 ,, 胡渝涵,, 曹悦岩, 朱强, 黄雨果, 李茜, 张霁四川大学华西基础医学与法医学院,成都 610041
,, 胡渝涵,, 曹悦岩, 朱强, 黄雨果, 李茜, 张霁四川大学华西基础医学与法医学院,成都 610041AI-SNPs screening based on the whole genome data and research on genetic structure differences of subcontinent populations
Haoyu Wang,, Yuhan Hu,, Yueyan Cao, Qiang Zhu, Yuguo Huang, Xi Li, Ji ZhangWest China School of Basic Medical Sciences and Forensic Medicine, Sichuan University, Chengdu 610041, China通讯作者: 张霁,博士,教授,研究方向:法医物证学。E-mail:zhangj@scu.edu.cn
编委: 朱波峰
收稿日期:2021-05-26修回日期:2021-07-23
| 基金资助: |
Received:2021-05-26Revised:2021-07-23
| Fund supported: |
作者简介 About authors
王浩宇,在读硕士研究生,专业方向:法医物证学。E-mail:
胡渝涵,在读硕士研究生,专业方向:法医物证学。E-mail:

摘要
在涉及多群体样本的医学研究中,群体遗传结构差异是不容忽视的影响因素之一。利用族源信息单核苷酸多态性遗传标记(ancestry-informative single nucleotide polymorphism, AI-SNP),通过分析群体遗传成分、推断个体遗传背景并对群体样本进行预筛选,可以有效降低群体遗传结构差异对医学研究影响。鉴于已发表的研究多为解析大陆间、大陆次级区域间的群体遗传结构差异,本研究拟基于千人基因组计划(GRCh37.p13)中东亚五群体:日本东京群体(Japanese in Tokyo, JPT)、北京汉族(Han Chinese in Beijing, CHB)、南方汉族(Southern Han Chinese, CHS)、西双版纳傣族(Chinese Dai in Xishuangbanna, CDX)、越南京族(Kinh in Ho Chi Minh City, KHV)的数据,以FST值为标准筛选AI-SNP并分析大陆次级区域内群体遗传结构差异。结果表明,研究涉及的东亚群体可分为三簇:JPT、CHB和CHS、CDX和KHV。利用AI-SNP可成功解析个体的遗传背景,而群体代表性遗传成分占比超过80%的个体具有良好的群体代表性。本研究表明,基于FST值筛选一组AI-SNP用于核验样本遗传背景、筛选群体代表性样本的方法在降低大陆次级区域内群体遗传结构差异对群体相关医学研究的影响中具有实际应用价值。
关键词:
Abstract
The genetic structure differences in population is one of the key elements in medical research involving multi-population samples. A set of ancestry-informative single nucleotide polymorphisms (AI-SNPs) can be utilized to analyze genetic component of a population, infer ancestral origin of individuals and pre-filter samples to reduce the impact of population genetic structure differences on medical research. However, most of the published studies were focused on revealing the differences between populations of continents or regions of a continent. In this paper, AI-SNPs were screened by calculating FST value in each pair of five East Asian populations: Japanese in Tokyo (JPT), Han Chinese in Beijing (CHB), Southern Han Chinese (CHS), Chinese Dai in Xishuangbanna (CDX) and Kinh in Ho Chi Minh City (KHV) in the 1000 Genomes Project phase 3 (GRCh37.p13) to analyze differences in subcontinent populations. The results demonstrate that the five East Asian populations in our study were assigned to three clusters: JPT, CHB and CHS, CDX and KHV. A set of AI-SNPs can be used for analysis of individual genetic composition and selection of representative individuals. Individuals with over 80% population representative genetic components have good representativeness of a population. This paper demonstrated the practical value of the method, which was performed to verify the ancestral composition and select representative samples with a panel of screened AI-SNPs by FST value, thereby reducing the influence of genetic structure differences in subcontinent populations on population-related medical research.
