10 | + | + | + | 纯合子 | c.986G>A | p.Cys329Tyr | ||||
| 男 | 44 | - | 11 | 9.3 | + | + | 纯合子 | c.344G>A | p.Arg94His | |||
| 男 | 31 | - | 9.2 | 7.1 | + | 纯合子 | ?c.77del GA | p.Cys5Thrfs | ||||
| 76 | 男 | 43 | 中国香港 | 16.0 | 14.3 | + | + | 复合杂合 | c.-44C>T, c.2054C>T | NA, p.Pro664Leu | 1998[75] | |
| 女 | 54 | 8.8 | 5.9 | + | 杂合子 | c.1432G>A | p.Gly478Arg | |||||
| 女 | 55 | 12 | 10.2 | + | 杂合子 | c.1285G>A | p.Val408Met | |||||
| 女 | 19 | 10.9 | 9.6 | + | 杂合子 | c.1880C>T | p.Ala606Val | |||||
| 男 | 42 | 9.5 | 7.6 | + | 杂合子 | c.1474G>A | p.Asp471Asn | |||||
| 男 | 47 | 10.3 | 8.7 | + | 杂合子 | c.1779delC | p.572fs | |||||
| 女 | 80 | 11.3 | 9.8 | + | 纯合子 | ?c.1706-1G3T | NA | |||||
| 男 | 44 | 9.5 | 7.9 | + | 杂合子 | c.1241T>G | p.Leu393Arg | |||||
| 女 | 39 | 8.8 | 6.8 | + | 纯合子 | c.364A>T | ?p.Ile101Phe | |||||
| 女 | 46 | 10.1 | 8.3 | 纯合子 | c.571C>T | ?p.Gly170* | ||||||
| 一般情况 | 治疗前血脂 | 临床表现 | 基因诊断 | 发表 时间 | ||||||||
| 序号 | 性别 (女/男) | 年龄 (岁) | 省/市 | TC (mmol/L) | LDL-C (mmol/L) | 冠心 病 | 黄色 瘤 | 角膜 弓 | 纯合子/ 复合杂合 | cDNA 突变 | 氨基酸突变 | |
| 77 | 9.33 | 7.63 | + | 杂合子 | c.550C>T | p.Cys163Arg | 1998[76] | |||||
| 78 | 7 | 9.3-12.0 | 6.3-10.2 | c.986G>A | p.Cys329Tyr | 1998[77] | ||||||
| c.1241T>G | p.Leu393Arg | |||||||||||
| c.1285G>A | p.Val408Met | |||||||||||
| 79 | 男 | 11 | 江苏 | 15.8 | 14.1 | + | 复合杂合 | Described as Exons 11-14 deleted, c.1747C > T | NA, p.His583Tyr | 1994[78] | ||
| 男 | 9 | 江苏 | 19.0 | 17.3 | + | 纯合子 | c.1448G>A | p.Trp483* | ||||
| 女 | 22 | 江苏 | 14.0 | 12.8 | + | + | 纯合子 | c.682G>T | p.Glu228* | |||
| 男 | 37 | 江苏 | 15.5 | 14.62 | + | + | 杂合子 | c.2015delT | p.615fs | |||
| 女 | 12 | 江苏 | 11.2 | 9.8 | + | 复合杂合 | c.2415delC, c.1664T>C | p.Gly805Glyfs, p.Leu555Pro | ||||
| 女 | 9 | 江苏 | 8.2# | 6.8# | + | 复合杂合 | c.1816G>A, c.809G>A | p.Ala606Thr, p.Cys270Tyr | ||||
| 男 | 37 | 江苏 | 15.5 | 14.6 | + | + | 复合杂合 | c.12G>A, c.1952delA | p.Trp4*, p.Asp651Aspfs | |||
| 女 | 34 | 江苏 | 20.1 | 18.7 | + | + | 复合杂合 | c.12G>A, c.1952delA | p.Trp4*, p.Asp651Aspfs | |||
| 女 | 13 | 江苏 | 15.4 | 13.8 | + | 杂合子 | NA | NA | ||||
| 女 | 34 | 江苏 | 20.08 | 18.72 | + | + | + | 复合杂合 | c.13G>A, c.1952delA | p.Trp18*, p.Asp651Aspfs | ||
新窗口打开|下载CSV
Supplementary Table 2
附表2
附表2中国FH患者Apo B基因突变致病位点总结
Supplementary Table 2
| 一般情况 | 治疗前血脂 | 临床表现 | 基因诊断 | 发表 时间 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 序 号 | 性别 (女/男) | 年龄 (岁) | 省/市 | TC (mmol/L) | LDL-C (mmol/L) | 冠心 病 | 黄色 瘤 | 角膜 弓 | 纯合子/ 复合杂合 | cDNA突变 | 氨基酸突变 | |
| 1 | 198/110 | 48.3?±?11.2 | 11.06?±?1.58 | 8.41?±?1.39 | 127 | 9 | c.10579C>T | p.Arg3527Trp | 2020[3] | |||
| 2 | 40/40 | 54.21±13.38 | 8.85 (7.95-10.00) | 5.52 (4.96-6.28) | 48 | 复合杂合 | c.C8216T,c.C1853T | p.Pro2739Leu,p.Ala618Val | 2019[79] | |||
| 复合杂合 | c.C10579T,c.C8216T, c.C1853T | p.Arg3527rp,p.Pro2739Leu,p.Ala618Val | ||||||||||
| 3 | 1/9 | 44.80 ± 6.36 | 5.04 (4.03-5.91) | 3.39 (2.58-4.08) | + | 杂合子 | c.10579C>T | p.Arg3527Trp | 2019[7] | |||
| 4 | 37/188 | 46.64?±?7.21 | 3.63 (2.97- 4.35) | 杂合子 | c.10579C>T | p.Arg3527Trp | 2019[6] | |||||
| 5 | 6/99 | 31.69±5.65 | 6.62±7.49 | 5.77±3.38 | 20 | 12 | c.1594C>T | p.Arg532Trp | 2018[80] | |||
| c.6110T>C | p.Ile2037Thr | |||||||||||
| c.7223C>T | p.Ser2408Phe | |||||||||||
| c.8267G>T | p.Gly2756Val | |||||||||||
| c.8462C>T | p.Pro2821Leu | |||||||||||
| c.889C>T | p.Arg297Cys | |||||||||||
| c.9164A>G | p.Asn3055Ser | |||||||||||
| 6 | 112/173 | ?49 ± 12 | ?7.03 ± 2.53 | ?5.22 ± 2.12 | 233 | 28 | c.10093C > G | p.His3365Asp | 2018[16] | |||
| 一般情况 | 治疗前血脂 | 临床表现 | 基因诊断 | 发表 时间 | ||||||||
| 序 号 | 性别 (女/男) | 年龄 (岁) | 省/市 | TC (mmol/L) | LDL-C (mmol/L) | 冠心 病 | 黄色 瘤 | 角膜 弓 | 纯合子/ 复合杂合 | cDNA突变 | 氨基酸突变 | |
| 7 | 115/104 | 31.15 ± 15.21 | 11.15 ± 3.96 | 8.32 ± 3.83 | 46 | 25 | c.11052A>T | p.Leu3684Phe | 2017[23] | |||
| c.11585T>C | p.Ile3862Thr | |||||||||||
| c.10579C>T | p.Arg3527Trp | |||||||||||
| c.10580G>A | p.Arg3527Gln | |||||||||||
| 8 | 女 | 31 | 北京 | 13.13 | 11.32 | + | + | 复合杂合 | c.1594C>T | p.Arg532Trp | 2017[18] | |
| 男 | 12 | 北京 | 10.92 | 9.26 | + | + | 复合杂合 | c.1594C>T | P.Arg532Trp | |||
| 男 | 21 | 北京 | 19.12 | 14.09 | + | + | 复合杂合 | c.581C>T | p.Thr194Met | |||
| 9 | 男 | 20 | 7.8 | 5.47 | 杂合子 | c.10579C>T | p.Arg3500Trp | 2016[26] | ||||
| 10 | 10/2 | 57(23-69) | 中国台湾 | 327(300-394) mg/dl | 235(200-318) mg/dl | 5 | 5 | 杂合子 | c.10579C>T | p.Arg3500Trp | 2012[46] | |
| 11 | 组1:26/15 组2:54/31 | 40±20 43±16 | 中国台湾 | 408±83mg/dl 338±49mg/dl | 311±78 mg/dl 250±45 mg/dl | c.10579C > T | p.Arg3500Trp | 2011[48] | ||||
| 12 | 女 | 5 | 16.8 | 13.1 | + | + | c.10707C>T | p.Arg3500Trp | 2010[52] | |||
新窗口打开|下载CSV
Supplementary Table 3
