 ,1, 徐涛,2, 王万鹏1, 张庆祝,1,3, 解莉楠,1,4
,1, 徐涛,2, 王万鹏1, 张庆祝,1,3, 解莉楠,1,4Role of epigenetic modifications in the development of crops essential traits
Ya'nan Wang,1, Tao Xu,2, Wanpeng Wang1, Qingzhu Zhang,1,3, Li'nan Xie,1,4通讯作者: 张庆祝,博士,教授,研究方向:植物表观遗传学。E-mail:qingzhu.zhang@nefu.edu.cn;解莉楠,博士,副教授,研究方向:植物抗逆生物学、植物表观遗传学。E-mail:linanxie@nefu.edu.cn
编委: 储成才
收稿日期:2021-05-11修回日期:2021-07-14
| 基金资助: |
Received:2021-05-11Revised:2021-07-14
| Fund supported: |
作者简介 About authors
王雅楠,本科,专业方向:生物科学。E-mail:
徐涛,博士,编辑,研究方向:植物遗传育种。E-mail:
王雅楠和徐涛并列第一作者。

摘要
表观遗传修饰是指染色体DNA和组蛋白上的化学修饰,主要包括DNA甲基化、组蛋白修饰和非编码RNA。在不改变DNA序列的情况下,这些修饰可以通过改变染色质状态来影响遗传信息的表达,并具有可遗传性,对植物的生长发育具有重要的调控作用。当特定的表观遗传修饰发生改变时,农作物可以获得优异的表型、更强的环境适应性,因此人为改变表观遗传修饰有望获得更适于农业生产的优质种质资源。本文综述了植物表观遗传修饰的主要类型及其在作物重要性状的形成和环境胁迫响应中的相关研究进展,总结了表观遗传学应用于作物改良必须解决的主要问题,以期在表观遗传修饰层面为作物的育种改良提供新思路。
关键词:
Abstract
Epigenetic modification refers to the chemical modifications of chromosomal DNA and histones, mainly including DNA methylation, histone modifications and non-coding RNAs. Without altering the DNA sequence, these heritable modifications can affect gene expression profiles by changing the chromatin state and play an important role in regulating the growth and development of plants. When the specific epigenetic modifications are changed, crops can obtain excellent phenotypes and stronger environmental adaptability. Therefore, artificially changing the epigenetic modifications are expected to obtain high-quality germplasm resources more suitable for agricultural production. In this review, we summarize the main types of plant epigenetic modifications, highlight the research progresses of functional plant epigenetic modifications on the important traits and responses to environmental stress, and identify the main problems that need be solved in the application of epigenetics in crop improvement, thereby providing new insights for the functional epigenetic modifications on crop breeding and improvement.
Keywords:
PDF (811KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
王雅楠, 徐涛, 王万鹏, 张庆祝, 解莉楠. 表观遗传修饰在作物重要性状形成中的作用. 遗传[J], 2021, 43(9): 858-879 doi:10.16288/j.yczz.21-170
Ya'nan Wang.
在真核生物的遗传系统中,除了DNA作为主要的遗传物质外,染色体DNA和组蛋白上的表观遗传修饰也携带着重要的遗传信息。表观遗传修饰主要包括DNA甲基化(DNA methylation)、组蛋白修饰(histone modification)和非编码RNA (non-coding RNA, ncRNAs)。这些修饰在不改变个体DNA序列的情况下使基因表达发生变化,生物体获得可遗传的表型变化[1]。目前用于研究植物表观遗传修饰基本规律的材料为模式植物拟南芥(Arabidopsis thaliana),研究相对深入的表观遗传修饰包括:DNA胞嘧啶上的甲基化修饰和组蛋白上的甲基化、乙酰化等修饰。前者大多与基因表达负相关;后者通过影响染色质状态诱导或抑制基因的表达,在转录水平上对基因表达进行调控。表观遗传修饰改变引起的表型变化既是个体正常发育程序的一部分,同时也受到环境影响[2],生物或非生物因子的刺激都可能引起表观遗传修饰的改变,进而使表型发生变化。不同类型表观遗传修饰的产生和维持需要依靠特定的酶来完成。同时,各修饰间存在密切的联系,调控机制复杂且受到环境影响,这也使得表观遗传修饰能够对植物基因的表达进行较为精细的调控,使植物体对环境胁迫做出一定反应,并可将这种反应在一定程度上遗传给后代。本文综述了表观遗传修饰在植物生长发育中的重要作用,同时对表观遗传修饰参与植物重要性状的形成及响应环境胁迫的机制进行了探讨,为进一步研究表观遗传修饰在作物改良育种中的作用提供了有力的支持。
1 植物表观遗传修饰的主要类型
DNA甲基化修饰是目前研究的比较清楚的表观遗传修饰之一,植物DNA甲基化主要发生在胞嘧啶第5位碳原子上,包括CG、CHG和CHH (H为碱基A、T或C) 3种不同类型[3]。植物DNA甲基化涉及从头合成和维持,其中RNA介导的DNA甲基化途径(RNA-directed DNA methylation, RdDM)完成DNA甲基化的从头合成。特定的甲基化酶维持不同类型的DNA甲基化,例如甲基转移酶MET1 (METHYLTRANSFERASE 1)负责维持CG甲基化,染色质甲基化酶CMT3 (CHROMOMETHY-LASE 3)是维持CHG甲基化所必需的,结构域重排甲基化酶DRM2 (DOMAINS REARRANGED METHYLASE 2)主要完成CHH位点甲基化的维持。其中CHG和CHH 是植物特有的甲基化类型,除了CMT3和DRM2的作用外,CMT2 (CHROMOMETHYLASE 2)也起到维持DNA甲基化的作用[4,5]。植物全基因组甲基化水平处于DNA甲基化与主动去甲基化(active demethylation)的动态调控中。在拟南芥DNA甲基化和主动去甲基化的调控系统中,DNA去甲基化酶基因ROS1启动子中具有DNA甲基化监测序列(methylation monitoring sequence, MEMS),RdDM途径使全基因组甲基化水平升高,包括MEMS,此时高甲基化水平的MEMS可以激活ROS1表达;DNA去甲基化酶ROS1则开始行使去甲基化功能,降低全基因组的DNA甲基化水平,其中也包括MEMS甲基化水平。这种DNA甲基化和主动去甲基化的相互协调机制能够防止DNA甲基化水平过高,从而实现动态平衡(图1)[6]。真核生物DNA甲基化修饰可以通过改变DNA构象、染色质结构来调控基因的表达,同时还可以改变DNA和蛋白质的相互作用方式。在植物中,DNA甲基化多发生在转座子(transposable element, TEs)和DNA重复序列上;而在动物中,DNA甲基化则集中发生在基因的启动子区,主要表现为抑制下游基因的表达[7]。拟南芥FWA (FLOWERING WAGENINGEN)编码开花抑制因子,当基因座上游两个重复序列被低甲基化修饰时,FWA基因表达,拟南芥开花受到抑制,表现为晚花(图2)[8]。
各类转座子和反转座子(retrotransposon)在大多数真核生物的基因组中都有分布,但转座子含量在不同物种中差异很大,例如水稻(Oryza sativa)基因组中转座子含量为35%[9],玉米(Zea mays)中可以接近85%[10]。转座子转座活性太高会造成基因组的不稳定,可能会对基因组造成损害[11]。这些具有活性、能够干扰正常DNA序列的转座子经常受甲基化作用而失去转座活性,特别是CHG和CHH的甲基化[12]。因此,DNA甲基化对于稳定基因组起到重要作用。
RdDM活性与常染色质中转座子的转录沉默相关,含有TEs的基因更容易受到RdDM途径的调节,其表达也可能随之受到影响[13]。环境胁迫常引起反转座子在基因组中的移动,并产生可稳定遗传的重排[14]。虽然过于活跃的转座子会造成基因组的不稳定,但是有活性的转座子和基因组的不稳定性在某种程度上对于植物体是有益的。例如,DNA甲基化引导的基因内转座子的插入有时会产生新基因,从而丰富植物表型和基因型的多态性[15,16],被环境胁迫激活的转座子也会帮助植物适应环境,在植物的生存、进化中起到积极作用。Ito等[17]在拟南芥中发现一个被热激活的反转座子ONSEN,当插入到脱落酸响应基因中后引起突变,表现为脱落酸不敏感,使植物体获得了更好的胁迫耐受性。相对于DNA甲基化在基因启动子区和转座子区的作用,转录区域的DNA甲基化修饰与基因表达的相关性不明显,但仍具有一定的规律。例如CG背景下转录区域的DNA甲基化水平在很多植物中与基因表达呈正相关,包括拟南芥[18]、水稻[19]、大豆(Glycine max)[20]等植物;CHG和CHH背景下转录区域DNA甲基化水平在番茄(Lycopersicon esculentum)[21]、菜豆(Phaseolus vulgaris)[22]等植物中与基因表达呈负相关[5]。
除DNA上发生的修饰外,包裹DNA的组蛋白也存在丰富的化学修饰。常见的组蛋白修饰包括组蛋白甲基化(methylation)、乙酰化(acetylation)、泛素化(ubiquitination)、磷酸化(phosphorylation)和糖基化(glycosylation)等[23]。组蛋白甲基化发生于组蛋白H3、H4的氨基酸侧链和氨基末端氮原子上,常在赖氨酸和精氨酸上发生单甲基化(me)、二甲基化(me2)或三甲基化(me3)。通常,H3K4和H3K36的二甲基化和三甲基化修饰与相关基因的激活有关,H3K9和H3K27甲基化通常与基因的抑制相关,其中H3K27me3具有较高的可逆性,在植物发育中起到重要的动态调节作用[24]。参与组蛋白修饰调控的表观因子种类繁多,例如拟南芥中发现的多梳蛋白家族(polycomb)重要组分CLF (Pc-G gene CURLY LEAF)编码一种甲基转移酶,对H3K27具有特异性,参与调控拟南芥多个器官的形态建成[25]。CLF可通过调控其下游基因的表达对叶片[26]、花、根和侧枝的生长产生影响[27,28],同时还参与种子、果实的发育。此外,Park等[29]还发现clf突变体对铁缺乏环境具有较高的耐受性。DNA甲基化修饰和组蛋白修饰通常密不可分,例如拟南芥H3K4me1与DNA的CG甲基化相关[30]。不同类型组蛋白修饰也常以多种方式相互作用,使其状态在一定程度上维持冗余和稳定[31]。