Keywords:
PDF (1108KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
王浩宇, 胡渝涵, 曹悦岩, 朱强, 黄雨果, 李茜, 张霁. 基于全基因组数据的AI-SNPs筛选及大陆次级区域内群体遗传结构差异研究. 遗传[J], 2021, 43(10): 938-948 doi:10.16288/j.yczz.21-185
Haoyu Wang.
不同群体间遗传结构的差异受到种群迁移、隔离、混合等人口学因素,以及基因突变、重组、自然选择、随机遗传漂变等遗传学因素影响[1,2]。涉及群体的医学领域研究中,往往需考虑由群体遗传结构差异带来的影响。如关联分析中,需排除与目标基因无关、由群体间结构差异导致的等位基因频率差异,才能提供标记与疾病间的真实关联[3,4]。而明确药物反应相关基因变异[5]在群体中的差异则有利于针对不同人群进行靶向药物的筛选并提供精准个性化用药建议。此外,族源信息遗传标记也被法医遗传学家用于生物样本的生物地理起源推断,并用于案件侦破[6]。
在排除群体结构差异对医学研究的影响时,需对纳入研究的个体和生物样本进行遗传背景分析以核验声明血统和实际血统的一致性,并选择具有群体代表性的样本进行后续研究。常用的遗传背景分析工具包括基因芯片[7]、全基因组测序[8]和使用族源信息遗传标记(ancestry informative marker, AIM)[9]。尽管基因组测序可得到最精确的分析结果,但其数据分析量巨大且成本较高。在当前大数据时代下,诸如国际基因组样本资源库(The International Genome Sample Resource, IGSR)[10]中千人基因组计划(1000 Genomes Project)[11]、人类基因组多样性计划(Human Genome Diversity Project)等数据库提供了大量不同人群的基因组参考数据。依托于公开数据库的大规模数据,以AIM为基础的族源分析可解析个体遗传背景,并作为应用基因芯片或全基因组测序前进行群体代表性样本预筛选的有效手段[12]。
分析个体遗传背景常用的方法包括主成分分析(principal component analysis, PCA)[13]、基因组控制(genomic control)[14]及结构化关联(structured association)[15]等。PCA分析是校正全基因组关联研究中群体分层的标准方法,但对如东亚群体这类遗传结构复杂的群体敏感性较差[16]。STRUCTURE[17]、ADMIXTURE[18]等结构化关联方法可依据族源成分和等位基因频率提供个体族源的最大似然估计,STRUCTURE还提供了基于相关等位基因频率的混合祖先模型用于复杂遗传结构群体的分析。同时,预筛选仅分析一组AIM,避免了结构化关联方法难以计算大型数据集的缺点[19],故结构化关联方法可在样本预筛选中发挥关键作用。
族源推断分析最初多以区分大陆群体为目标[20]。近来也有不少研究者针对大陆内特定区域群体的区分开发了多类次级体系。以亚洲为例,主要包括亚洲内次级区域群体[21]、大陆次级区域内群体与全球其他群体区分[22]、亚洲内次级区域群体间的区分[23]和国家内民族的区分[24],而大陆次级区域内群体间区分的相关研究则相对较少[25]。由于大陆次级区域内群体间遗传结构的相似性,以及人口迁移、通婚带来的基因流动等因素,此类区分最为困难,但也是最为必要的。