附表3
附表3中国FH患者PCSK9基因突变致病位点总结
Supplementary Table 3
| 一般情况 | 治疗前血脂 | 临床表现 | 基因诊断 | 发表 时间 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 序号 | 性别 (女/男) | 年龄 (岁) | 省/市 | TC (mmol/L) | LDL-C (mmol/L) | 冠心 病 | 黄色 瘤 | 角膜 弓 | 纯合子/ 复合杂合 | cDNA突变 | 氨基酸突变 | |
| 1 | 37/188 | 46.64±7.21 | 3.63 (2.97- 4.35) | 杂合子 | c.277C > T | p.Arg93Cys | 2019[6] | |||||
| 杂合子 | c.1792G > A | p.Ala598Thr | ||||||||||
| 杂合子 | c.517C > T | p.Pro173Ser | ||||||||||
| 杂合子 | c.1954A > G | p.Asn652Asp | ||||||||||
| 2 | 6/99 | 31.69±5.65 | 6.62±7.49 | 5.77±3.38 | 20 | 12 | c.10G>A | p.Val4Ile | 2018[80] | |||
| c.644G>A | p.Arg215His | |||||||||||
| c.626C>T | p.Pro209Leu | |||||||||||
| 3 | 115/104 | 31.15±15.21 | 11.15±3.96 | 8.32 ± 3.83 | 46 | 25 | c.287G>T | p.Arg96Leu | 2017[23] | |||
| c.313C>T | p.Arg105Trp | |||||||||||
| 4 | 男 | 12 | 北京 | 10.92 | 9.26 | + | + | 复合杂合 | c.63-65insCTG | p.Leu21dup | 2017[18] | |
| 5 | 女 | 14 | 16.32 | 14.97 | + | + | + | 杂合子 | c.918G>T | p.Arg306Ser | 2010[81] | |
新窗口打开|下载CSV
Supplementary Table 4
附表4
附表4中国FH患者其他基因突变致病位点总结
Supplementary Table 4
| 一般情况 | 治疗前血脂 | 临床表现 | 基因诊断 | 发表时间 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 序 号 | 性别 (女/男) | 年龄 (岁) | 省/市 | TC (mmol/L) | LDL-C (mmol/L) | 冠心 病 | 黄色 瘤 | 角膜 弓 | 突变基因 | 纯合子/ 复合杂合 | cDNA突变 | 氨基酸突变 | |
| 1 | 1/9 | 44.80±6.36 | 5.04 (4.03-5.91) | 3.39 (2.58-4.08) | + | LDLRAP1 | 杂合子 | c.65G>C | p.Trp22Ser | 2019[7] | |||
| 2 | 男 | 8 | 北京 | 18.96 | 15.96 | + | ABCA1 | 复合杂合 | c.2170G>A | p.Val724Met | 2017[18] | ||
| 男 | 27 | 北京 | 13.81 | 12.56 | + | + | APOE | 纯合子 | c.149G>A | p.Arg50His | |||
| 男 | 27 | 北京 | 13.81 | 12.56 | + | + | GHR | 杂合子 | c.1483C>A | p.Pro495Thr | |||
| 女 | 32 | 北京 | 17.90 | 13.54 | + | + | EPHX2 | 杂合子 | c.461G>A | p.Cys154Tyr | |||
| 男 | 21 | 北京 | 19.12 | 14.09 | + | + | GHR | 复合杂合 | c.1735C>A | p.Pro579Thr | |||
| 3 | 女 | 6 | 北京 | 17.06 | 13.77 | + | EPHX2 | 纯合子 | c.860G>A | p.Arg287Gln | 2016[82] | ||
新窗口打开|下载CSV
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URL [本文引用: 2]
DOI:10.1016/j.jacl.2016.08.013URL [本文引用: 2]

To analyze the genetic characteristics of a five generations pedigree with homozygous familial hypercholesterolemia (HoFH). Prospective study. Twenty family members included a proband diagnosed as familial hyperlipidemia at the cardiology Department of Xi'an Children's Hospital in October 2018 were research object. Clinical data were collected. Genome DNAs were extracted. Whole exons sequencing was performed on the proband using target capture next generation sequencing. Candidate gene mutation sites identified by bioinformatics were verified by Sanger sequencing in the family members. The genotype-phenotype correlation of the pedigree was analyzed between heterozygous mutation carriers and non-carriers. The proband was a 7-years and 10-month-old boy. He was born with a roundgreen bean size yellow skin protuberance in the skin of the coccyx. Since the age of 3-4 years old, xanthoma-like lesions with a diameter of 0.5-1.5 cm gradually appeared in the skin of bilateral elbow joints, knee joints and Achilles tendon. The height, weight and intellectual development of the child were the same as those of normal children at the same age. No similar xanthoma-like lesion was found in the other family members. The proband's total cholesterol (TC) reached 18.16-21.24 mmol/L, and his low density lipoproteincholesterol (LDL-C) was 14.08-15.51 mmol/L. Carotid ultrasonography showed diffuse sclerotic plaques in bilateral carotid and vertebral arteries, and color Doppler echocardiography revealed aortic valve thickening and calcification. Gene testing identified that the proband carried a homozygous mutation C. 418G>A (p. E140K) in LDLR gene inherited from his parents who had a consanguineous marriage and carried a heterozygous mutation of LDLR-E140K, respectively.The TC, LDL-C and apolipoproteinB (ApoB) of LDLR-E140K gene heterozygous carriers ((8.40±0.13), (6.79±0.01) and (1.95±0.05) mmol/L, respectively) were significantly higher than those of non-carriers ((4.59±0.28), (3.35±0.39) and (0.86±0.10) mmol/L, 7.269, 4.595, 6.311, respectively, 0.05). LDLR-E140K gene homozygous mutation is first reported to be associated with most severe phenotype HoFH. The genotype-phenotype analysis of the pedigree shows that the clinical phenotype of the proband with homozygous mutation is the most serious, and all the heterozygous mutation carriers present with hypercholesterolemia phenotype. The investigation confirms that LDLR-E140K is the pathogenic variation of familial hyperlipidemia.