图1
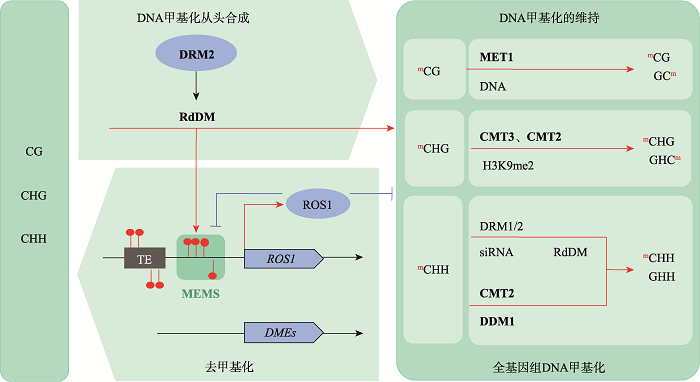 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1拟南芥DNA甲基化的从头合成、维持和去甲基化
图根据参考文献[4]修改绘制。DRM2:DOMAINS REARRANGED METHYLTRANSFERASE 2,甲基化转移酶2;RdDM:RNA directed DNA methylation,RNA介导的DNA甲基化;MEMS:methylation monitoring sequence,甲基化监测序列;MET1: METHYLTRANSFERASE 1,甲基转移酶1;CMT3:CHROMOMETHYLASE 3,甲基转移酶3;CMT2:CHROMOMETHYLASE 2,甲基转移酶2;DDM1:DECREASED DNA METHYLATION 1,染色质重塑因子。基因上的红色圆圈标记代表DNA甲基化修饰;DNA主动去甲基化过程主要涉及2类主要的DNA去甲基化酶—ROS1和DMEs。
Fig. 1De novo synthesis, maintenance of DNA methylation and demethylation in Arabidopsis thaliana
在表观遗传学研究中,除了DNA甲基化和组蛋白甲基化、乙酰化等具体的化学修饰外,染色质重塑也是表观遗传学的重要内容。因为化学修饰所引发的基因表达的变化,通常需要通过染色质重塑来实现。染色质重塑广泛地包括真核细胞基因表达调控过程中出现的一系列染色质结构变化。目前研究的主要类型是ATP依赖型染色质重组,即染色质重塑复合物通过ATP水解释放的能量改变染色质结构[32],根据ATP酶类型分为SWI/SNF、ISWI、CHD和INO80/SWR[33]。染色质重塑在植物生长发育过程中起着重要的调控作用,如SWF/SNF染色质重组复合物中的负调控因子CHR12参与植物在胁迫环境中的应答。处于胁迫条件下时,过表达突变体的初生芽和茎会停止生长[34]。
表观遗传修饰的遗传与变异相对于经典遗传信息来说更为复杂,其中DNA甲基化修饰具有相对稳定的遗传性,其他表观遗传修饰的遗传规律还有待探索。另外,与碱基排列编码的遗传信息相比,各类型的表观遗传修饰变异更易受到环境的影响。植物的整个生命过程基本是固着生长,在其生活周期中,不论是形成种子的有性生殖方式还是无性繁殖,都容易受到环境影响,能够对胁迫进行响应的表观遗传修饰变化具有一定的遗传性,甚至有些能够积累,这也是植物在逆境胁迫下建立的自我保护机制和适应环境的生存策略。虽然表观遗传修饰能伴随着细胞增殖传递,但并不总是在每个细胞周期中都精确地以亲代细胞为模板复制,同时在其遗传过程中存在很多偶然性事件。如各种情况下形成的分生组织、植物体不同细胞对生物和非生物胁迫的不同反应、以及易产生杂种优势的杂交现象等,都为表观基因组的变异提供了机会。分生组织形成时,植物体细胞间的竞争有助于选择出最有利的变异体,分生组织的遗传镶嵌产生表型丰富多变的幼苗,这些幼苗将在下一代竞争中产生最适合当前生存需求的变种,且变异后的DNA甲基化修饰大多数情况下也可以遗传。植物受到环境胁迫时,DNA甲基化修饰的遗传稳定性会适当降低,即在表观遗传修饰总体稳定遗传的情况下,后代中一小部分表观修饰的状态与亲代相似,但不完全一致。Minow等[31]以玉米为材料,针对这一遗传现象提出了一个观点,他们认为随着植物的世代繁衍,少数亲代的DNA甲基化修饰状态将发生漂移,但也存在偏离后的偶尔回归。从整体上看,在植物生长繁殖过程中,相对于变异,表观遗传修饰遗传的部分远大于变异。总之,相对于DNA序列信息,表观修饰在稳定遗传的同时具有可塑性的优势,在植物世代间和生命周期中能体现出一定程度的逆境胁迫响应。Wang等[35]通过对拟南芥的研究,揭示了植物表观遗传修饰的类型、形成和去除机制,以及不同类型修饰间的联系,还发现了表观遗传修饰对植物表型的重要影响,这对作物的表观研究提供了重要的参考与指导。
2 表观遗传修饰在调控作物表型中的作用
随着表观遗传学研究在拟南芥、玉米、水稻中的开展,越来越多的研究成果体现了表观遗传修饰在调控农作物重要品质性状、生产性状和环境适应性等方面的作用。表观遗传机制的深入研究也将有助于将表观遗传分子育种推广到更多作物上,获得性状突破性作物种质资源。2.1 表观遗传修饰对植物生殖生长的作用
2.1.1 开花植物开花容易受温度等环境因素的影响,表观遗传修饰在其中起到了重要作用。拟南芥开花抑制因子FLC (FLOWERING LOCUS C)的表达在春化过程中受到组蛋白甲基化修饰的负调控,低温会使激活型组蛋白的修饰H3K4me3和HAC被替换为抑制型H3K9me3和H3K27me3修饰,解除FLC对下游开花基因的抑制作用,促进开花(图2)[36]。拟南芥H3K36me3是温度诱导开花所必需的组蛋白甲基化修饰,Pajoro等[37]发现拟南芥甲基化酶突变体开花时对温度的响应不同于野生型,体现了组蛋白修饰在植物响应环境变化中的重要作用。同时,低温诱导与开花之间具有一定的时间间隔,拟南芥经过春化处理并解除低温作用后仍保持较低的FLC mRNA水平,并未随春化处理的停止或植物体有丝分裂的发生而改变,在一定程度上体现了表观修饰的记忆性。利用这一表观遗传修饰调控机制有可能实现对拟南芥开花的人为干预。含有FLC同源基因的白菜(Brassica rapa)和甘蓝(Brassica oleracea)[38]等芸薹属(Brassica)作物与拟南芥同属十字花科[39],该策略有望被应用于培育耐未熟抽薹的高产优质品种[36]。
图2
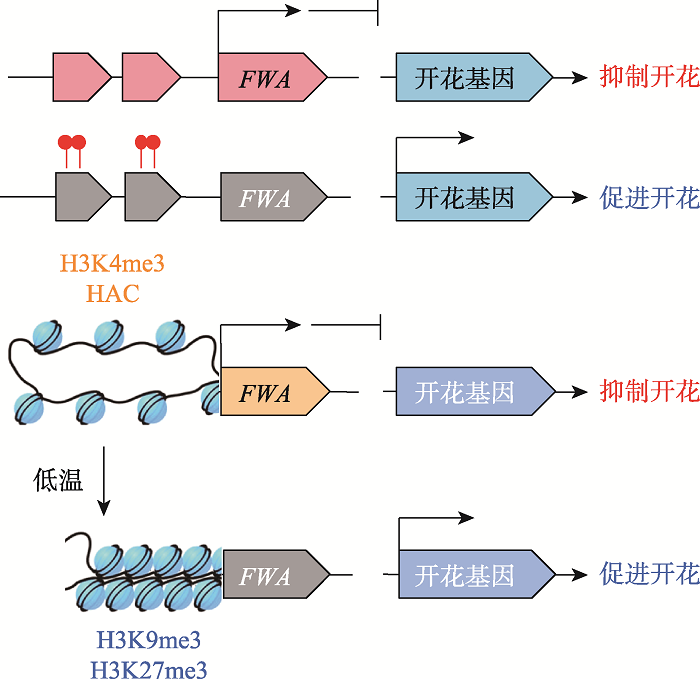 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2表观遗传修饰参与调控拟南芥开花活动
Fig. 2Epigenetic modifications are involved in the regulation of Arabidopsis thaliana flowering activity
组蛋白赖氨酸上的甲基化是参与基因表达调控的重要表观遗传修饰,由SDG (SET domain group)基因家族调控[40]。拟南芥SDG27突变体的花期显著提前;SDG8介导的H3K36me3和HUB2介导的组蛋白H2B单泛素化(H2Bub1)可以相互增强,Shafiq等[41]对开花基因进行检测后发现,其染色质处于活跃状态,且H3K36me3在其中起到的作用比H2Bub1更大。
在水稻中,SDG701可增强开花基因座T1 (RICE FLOWERING LOCUS T1, RFT1)的表达,促进植物光周期非依赖型开花[42]。SDG711和SDG718是水稻中H3K27me3复合物PRCs (polycomb group repressive complexes)的两个组份,分别在长日照和短日照光周期中介导H3K27me3积累,从而抑制开花抑制基因OsLF的表达,使编码锌指蛋白的基因Hd1表达升高,导致水稻在长日照光周期中延迟开花,在短日照中提前开花[43]。SDG725则参与促进水稻开花和油菜素类固醇相关基因的表达[44]。类似的,染色质重塑因子4 (CHROMATIN REMODELING4, CHR4)通过调节H3K4me3和H3K27me3水平来调控开花基因的表达,作用于花发育的不同阶段[45]。
2.1.2 果实成熟
DNA甲基化和去甲基化在番茄果实发育中会建立一种平衡,但在果实的成熟过程中,这种平衡会被DNA甲基化总体水平急剧下降所打破。研究发现,在果实成熟期间,果皮中rDNA基因座处的DNA甲基化水平降低[46],引起这种改变的动力为主动去甲基化,是触发果实成熟的必需活动,也是控制番茄成熟的中心环节[47]。Yao等[48]研究发现,MET1的沉默能够促进番茄无色不成熟突变体cnr果实的早熟、种子萌发和幼苗生长,证实了DNA去甲基化活动在番茄果实成熟中的作用;另一个番茄果实成熟关键基因是与去甲基化酶基因AtROS1同源的SlDML2,具有DNA 主动去甲基化功能,参与激活成熟诱导基因、抑制成熟抑制基因的过程。这些成熟诱导基因包括参与合成果实色素、香味、乙烯的基因以及与信号转导、细胞壁水解有关的基因。另一方面可通过造成启动子区低甲基化来抑制成熟抑制基因,例如参与光合作用和细胞壁合成与组织的基因,从而促进果实成熟[49,50],随着SlDML2表达增加,番茄果实会发生全基因组低甲基化以促进其成熟。通过对番茄的研究,科研人员揭示了全基因组DNA甲基化水平下降诱发果实成熟的表观调控机制。除番茄外,还有一些植物的果实成熟过程也由DNA去甲基化活动的改变引起。Huang等[51]发现,甜橙(Citrus sinensis)成熟时发生的全基因组高甲基化与甜橙去甲基化相关基因的表达量降低有关;Cheng等[52]在非跃变型水果草莓(Fragaria×ananassa)的成熟过程中也观察到了相同现象,特别是在非CG类型中,成熟期草莓的全基因组DNA甲基化水平降低,不是由于DNA去甲基化酶基因的诱导表达,而是RdDM途径减弱导致的。由此可以推测,果实成熟的诱导调控机制可能具有物种特异性,但与表观遗传修饰水平密切相关。此外,Quadrana等[53]研究表明,番茄果实中维生素E含量改变的自然变异也与DNA甲基化的变化有关。
组蛋白修饰对于果实的成熟也有一定的调控作用。在番茄中,组蛋白去乙酰化酶HDA1和HDA2的下调可促进乙烯合成和类胡萝卜素的积累,进而促进果实成熟;而另一种去乙酰化酶HDT3的下调作用则与之相反[54]。在香蕉(Musa nana)中,HDA1与转录因子ERF11 (ethylene response factor11)互作,能够抑制部分成熟基因的表达[55]。除以上调控机制外,番茄果实成熟时,组蛋白去甲基化酶JMJ6也参与其中,并且可以通过多种途径参与调控,例如直接靶向成熟相关基因,还可以通过降低DML2的H3K27甲基化修饰来进行调控[56]。关于植物果实成熟过程中DNA甲基化和组蛋白乙酰化修饰的关系已有初步进展,组蛋白H2A突变体H2A.Z特异性地定位于常染色质,与DNA甲基化在全基因组水平上反向关联[57,58],这是ROS1被招募至特定DNA去甲基化靶位点所必需的[57],在类胡萝卜素的调控中也起到了重要作用[59]。拟南芥甲基转移酶PRCs中的重要组分CLF蛋白作用于植物的生长发育,在番茄中也存在其同源物质,如SlEZ2。有研究已发现其对植物果实成熟具有的重要调控作用[25],该结果有望为番茄增产做出贡献。表观遗传修饰调控果实成熟的机制能够体现出一定程度的种间保守性,解析番茄和其他水果成熟时的DNA甲基化水平变化和组蛋白调控机制,进行相关表观遗传修饰基因的关联性分析,将有助于探索发现作物调控果实发育的目标等位基因[60]。
2.2 表观遗传修饰对作物产量和品质的作用