本研究拟以FST值大小为标准,从千人基因组计划东亚五群体的数据中筛选一组AIM对东亚五个群体进行群体结构分析,从各个群体中找到具有群体代表性的个体。并以结果评估使用AIM方法对遗传结构复杂群体中个体遗传背景的解析能力,为其实际应用于核实样本的声明血统和实际血统、准确排除群体遗传结构对群体相关医学研究的影响提供理论依据和方法参考。
1 材料与方法
1.1 研究对象
本研究使用的东亚五个群体共504个无关个体均来自千人基因组计划第三阶段(GRCh37.p13)数据库(http://www.1000genomes.org)[11],包括104个日本东京(Japanese in Tokyo, JPT)个体、103个中国北京汉族(Han Chinese in Beijing, CHB)个体、105个中国南方汉族(Southern Han Chinese, CHS)个体、93个中国西双版纳傣族(Chinese Dai in Xishuangbanna, CDX)个体和99个越南胡志明市京族(Kinh in Ho Chi Minh City, KHV)个体。1.2 位点筛选
基于千人基因组数据库(GRCh37.p13)的整体数据,使用VCFtools[26]筛选1~22号常染色体上最小等位基因频率大于0.01、P>0.05阈值下满足Hardy- Weinberg平衡的二等位基因SNP。按Weir和Cockerham等[27]的方法计算东亚五个群体两两之间,即10个群体对中所有保留SNP的FST值。本研究保留FST> 0.05的SNP,并使用VCFtools进行同染色体上的连锁不平衡计算。目前在族源推断体系中加入连锁不平衡位点是否会对体系区分具体群体的效能产生影响尚无定论,但研究者们在进行AIM筛选时会避免使用强连锁不平衡的基因座[21]。此外,STRUCTURE软件也建议在体系中尽可能只使用弱连锁不平衡的位点[28]。因此本研究进行连锁不平衡计算时根据前人经验将阈值设置为r2>0.2,并将检测窗口设置为50 Mb。当一个位点与任意位点满足r2>0.2时,将它们分为同一连锁不平衡组,否则分至弱连锁不平衡组。1.3 数据集构建
基于前述分组结果,10个群体对分别建立数据集。各个群体对的每个连锁不平衡组中仅保留FST值最高的SNP,将连锁不平衡组中筛选出来的SNP与该弱连锁不平衡组合并后确定最终的数据集A1~A10。各数据集分别从FST值最高的10个SNP开始,使用Snipper在线分析应用套件进行分析(后简称为Snipper分析)。自该体系开始,每次按FST值大小逐步顺序增加10个SNP并进行Snipper分析。为了保证结果的稳定性,此步骤将持续到连续三组体系(如分别由60、70、80个SNP组成的体系)均能将两个群体的个体均正确分配至原所属群体,也即分配正确率达到100%时停止。经STRUCTURE分析和PCA分析验证后,认为该三组体系中的第一组(上述例子中由60个SNP组成的体系)所包含的SNP数是完全区分该群体对所需的最少SNP数。基于此结果,本研究筛选了包含尽可能多SNP(975个)的数据集B分析东亚五个群体的遗传结构。在筛选数据集B时,综合考虑了SNP的如下信息:在10个数据集中出现的次数、在各数据集中对应的FST值大小、FST值在该数据集中的排序、是否涉及较难区分的群体(数据集内SNP数目较少或SNP的FST值普遍较低)等因素。