DOI:10.1038/nrdp.2017.93URLPMID:29219151 [本文引用: 3]
Familial hypercholesterolaemia is a common inherited disorder characterized by abnormally elevated serum levels of low-density lipoprotein (LDL) cholesterol from birth, which in time can lead to cardiovascular disease CVD). Most cases are caused by autosomal dominant mutations in LDLR, which encodes the LDL receptor, although mutations in other genes coding for proteins involved in cholesterol metabolism or LDLR function and processing, such as APOB and PCSK9, can also be causative, although less frequently. Several sets of diagnostic criteria for familial hypercholesterolaemia are available; common diagnostic features are an elevated LDL cholesterol level and a family history of hypercholesterolaemia or (premature) CVD. DNA-based methods to identify the underlying genetic defect are desirable but not essential for diagnosis. Cascade screening can contribute to early diagnosis of the disease in family members of an affected individual, which is crucial because familial hypercholesterolaemia can be asymptomatic for decades. Clinical severity depends on the nature of the gene that harbours the causative mutation, among other factors, and is further modulated by the type of mutation. Lifelong LDL cholesterol-lowering treatment substantially improves CVD-free survival and longevity. Statins are the first-line therapy, but additional drugs, such as ezetimibe, bile acid sequestrants, PCSK9 inhibitors and other emerging therapies, are often required.
DOI:10.1093/eurheartj/eht273URL [本文引用: 2]
Aims The first aim was to critically evaluate the extent to which familial hypercholesterolaemia (FH) is underdiagnosed and undertreated. The second aim was to provide guidance for screening and treatment of FH, in order to prevent coronary heart disease (CHD).;Methods and results Of the theoretical estimated prevalence of 1/500 for heterozygous FH, < 1% are diagnosed in most countries. Recently, direct screening in a Northern European general population diagnosed approximately 1/200 with heterozygous FH. All reported studies document failure to achieve recommended LDL cholesterol targets in a large proportion of individuals with FH, and up to 13-fold increased risk of CHD. Based on prevalences between 1/500 and 1/200, between 14 and 34 million individuals worldwide have FH. We recommend that children, adults, and families should be screened for FH if a person or family member presents with FH, a plasma cholesterol level in an adult >= 8 mmol/L(>= 310 mg/dL) or a child >= 6 mmol/L(>= 230 mg/dL), premature CHD, tendon xanthomas, or sudden premature cardiac death. In FH, low-density lipoprotein cholesterol targets are < 3.5 mmol/L(< 135 mg/dL) for children, < 2.5 mmol/L(< 100 mg/dL) for adults, and < 1.8 mmol/L(< 70 mg/dL) for adults with known CHD or diabetes. In addition to lifestyle and dietary counselling, treatment priorities are (i) in children, statins, ezetimibe, and bile acid binding resins, and (ii) in adults, maximal potent statin dose, ezetimibe, and bile acid binding resins. Lipoprotein apheresis can be offered in homozygotes and in treatment-resistant heterozygotes with CHD.;Conclusion Owing to severe underdiagnosis and undertreatment of FH, there is an urgent worldwide need for diagnostic screening together with early and aggressive treatment of this extremely high-risk condition.
DOI:10.1186/s12881-019-0901-0URL [本文引用: 3]
DOI:10.1093/eurheartj/ehu274URL [本文引用: 4]
Aims Homozygous familial hypercholesterolaemia (HoFH) is a rare life-threatening condition characterized by markedly elevated circulating levels of low-density lipoprotein cholesterol (LDL-C) and accelerated, premature atherosclerotic cardiovascular disease (ACVD). Given recent insights into the heterogeneity of genetic defects and clinical phenotype of HoFH, and the availability of new therapeutic options, this Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society (EAS) critically reviewed available data with the aim of providing clinical guidance for the recognition and management of HoFH. Methods and results Early diagnosis of HoFH and prompt initiation of diet and lipid-lowering therapy are critical. Genetic testing may provide a definitive diagnosis, but if unavailable, markedly elevated LDL-C levels together with cutaneous or tendon xanthomas before 10 years, or untreated elevated LDL-C levels consistent with heterozygous FH in both parents, are suggestive of HoFH. We recommend that patients with suspected HoFH are promptly referred to specialist centres for a comprehensive ACVD evaluation and clinical management. Lifestyle intervention and maximal statin therapy are the mainstays of treatment, ideally started in the first year of life or at an initial diagnosis, often with ezetimibe and other lipid-modifying therapy. As patients rarely achieve LDL-C targets, adjunctive lipoprotein apheresis is recommended where available, preferably started by age 5 and no later than 8 years. The number of therapeutic approaches has increased following approval of lomitapide and mipomersen for HoFH. Given the severity of ACVD, we recommend regular follow-up, including Doppler echocardiographic evaluation of the heart and aorta annually, stress testing and, if available, computed tomography coronary angiography every 5 years, or less if deemed necessary. Conclusion This EAS Consensus Panel highlights the need for early identification of HoFH patients, prompt referral to specialized centres, and early initiation of appropriate treatment. These recommendations offer guidance for a wide spectrum of clinicians who are often the first to identify patients with suspected HoFH.
URL [本文引用: 4]
URL [本文引用: 4]
[本文引用: 2]
[本文引用: 2]
URL [本文引用: 5]
DOI:10.1038/ng1161URL [本文引用: 4]
[本文引用: 2]
Atherogenic low density lipoproteins are cleared from the circulation by hepatic low density lipoprotein receptors (LDLR). Two inherited forms of hypercholesterolemia result from loss of LDLR activity: autosomal dominant familial hypercholesterolemia (FH), caused by mutations in the LDLR gene, and autosomal recessive hypercholesterolemia (ARH), of unknown etiology. Here we map the ARH locus to an approximately 1-centimorgan interval on chromosome 1p35 and identify six mutations in a gene encoding a putative adaptor protein (ARH). ARH contains a phosphotyrosine binding (PTB) domain, which in other proteins binds NPXY motifs in the cytoplasmic tails of cell-surface receptors, including the LDLR. ARH appears to have a tissue-specific role in LDLR function, as it is required in liver but not in fibroblasts.
[本文引用: 3]
URL [本文引用: 6]
URL [本文引用: 6]
DOI:10.1016/j.atherosclerosissup.2019.01.003URL [本文引用: 3]
DOI:10.11909/j.issn.1671-5411.2018.06.006PMID:30108616 [本文引用: 2]
Familial hypercholesterolemia (FH) is an autosomal dominant disorder of lipoprotein metabolism which can lead to premature coronary heart disease (pCHD). There are about 3.8 million potential FH patients in China, whereas the clinical and genetic data of FH are limited.Dutch Lipid Clinic Network (DLCN) criteria was used to diagnose FH in outpatients with hypercholesterolemia. Resequencing chip analysis combined with Sanger sequencing validation were used to identify mutations in the definite FH patients according to DLCN criteria. In silico analysis was conducted in mutations with previously unknown pathogenicity. Then, the novel mutant receptors were transfected into human embryo kidney 293 (HEK-293) cells. The binding and the internalization activities of the mutant receptors were analyzed by flow cytometry.The prevalence of definite FH in outpatients with hypercholesterolemia in this study is 3.2%. Using genetic testing, one homozygous FH (HoFH), one heterozygous FH (HeFH) and three compound heterozygous FH patients were confirmed. Eight mutations in low-density lipoprotein receptor (LDLR) gene were identified, in which c.357delG was a novel mutation and co-segregated with the FH phenotype. Bioinformatic analysis confirmed that c.357delG was a pathogenic mutation. Furthermore, when compared with the wild-type LDLRs by flow cytometry analysis, the binding and internalization activities of c.357delG mutant LDLRs were reduced by 35% and 49%, respectively.This study identified eight LDLR gene mutations in five patients with definite FH, in which c.357delG is a novel pathogenic mutation. These findings increase our understanding of the genetic spectrum of FH in China.