表观遗传修饰对作物的产量性状和品质性状起重要的调控作用。作物株型、子粒大小和重量等都是决定产量的关键性状。水稻RAV6 (RELATED TO ABSCISIC ACID INSENSITIVE3/VIVIPAROUS1 6)基因启动子区低甲基化会导致其过度表达,造成水稻叶片角度和种子大小改变,从而影响作物产量[61]。RdDM通过影响小型反向重复转座子MITEs (miniature inverted-repeat transposable elements)的甲基化来调控周边的OsMIR156d/j和D14 (DWARF14)等多个下游关键基因的表达,进而影响水稻分蘖/株型[62]。大豆种子从球状胚、成熟种子及萌发的全过程中,CHH甲基化发生了明显的动态变化,并主要靶向TEs,很多已知的重要编码种子蛋白的关键基因都位于低甲基化区域[63,64]。这些结果都暗示,在作物种子的发育中存在重要的表观遗传学调控机制,表观遗传调控因子控制下游多个基因的表达,使得复杂的数量性状转变为单基因控制的质量性状,显现出表观遗传在复杂数量性状分子育种中的巨大潜力。表观遗传修饰除了在水稻产量决定中起到重要作用外,还具有改善水稻营养的潜力。为了得到多细胞层糊粉谷物,传统的改良方法是突变转录因子,但得到的突变体容易出现严重缺陷,Liu等[65]在表观遗传研究中发现去甲基化酶OsROS1参与水稻胚乳中CG和CHG类型DNA去甲基化,使限制胚乳糊粉细胞层数的基因能表达。而OsROS1基因的第14个内含子出现点突变后,转录出的产物除了OsROS1外,还有去甲基功能失调的mOsROS1,当后者的比例占据50%以上时,水稻种子胚乳的CHH和CHG甲基化水平明显升高。这时,突变体表现为粒重和结实率降低,糊粉部位变稠,糊粉细胞层数增加,营养因子的含量和膳食纤维都有所增加,这使得“利用表观遗传修饰增厚糊粉层”有望成为改善水稻营养的新途径。
表观遗传修饰在其他经济作物的产量和品质的调控中起着重要的作用。Rajkumar等[66]在鹰嘴豆(Cicer arietinum)中发现,CG的超甲基化参与其种子大小和重量相关基因的高表达。Telias等[67]发现苹果(Malus pumila)和番茄的DNA甲基化模式与果实大小相关。苹果属植物中的MYB10启动子存在甲基化差异时,苹果表皮上花青苷差异性积累,产生红条斑和黄条斑,在梨(Pyrus spp)中则形成表皮的红斑点和条纹[68]。油棕(Elaeis guineensis)的优良杂交种在繁殖中出现体细胞无性系变异,极大地降低了产量。Ong-Abdullah等[69]通过表观基因组广泛关联研究发现,Karma剪接位点附近的低甲基化影响了mRNA的剪切,该变化导致油棕同源异型转化、孤雌结实和产量的急剧下降。Raju等[70]在大豆中发现,植物特异性基因MutS同系物1 (MutS HOMOLOG1, MSH1)衍生的表观遗传变异也具有提高大豆产量及产量稳定性的潜力。组蛋白修饰对作物品质也有影响,如拟南芥中作用广泛的H3K27特异性甲基转移酶可调控种子合成相关的基因,其突变体clf-28具有种子更大、含油量更高的优良性状[71]。
关于DNA甲基化导致基因表达沉默这一规律,水稻中存在一些非普遍模式和更复杂的调控机制。例如水稻胚乳中的低甲基化通过RNAi途径增强了胚胎中的转座子沉默,而CG和CHG低甲基化则成为其基因激活的主要机制[10],这与在拟南芥中发现的相关机制有所不同。这些发现为通过表观基因组编辑提高水稻农艺性状提供了可能,也为其他作物的相关研究提供了参考。
2.3 表观遗传修饰在植物胁迫响应中的作用
环境条件对植物生长发育起到重要调控作用,植物对环境有敏感的响应。生物和非生物胁迫能引起植物表观遗传修饰变异。目前虽然无法证明由此产生的所有变异都利于植物适应环境,但已有大量的研究表明,响应胁迫的表观遗传修饰变异能在一定程度上改变植物表型,使其处于更适应当前环境的生理状态,在植物适应和进化过程中起着重要调节作用。DNA甲基化修饰变异具有一定的遗传稳定性,植物长时间不再受到胁迫后,相应的DNA甲基化修饰的状态也有可能改变[4]。不同胁迫下,不同物种所建立的表观遗传修饰的规律并不一致[72]。当水稻植株暴露于脱落酸、盐和冷胁迫等不同的非生物胁迫下时,其组蛋白乙酰转移酶基因表达模式也会不同[73];参与抗白叶枯菌(Xanthomonas oryzae)的表观遗传机制,涉及到DNA去甲基化和多个通路中的组蛋白去甲基化行为[74,75,76]。植物在胁迫条件下的表观遗传变异有些也具有一定的变异规律,其中最常见的组蛋白修饰为H3K4me3,这种修饰可能是胁迫条件下高表达基因的标记。随着人们对植物表观等位基因的鉴定和相关表型调控机制的解析,表观遗传修饰对胁迫的响应为改善植物的适应性、实现品种优化提供了一个新的策略,而通过胁迫等方式激发一定程度的表观遗传修饰变异有望成为实现这一策略的有效途径。2.3.1 表观遗传修饰在植物温度胁迫响应中的作用
温度的细微变化能对植物的生长发育产生很大影响,一定程度的冷、热胁迫有助于植物调整生理状态和生长发育进程,例如促进植物进入下一生长阶段或增强环境适应能力,还可能产生新型优良的作物品种。对环境变化具有灵敏响应的表观遗传修饰在响应温度变化中起到了重要的作用。植物表观遗传修饰可对短期的低温胁迫产生精确的感知,帮助植物应对未来更强的低温,即对低温胁迫有一定的记忆性,称应激记忆[77]。Zuther等[78]对拟南芥进行冷驯化后发现,拟南芥中的脂质和代谢物的组成发生改变,能够在一定程度上帮助拟南芥提升抗冻性,这些变化受到表观遗传修饰的调控。DNA甲基化修饰可通过调控赖氨酸代谢途径以多种方式参与胁迫响应[79]。苦荞(Fagopyrum tataricum)植株在低温条件下,赖氨酸积累水平受到影响,且不同的冷处理方式所导致的赖氨酸水平改变机制也不同[80]。血橙(Citrus sinensis)受到低温胁迫后,反转座子的转录被去甲基化活动所激活,下游Ruby基因随之表达,影响了花色苷合成[81];且不同品种的血橙对Ruby的表达需求不同,这种特性有助于实现不同目标性状作物的生产[82]。Liu等[83]在芜菁(Brassica rapa)冷驯化实验中发现,启动子去甲基化导致BramMDH1表达升高,能够增强耐寒芜菁的耐热性,并促进其提高生长率。这种现象称为“交叉适应”,即植物被暴露在某种中度胁迫下,可能诱导产生出一定的对其他胁迫的耐受性。
植物中参与响应低温胁迫调控的表观遗传修饰还有组蛋白修饰,如组蛋白乙酰化酶HAT和HDAC能对低温胁迫转录因子的表达进行动态调节[82]。在植物低温应答机制中,CBF(C repeat binding factor)- COR (cold responsive)为核心组分,Kwon等[84]研究发现,COR-15A和AtGOLS3 (galactinol synthase 3)中H3K27me3修饰在拟南芥受到短暂低温胁迫后减少,而且这种低甲基化状态可维持超过3天,表明此处的组蛋白甲基化修饰可能是一种低温胁迫记忆的标记。拟南芥中PKL(PICKLE)蛋白是CHD3型染色质重塑因子,作为转录阻遏物起作用[85],Yang等[86]发现PKL蛋白在低温胁迫时通过调控H3K27me3水平来影响其染色质状态,调控COR基因表达,从而帮助植物响应低温胁迫。在高温诱导下,拟南芥组蛋白去乙酰化酶HDA15靶向抑制一系列下胚轴伸长相关基因的启动子[87],热应激记忆则需要热休克因子A2 (HEAT SHOCK FACTOR A2, HSFA2)维持高水平的H3K4me3/2修饰[88]。此后,Liu等[89]还发现HSFA2能靶向激活H3K27me3去甲基化酶REF6 (RELATIVE OF EARLY FLOWERING 6),REF6参与激活基因HTT5 (HEAT-INDUCED TAS1 TARGET 5)的表达,最终使植物提前开花,同时感病性增加。研究还揭示了REF6和HSFA2可形成正反馈回路来维持植物对高温的传代记忆,即REF6上调表达能够降低HSFA2位点H3K27me3修饰水平,从而减弱H3K27me3对HSFA2转录的抑制作用。非编码RNA也参与植物对温度胁迫的响应,例如Guan等[90]发现拟南芥受到高温胁迫时,miR398将激活热休克因子HSFA1b (HEAT SHOCK FACTOR 1b)和HSFA7b (HEAT SHOCK FACTOR 7b),并形成可增强耐热性的正反馈调节。
2.3.2 表观遗传修饰在植物干旱胁迫响应中的作用
植物对干旱胁迫的响应和适应通常通过转录重编程网络实现,其中应激信号转导会改变转录因子基因特定的表观遗传修饰水平。干旱胁迫下,作物耐旱性状多与DNA甲基化水平降低有关。González等[12]发现,为适应干旱环境,番茄ABA胁迫-成熟响应蛋白(Abscisic Acid Stress Ripening1) CHH甲基化水平下调。Gayacharan等[91]发现,干旱条件下,干旱敏感的水稻品系IR20和CO43主要表现为全基因组高甲基化,耐旱品系PL和PMK3及其杂交子代表现为全基因组低甲基化,且这样的甲基化水平改变对产量有一定的影响,水稻小穗不育率与甲基化水平正相关,而穗长、穗粒数、穗重、百粒重和单株产量与甲基化水平负相关,即受到干旱胁迫的干旱敏感品系全基因组DNA甲基化水平升高,不育率升高,与优良农艺性状相关的基因表达量降低。这体现了干旱胁迫下水稻在减少有效分蘖数以减少耗水量的“生存策略”和增加结实率以保持产量的“生殖策略”之间的平衡。这些微量的表达变化由表观遗传修饰变异介导,具有富集效应,可以导致明显的表型变化。进一步研究表明这样的胁迫反应可多代响应[92],后代的生理和形态变化不断优化着这种平衡,更为直接地助力于农业生产。Herman等[93]在桃叶蓼(Polygonum persicaria)中发现DNA甲基化介导的干旱胁迫响应也具有“跨代胁迫记忆”。除了通过转录重编程网络,胁迫因子可能也会直接影响甲基化酶和去甲基化酶来调节甲基化水平。在水稻灌浆的中后期至后期,Wang等[94]进行土壤中度干燥胁迫(moderate soil drying, MD)时发现,水稻会发生去甲基化活动,应激反应敏感基因被激活并表达,改善淀粉降解与蔗糖合成来增强碳储备迁移,并控制叶片衰老。此外,Benoit等[95]还发现反转座子Rider广泛存在于多种植物中,具有较强的表观遗传沉默作用,转录水平受环境因素和RdDM途径的调节,涉及代偿途径,可通过干旱诱导产生的遗传积累和新的胁迫响应形成变异表型。此外,组蛋白修饰也参与植物抗旱响应,Sukiran等[96]在拟南芥干旱胁迫研究中发现,当干旱胁迫应答因子的H3K27me3 水平下调时,其结合蛋白PRC1复合物水平降低,可以解除对干旱响应和花发育基因ANAC019、ANAC055的表达抑制。干旱胁迫下,拟南芥组蛋白脱乙酰酶HDA9参与ABA的积累,HDA9与Powerdress (PWR)形成复合物,通过协助AP2/ERF超家族成员ABI4作用来降低CYP707A1基因的启动子乙酰化水平,导致拟南芥ABA积累,从而响应胁迫[97]。参与调控ABA耐旱响应通路的表观遗传因子还有H3K4甲基转移酶ATX1[98],ATX4和ATX5也起到一定的作用[99]。Ding等[100]发现,干旱胁迫下拟南芥耐旱突变体atx-1的H3K4me3水平下降;而在野生型拟南芥中,与DNA甲基化修饰响应干旱胁迫时出现的胁迫记忆类似,高水平的H3K4me3修饰和Ser5P聚合酶II能够使拟南芥再次遭受干旱胁迫时快速表达相应的响应基因[101]。在水稻的研究中,Guo等[102]发现OsCHR4基因除了对产量和质量有关的性状具有调控作用以外,还通过影响组蛋白修饰调控生长素和蜡质生物合成基因的表达,调控叶形发育和角质层蜡质形成,使植物适应干旱。Li等[103]揭示了杨树(Populus)组蛋白乙酰化酶复合体GCN5-ADA2和转录因子AREB1之间的相互作用和共同靶向作用。DNA甲基化修饰和组蛋白修饰常共同作用于植物基因的表达调控。Mao等[104]推断,玉米耐旱正相关基因NAC111在被驯化后插入了转座元件MITE,它能够通过RdDM途径和H3K9me2抑制NAC111的表达。番茄响应干旱胁迫时,H3K9me2与CHH甲基化共同负调控Ars2的表达[105]。而毛果杨(Populus trichocarpa)受到干旱胁迫的信号后,其DNA甲基化水平会随之波动,进而改变许多干旱胁迫基因的表达模式[106],同时组蛋白乙酰转移酶会被招募至耐旱基因NAC家族成员的启动子处,H3K9ac富集会促进其表达[103]。染色体重塑因子也参与植物胁迫响应。拟南芥受干旱胁迫后,SWI2/SNF2染色质重塑因子ATPaseBRM (BRAHNA)突变体耐性提高[107],SWI/SNF核心组分CHR12作用于拟南芥使其瞬时生长停滞。Mlynárová等[34]在拟南芥中发现,SWI/SNF重塑复合物可通过调节染色体结构来帮助拟南芥产生耐旱性。作物中SWI/ SNF参与干旱响应的现象和调控机制逐渐被发现,如Yang等[108]揭示了水稻中核基质组成蛋白NMCP1 (nuclear matrix constituent protein 1)与SWI/SNF的染色质重构复合物SWI3C相互作用,而它们在干旱胁迫下对于抗旱性或根系生长相关的基因分别起到正调控和负调控的作用。