依据数据集B的STRUCTURE分析结果,筛选群体代表性遗传成分占个体总遗传成分分别达到70%~80% (C7)、80%~90% (C8)和90% (C9)以上的个体作为数据集C,各数据集内群体则按照群体编号(如数据集C7中JPT编号为JPT7)。对数据集C进行STRUCTURE分析和PCA分析,验证筛选群体代表性个体的可靠性、评估群体代表性遗传成分占比对判断群体代表性个体的影响。
1.4 群体遗传结构分析
使用STRUCTURE v2.3.4[17]基于相关等位基因频率的混合祖先模型对每个数据集进行群体基因结构分析,数据集A1~A10设置K=2,数据集B、C设置K=2~7,均运行10次。利用STRUCTURE HARVESTER[29]计算最佳K值,CLUMPP v.1.1.2[30]和Distruct v.1.1[31]用于构建结果图。Python脚本用于基于个体基因型的PCA分析和结果图构建。Snipper 2.5在线贝叶斯二分类分析应用套件(2 结果与分析
2.1 数据集A、B中SNP概况
数据集A1~A10中SNP的FST值分布情况见表1。FST值最高的20个SNP分别来自A3 (JPT-CDX,12个)、A4(JPT-KHV,3个)、A2(JPT-CHS,3个)和A6(CHB-CDX,2个);而FST值最小的20个SNP均来自A5 (CHB-CHS)。除A3 (JPT-CDX)、A4 (JPT-KHV)中SNP的FST值主要分布于0.15~0.25外,其余数据集内绝大多数SNP的FST值均小于0.15,其中A5 (CHB-CHS)所有SNP的FST值均小于0.15。Table 1
表1
表1数据集A中SNP的FST值分布情况
Table 1
| 数据集 | 最大FST值 | 最小FST值 | 总位点数 | FST≥0.25位点数 | 0.25>FST≥0.15位点数 | 0.15>FST≥0.05位点数 |
|---|---|---|---|---|---|---|
| A1(JPT-CHB) | 0.266925 | 0.095475 | 591 | 1 | 19 | 571 |
| A2(JPT-CHS) | 0.407479 | 0.111107 | 598 | 10 | 87 | 501 |
| A3(JPT-CDX) | 0.788409 | 0.16243 | 630 | 46 | 584 | 0 |
| A4(JPT-KHV) | 0.583637 | 0.141417 | 623 | 19 | 435 | 169 |
| A5(CHB-CHS) | 0.146048 | 0.05001 | 723 | 0 | 0 | 723 |
| A6(CHB-CDX) | 0.517659 | 0.109475 | 563 | 9 | 84 | 470 |
| A7(CHB-KHV) | 0.25611 | 0.087147 | 670 | 1 | 18 | 651 |
| A8(CHS-CDX) | 0.310909 | 0.081595 | 787 | 3 | 18 | 766 |
| A9(CHS-KHV) | 0.192399 | 0.069052 | 631 | 0 | 7 | 624 |
| A10(CDX-KHV) | 0.256551 | 0.067479 | 461 | 1 | 6 | 454 |
新窗口打开|下载CSV