DOI:10.1161/CIRCULATIONAHA.110.979450URL [本文引用: 5]
BackgroundAlthough there have been many reports in the genetics of familial hypercholesterolemia (FH) worldwide, studies in regard of Chinese population are lacking. In this multi-center study, we aim to characterize the genetic spectrum of FH in Chinese population, and examine the genotype-phenotype correlations in detail.MethodsA total of 285 unrelated index cases from China with clinical FH were consecutively recruited. Next-generation sequencing and bioinformatics tools were used for mutation detection of LDLR, APOB and PCSK9 genes and genetic analysis.ResultsOverall, the detection rate is 51.9% (148/285) in the unrelated index cases with a total of 119 risk variants identified including 84 in the LDLR gene, 31 in APOB and 4 in PCSK9 gene. Twenty-eight variants were found in more than one individual and LDLR c.1448G>A (p. W483X) was most frequent one detected in 9 patients. Besides, we found 8 (7 LDLR and 1 APOB) novel variants referred as pathogenic (or likely pathogenic) variants according to in silico analysis. In the phenotype analysis, patients with LDLR null mutation had significantly higher LDL cholesterol level than LDLR defective and APOB/PCSK9 mutation carriers and those with no mutations (p<0.001). Furthermore, 13 double heterozygotes, 16 compound heterozygotes and 5 true LDLR homozygotes were identified and the true LDLR homozygotes had the most severe phenotypes.ConclusionsThe present study confirmed the heterogeneity of FH genetics in the largest Chinese cohort, which could replenish the knowledge of mutation spectrum and contribute to early screening and disease management.
DOI:10.1016/j.atherosclerosis.2018.08.022URL [本文引用: 2]
DOI:10.1186/s40064-016-3763-3URL [本文引用: 4]
DOI:10.1371/journal.pone.0189316URL [本文引用: 2]
Familial hypercholesterolemia (FH) is the most common and severe autosomal dominant lipid metabolism dysfunction, which causes xanthoma, atherosclerosis and coronary heart disease. Earlier studies showed that mutations in LDLR, APOB and PCSK9 cause FH. Although more than 75% of the population in Europe has been scrutinized for FH-causing mutations, the genetic diagnosis proportion among Chinese people remains very low (less than 0.5%). The aim of this study was to perform a survey and mutation detection among the Chinese population.219 FH patients from the central south region of China were enrolled. After extracting DNA from circulating lymphocytes, we used direct DNA sequencing to screen each exon of LDLR, APOB and PCSK9. All detected variants were predicted by Mutationtaster, Polyphen-2 and SIFT to assess their effects.In total, 43 mutations were identified from 158 FH patients. Among them, 11 novel mutations were found, including seven LDLR mutations, two APOB mutations and two PCSK9 mutations. Moreover, five common mutations in LDLR were detected. We geographically marked their distributions on the map of China.The spectrum of FH-causing mutations in the Chinese population is refined and expanded. Along with future studies, our study provides the necessary data as the foundation for the characterization of the allele frequency distribution in the Chinese population. The identification of more LDLR, APOB and PCSK9 novel mutations may expand the spectrum of FH-causing mutations and contribute to the genetic diagnosis and counseling of FH patients.Copyright © 2017 Elsevier B.V. All rights reserved.
DOI:S0021-9150(17)30263-0PMID:28645073 [本文引用: 1]
Familial hypercholesterolemia (FH) is an autosomal dominant disease with widespread global prevalence that partially accounts for the high prevalence of premature coronary heart disease. Although the majority of research on FH has focused on single heterozygous LDLR mutations, there have been limited reports of double LDLR mutations on the same chromosome. The aim of this study was to gain insight into the clinical consequences of the presence of multiple mutations in the LDLR gene.DNA from two clinical homozygous FH patients and their relatives was analysed using targeted exome sequencing and DNA resequencing. Functional characterization of novel variants was performed by Western blot, flow cytometry and confocal microscopy.Proband 1 carried p.Q12X, NTDA (p.N276T and c.892delA) mutations in LDLR, and Proband 2 carried c.971delG, GSDN (p.G77S + D601N). Results showed that p.Q12X, c.892delA, and c.971delG are non-functional LDLR variants. Conversely, N276T and G77S are non-pathogenic variants. Interestingly, while D601N alone only slightly diminishes LDLR activity, its co-presence with the non pathogenic p.G77S mutation results in a more strongly pathogenic variant with LDLR activity reduced by 40%. One of the double mutants, NTDA, is as non functional as c.892delA alone. The other double mutant, GSDN, is more severe than either of the component single mutants.An early gene screening and laboratory functional verification of LDLR activity is of vital importance to enable a definite FH diagnosis. Functional verification is also necessary for prenatal and postnatal care in patients with FH.Copyright © 2017 Elsevier B.V. All rights reserved.
URL [本文引用: 2]
DOI:10.1161/ATVBAHA.116.308456URLPMID:27932355 [本文引用: 2]
Familial hypercholesterolemia (FH) is characterized by an elevated low-density lipoprotein cholesterol and increased risk of premature coronary artery disease. However, the general picture and mutational spectrum of FH in China are far from recognized, representing a missed opportunity for the investigation.A total of 8050 patients undergoing coronary angiography were enrolled. The diagnosis of clinical FH was made using Dutch Lipid Clinic Network criteria, and the information of relatives was obtained by inquiring for the probands or from their own medical records of certain clinics/hospitals. Molecular analysis of FH was performed using target exome sequencing in (low-density lipoprotein cholesterol receptor gene), (apolipoprotein B gene), and (proprotein convertase subtilisin/kexin type 9 gene). As a result, 3.5% of the patients with definite/probable FH phenotype (definite 1.0% and probable 2.5%) were identified. Women FH had fewer premature coronary artery disease (women <60, or men <55 years of age) when compared with men FH (70.6% versus 82.7%; <0.001), whereas angiographic extension of coronary artery disease was significantly increased with FH diagnosis in both men and women (<0.001). Patterns of medication use in definite/probable FH were as follows: nontreated, 20.6%; low intensity, 6.0%; moderate intensity, 68.3%; and high intensity, 5.0%. However, none of them had achieved the low-density lipoprotein cholesterol <100 mg/dL. Additionally, mutational analysis was performed in 245 definite/probable FH cases, and risk variants were identified in 115 patients, giving a detection rate of 46.9%.We showed firsthand a common identification but poor treatment of patients with FH phenotype in Chinese coronary angiography patients. Genetic data in our FH cases might contribute to update the frequency and spectrum of Chinese FH scenarios.© 2016 American Heart Association, Inc.