2.3.3 表观遗传修饰在调控植物耐盐中的作用
高盐条件会对植物产生渗透胁迫和离子胁迫,研究发现,盐胁迫通常与干旱胁迫伴随发生,耐盐性也是植物迫切需求改良的育种性状之一。Baek等[109]发现拟南芥DNA甲基化缺陷突变体met1-3对盐具有超敏性,原因是一个小RNA靶位点的大量胞嘧啶甲基化缺失,导致钠离子转运相关的高亲和性K转运蛋白AtHKT1 (high-affinity potassium transporter1)基因表达量升高。Dyachenko等[110]发现,在干旱和高盐胁迫下,冰叶日中花(Mesembryanthemum crystallinum)卫星DNA会出现超甲基化,可能参与调控植物光合模式从C3至CAM的转变,从而更适应环境。Wang等[111]研究表明,小麦(Triticum aestivum)耐盐品种SR3与亲本JN177的甲基化水平不同,但在盐胁迫后两品种的基因组甲基化水平都有所降低,SR3背景下甲基化改变的体细胞在杂交诱导后表现出更强的耐盐性。Ogneva等[112]发现DNA甲基化抑制剂5-氮杂胞苷(5-azadeoxycytidine, 5-AZ)处理过的水稻植株中,氧化应激有关的基因表达上调,耐盐性增强。Bharti等[113]发现过表达AtROS1基因的烟草(Nicotiana tabacum)中类黄酮和抗氧化相关基因的甲基化水平降低,转基因烟草盐耐受性增加。
受到盐胁迫时,组蛋白修饰也是植物响应产生过程中的重要因子。Duan等[114]发现拟南芥中H3K4me3去甲基酶JMJ15缺失的突变体对盐胁迫更加敏感,而Sani等[115]结果表明拟南芥中低水平的H3K27me3会激活HKT1基因表达,其表达量急剧上升,控制根茎中的Na+浓度以抵抗盐胁迫。组蛋白去乙酰化有关的基因也参与调控植物对ABA和盐的胁迫响应,如拟南芥组蛋白去乙酰化酶突变体hda-19表现出对ABA和盐胁迫高度敏感[116]。此外,还有研究显示,组蛋白上一些不常见的修饰如类泛素化,会有抑制基因表达的作用[117],受到干旱和盐胁迫刺激信号的植物会通过类泛素化相关蛋白增加抵抗能力[118]。
2.3.4 表观遗传修饰在植物抗病机制中的作用
高等植物中的DNA甲基化能够帮助个体对生物胁迫做出响应,调控抗病性相关基因的表达,并具有保护基因完整性的作用[119]。在免疫应答途径中,DNA甲基化的调控机制具有一定的特异性[82]。Yu等[120]揭示了拟南芥RdDM途径参与茉莉酸信号通路的正调节,能够对灰葡萄孢霉(Botrytis cinerea)和黄瓜枯萎菌(Plectosphaerella cucumerina)等死体营养性真菌产生抗性。大豆应对印度绿豆黄花叶病毒(mungbean yellow mosaic India virus, MYMIV)时,通过在基因间隔区进行DNA高甲基化修饰将其沉默[121],来提高抗病性,但病毒互补链上出现的AC5基因会抑制甲基转移酶DRM2,从而抑制植物通过高甲基化引起的抗病毒防卫反应[122]。
DNA去甲基化机制在植物抗病反应中也能起到重要的激活作用。拟南芥在受到病毒入侵时,去甲基化机制可激活TEs及重复序列中的抗病相关基因,参与抗病反应。例如抗病基因RMG1主动去甲基化过程增强了拟南芥对丁香假单胞菌(Pseudomonas syringae) DC3000的免疫反应,其中接收病毒刺激的转录诱导和去甲基活动都依赖于去甲基化酶ROS1的活性[120],而拟南芥抗性的遗传依赖于RdDM途径调控的非CG位点去甲基化[123]。除此之外,AGO4也具有介导植物抵抗丁香假单胞菌DC3000的作用,且独立于RdDM通路的其他组成部分[124]。Geng等[125]在接种白粉菌(Blumeria graminis)的小麦中发现一些个体抗病性增强,研究发现这些植株的CHH甲基化水平降低,AeGlu/PR2-like等防御基因的表达增强。烟草受病毒感染时,抗病基因相关的亮氨酸重复序列区域发生去甲基化,抗病基因表达增强[126]。对表观遗传调节的深入理解将为选育和改良植物抗病品种提供理论依据[120],例如水稻R基因Xa21G去甲基化能够使个体对白叶枯菌产生抗性,并遗传给后代,产生抗病性增强的突变株系Line-2,用DNA甲基化抑制剂5-氮杂胞苷处理可增强水稻抗白叶枯病[74]。在水稻响应白叶枯病反应中,除了DNA去甲基化参与以外,组蛋白去乙酰化酶HDT701直接与防御相关基因结合,上调其转录水平来增强水稻对白叶枯病菌的抗性[75];H3K27去甲基化酶JMJ705通过激活防御调节因子参与提高水稻对白
叶枯病菌的抗性,JMJ704则参与H3K4me2/3去甲基化反应,从而抑制负防御调节因子,同样帮助植物体抵抗白叶枯病菌[76]。Lee等[127]和Berr等[128]发现,在拟南芥对真菌病原体的抵抗中,组蛋白甲基转移酶SDG8(SET domain group 8)和SDG25(SET domain group 25)通过调节H3K4me和H3K36me水平来激活免疫所需的病菌抵御基因的表达;另一个调节H3K4me的甲基转移酶ATXR7则参与抵抗真菌病原体拟南芥霜霉病菌Emwa1[129]。Zhou等[130]在拟南芥中鉴定出了酵母组蛋白去乙酰化酶HDA19。HDA19可由病原菌链格孢菌(Alternaria brassicicola)诱导表达,其过表达会降低拟南芥基因组的组蛋白乙酰化水平,并增加其对链格孢菌的抗性。玉米圆斑病菌(Cochiobolus carbonum)感染玉米后,病菌产生的HC毒素(HC-toxin)将抑制植株中的去乙酰化酶HDAC,植株组蛋白乙酰化水平降低并表现出对此病原菌更强的抗性[131]。Shi等[132]发现甜橙对柑橘亚洲韧皮杆菌(Liberbacter asianticumjagoueix)侵染的响应可能受到组蛋白乙酰转移酶1基因(CsHAC1)和其互作蛋白的调控。此外,相关基因的过度表达也会产生一定的影响,例如HDA19的过表达会导致拟南芥子叶和叶片异常[133]、开花延迟和育性降低[134,135]等表型。
通过改变表观修饰可以调控抗病基因的表达,具有应用于作物抗病性改良育种的潜力。Deng等[136]在水稻中发现了包含一组编码核苷酸结合富亮氨酸重复受体(nucleotide-binding domain leucine-rich repeat containing, NLR)的Pigm基因座,使水稻对稻瘟病菌具有持久抗性且不影响产量,这两个性状间是通过成对拮抗NLR受体从而受到严格的表观遗传机制调控,利用Pigm培育的新水稻优良品种(龙两优3189)种植在不同地点均表现出较高的抗瘟性和产量。
非编码RNA也是植物响应病原菌反应的重要参与者。Navarro等[137]首次发现拟南芥miR393通过靶向生长素受体TIR1、ABF2和ABF3调节生长素信号通路,使植物增强了对丁香假单胞菌DC3000的基础抗性。Katiyar-Agarwal等[138]发现致病菌假单胞菌侵染拟南芥后,诱导出的拟南芥内源分子na-siRNA ATGB2可调节抗病基因RPS2的表达。烟草和小麦只有在低温16℃的条件下才会感染中国小麦花叶病毒(Chinese wheat mosaic virus, CWMV),Andika等[139]研究发现,在高温环境中,烟草RNA聚合酶RDR6蛋白(RNA-dependent RNA polymerase 6, NbRDR6)在根部促进CWMV siRNAs的生成以抑制CWMV积累。
2.4 表观遗传修饰在植物杂种优势现象中的作用
杂种优势(heterosis)是杂交子代优势表型突出的现象,是作物育种中需要特别关注的因素。这些表型包括植物的生长力、抗逆性、果实产量和质量等,是能够为人类带来更好农业生产收益的表型,表观遗传修饰是杂种优势现象的关键决定因素之一。相比于染色体加倍,杂交更能引起DNA甲基化模式重新整合或编排[140,141],而这种亲本系之间的基因座特异性表观遗传特征可以直接或间接触发拟南芥杂种优势的形成,与基因组序列变化无关[142]。对高粱(Sorghum bicolor)[143]、杉木(Cunninghamia Lanceolata)[144]和籼稻(Oryza sativa. subsp. indica)[145]的研究发现,在亲本与杂交种之间,特异位点的甲基化变异大于DNA序列变异,同时杂交前亲本间的表观遗传变异也可为育种提供新的资源。Seifert等[146]对21个玉米亲本自交系的小RNA文库测序表明,ha-sRNAs可靶向调控表观遗传修饰标记,最终影响杂交玉米的表型。DNA甲基化的从头合成途径RdDM会参与杂交种F1等位基因表达的调控,在拟南芥和水稻杂交种中都发现siRNA对杂种优势的表型有重要作用[147,148]。Barber等[149]对玉米的研究表明,可能有另一套DNA甲基化修饰,同样起作用但不依赖于RdDM途径。siRNA在不同物种甚至品系之间的表达水平都有较大的差异,双亲转座子的区域不同也会导致siRNA的变化。Ng等[150]发现,杂交种与其亲本之间的差异能够引起等位基因表达模式的改变,并影响基因组的稳定性。此外,非加性miRNA常与亲本和杂种F1之间的差异表达关系密切[151,152],且因亲本间的亲缘关系远近不同miRNA和siRNA的表达模式上也会出现差异[153]。组蛋白修饰对杂种优势也有一定的作用。Ha等[154]研究发现,拟南芥中生物钟调控基因CCA和LHY启动子区域的H3K9ac和H3K4me2修饰发生改变,影响了光合作用和淀粉代谢相关基因的表达,使杂种后代表现出生长优势。Wang等[155]对人工拟南芥异源四倍体的研究也发现,FLC基因启动子区域出现了一系列的组蛋白修饰变化,例如H3K9乙酰化和H3K4me3水平提高、H3K9me3降低,使得四倍体表现出明显的开花延迟。He等[151]和Moghaddam等[156]通过进一步研究发现,不同生态型和亚种中的H3K4me3和H3K27me3水平有较大的差异,拟南芥和水稻各自的杂交后代中分别表现出加性和非加性的作用模式。对于各种表观遗传修饰在杂种优势中的作用、机制的研究有助于人们深入理解杂交与进化的关系,为利用杂交培育驯化新品种提供了有效途径。