数据集B中975个SNP在染色体上的分布情况如图1所示,整体分布较为均匀。来自1号染色体和6号染色体的SNP最多,分别为109个和95个,而来自22号染色体的SNP最少,为12个。此外,本研究也统计了此975个SNP在10个群体对中出现的情况,结果如图2所示。975个SNP中,大多数SNP只在一个(470/975)、两个(296/975)或三个(132/975)群体对中出现,只有极少数SNP在五个及以上(25/975)群体对中出现。其中rs11850206和rs28558239在除了CHS-KHV以外的九个群体对中均有出现,rs28498529则在除了JPT-CHB、CHS-KHV、CDX-KHV以外的七个群体对中出现。此三个SNP均来自于14号染色体。
图1
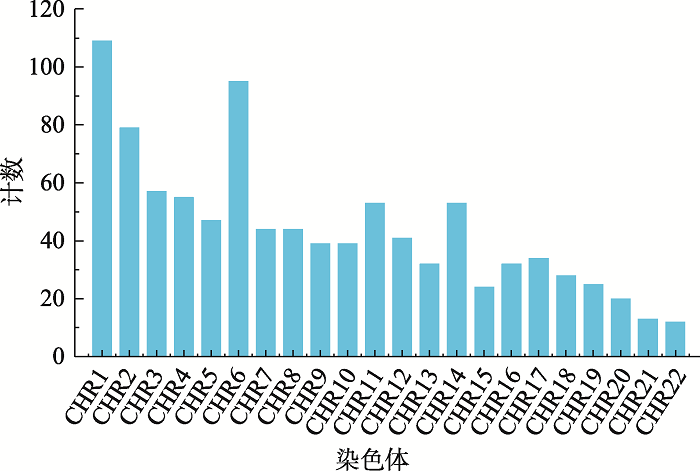 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1数据集B中SNP在染色体上的分布情况
Fig. 1The chromosome distribution of SNPs in dataset B
图2
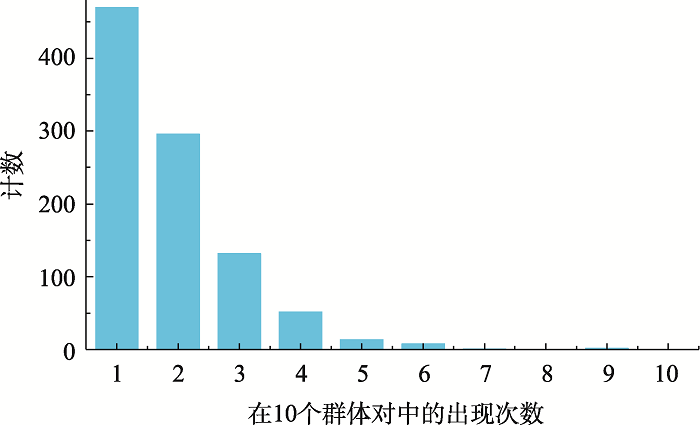 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2数据集B中SNP在10个群体对中出现次数的分布情况
Fig. 2The distribution of SNPs in dataset B in ten paired population
此外,本研究将数据集B与部分此前研究东亚群体遗传结构差异的文献[21,23,25]所使用的SNP进行了比较,发现数据集B未包含此三文献中报道的任一SNP。
2.2 东亚五群体的遗传结构差异分析
对数据集A1~A10进行Snipper交叉验证分析,测试集分配完全正确所需最少SNP数结果见表2。群体对中个体祖先分配完全正确所需的最少位点数可反映出群体两两之间遗传关系的远近。结果表明JPT-CDX、JPT-KHV群体对最易区分,而CHB-CHS、CHS-KHV、CDX-KHV较难区分。各群体对中的群体与STRUCTURE计算得到的聚类高度符合,而PCA分析中各个群体对均能在使用最少位点数时分别聚类且彼此分离(结果未列出)。Table 2
表2
表2数据集A中两两群体完全区分所需最少SNP数
Table 2
| 子数据集 | A1 | A2 | A3 | A4 | A5 | A6 | A7 | A8 | A9 | A10 |
|---|---|---|---|---|---|---|---|---|---|---|
| SNP数目 | 40 | 110 | 30 | 40 | 150 | 50 | 90 | 100 | 280 | 230 |
新窗口打开|下载CSV
使用数据集B对东亚五群体进行STRUCTURE分析的结果如图3所示。K值设置为2~7,STRUCTURE HARVESTER计算得到的最佳K值为3。各个K值下JPT均表现出与其余群体不同的遗传成分。在最佳K值时,各群体均表现为混合遗传成分,975 SNPs可将东亚五群体分为三簇:JPT一簇、CHB和CHS一簇、CDX和KHV一簇,其中CHB和CHS还可依据遗传成分的比例区分。自K=4开始,CDX和KHV也表现出主要遗传成分的差异,这一差异在K=5时更加显著。而自K=6开始,各群体混合遗传成分中的主要遗传成分各不相同,即主要遗传成分可与STRUCTURE计算得到的聚类匹配,可据此将五个群体分为五簇。