DOI:S0021-9150(17)30059-XURLPMID:28235710 [本文引用: 4]
Familial hypercholesterolemia (FH) is the most common and severe autosomal dominant lipid metabolism dysfunction, which causes xanthoma, atherosclerosis and coronary heart disease. Earlier studies showed that mutations in LDLR, APOB and PCSK9 cause FH. Although more than 75% of the population in Europe has been scrutinized for FH-causing mutations, the genetic diagnosis proportion among Chinese people remains very low (less than 0.5%). The aim of this study was to perform a survey and mutation detection among the Chinese population.219 FH patients from the central south region of China were enrolled. After extracting DNA from circulating lymphocytes, we used direct DNA sequencing to screen each exon of LDLR, APOB and PCSK9. All detected variants were predicted by Mutationtaster, Polyphen-2 and SIFT to assess their effects.In total, 43 mutations were identified from 158 FH patients. Among them, 11 novel mutations were found, including seven LDLR mutations, two APOB mutations and two PCSK9 mutations. Moreover, five common mutations in LDLR were detected. We geographically marked their distributions on the map of China.The spectrum of FH-causing mutations in the Chinese population is refined and expanded. Along with future studies, our study provides the necessary data as the foundation for the characterization of the allele frequency distribution in the Chinese population. The identification of more LDLR, APOB and PCSK9 novel mutations may expand the spectrum of FH-causing mutations and contribute to the genetic diagnosis and counseling of FH patients.Copyright © 2017 Elsevier B.V. All rights reserved.
DOI:10.1016/j.gene.2012.01.092URL [本文引用: 2]
Familial hypercholesterolemia (FH) is an autosomal dominant disorder. Although genetic testing is an important tool for detecting FH-causing mutations in patients, diagnostic methods for young patients with severe hypercholesterolemia are understudied. This study compares the target exome sequencing (TES) technique with the DNA resequencing array technique on young patients with severe hypercholesterolemia. A total of 20 unrelated patients (mean age 14.8 years) with total cholesterol > 10 mmol/L were included. 12 patient samples were processed by DNA resequencing array, 14 patient samples were processed by TES, and 6 patient samples were processed by both methods. Functional characterization of novel mutations was performed by flow cytometry. The mutation detection rate (MDR) of DNA resequencing array was 75%, while the MDR of TES was 100%. A total of 27 different mutations in the LDLR were identified, including 3 novel mutations and 8 mutations with previously unknown pathogenicity. Functional characterization of c. 673delA, c. 1363delC, p. Leu575Phe and p. Leu582Phe variants found that all of them are pathogenic. Additionally, 7 patients were diagnosed with Heterozygous FH (HeFH) in which lipid levels were significantly higher than common HeFH patients. This data indicates that TES is a very efficient tool for genetic diagnosis in young patients with severe hypercholesterolemia.
DOI:10.1097/HCO.0000000000000517PMID:29561319 [本文引用: 2]
This review describes the pivotal role of genetic insights and technologies in the discovery of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the rapid development of PCSK9 inhibitors - a revolutionary new class of lipid-lowering agents.PCSK9 was discovered as a the third gene implicated in familial hypercholesterolemia. Population genetics studies, enabled by technological advances, were instrumental in validating PCSK9 as a therapeutic target. Monoclonal antibodies against PCSK9 were introduced in the clinic after an unprecedently rapid development path, in which clinical trial results confirmed that these drugs robustly lower cholesterol and improve clinical outcomes regardless of disease indication or background therapy. New strategies to PCSK9 inhibition are underway and have delivered promising preliminary results, including inhibition of PCSK9 synthesis by targeting the cellular gene expression machinery and vaccination. The future will tell whether directly targeting the genome through editing techniques will ultimately enable us to virtually eliminate many of the traditional CVD risk factors.The extraordinary PCSK9 narrative highlights the opportunities offered by genetics-driven drug development and holds valuable lessons for future development programs.
DOI:10.1186/s40064-016-3763-3URL [本文引用: 3]
DOI:10.1016/j.jacl.2015.12.016PMID:27206941 [本文引用: 2]
Recent guidelines suggest that more attention should be focused on children with homozygous familial hypercholesterolemia (HoFH). China may have 3.8 million potential FH patients, but there are limited data focused on HoFH children.We systematically analyzed the characteristic phenotype and the relationship between the genotype and the phenotype in HoFH children with the unique Chinese W483X mutation in the low-density lipoprotein (LDL)-receptor gene.A systematic retrospective analysis of the lipid and cardiovascular characteristics of HoFH patients in the atherosclerosis clinic of Beijing Anzhen Hospital was performed. The W483X mutation was confirmed using DNA sequencing of the patients and their parents.Two HoFH and 9 compound heterozygous patients (mean age = 14.7 years) with 2 novel mutations, Q254X and c.1363delC, were found. In total, 81.8% of the patients were from southern China. All the patients had xanthoma, and the average TC and LDL-C levels were 16.8 and 14.4 mmol/L, respectively. Echocardiography showed that 63.6% of the patients had aortic calcification, and 54.5% had mild regurgitation of the aortic valve. The coronary flow velocity reserve had a mean value of 2.12, and the cIMT was 0.17 cm. The follow-up period was between 3 months and 8 years. Although all the patients began the lipid-lowering treatment, 2 patients died because of severe cardiovascular disease. The LDL-C levels of 6 patients were slightly decreased by approximately 21% and remained far from the target values, and the other 3 patients' LDL-C levels increased by 13%.The results suggest that younger HoFH patients with W483X mutations had a severe phenotype and should receive more aggressive treatment.Copyright © 2015 National Lipid Association. Published by Elsevier Inc. All rights reserved.
DOI:10.1016/j.ijcard.2016.01.087URL [本文引用: 3]
DOI:10.1016/j.ijcard.2016.01.087URL [本文引用: 3]
DOI:10.1017/S1047951115000591URL [本文引用: 2]
Mammalian soluble epoxide hydrolase (sEH) converts epoxides to their corresponding diols through the addition of a water molecule. sEH readily hydrolyzes lipid signaling molecules, including the epoxyeicosatrienoic acids (EETs), epoxidized lipids produced from arachidonic acid by the action of cytochrome p450s. Through its metabolism of the EETs and other lipid mediators, sEH contributes to the regulation of vascular tone, nociception, angiogenesis and the inflammatory response. Because of its central physiological role in disease states such as cardiac hypertrophy, diabetes, hypertension, and pain sEH is being investigated as a therapeutic target. This review begins with a brief introduction to sEH protein structure and function. sEH evolution and gene structure are then discussed before human small nucleotide polymorphisms and mammalian gene expression are described in the context of several disease models. The review ends with an overview of studies that have employed the sEH knockout mouse model. Copyright © 2013 Elsevier B.V. All rights reserved.