3 可塑性和遗传性使表观遗传修饰有望应用于作物改良育种
如果说DNA信息是遗传密码,那么表观遗传修饰就是DNA的外加锁,这些锁的钥匙通常不止一把,其中很多受到生物体外界环境的影响。尽管表观遗传修饰种类多样,但它们共同的作用都是尽可能地帮助生物体适应环境并更好的生存。这些锁可以灵活精巧地打开或关闭,影响DNA密码的读取方式、读取时间以及读取的程度,并且传递给子代。得到表观遗传修饰的子代既能够获取上一代的生存经验,同时依然具有控制锁开关的能力。因此,掌握密码锁的钥匙,即表观遗传修饰在植物特定表型调控中的机制和规律,提供了新的具有更广泛调控手段的改良育种思路。表观遗传修饰的类型和其产生、维持、调控等机制在植物中具有一定的共性,在植物个体层面上还具有组织特异性、发育阶段特异性等;同时在不同的植物种属背景中具有针对生命活动的特异性调控机制。当植物的亲缘关系相近、调控机制相似或具有相同特征的表观修饰时,模式植物的表观调控机制具有一定的参考性和导向性。这些优势特征有利于人们通过靶向诱导等方式培育优良的农作物品种,改善农业生产。3.1 表观遗传修饰在植物中的胁迫跨代记忆
表观遗传修饰具有遗传稳定性,但相对于传统的遗传信息来说,更能对环境胁迫做出反应,具有一定的可塑性来应对环境改变。有研究表明DNA甲基化修饰改变在世代胁迫中产生稳定的表型改变,在生物胁迫下,这些改变包括DNA甲基化修饰的模式和水平。在多种植物中,即使后代没有受到胁迫,但还是有可能延续亲本应对逆境胁迫做出的表观遗传修饰改变[157]。这样的延续可伴随着有丝分裂和减数分裂进行遗传,分别称为“体细胞胁迫记忆”和“胁迫跨代记忆”[158]。Liu等[49]研究表明番茄受DNA去甲基化酶DMLs调控,与花、果实相关的表型会随世代的增加而积累。水稻受到干旱诱导后,产生了适应干旱的非随机DNA甲基化突变,有很大比例的后代保持了这种改变的DNA甲基化状态。Zheng等[92]基于一组干旱响应基因进行了分析,其结果也从机制上体现了它们的DNA甲基化模式受到多代干旱的影响。Mathieu等[159]在拟南芥胁迫跨代记忆研究中发现,起到维持遗传稳定中心作用的是CG类型的DNA甲基化。同时也存在这种胁迫记忆消失的现象,不再长期受到某胁迫后,植物将移除相应的表观遗传修饰以获取更高的环境适应性[160]。另有研究表明,组蛋白修饰等其他类型的表观遗传修饰在有丝分裂中也可以一定程度上稳定遗传,并且可能在调节植物对环境条件和发育的反应中起关键作用[161]。环境胁迫下的表观遗传修饰改变及其遗传稳定性对于农作物品种优化十分重要,使植物体更好地适应环境的表观遗传修饰是否能稳定遗传,以及能否在环境再度发生改变时产生新的改变、以适应新的环境,仍有待进一步研究。3.2 表观遗传修饰在植物中的保守性
表观遗传修饰在不同物种之间存在一定的保守性,一些植物中相对成熟的研究成果可为相关作物的表观遗传研究和改良育种提供参考。不同物种间表观遗传修饰的保守性首先与植物中普遍存在的TEs相关。TEs是DNA甲基化的调控对象热点区,越来越多的研究表明,植物表型的产生和受到胁迫后的环境适应性改变与基于TEs的基因多态性相关[162]。尽管不同植物的TE含量差异很大,但各物种中TE都占基因组序列的重要组成部分,因此TE成为决定表观遗传修饰保守性的基础条件之一。不同物种之间的表观遗传修饰类型和调控机制也具有保守性,单子叶植物水稻和双子叶植物拟南芥中,其胚乳与最初的胚相比都呈现低甲基化状态。Zemach等[11]发现去甲基化活动机制可能具有共同的进化起源,这在一定程度上提示该过程在单子叶植物和双子叶植物之间可能是保守的。在亲缘关系相近的植物中,表观遗传修饰的机制具相对更强的保守性。如番茄果实成熟过程中,DNA去甲基化反应起到了重要的作用,回顾一系列在番茄成熟中受DNA甲基化调控的基因,其中很多基因的发现都来自于对模式植物拟南芥DNA甲基转移酶的序列同源性筛选。将果实成熟期间基因组去甲基化机制的调控应用于番茄优良品种的培育,是一种新颖的策略,可用于改善缺乏遗传多样性品种的农艺性状[163]。而番茄作为研究肉质果实发育的模式生物,其表观遗传修饰的研究成果又将为其他植物的相关探究提供参考[164,165]。因此,选取典型作物探究与重要农艺性状有关的表观遗传机制,可帮助人们在与其存在亲缘关系植物中发现潜在的表观遗传作用。从全基因组的DNA甲基化修饰情况分析,甲基化差异区域(differentially methylated regions, DMRs)和单甲基化多样性位点(single methylation polymorphism, SMPs)与植物表型都有显著的相关性,说明DNA甲基化位点可作为稳定的表观遗传标记。在基因组亲缘关系相近的植物中,甲基化的分布和程度具有相似性,其从头合成和维持途径在物种间也具有保守性,这为探究表观修饰参与各物种基因表达调控机制的关联性提供了基础。Lin等[163]进行的表观全基因组关联分析(epigenome-wide association study, EWAS)可将DNA甲基化变异与植物体各层面的性状对应并分析,有望探究出具有关联性和参考性的机制,以应用于植物生长发育的全面研究。应用EWAS分析,Ong-Abdullah等[69]在油棕中发现一种水稻的LINE类反转座子Karma低甲基化,导致油棕形成厚壳无性系,严重影响生产。
3.3 表观遗传变异模拟模型的初步应用
Morrell等[166]建立了一种表观遗传变异模拟模型。该模型基于现有的表观遗传调节机制,整合表观遗传标记和目标生理过程,通过模拟变异,评估和量化它们对植物表型的影响,预测某种/些表观遗传变异对各种环境情景下可能出现的性状进行量化,并获得这些性状的动态变化。该模型在番茄成熟代谢物预测中取得了初步的成果。Liu[49]等通过对番茄基因组DNA甲基化、基因表达和代谢之间的相互作用进行定量建模,精确地模拟了野生型和RNAi DML2敲除型果实中NOR和PSY1启动子的甲基化水平、NOR和PSY1基因表达水平,以及番茄成熟过程中关键代谢物番茄红素的积累。在多种植物中都存在表观修饰在遗传过程中出现的变异能够产生新的表型,由此发现了一些新的“表观等位基因”。例如无色不成熟基因座、甜瓜性别决定[167]、水稻植株矮化[168]、坏Karma导致的低油油棕[69],以及番茄果实中维生素E含量等,也都是由表观遗传变异引起[53]。基于对不同类型表观修饰的研究,也可根据分析不同序列环境下的表观遗传修饰水平、比较表观遗传学研究等方法进行建模,对新的表观遗传变异的产生、表观变异导致的新表型和表型对植物生存的作用等方面做出预测。虽然这种模型反馈对于开发模型驱动的育种方法至关重要,但在具体实践中,还有很多因素需要考虑,如植物不同的繁殖方式等。因为繁殖方式(无性繁殖或有性繁殖)是引发表观遗传变异的重要途径之一,故培育对环境变化具有足够表观遗传响应的作物品种时,也需要考虑到作物的繁殖方式对后代表观遗传变异的遗传性和环境适应性产生的影响[165]。
4 结语与展望
具有遗传稳定性和可塑性的表观遗传修饰在调控植物的生长发育中起到了至关重要的作用,在作物的改良和育种方面具有很大的潜质。目前,植物表观遗传学研究已经拓展到作物重要性状的调控机理,如表观遗传修饰在番茄果实成熟中的作用,DNA甲基化在水稻产量调控中的突破等研究成果。表观遗传调控因子可控制多个相关基因的表达,使得复杂的数量性状转变为单基因控制的质量性状,从而简化育种过程,大大提高育种效率,显现出表观遗传在复杂数量性状分子育种中的巨大潜力。然而,将表观遗传学用于作物改良必须解决如下问题:(1)掌握植物表观遗传变异产生的机制。深入解析物种的表观遗传变异水平与外部环境、内部基因组环境等因素影响表观遗传变异的机制,明确杂交、无性繁殖和组织培养等植物育种和农业中的常规做法对表观遗传变异产生的影响。(2)开发可定向改变表观遗传修饰的技术。开发并应用表观遗传基因编辑等技术,为通过重塑植物表观遗传修饰来改变植物性状提供可能,实现将表观遗传修饰和调控机制应用到作物改良和育种中等可操作性。(3)明确植物表观遗传修饰在作物育种中的应用范围。虽然研究成果显示表观遗传修饰广泛调节着植物的重要性状,如提高作物在生物和非生物环境胁迫中的耐受性,以拓宽生态幅进行广泛种植;促使植物提早开花或打破休眠等,增强作物的经济优势,包括产量、品质和生长周期等。但表观遗传修饰对于作物表型的影响程度和可应用程度仍无法预测,表观遗传变异模拟模型的建立和探索有望加快这一进程。(4)充分了解作物改良育种应用中表观遗传变异的稳定性和可遗传性。因为作物繁殖方式多种多样,可利用的表观遗传修饰在有丝分裂、减数分裂中的遗传稳定性如何,多代后的表观遗传信息稳定性能否满足改良和育种需求,如何稳定益于农业生产的表观遗传修饰等都是需要考虑的问题。(责任编委: 储成才)
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.bse.2015.09.009URL [本文引用: 1]
DOI:10.1038/nrg2719URL [本文引用: 1]
DOI:10.1038/s41580-018-0016-zURL [本文引用: 3]
DOI:10.1016/j.tplants.2019.08.007URL [本文引用: 2]
DOI:10.1073/pnas.1502279112URL [本文引用: 1]
[本文引用: 1]
PMID:11090618 [本文引用: 1]

The transition to flowering in Arabidopsis thaliana is delayed in fwa mutant plants. FWA was identified by loss-of-function mutations in normally flowering revertants of the fwa mutant, and it encodes a homeodomain-containing transcription factor. The DNA sequence of wild-type and fwa mutant alleles was identical in the genomic region of FWA. Furthermore, the FWA gene is ectopically expressed in fwa mutants and silenced in mature wild-type plants. This silencing is associated with extensive methylation of two direct repeats in the 5' region of the gene. The late flowering phenotype, ectopic FWA expression, and hypomethylation of the repeats were also induced in the ddm1 hypomethylated background. Mechanisms for establishment and maintenance of the epigenetic mark on FWA are discussed.