使用数据集B对东亚五群体进行PCA分析的结果如图4所示。前三个主成分分别占总方差的3.21%、2.12%、1.36%。JPT、CHB、CDX群体的个体紧密聚集,而CHS、KHV群体的聚类较分散。整体上,JPT、CHB、CHS之间较为接近,其可与互相接近的CDX、KHV区分。PC1维度可进一步将JPT与CHB、CHS区分,其中CHB和CHS个体相互重叠,表明二者的遗传关系十分接近(图4),而PC3维度可将CDX和KHV区分(图4B)。
图3
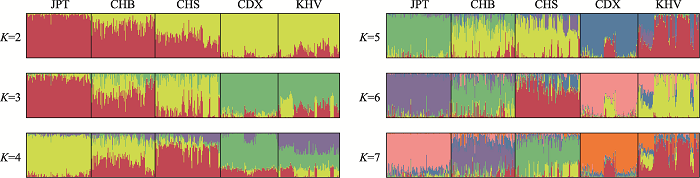 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3975 SNPs (数据集B)的东亚五群体STRUCTURE分析结果
Fig. 3The STRUCTURE analysis of 975 SNPs (dataset B) for five East Asian populations
图4
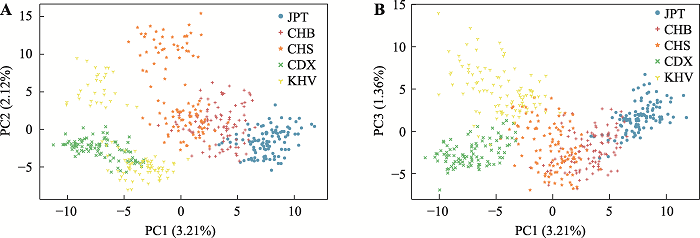 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4975 SNPs (数据集B)的东亚五群体PCA分析结果
各颜色代表群体:JPT(蓝色),CHB(红色),CHS(橙色),CDX(绿色),KHV(黄色)。A:975 SNPs的东亚五群体PCA分析(PC1-PC2),PC1=3.21%,PC2=2.12%;B:975 SNPs的东亚五群体PCA分析(PC1-PC3),PC1=3.21%,PC3=1.36%。
Fig. 4The PCA analysis of 975 SNPs (dataset B) for five East Asian populations
2.3 东亚五群体代表性个体筛选及分析
以数据集B进行STRUCTURE分析时K=6的结果为参考,按1.3的方法判断五个群体的群体代表性遗传成分并构建数据集C (表3)。数据集C中共包括317个个体,JPT中群体代表性遗传成分占总体遗传成分超过70%的个体最多,达93%,其次是CDX和KHV,分别为78%和59%,CHB和CHS均未超过50%。JPT、CDX、KHV的筛选个体中大部分群体代表性遗传成分占比超过80%,CHB和CHS只有较少个体的群体代表性遗传成分占比超过90%。Table 3
表3
表3数据集C中C7、C8、C9组个体数目
Table 3
| 组别 | JPT | CHB | CHS | CDX | KHV |
|---|---|---|---|---|---|
| C7 | 18 | 20 | 13 | 9 | 6 |
| C8 | 41 | 20 | 21 | 20 | 11 |
| C9 | 38 | 6 | 9 | 44 | 41 |
| 合计/总样本数 | 97/104 | 46/103 | 43/105 | 73/93 | 58/99 |
新窗口打开|下载CSV
使用数据集B的975个SNP对筛选个体进行STRUCTURE分析的结果如图5所示。在各个K值下,筛选个体均表现为混合遗传成分。计算得到的最佳K值为4,此时筛选出的个体可被分为四簇:JPT一簇、CHB和CHS一簇、CDX一簇、KHV一簇。自K=5开始,317个个体可被分为五簇,各簇几乎都完全由其主要遗传成分组成,且其比例随着群体代表性遗传成分占比的增加而增加,但占比达到80%后趋于稳定。STRUCTURE的结果表明体系能够很好地区分筛选出的个体,即筛选个体能有效代表其所属群体。此外,群体代表性遗传成分占比更高的个体具有更强的群体代表性。
图5