DOI:10.1016/j.ijcard.2015.07.092PMID:26298359 [本文引用: 2]
Epoxide hydrolases (EH) are ubiquitously expressed in all living organisms and in almost all organs and tissues. They are mainly subdivided into microsomal and soluble EH and catalyze the hydration of epoxides, three-membered-cyclic ethers, to their corresponding dihydrodiols. Owning to the high chemical reactivity of xenobiotic epoxides, microsomal EH is considered protective enzyme against mutagenic and carcinogenic initiation. Nevertheless, several endogenously produced epoxides of fatty acids function as important regulatory mediators. By mediating the formation of cytotoxic dihydrodiol fatty acids on the expense of cytoprotective epoxides of fatty acids, soluble EH is considered to have cytotoxic activity. Indeed, the attenuation of microsomal EH, achieved by chemical inhibitors or preexists due to specific genetic polymorphisms, is linked to the aggravation of the toxicity of xenobiotics, as well as the risk of cancer and inflammatory diseases, whereas soluble EH inhibition has been emerged as a promising intervention against several diseases, most importantly cardiovascular, lung and metabolic diseases. However, there is reportedly a significant overlap in substrate selectivity between microsomal and soluble EH. In addition, microsomal and soluble EH were found to have the same catalytic triad and identical molecular mechanism. Consequently, the physiological functions of microsomal and soluble EH are also overlapped. Thus, studying the biological effects of microsomal or soluble EH alterations needs to include the effects on both the metabolism of reactive metabolites, as well as epoxides of fatty acids. This review focuses on the multifaceted role of EH in the metabolism of xenobiotic and endogenous epoxides and the impact of EH modulations.
DOI:10.1038/srep11380URL [本文引用: 2]
URLPMID:7649083 [本文引用: 2]
Human GH (hGH) has been shown to stimulate hepatic low density lipoprotein (LDL) receptor expression in man in vivo. To further characterize this effect in vitro, we determined the expression of LDL receptors in cultured human hepatoma (HepG2) cells exposed to hGH. After incubation with hGH, stimulation of LDL receptors appeared at a concentration of 0.25 nM hGH. The presence of hGH receptors on HepG2 cells could be demonstrated by immunocytochemistry using a hGH receptor-specific monoclonal antibody. Binding studies, using 125I-labeled hGH, revealed high affinity binding with the appropriate somatogenic specificity. The LDL receptor induction was specific for hGH, as both bovine GH and recombinant human PRL were without effect. The LDL receptor stimulation occurred in parallel with increased levels of LDL receptor messenger RNA. Inclusion of dexamethasone and thyroid hormone in the incubation medium enhanced the LDL receptor stimulation by hGH. Although incubation with insulin-like growth factor-I (IGF-I) stimulated LDL receptor expression, the hGH-induced stimulation was unaltered after preincubation of cells with a monoclonal mouse anti-IGF-I antibody, suggesting that the release of IGF-I is not involved in LDL receptor stimulation by hGH. We conclude that hGH specifically induces the LDL receptor in cultured HepG2 cells at both the protein and the messenger RNA level, and that the induction is independent of IGF-I release.
DOI:10.1016/j.ijporl.2015.04.020URL [本文引用: 2]
Apolipoprotein E (apoE) is a major apolipoprotein involved in lipoprotein metabolism. It is a polymorphic protein and different isoforms are associated with variations in lipid and lipoprotein levels and thus cardiovascular risk. The isoform apoE4 is associated with an increase in LDL-cholesterol levels and thus a higher cardiovascular risk compared to apoE3. Whereas, apoE2 is associated with a mild decrease in LDL-cholesterol levels. In the presence of other risk factors, apoE2 homozygotes could develop type III hyperlipoproteinemia (familial dysbetalipoproteinemia or FD), an atherogenic disorder characterized by an accumulation of remnants of triglyceride-rich lipoproteins. Several rare APOE gene variants were reported in different types of dyslipidemias including FD, familial combined hyperlipidemia (FCH), lipoprotein glomerulopathy and bona fide autosomal dominant hypercholesterolemia (ADH). ADH is characterized by elevated LDL-cholesterol levels leading to coronary heart disease, and due to molecular alterations in three main genes: LDLR, APOB and PCSK9. The identification of the APOE-p.Leu167del variant as the causative molecular element in two different ADH families, paved the way to considering APOE as a candidate gene for ADH. Due to non mendelian interacting factors, common genetic and environmental factors and perhaps epigenetics, clinical presentation of lipid disorders associated with APOE variants often strongly overlap. More studies are needed to determine the spectrum of APOE implication in each of the diseases, notably ADH, in order to improve clinical and genetic diagnosis, prognosis and patient management. The purpose of this review is to comment on these APOE variants and on the molecular and clinical overlaps between dyslipidemias.Copyright © 2021 The Author(s). Published by Elsevier B.V. All rights reserved.
DOI:10.1007/s12010-015-1554-xURL [本文引用: 2]
Studies in ctltured fibroblasts indicate that the primary genetic abnormality in familial hypercholesterolemia involves a deficiency in a cell surface receptor for low density lipoproteins (LDL). In normal cells, binding of LDL to this receptor regulates cholesterol metabolism by suppressing cholesterol synthesis and increasing LDL degradation. In cells from heterozygotes, a 60 percent reduction in LDL receptors leads to a concentration-dependent defect in regulation, so that attainment of equal rates of cholesterol synthesis and LDL degradation in normal and heterozygous cells requires a two- to threefold higher concentration of LDL in the heterozygote. The identification of this genetic regulatory defect in fibroblasts of heterozygotes makes available an in vitro system for studying the effects of a dominant mutation on gene expression in mammalian cells.
DOI:S0735-1097(16)33132-1PMID:27417002 [本文引用: 2]
A statin-induced reduction of coronary artery disease (CAD) events and mortality has not been adequately quantified in patients with heterozygous familial hypercholesterolemia (FH).This study estimated the relative risk reduction for CAD and mortality by statins in heterozygous FH patients.The authors included all adult heterozygous FH patients, identified by the Dutch screening program for FH between 1994 and 2013, who were free of CAD at baseline. Hospital, pharmacy, and mortality records between 1995 and 2015 were linked to these patients. The primary outcome was the composite of myocardial infarction, coronary revascularization, and death from any cause. The effect of statins (time-varying) was determined using a Cox proportional hazard model, while correcting for the use of other lipid-lowering therapy, thrombocyte aggregation inhibitors, and antihypertensive and antidiabetic medication. The authors applied inverse-probability-for-treatment weighting (IPTW) to account for differences at baseline between statin users and never-users.The authors obtained medical records of 2,447 patients, of whom 888 were excluded on the basis of age <18 years or previous CAD. Simvastatin 40 mg and atorvastatin 40 mg accounted for 23.1% and 22.8% of all prescriptions, respectively. Statin users (n = 1,041) experienced 89 CAD events and 17 deaths during 11,674 person-years of follow-up versus statin never-users (n = 518), who had 22 CAD events and 9 deaths during 4,892 person-years (combined rates 8.8 vs. 5.3 per 1,000 person-years, respectively; p < 0.001). After applying IPTW and adjusting for other medications, the hazard ratio of statin use for CAD and all-cause mortality was 0.56 (95% confidence interval: 0.33 to 0.96).In patients with heterozygous FH, moderate- to high-intensity statin therapy lowered the risk for CAD and mortality by 44%. This is essential information in all cost-effectiveness studies of this disorder, such as when evaluating reimbursement of new lipid-lowering therapies.Copyright © 2016 American College of Cardiology Foundation. Published by Elsevier Inc. All rights reserved.