DOI:10.1186/gb-2007-8-2-r28URL [本文引用: 1]
DOI:10.1126/science.1178534PMID:19965430 [本文引用: 2]
We report an improved draft nucleotide sequence of the 2.3-gigabase genome of maize, an important crop plant and model for biological research. Over 32,000 genes were predicted, of which 99.8% were placed on reference chromosomes. Nearly 85% of the genome is composed of hundreds of families of transposable elements, dispersed nonuniformly across the genome. These were responsible for the capture and amplification of numerous gene fragments and affect the composition, sizes, and positions of centromeres. We also report on the correlation of methylation-poor regions with Mu transposon insertions and recombination, and copy number variants with insertions and/or deletions, as well as how uneven gene losses between duplicated regions were involved in returning an ancient allotetraploid to a genetically diploid state. These analyses inform and set the stage for further investigations to improve our understanding of the domestication and agricultural improvements of maize.
DOI:10.1073/pnas.1009695107URL [本文引用: 2]
DOI:10.1126/science.1186366PMID:20395474 [本文引用: 2]
Eukaryotic cytosine methylation represses transcription but also occurs in the bodies of active genes, and the extent of methylation biology conservation is unclear. We quantified DNA methylation in 17 eukaryotic genomes and found that gene body methylation is conserved between plants and animals, whereas selective methylation of transposons is not. We show that methylation of plant transposons in the CHG context extends to green algae and that exclusion of histone H2A.Z from methylated DNA is conserved between plants and animals, and we present evidence for RNA-directed DNA methylation of fungal genes. Our data demonstrate that extant DNA methylation systems are mosaics of conserved and derived features, and indicate that gene body methylation is an ancient property of eukaryotic genomes.
DOI:10.3389/fpls.2018.00600URL [本文引用: 1]
DOI:S0168-9452(19)31549-3PMID:32234231 [本文引用: 1]
Recent progress in "omics" methodologies allow us to gain insight into the complex molecular regulatory networks underlying plant responses to environmental stresses. Among the different genome-wide analysis, epigenomics is the most under-investigated "omic" approach requiring more critical and speculative discussion about approaches, methods and experimental designs. Epigenomics allows us to gain insight into the molecular adaptation of plants in response to environmental stresses. The identification of epigenetic marks transmitted during filial generations enables new theories to be developed on the evolution of living organisms in relation to environmental changes. The molecular mechanisms driving the capacity of plants to memorize a stress and to generate stress-resistant progenies are still unclear and scarcely investigated. The elucidation of these cryptic molecular switches will assist breeders in designing crops characterized by minimally compromised productivity in relation to stresses caused by climate change. The aim of this review is to briefly describe the most uptodate epigenomic approaches, update recent progresses in crop epigenomics in plant stress biology, and to stimulate the discussion of new epigenomic methods and approaches in the new era of "omic" sciences.Copyright © 2019 Elsevier B.V. All rights reserved.
DOI:10.1038/hdy.2009.36PMID:19337304 [本文引用: 1]
DOI:10.1016/j.gene.2013.01.034URL [本文引用: 1]
DOI:10.1038/srep23181URL [本文引用: 1]
DOI:10.1038/nature11968URL [本文引用: 1]
DOI:10.1038/s41598-017-02860-4URL [本文引用: 1]
DOI:10.1104/pp.15.00408 [本文引用: 1]
Soybean (Glycine max) and common bean (Phaseolus vulgaris) share a paleopolyploidy (whole-genome duplication WGD) event, approximately 56.5 million years ago, followed by a genus Glycine-specific polyploidy, approximately 10 million years ago. Cytosine methylation is an epigenetic mark that plays an important role in the regulation of genes and transposable elements (TEs); however, the role of DNA methylation in the fate/evolution of genes following polyploidy and speciation has not been fully explored. Whole-genome bisulfite sequencing was used to produce nucleotide resolution methylomes for soybean and common bean. We found that, in soybean, CG body-methylated genes were abundant in WGD genes, which were, on average, more highly expressed than single-copy genes and had slower evolutionary rates than unmethylated genes, suggesting that WGD genes evolve more slowly than single-copy genes. CG body-methylated genes were also enriched in shared single-copy genes (single copy in both species) that may be responsible for the broad and high expression patterns of this class of genes. In addition, diverged methylation patterns in non-CG contexts between paralogs were due mostly to TEs in or near genes, suggesting a role for TEs and non-CG methylation in regulating gene expression post polyploidy. Reference methylomes for both soybean and common bean were constructed, providing resources for investigating epigenetic variation in legume crops. Also, the analysis of methylation patterns of duplicated and single-copy genes has provided insights into the functional consequences of polyploidy and epigenetic regulation in plant genomes.
DOI:10.1186/1471-2229-11-94PMID:21599976 [本文引用: 1]
Background: Eukaryotic DNA methylation is one of the most studied epigenetic processes, as it results in a direct and heritable covalent modification triggered by external stimuli. In contrast to mammals, plant DNA methylation, which is stimulated by external cues exemplified by various abiotic types of stress, is often found not only at CG sites but also at CNG (N denoting A, C or T) and CNN (asymmetric) sites. A genome-wide analysis of DNA methylation in Arabidopsis has shown that CNN methylation is preferentially concentrated in transposon genes and non-coding repetitive elements. We are particularly interested in investigating the epigenetics of plant species with larger and more complex genomes than Arabidopsis, particularly with regards to the associated alterations elicited by abiotic stress.;Results: We describe the existence of CNN-methylated epialleles that span Asr1, a non-transposon, protein-coding gene from tomato plants that lacks an orthologous counterpart in Arabidopsis. In addition, to test the hypothesis of a link between epigenetics modifications and the adaptation of crop plants to abiotic stress, we exhaustively explored the cytosine methylation status in leaf Asr1 DNA, a model gene in our system, resulting from water-deficit stress conditions imposed on tomato plants. We found that drought conditions brought about removal of methyl marks at approximately 75 of the 110 asymmetric (CNN) sites analysed, concomitantly with a decrease of the repressive H3K27me3 epigenetic mark and a large induction of expression at the RNA level. When pinpointing those sites, we observed that demethylation occurred mostly in the intronic region.;Conclusions: These results demonstrate a novel genomic distribution of CNN methylation, namely in the transcribed region of a protein-coding, non-repetitive gene, and the changes in those epigenetic marks that are caused by water stress. These findings may represent a general mechanism for the acquisition of new epialleles in somatic cells, which are pivotal for regulating gene expression in plants.
DOI:10.1093/dnares/dsx046PMID:29149287 [本文引用: 1]
In plants, a key class of genes comprising most of disease resistance (R) genes encodes Nucleotide-binding leucine-rich repeat (NL) proteins. Access to common bean (Phaseolus vulgaris) genome sequence provides unparalleled insight into the organization and evolution of this large gene family (~400 NL) in this important crop. As observed in other plant species, most common bean NL are organized in cluster of genes. However, a particularity of common bean is that these clusters are often located in subtelomeric regions close to terminal knobs containing the satellite DNA khipu. Phylogenetically related NL are spread between different chromosome ends, suggesting frequent exchanges between non-homologous chromosomes. NL peculiar location, in proximity to heterochromatic regions, led us to study their DNA methylation status using a whole-genome cytosine methylation map. In common bean, NL genes displayed an unusual body methylation pattern since half of them are methylated in the three contexts, reminiscent of the DNA methylation pattern of repeated sequences. Moreover, 90 NL were also abundantly targeted by 24?nt siRNA, with 90% corresponding to methylated NL genes. This suggests the existence of a transcriptional gene silencing mechanism of NL through the RdDM (RNA-directed DNA methylation) pathway in common bean that has not been described in other plant species.
DOI:10.1016/j.cub.2004.07.007URL [本文引用: 1]
DOI:10.1038/s41580-019-0151-1URL [本文引用: 1]
[本文引用: 2]
[本文引用: 2]
DOI:10.1371/journal.pone.0030715URL [本文引用: 1]
DOI:10.1007/s11738-015-2023-4URL [本文引用: 1]
DOI:10.1242/dev.104687PMID:24496620 [本文引用: 1]
In plants, the shoot apical meristem (SAM) serves as a reservoir of pluripotent stem cells from which all above ground organs originate. To sustain proper growth, the SAM must maintain homeostasis between the self-renewal of pluripotent stem cells and cell recruitment for lateral organ formation. At the core of the network that regulates this homeostasis in Arabidopsis are the WUSCHEL (WUS) transcription factor specifying stem cell fate and the CLAVATA (CLV) ligand-receptor system limiting WUS expression. In this study, we identified the ERECTA (ER) pathway as a second receptor kinase signaling pathway that regulates WUS expression, and therefore shoot apical and floral meristem size, independently of the CLV pathway. We demonstrate that reduction in class III HD-ZIP and ER function together leads to a significant increase in WUS expression, resulting in extremely enlarged shoot meristems and a switch from spiral to whorled vegetative phyllotaxy. We further show that strong upregulation of WUS in the inflorescence meristem leads to ectopic expression of the AGAMOUS homeotic gene to a level that switches cell fate from floral meristem founder cell to carpel founder cell, suggesting an indirect role for ER in regulating floral meristem identity. This work illustrates the delicate balance between stem cell specification and differentiation in the meristem and shows that a shift in this balance leads to abnormal phyllotaxy and to altered reproductive cell fate.
DOI:10.3389/fpls.2019.00627URL [本文引用: 1]
DOI:10.1186/gb-2009-10-6-r62URL [本文引用: 1]
DOI:10.1139/gen-2019-0190PMID:32053387 [本文引用: 2]
Epigenetic changes influence gene expression and contribute to the modulation of biological processes in response to the environment. Transgenerational epigenetic changes in gene expression have been described in many eukaryotes. However, plants appear to have a stronger propensity for inheriting novel epialleles. This mini-review discusses how plant traits, such as meristematic growth, totipotency, and incomplete epigenetic erasure in gametes promote epiallele inheritance. Additionally, we highlight how plant biology may be inherently tailored to reap the benefits of epigenetic metastability. Importantly, environmentally triggered small RNA expression and subsequent epigenetic changes may allow immobile plants to adapt themselves, and possibly their progeny, to thrive in local environments. The change of epigenetic states through the passage of generations has ramifications for evolution in the natural and agricultural world. In populations containing little genetic diversity, such as elite crop germplasm or habitually self-reproducing species, epigenetics may provide an important source of heritable phenotypic variation. Basic understanding of the processes that direct epigenetic shifts in the genome may allow for breeding or bioengineering for improved plant traits that do not require changes to DNA sequence.
DOI:10.1128/MCB.20.6.1899-1910.2000PMID:10688638 [本文引用: 1]
DOI:10.1242/dev.128892URL [本文引用: 1]
PMID:17605754 [本文引用: 2]
One of the earliest responses of plants to environmental stress is establishing a temporary growth arrest that allows adaptation to adverse conditions. The response to abiotic stress requires the modulation of gene expression, which may be mediated by the alteration of chromatin structures. This alteration can be accomplished with the help of chromatin-remodeling enzymes, such as the various SWI/SNF classes of ATPases. Here, we investigate the role of the Arabidopsis SNF2/Brahma-type AtCHR12 chromatin-remodeling gene in plant growth and development in reaction to adverse environmental conditions. We show that the AtCHR12 chromatin-remodeling gene plays a vital role in mediating the temporary growth arrest of Arabidopsis that is induced upon perception of stress. Exposing an AtCHR12 overexpressing mutant to stress conditions leads to growth arrest of normally active primary buds, as well as to reduced growth of the primary stem. In contrast, the AtCHR12 knockout mutant shows less growth arrest than the wild-type when exposed to moderate stress. Without stress, mutant plants are indistinguishable from the wild-type, and the growth arrest response seems to depend on the severity of the stress applied. Modulation of AtCHR12 expression correlates with changes in expression of dormancy-associated genes. This is in agreement with the concept of AtCHR12 participation in priming the plants for the growth arrest response. Our data indicate that AtCHR12-associated growth arrest differs from DELLA-mediated growth restraint. This establishes AtCHR12 as a novel gene involved in the response repertoire of plants that permits flexible modulation of growth in adverse and/or otherwise limiting environments.