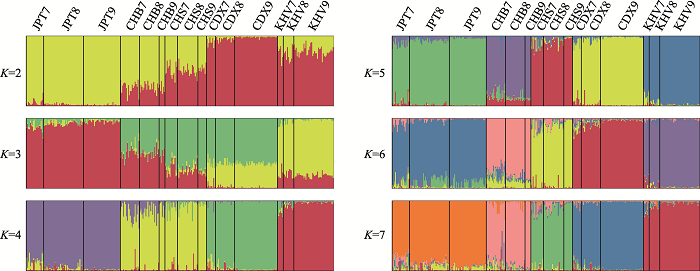 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5975 SNPs的数据集C STRUCTURE分析结果
Fig. 5The STRUCTURE analysis of 975 SNPs for dataset C
将数据集C7、C8、C9在前述PCA分析中分别高亮表示的结果如图6所示。在全部个体中,筛选个体之间区分度更高,并随着个体的群体代表性遗传成分增加而增强。数据集C7 (图6A)和C8 (图6B)中的五个群体聚类为四簇,数据集C7中仅JPT和CHB、CHB和CHS的个体仍有少部分重叠,数据集C8中仅有个别CHB、CHS的个体重叠。群体代表性遗传成分增加至90%以上后(图6C)五个群体可分别单独聚类。
图6
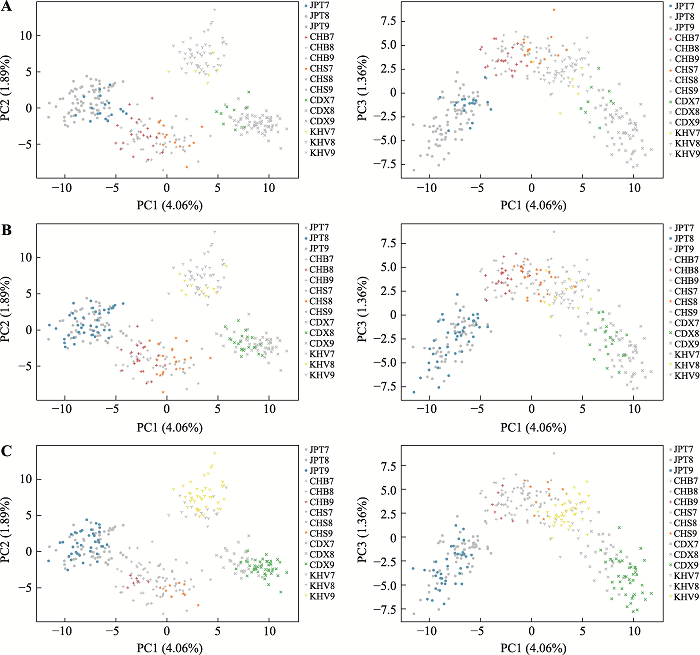 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6975 SNPs的数据集C PCA分析结果
数据集C7、C8、C9中个体分别依次标记为彩色,各数据集以外的个体标记为灰色。各颜色代表群体:JPT(蓝色),CHB(红色),CHS(橙色),CDX(绿色),KHV(黄色)。前三个主成分分别为:PC1=3.21%,PC2=2.12%,PC3=1.36%。A:标记数据集C7;B:标记数据集C8;C:标记数据集C9。
Fig. 6The PCA analysis of 975 SNPs for dataset C
依据上述STRUCTURE分析和PCA分析结果,本研究认为群体代表性遗传成分超过个体总遗传成分80%的个体具有很好的群体代表性,可用于排除群体结构对医学研究的影响。
3 讨论
涉及群体的医学研究中,群体遗传结构的差异可影响结果的正确性和准确性,进行研究时需排除这种影响。而明确采集的样本能否真正代表群体、反映群体遗传结构则是准确排除这种影响的关键。因此,对采集的样本进行遗传结构分析、判断个体声明血统和实际血统的吻合度、筛选群体代表性个体对于获取正确、准确的研究结果十分必要。一般而言,研究者们多直接在研究过程中对样本的群体遗传结构进行质控。此方法在有较少特定目标基因片段的研究[32]中十分合理且高效。然而,对于目标基因片段较多,或应用基因芯片或全基因组测序进行大规模基因筛查的研究[33],不合格的样本可能会导致测序成本的损耗。近年来,公开的多群体全基因组数据库为研究者们提供了新的思路:通过对大量数据进行分析、按照一定标准(如本研究所使用的FST值)进行筛选,找到一组可以反映特定群体之间遗传结构差异、区分群体来源的AIM,将其作为测序前对群体样本进行预筛选的手段。
本研究使用FST值作为筛选AI-SNP的标准。Wright[34]提出的FST值是最常用于表征群体间遗传分化程度的指标之一[27],其也可应用于控制遗传结构对关联分析的影响[35]。一组高FST值的AIM是进行群体遗传结构和遗传关系分析的有力工具。基于FST值筛选的SNP进行Snipper分析、STRUCTURE分析和PCA分析的结果揭示了东亚群体中的亚结构。结果表明,虽然东亚五个群体两两之间遗传结构复杂,遗传分化程度并不显著,但仍可使用一组包含较多AIM的体系加以解析。