DOI:10.1111/ced.12644PMID:25807990 [本文引用: 2]
Xanthomas are important clinical manifestations of disordered lipid metabolism, which are mostly found in patients with familial hypercholesterolaemia (FH), an inherited disorder that is predominantly caused by mutations in the low-density lipoprotein receptor gene (LDLR). Tuberous and tendinous xanthomas with wide distribution and large size are rare; however, they may indicate the severity of FH, and tend to be found in homozygous FH. In this study, we investigated the clinical and genetic aspects of a young patient with FH presenting with multiple large masses in various locations. The lesions on the elbows and buttocks were locally excised and subsequently confirmed by biopsy to be xanthomas. Genetic analysis further confirmed that the patient was compound heterozygous for two mutations in both alleles of the LDLR gene. This rare case of compound heterozygous FH presenting with multiple large and widely distributed xanthomas provides a better understanding of FH and xanthomas. © 2015 British Association of Dermatologists.
DOI:10.1016/j.jacl.2014.11.011URL [本文引用: 2]
Proprotein convertase subtilisin/kexin9 monoclonal antibodies (PCSK9-mAb) have been studied intensively to identify their effect in lowering levels of low density lipoprotein cholesterol (LDL-C). However, the applicable target of PCSK9-mAbs remains inconclusive so far. Therefore, this first metaanalysis was carried out to clarify the therapeutic efficacy and safety of PCSK9-mAbs on the potential patients: familial hypercholesterolemia and statin-intolerant patients. All randomized controlled trials that met the search terms were retrieved in multiple databases. Efficacy outcomes included parameter changes from baseline in LDL-C and other lipid levels. Therapeutic safety were evaluated by rates of common adverse events. A total of 15 studies encompassing 4,288 patients with at least 8 weeks duration were selected. Overall, the therapeutic efficacy was achieved with significant reduction in LDLC, TC, TG, Lp(a), Apo-B versus placebo. The decline in familial hypercholesterolemia patients (-53.28%, 95% CI: -59.88 to -46.68%) was even more obvious than that in statin-intolerant patients (-34.95%, 95% CI: -41.46 to -28.45%). No obvious safety difference was found out in the rates of common and serious adverse events. To conclude, PCSK9-mAb contributes to the decreased level of LDL-C and other lipids in familial hypercholesterolemia and statin-intolerant patients with satisfactory safety and tolerability.
DOI:10.1016/S2213-8587(16)30041-9URL [本文引用: 3]
URL [本文引用: 2]
DOI:10.1016/j.ijcard.2014.06.083URL [本文引用: 2]
URL [本文引用: 2]
URL [本文引用: 2]
DOI:10.1371/journal.pone.0092703URL [本文引用: 2]
DOI:10.4103/0301-4738.118456PMID:24088637 [本文引用: 2]
We report the case of a 12-year-old male who developed corneal arcus and multiple skin lesions with a 10-year history of xanthomas. The lesions appeared over his fingers, hands, elbows, knees, buttocks and feet. Laboratory studies showed a total serum cholesterol level of 752.1 mg/dL; a triglyceride level of 96.6 mg/dL; a low-density lipoprotein cholesterol level of 661.3 mg/dL. Findings were consistent with homozygous familial hypercholesterolemia. To our knowledge, this is the first such case to be reported from China.
[本文引用: 2]
[本文引用: 2]
J Clin Lipidol,
[本文引用: 3]
DOI:10.1016/j.gene.2012.01.092URL [本文引用: 3]
DOI:10.1161/ATVBAHA.116.308456PMID:27932355 [本文引用: 2]
Familial hypercholesterolemia (FH) is characterized by an elevated low-density lipoprotein cholesterol and increased risk of premature coronary artery disease. However, the general picture and mutational spectrum of FH in China are far from recognized, representing a missed opportunity for the investigation.A total of 8050 patients undergoing coronary angiography were enrolled. The diagnosis of clinical FH was made using Dutch Lipid Clinic Network criteria, and the information of relatives was obtained by inquiring for the probands or from their own medical records of certain clinics/hospitals. Molecular analysis of FH was performed using target exome sequencing in (low-density lipoprotein cholesterol receptor gene), (apolipoprotein B gene), and (proprotein convertase subtilisin/kexin type 9 gene). As a result, 3.5% of the patients with definite/probable FH phenotype (definite 1.0% and probable 2.5%) were identified. Women FH had fewer premature coronary artery disease (women <60, or men <55 years of age) when compared with men FH (70.6% versus 82.7%; <0.001), whereas angiographic extension of coronary artery disease was significantly increased with FH diagnosis in both men and women (<0.001). Patterns of medication use in definite/probable FH were as follows: nontreated, 20.6%; low intensity, 6.0%; moderate intensity, 68.3%; and high intensity, 5.0%. However, none of them had achieved the low-density lipoprotein cholesterol <100 mg/dL. Additionally, mutational analysis was performed in 245 definite/probable FH cases, and risk variants were identified in 115 patients, giving a detection rate of 46.9%.We showed firsthand a common identification but poor treatment of patients with FH phenotype in Chinese coronary angiography patients. Genetic data in our FH cases might contribute to update the frequency and spectrum of Chinese FH scenarios.© 2016 American Heart Association, Inc.
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 3]
[本文引用: 2]
[本文引用: 3]
[本文引用: 4]
[本文引用: 4]
[本文引用: 2]
[本文引用: 2]
[本文引用: 5]
[本文引用: 4]
[本文引用: 2]
[本文引用: 3]
[本文引用: 6]
[本文引用: 3]
[本文引用: 3]
[本文引用: 2]
[本文引用: 5]
[本文引用: 2]
[本文引用: 4]
[本文引用: 2]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 4]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 3]
[本文引用: 2]
[本文引用: 3]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 3]
[本文引用: 3]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 3]
[本文引用: 3]
[本文引用: 2]
DOI:10.1016/j.atherosclerosis.2011.02.006URL [本文引用: 2]
[本文引用: 1]
[本文引用: 1]
DOI:10.1002/humu.v31:11URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
DOI:10.1016/j.numecd.2008.07.011PMID:19073363 [本文引用: 1]
Familial hypercholesterolemia (FH) is an autosomal dominant disorder of lipoprotein metabolism caused by mutations in the low-density lipoprotein receptor (LDL-R) gene, leading to elevated levels of cholesterol and an increased risk of coronary heart disease. In this article, from four homozygous FH phenotype probands we identified disease causing mutations and analyzed the relationship between genotype and phenotype.DNA sequencing identified five LDL-R point mutations in four unrelated families. We found a novel homozygous mutation (C210R), a homozygous mutation at W462X, a compound heterozygous mutation of C122Y and T383I, and a G>A intron 3 splice site homozygous mutation. The functional alteration caused by the novel C210R mutation was confirmed by FACS analysis. Four probands have high low-density lipoprotein cholesterol (LDL-C) levels, ranging from 14.65 to 27.66 mmol/L. Their heterozygous parents had relatively low levels. B-mode ultrasound supplemented by Doppler was used to examine aortic/mitral valve structural alterations and carotid intima-media thickness (ITM) in all probands. The ITM values were between 1.2 and 2.3mm, much higher than the normal value of <0.8mm.Our data demonstrated that all the probands were associated with severe hypercholesterolemia, thick carotid IMT and a low CFVR (coronary flow velocity reserve) value. The novel mutation (C120Y) is a disease causing mutation.