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
DOI:10.1186/s13059-017-1235-xPMID:28566089 [本文引用: 1]
Background: Global warming severely affects flowering time and reproductive success of plants. Alternative splicing of pre-messenger RNA (mRNA) is an important mechanism underlying ambient temperature-controlled responses in plants, yet its regulation is poorly understood. An increase in temperature promotes changes in plant morphology as well as the transition from the vegetative to the reproductive phase in Arabidopsis thaliana via changes in splicing of key regulatory genes. Here we investigate whether a particular histone modification affects ambient temperature-induced alternative splicing and flowering time.Results: We use a genome-wide approach and perform RNA-sequencing (RNA-seq) analyses and histone H3 lysine 36 tri-methylation (H3K36me3) chromatin immunoprecipitation sequencing (ChIP-seq) in plants exposed to different ambient temperatures. Analysis and comparison of these datasets reveal that temperature-induced differentially spliced genes are enriched in H3K36me3. Moreover, we find that reduction of H3K36me3 deposition causes alteration in temperature-induced alternative splicing. We also show that plants with mutations in H3K36me3 writers, eraser, or readers have altered high ambient temperature-induced flowering.Conclusions: Our results show a key role for the histone mark H3K36me3 in splicing regulation and plant plasticity to fluctuating ambient temperature. Our findings open new perspectives for the breeding of crops that can better cope with environmental changes due to climate change.
DOI:10.1007/s00299-006-0243-1URL [本文引用: 1]
DOI:10.1104/pp.104.058974URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1111/nph.2013.201.issue-1URL [本文引用: 1]
DOI:10.1111/nph.2017.215.issue-2URL [本文引用: 1]
DOI:10.1016/j.tig.2012.10.009PMID:23128009 [本文引用: 1]
There is considerable excitement about the potential for epigenetic information to contribute to heritable variation in many species. Our understanding of the molecular mechanisms of epigenetic inheritance is rapidly growing, and it is now possible to profile the epigenome at high resolution. Epigenetic information plays a role in developmental gene regulation, response to the environment, and in natural variation of gene expression levels. Because of these central roles, there is the potential for epigenetics to play a role in crop improvement strategies including the selection for favorable epigenetic states, creation of novel epialleles, and regulation of transgene expression. In this review we consider the potential, and the limitations, of epigenetic variation in crop improvement.Copyright © 2012 Elsevier Ltd. All rights reserved.
DOI:10.1111/nph.2016.210.issue-2URL [本文引用: 1]
DOI:10.1105/tpc.19.00992URL [本文引用: 1]
DOI:10.1007/s00425-008-0743-zPMID:18488247 [本文引用: 1]
Tomato fruit cells are characterized by a strong increase in nuclear ploidy during fruit development. Average ploidy levels increased to similar levels (above 50C) in two distinct fruit tissues, pericarp and locular tissue. However, ploidy profiles differed significantly between these two tissues suggesting a tissue-specific control of endoreduplication in tomato fruit. To determine possible relationships between endoreduplication and epigenetic mechanisms, the methylation status of genomic DNA from pericarp and locular tissue of tomato fruit was analysed. Pericarp genomic DNA was characterized by an increase of CG and/or CNG methylation at the 5S and 18S rDNA loci and at gyspsy-like retrotransposon sequences during fruit growth. A sharp decrease of the global DNA methylation level together with a reduction of methylation at the rDNA loci was also observed in pericarp during fruit ripening. Inversely, no major variation of DNA methylation either global or locus-specific, was observed in locular tissue. Thus, tissue-specific variations of DNA methylation are unlikely to be triggered by the induction of endoreduplication in fruit tissues, but may reflect tissue-specific ploidy profiles. Expression analysis of eight putative tomato DNA methyltransferases encoding genes showed that one chromomethylase (CMT) and two rearranged methyltransferases (DRMs) are preferentially expressed in the pericarp during fruit growth and could be involved in the locus-specific increase of methylation observed at this developmental phase in the pericarp.
[本文引用: 1]
DOI:10.1104/pp.20.00499URL [本文引用: 1]
DOI:10.1073/pnas.1503362112URL [本文引用: 3]
DOI:10.1073/pnas.1705233114URL [本文引用: 1]
DOI:10.1073/pnas.1815441116URL [本文引用: 1]
DOI:10.1186/s13059-018-1587-xURL [本文引用: 1]
DOI:10.1038/ncomms5027PMID:24967512 [本文引用: 2]
Vitamin E (VTE) content is a low heritability nutritional trait for which the genetic determinants are poorly understood. Here, we focus on a previously detected major tomato VTE quantitative trait loci (QTL; mQTL(9-2-6)) and identify the causal gene as one encoding a 2-methyl-6-phytylquinol methyltransferase (namely VTE3(1)) that catalyses one of the final steps in the biosynthesis of gamma- and alpha-tocopherols, which are the main forms of VTE. By reverse genetic approaches, expression analyses, siRNA profiling and DNA methylation assays, we demonstrate that mQTL(9-2-6) is an expression QTL associated with differential methylation of a SINE retrotransposon located in the promoter region of VTE3(1). Promoter DNA methylation can be spontaneously reverted leading to different epialleles affecting VTE3(1) expression and VTE content in fruits. These findings indicate therefore that naturally occurring epialleles are responsible for regulation of a nutritionally important metabolic QTL and provide direct evidence of a role for epigenetics in the determination of agronomic traits.
DOI:10.1038/s41598-017-08512-xURL [本文引用: 1]
[本文引用: 1]
DOI:10.1111/nph.v227.4URL [本文引用: 1]
DOI:10.1073/pnas.1906023116URL [本文引用: 2]
DOI:10.1016/j.cell.2014.06.006PMID:24995981 [本文引用: 1]
Histone variants play crucial roles in gene expression, genome integrity, and chromosome segregation. We report that the four H2A variants in Arabidopsis define different genomic features, contributing to overall genomic organization. The histone variant H2A.W marks heterochromatin specifically and acts in synergy with heterochromatic marks H3K9me2 and DNA methylation to maintain transposon silencing. In vitro, H2A.W enhances chromatin condensation by promoting fiber-to-fiber interactions via its conserved C-terminal motif. In vivo, H2A.W is required for heterochromatin condensation, demonstrating that H2A.W plays critical roles in heterochromatin organization. Similarities in conserved motifs between H2A.W and another H2A variant in metazoans suggest that plants and animals share common mechanisms for heterochromatin condensation.Copyright © 2014 Elsevier Inc. All rights reserved.
DOI:10.1038/s41438-021-00520-3URL [本文引用: 1]
[本文引用: 1]
DOI:10.1104/pp.15.00836URL [本文引用: 1]
DOI:10.1016/j.molp.2020.02.009URL [本文引用: 1]
DOI:10.1073/pnas.1811017115URL [本文引用: 1]
DOI:10.1073/pnas.1716758114URL [本文引用: 1]
DOI:10.1073/pnas.1806304115URL [本文引用: 1]
DOI:10.1038/s42003-020-1059-1PMID:32620865 [本文引用: 1]
Seed development is orchestrated via complex gene regulatory networks and pathways. Epigenetic factors may also govern seed development and seed size/weight. Here, we analyzed DNA methylation in a large-seeded chickpea cultivar (JGK 3) during seed development stages. Progressive gain of CHH context DNA methylation in transposable elements (TEs) and higher frequency of small RNAs in hypermethylated TEs during seed development suggested a role of the RNA-dependent DNA methylation pathway. Frequency of intragenic TEs was higher in CHH context differentially methylated region (DMR) associated differentially expressed genes (DEGs). CG context hyper/hypomethylation within the gene body was observed for most of DMR-associated DEGs in JGK 3 as compared to small-seeded chickpea cultivar (Himchana 1). We identified candidate genes involved in seed size/weight determination exhibiting CG context hypermethylation within the gene body and higher expression in JGK 3. This study provides insights into the role of DNA methylation in seed development and seed size/weight determination in chickpea.
DOI:10.1186/1471-2229-11-93URL [本文引用: 1]
DOI:10.1016/j.phytochem.2014.08.001URL [本文引用: 1]
DOI:10.1038/nature15365URL [本文引用: 3]
DOI:10.1111/pbi.2018.16.issue-11URL [本文引用: 1]
[本文引用: 1]
DOI:10.3389/fpls.2015.00699PMID:26442015 [本文引用: 1]
The transgenerational inheritance of stress-induced epigenetic modifications is still controversial. Despite several examples of defense "priming" and induced genetic rearrangements, the involvement and persistence of transgenerational epigenetic modifications is not known to be general. Here I argue that non-transmission of epigenetic marks through meiosis may be regarded as an epigenetic modification in itself, and that we should understand the implications for plant evolution in the context of both selection for and selection against transgenerational epigenetic memory. Recent data suggest that both epigenetic inheritance and resetting are mechanistically directed and targeted. Stress-induced epigenetic modifications may buffer against DNA sequence-based evolution to maintain plasticity, or may form part of plasticity's adaptive potential. To date we have tended to concentrate on the question of whether and for how long epigenetic memory persists. I argue that we should now re-direct our question to investigate the differences between where it persists and where it does not, to understand the higher order evolutionary methods in play and their contribution.
DOI:10.1186/1471-2229-12-145URL [本文引用: 1]
DOI:10.1093/aob/mcm110URL [本文引用: 2]
DOI:10.1105/tpc.112.101972URL [本文引用: 2]
DOI:10.1186/s12870-015-0674-3URL [本文引用: 2]
DOI:10.1111/jipb.v62.1URL [本文引用: 1]
DOI:10.1111/pce.13502 [本文引用: 1]
Alternating temperatures require fast and coordinated adaptation responses of plants. Cold acclimation has been extensively investigated and results in increased freezing tolerance in Arabidopsis thaliana. Here, we show that the two Arabidopsis accessions, Col-0 and N14, which differ in their freezing tolerance, showed memory of cold acclimation, that is, cold priming. Freezing tolerance was higher in plants exposed to cold priming at 4 degrees C, a lag phase at 20 degrees C, and a second triggering cold stress (4 degrees C) than in plants that were only cold primed. To our knowledge, this is the first report on cold memory improving plant freezing tolerance. The triggering response was distinguishable from the priming response at the levels of gene expression (RNA-Seq), lipid (ultraperformance liquid chromatography-mass spectrometry), and metabolite composition (gas chromatography-mass spectrometry). Transcriptomic responses pointed to induced lipid, secondary metabolism, and stress in Col-0 and growth-related functions in N14. Specific accumulation of lipids included arabidopsides with possible functions as signalling molecules or precursors of jasmonic acid. Whereas cold-induced metabolites such as raffinose and its precursors were maintained in N14 during the lag phase, they were strongly accumulated in Col-0 after the cold trigger. This indicates genetic differences in the transcriptomic and metabolic patterns during cold memory.
DOI:10.3389/fpls.2019.00246PMID:30881369 [本文引用: 1]
In recent years enormous progress has been made in understanding the role of epigenetic regulation response to environmental stimuli, especially in response to stresses. Molecular mechanisms involved in chromatin dynamics and silencing have been explained, leading to an appreciation of how new phenotypes can be generated quickly in response to environmental modifications. In some cases, it has also been shown that epigenetic modifications can be stably transmitted to the next generations. Despite this, the vast majority of studies have been carried out with model plants, particularly with Arabidopsis, and very little is known on how native plants in their natural habitat react to changes in their environment. Climate change has been affecting, sometimes drastically, the conditions of numerous ecosystems around the world, forcing populations of native species to adapt quickly. Although part of the adaptation can be explained by the preexisting genetic variation in the populations, recent studies have shown that new stable phenotypes can be generated through epigenetic modifications in few generations, contributing to the stability and survival of the plants in their natural habitat. Here, we review the recent data that suggest that epigenetic variation can help natural populations to cope to with change in their environments.