STRUCTURE分析可计算各个聚类中每个个体的遗传成分比例。当定义的群体与其计算得到的聚类十分匹配(或相似)时,各聚类中的血统比例可看作群体的血统比例[36]。此时,STRUCTURE聚类对应的遗传成分在整个群体的总体成分中占比最大,在每个个体中稳定存在,且与其他群体无关,这种成分可看作该群体的群体代表性遗传成分。高群体代表性遗传成分的个体遗传背景相对单一,可作为该群体一种较固定的遗传背景模式。同时,本研究中具有这类遗传背景模式的个体出现频率也较高,具有一定的群体代表性。综上,本研究设定此类个体作为潜在的群体代表性样本,按群体代表性遗传成分的占比设定了三个阈值:70%、80%、90%,并筛选出相应个体进行STRUCTURE分析和PCA分析验证。PCA分析是目前最常用于校正研究中群体分层的方法[13],可用于验证基于STRUCTURE筛选的群体代表性个体是否可靠,同时评估并确定筛选标准。结果表明筛选的个体具有群体代表性,群体代表性遗传成分超过个体总遗传成分80%可作为筛选群体代表性个体的标准。
需要注意的是,筛选AIM、分析群体遗传结构以及筛选群体代表性个体依赖于实际群体样本的组成。本研究的样本来自被广泛应用于各类研究的千人基因组数据库,分析这些群体、筛选具有群体代表性的个体可提供更大的实际应用价值。而为了弥补在大陆次级区域内AIM分析群体间遗传结构差异研究的缺失,同时证明使用AIM核验样本血统的实际应用可行性,本研究选取遗传结构非常复杂的东亚群体作为研究对象。在分析时,尽可能使用更多的AIM以得到更准确的群体结构信息,以夯实后续筛选群体代表性个体的数据基础。与既往区分全球群体的研究[20]相比,本研究所使用的AIM数量更多,但与同样对大陆次级区域内(欧洲)人口亚结构进行的研究[7]相比,本研究所使用AIM的数量则要更少。研究结果表明,即使是遗传背景高度混杂的多个群体,也可使用一组AIM解析群体遗传结构并成功筛选出具有群体代表性的个体,这充分说明了本研究方法的可行性,也证明了其应用于各类涉及群体的医学研究中以排除群体结构对医学研究影响的实际价值。
如上所述,此类研究的结论高度依赖于实际群体样本的组成。本研究证明了基于公开数据库中东亚五群体数据筛选的一组AI-SNP能在理论上解析遗传结构复杂的群体间遗传结构的差异,并成功依据个体血统差异筛选出群体代表性个体。然而,受到众多的族群种类、庞大的人口基数,以及复杂的人口流动等因素的影响,东亚地区实际的群体遗传结构极端复杂。因此,使用更多不同来源的族群个体真实样本对研究东亚群体间遗传结构的差异是十分迫切且必要的。对于本研究中筛选出的此组AI-SNP,后续将构建体系并进一步使用来源于各个群体的真实样本进行验证。此外,今后的研究也将基于该体系尽可能补充更多不同群体的样本,以进一步将研究结果扩大化,使其能真正在实际应用中发挥价值。
综上所述,本研究使用FST值筛选的一组AI-SNP对遗传结构复杂的东亚五群体进行了遗传结构分析,基于STRUCTURE的结果成功从各个群体中筛选了具有潜在群体代表性的个体。经STRUCTURE分析和PCA分析的验证,群体代表性遗传成分占个体总遗传成分超过80%的个体具备良好的群体代表性。本研究的结果表明,使用一组筛选的AIM可对研究群体中个体的遗传结构进行解析,可核实样本的声明血统和实际血统的吻合度并成功筛选具有群体代表性的个体,这一方法在排除群体遗传结构差异对医学研究的影响时具备实际应用价值。
(责任编委: 朱波峰)
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 3]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}