DOI:10.1007/s11033-008-9416-zURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.metabol.2006.12.011URL [本文引用: 2]
[本文引用: 1]
PMID:17087781 [本文引用: 1]
Familial hypercholesterolaemia (FH) is an autosomal dominant disease associated with a very high risk of coronary vascular disease. The study objective was to identify patients with FH in Taiwan and characterize novel mutations.Fifty-one patients with suspected FH living in Taiwan were screened for mutations in both the low-density lipoprotein (LDL) receptor and the apolipoprotein (apoB) genes using the multiplex polymerase chain reaction and exon-by-exon DNA sequencing technique. Functional consequences on LDL receptor activity were characterized in vitro for novel mutations and family pedigree was also analyzed.Thirteen different functional mutations in the LDL receptor gene and one mutation in the apoB gene were found in 21 patients. Among the 13 mutations in the LDL receptor gene, 10 were single-point missense mutations, one was a two-point mutation in the same allele, one was a non-sense mutation and one was a frame-shift mutation. There were three novel mutations, including two missense mutations (M510K and W512R) and one frame-shift mutation (1953 delTA mutation). The characterization of missense M510K retained 36.2% of the activity of the normal receptor. Conversely, frame-shift 1953 delTA and missense W512R led to defective proteins, with only 0-6% of normal receptor activity.The study identified 13 LDL receptor gene mutations and characterized three novel mutations causing FH in Taiwan. This facilitated a better understanding of FH among the Chinese population and may enable diagnosis of FH at the molecular level at a presymptomatic, early age.
PMID:15701167 [本文引用: 1]
Familial hypercholesterolemia is a human monogenic disease caused by population-specific mutations in the low density lipoprotein (LDL) receptor gene. Despite thirteen different mutations of the LDL receptor gene were reported from Russia prior to 2003, the whole spectrum of disease-causing gene alterations in this country is poorly known and requires further investigation provided by the current study.Forty-five patients with clinical diagnosis of FH were tested for the apolipoprotein B (apoB) mutation R3500Q by restriction fragment length analysis. After exclusion of R3500Q mutation high-sensitive fluorescent single-strand conformation polymorphism (SSCP) analysis and automatic DNA sequencing were used to search for mutations in the LDL receptor gene.We found twenty one rare sequence variations of the LDL receptor gene. Nineteen were probably pathogenic mutations, and two (P518P, T705I) were considered as neutral ones. Among the mutations likely to be pathogenic, eight were novel (c.670-671insG, C249X, c.936-940del5, c.1291-1331del41, W422X, c.1855-1856insA, D601N, C646S), and eleven (Q12X, IVS3+1G>A, c.651-653del3, E207X, c.925-931del7, C308Y, L380H, c.1302delG, IVS9+1G>A, V776M, V806I) have already been described in other populations. None of the patients had the R3500Q mutation in the apoB gene.Nineteen pathogenic mutations in the LDL receptor gene in 23 probands were identified. Two mutations c.925-931del7 and L380H are shared by St.-Petersburg population with neighbouring Finland and several other mutations with Norway, Sweden or Denmark, i.e. countries from the Baltic Sea region. Only four mutations (c.313+1G>A, c.651-653del3, C308Y and W422X) were recurrent as all those were found in two unrelated families. By this study the number of known mutations in the LDL receptor gene in St.-Petersburg area was increased nearly threefold. Analysis of all 34 low density lipoprotein receptor gene mutations found in St.-Petersburg argues against strong founder effect in Russian familial hypercholesterolemia.
DOI:10.1016/j.metabol.2005.03.012URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
PMID:11310584 [本文引用: 1]
Familial hypercholesterolemia (FH) is an autosomal dominant disorder caused by mutations in the low-density lipoprotein receptor (LDLR) gene; it is characterized by a high concentration of LDL, which frequently gives rise to tendon xanthomas and premature coronary artery disease (CAD). Individuals with heterozygous FH in China often exhibit a milder phenotype than those in other countries. The diagnosis of heterozygous FH relies on the clinical phenotype and this does not always permit unequivocal diagnosis of the disease. In the course of investigation of FH in a Chinese population sample, we found a family whose proband showed a markedly raised concentration of LDL cholesterol in plasma, and the presence of skin and tendon xanthomata. We used single-strand conformation polymorphism (SSCP) analysis to screen all the 18 exons and the exon-intron boundaries of the LDLR gene. One novel homozygous mutation, replacing T by C at nucleotide 850 in exon 6 was identified. This change substituted cysteine for arginine at codon 263 (C263R) of the LDLR. By means of mutant allele-specific amplification, we unequivocally diagnosed six heterozygotes with this novel mutation in the proband's family. forms of heterozygous FH are characterized by high serum LDL cholesterol levels, which are usually associated with premature coronary heart disease (CHD) and tendon xanthomas in early middle age, while homozygous FH individuals frequently suffer fatal CHD by their third decade (Goldstein et al. 1995). To date, more than 200 different mutations of the LDLR gene have been characterized worldwide (Hobbs et al. 1990; Hobbs et al. 1992; Varret et al. 1997). Chinese individuals with heterozygous FH are often recognized by virtue of their being parents of offspring with mutations in both alleles of the LDLR gene (Mak et al. 1998). Unlike their heterozygous parents, the Chinese homozygous FH patients are as severely affected, as are those elsewhere (Sun et al. 1994). In this article, we present an investigation of a Chinese family whose proband, with severe hypercholesterolemia, carried a novel mutation of the LDLR gene in a homozygous form, while the other six patients in the family, who carried this mutation in a heterozygous form, have a milder phenotype of FH. The main objective of our analysis was to delineate the full spectrum of mutations that underline FH in the Chinese population; our work represents a first step towards the implementation of nationwide DNA testing for this disease.
[本文引用: 1]
[本文引用: 1]
PMID:11005141 [本文引用: 1]
The aim of this study was to detect mutations in the genes coding for the low-density lipoprotein receptor and apolipoprotein B in patients of Southeast Asian origin with clinically diagnosed familial hypercholesterolemia (FH) and to relate these findings with the observed lower incidence of coronary heart disease in this part of the world. A total of 86 unrelated patients with FH were selected on clinical grounds, and complete DNA analysis of the low-density lipoprotein (LDL)-receptor and apolipoprotein B (apoB) genes by DGGE and DNA-sequencing was performed. In the majority (73%) of the cohort studied, no mutations could be detected, even after extensive analysis of the LDL-receptor and apoB genes. However, the 22 patients with a mutation had significantly more xanthomas and a higher incidence of coronary heart disease and levels of low-density lipoproteins were also significantly different. There was no correlation between the type of the mutation and lipoprotein levels or clinical signs of atherosclerosis. The fact that the majority of the FH patients studied had no detectable mutation and that this group had a significant milder phenotype, suggests the presence of a third gene in the Southeast Asian population, predominantly leading to a disorder resembling a milder form of FH. A similar, but less frequent, trait has recently been described in a number of European families.
DOI:10.1161/01.ATV.18.10.1600URL [本文引用: 1]
DOI:10.1161/01.ATV.18.2.309URL [本文引用: 1]
[本文引用: 1]
DOI:10.1161/01.ATV.14.1.85URL [本文引用: 1]
DOI:10.2174/1381612825666190228000932URL [本文引用: 1]
DOI:10.1186/s12967-018-1737-7URL [本文引用: 2]
[本文引用: 1]
?
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}