DOI:10.3389/fpls.2020.608540URL [本文引用: 1]
DOI:10.1105/tpc.111.095232URL [本文引用: 1]
[本文引用: 3]
[本文引用: 3]
DOI:10.1093/jxb/erw496URL [本文引用: 1]
DOI:10.1111/tpj.2009.60.issue-1URL [本文引用: 1]
PMID:17344210 [本文引用: 1]
The c-Myb protein belongs to a group of early hematopoietic transcription factors that are important for progenitor generation and proliferation. These factors have been hypothesized to participate in establishing chromatin patterns specific for hematopoietic genes. In a two-hybrid screening we identified the chromatin remodeling factor Mi-2alpha as an interaction partner for human c-Myb. The main interacting domains were mapped to the N-terminal region of Mi-2alpha and the DNA-binding domain of c-Myb. Surprisingly, functional analysis revealed that Mi-2alpha, previously studied as a subunit in the NuRD co-repressor complex, enhanced c-Myb-dependent reporter activation. Consistently, knock-down of endogenous Mi-2alpha in c-Myb-expressing K562 cells, led to down-regulation of the c-Myb target genes NMU and ADA. When wild-type and helicase-dead Mi-2alpha were compared, the Myb-Mi-2alpha co-activation appeared to be independent of the ATPase/DNA helicase activity of Mi-2alpha. The rationale for the unexpected co-activator function seems to lie in a dual function of Mi-2alpha, by which this factor is able to repress transcription in a helicase-dependent and activate in a helicase-independent fashion, as revealed by Gal4-tethering experiments. Interestingly, desumoylation of c-Myb potentiated the Myb-Mi-2alpha transactivational co-operation, as did co-transfection with p300.
DOI:10.3389/fpls.2019.00900URL [本文引用: 1]
DOI:10.1111/tpj.v100.5URL [本文引用: 1]
DOI:10.1111/tpj.2018.95.issue-3URL [本文引用: 1]
DOI:10.1038/s41422-019-0145-8URL [本文引用: 1]
DOI:10.1111/tpj.2013.74.issue-5URL [本文引用: 1]
DOI:10.1007/s12298-013-0176-4URL [本文引用: 1]
DOI:10.1038/srep39843URL [本文引用: 2]
[本文引用: 1]
DOI:10.1104/pp.20.00141URL [本文引用: 1]
DOI:10.1371/journal.pgen.1008370URL [本文引用: 1]
DOI:10.1007/s11103-018-0810-1PMID:30604322 [本文引用: 1]
Morphological and transcriptomic evidences provide us strong support for the function of ANAC019 in reproductive development under drought stress. Plants are sensitive to drought conditions, particularly at the reproductive stage. Several studies have reported drought effects on crop reproductive development, but the molecular mechanism underlying drought response during reproduction is still unclear. A recent study showed that drought induces in Arabidopsis inflorescence increased expression of many genes, including ANAC019. However, the function of ANAC019 in drought response during reproductive development has not been characterized. Here, we report an investigation of the ANAC019 function in the response to drought during reproduction. ANAC019 is preferentially expressed in the inflorescence compared with the leaf, suggesting possible roles in regulating both stress response and flower development. The anac019 mutant was more sensitive to drought than WT plant, and exhibited a delay in recovery of floral organ development under prolonged drought stress. Moreover, many fewer genes were differentially expressed in the anac019 inflorescence under drought than that of WT, suggesting that the mutant was impaired in drought-induced gene expression. The genes affected by ANAC019 were associated with stress and hormone responses as well as floral development. In particular, the expression levels of several key drought-induced genes, DREB2A, DREB2B, ARF2, MYB21 and MYB24, were dramatically reduced in the absence of ANAC019, suggesting that ANAC019 is an upstream regulator these genes for drought response and flower development. These results provide strong support for the potential function of ANAC019 in reproductive development under drought stress.
DOI:10.3389/fpls.2020.00623URL [本文引用: 1]
DOI:10.1111/j.1365-313X.2011.04534.xURL [本文引用: 1]
DOI:10.1111/nph.14933URL [本文引用: 1]
DOI:10.4161/psb.4.11.10103URL [本文引用: 1]
DOI:10.1038/ncomms1732URL [本文引用: 1]
DOI:10.3390/ijms20102567URL [本文引用: 1]
DOI:10.1105/tpc.18.00437URL [本文引用: 2]
DOI:10.1038/ncomms9326URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1105/tpc.112.105114URL [本文引用: 1]
DOI:10.1111/nph.16518PMID:32129897 [本文引用: 1]
Lamin proteins in animals are implicated in important nuclear functions, including chromatin organization, signalling transduction, gene regulation and cell differentiation. Nuclear Matrix Constituent Proteins (NMCPs) are lamin analogues in plants, but their regulatory functions remain largely unknown. We report that OsNMCP1 is localized at the nuclear periphery in rice (Oryza sativa) and induced by drought stress. OsNMCP1 overexpression resulted in a deeper and thicker root system, and enhanced drought resistance compared to the wild-type control. An assay for transposase accessible chromatin with sequencing (ATAC-seq) analysis revealed that OsNMCP1-overexpression altered chromatin accessibility in hundreds of genes related to drought resistance and root growth, including OsNAC10, OsERF48, OsSGL, SNAC1 and OsbZIP23. OsNMCP1 can interact with SWITCH/SUCROSE NONFERMENTING (SWI/SNF) chromatin remodelling complex subunit OsSWI3C. The reported drought resistance or root growth-related genes that were positively regulated by OsNMCP1 were negatively regulated by OsSWI3C under drought stress conditions, and OsSWI3C overexpression led to decreased drought resistance. We propose that the interaction between OsNMCP1 and OsSWI3C under drought stress conditions may lead to the release of OsSWI3C from the SWI/SNF gene silencing complex, thus changing chromatin accessibility in the genes related to root growth and drought resistance.© 2020 The Authors. New Phytologist © 2020 New Phytologist Trust.
DOI:10.1093/pcp/pcq182URL [本文引用: 1]
[本文引用: 1]
DOI:10.1093/pcp/pcu059URL [本文引用: 1]
[本文引用: 1]
DOI:10.1093/jxb/erv304URL [本文引用: 1]
DOI:10.1016/j.jgg.2018.09.004URL [本文引用: 1]
DOI:10.1186/gb-2013-14-6-r59URL [本文引用: 1]
DOI:10.4161/psb.5.10.13168URL [本文引用: 1]
DOI:10.1371/journal.pgen.1001325URL [本文引用: 1]
DOI:10.1007/s10059-011-0122-7PMID:21912873 [本文引用: 1]
The traditional focus on the central dogma of molecular biology, from gene through RNA to protein, has now been replaced by the recognition of an additional mechanism. The new regulatory mechanism, post-translational modifications to proteins, can actively alter protein function or activity introducing additional levels of functional complexity by altering cellular and sub-cellular location, protein interactions and the outcome of biochemical reaction chains. Modifications by ubiquitin (Ub) and ubiquitin-like modifiers systems are conserved in all eukaryotic organisms. One of them, small ubiquitin-like modifier (SUMO) is present in plants. The SUMO mechanism includes several isoforms of proteins that are involved in reactions of sumoylation and de-sumoylation. Sumoylation affects several important processes in plants. Outstanding among those are responses to environmental stresses. These may be abiotic stresses, such as phosphate deficiency, heat, low temperature, and drought, or biotic stressses, as well including defense reactions to pathogen infection. Also, the regulations of flowering time, cell growth and development, and nitrogen assimilation have recently been added to this list. Identification of SUMO targets is material to characterize the function of sumoylation or desumoylation. Affinity purification and mass spectrometric identification have been done lately in plants. Further SUMO noncovalent binding appears to have function in other model organisms and SUMO interacting proteins in plants will be of interest to plant biologists who dissect the dynamic function of SUMO. This review will discuss results of recent insights into the role of sumoylation in plants.
[本文引用: 1]
[本文引用: 1]
DOI:10.1073/pnas.1211757110URL [本文引用: 3]
DOI:10.1128/JVI.01474-08URL [本文引用: 1]
DOI:10.1111/nph.2015.208.issue-2URL [本文引用: 1]
DOI:10.1104/pp.111.187468URL [本文引用: 1]
DOI:10.1105/tpc.107.054494URL [本文引用: 1]
DOI:10.1111/nph.2019.221.issue-2URL [本文引用: 1]
PMID:15148604 [本文引用: 1]
Transcripts that specifically accumulate in transgenic tobacco plants expressing an anti-sense construct for a tobacco type I DNA methyltransferase, NtMET1, were screened by the differential display method. Of the 31 genes identified, 16 encoded proteins with known functions; ten of these were related to biotic and abiotic stress responses, and the other six to cellular functions. In order to examine whether expression of these genes is correlated with DNA methylation status under natural stress conditions, a pathogen-responsive gene (NtAlix1) was selected as representative, and assayed for transcript induction and genomic methylation in tobacco plants infected with tobacco mosaic virus (TMV). In inoculated leaves of wild-type plants, NtAlix1 transcripts began to accumulate 12 h after the onset of the hypersensitive response (HR), and levels remained high for up to 24 h. Changes in the methylation status at the locus became obvious 24 h later, as detected by digestion of genomic DNA with a methylation-sensitive restriction enzyme. The results suggest that the level of DNA methylation may change in response to external stresses, and that this is closely related to the activation of stress-responsive genes.
[本文引用: 1]
DOI:10.1104/pp.110.161497URL [本文引用: 1]
DOI:10.1104/pp.113.214551URL [本文引用: 1]
DOI:10.1105/tpc.104.028514URL [本文引用: 1]
DOI:10.1111/j.1364-3703.2010.00621.xPMID:20618712 [本文引用: 1]
In eukaryotic genomes, gene expression and DNA recombination are affected by structural chromatin traits. Chromatin structure is shaped by the activity of enzymes that either introduce covalent modifications in DNA and histone proteins or use energy from ATP to disrupt histone-DNA interactions. The genomic 'marks' that are generated by covalent modifications of histones and DNA, or by the deposition of histone variants, are susceptible to being altered in response to stress. Recent evidence has suggested that proteins generating these epigenetic marks play crucial roles in the defence against pathogens. Histone deacetylases are involved in the activation of jasmonic acid- and ethylene-sensitive defence mechanisms. ATP-dependent chromatin remodellers mediate the constitutive repression of the salicylic acid-dependent pathway, whereas histone methylation at the WRKY70 gene promoter affects the activation of this pathway. Interestingly, bacterial-infected tissues show a net reduction in DNA methylation, which may affect the disease resistance genes responsible for the surveillance against pathogens. As some epigenetic marks can be erased or maintained and transmitted to offspring, epigenetic mechanisms may provide plasticity for the dynamic control of emerging pathogens without the generation of genomic lesions.
[本文引用: 1]
[本文引用: 1]
DOI:10.1073/pnas.98.1.200URL [本文引用: 1]
DOI:10.1016/j.molp.2020.11.002URL [本文引用: 1]
PMID:10792817 [本文引用: 1]
Post-translational modification of histones, in particular acetylation, is an important mechanism in the regulation of eukaryotic gene expression. Histone deacetylases are enzymes that remove acetyl groups from the core histones and play a key role in the repression of transcription. HD2 is a maize histone deacetylase, which shows no sequence homology to the histone deacetylases identified from other eukaryotes. We have identified two putative HD2-like histone deacetylase cDNA clones, AtHD2A and AtHD2B, from Arabidopsis thaliana by screening the expressed sequence tag database. AtHD2A and AtHD2B encode putative proteins of 246 and 305 amino acids, and share 44% and 46% amino acid identity to the maize HD2, respectively. Northern blot analysis indicated that AtHD2A was highly expressed in flowers and young siliques of Arabidopsis plants, whereas AtHD2B was widely expressed in stems, leaves, flowers and young siliques. AtHD2A repressed transcription when directed to a promoter containing GAL4-binding sites as a GAL4 fusion protein. Deletion of the extended acidic domain or the domain containing predicted catalytic residues of AtHD2A resulted in the loss of gene repression activity, revealing the importance of both domains to AtHD2A function. Arabidopsis plants were transformed with a gene construct comprising an AtHD2A cDNA in the antisense orientation driven by a strong constitutive promoter, -394tCUP. Silencing of AtHD2A expression resulted in aborted seed development in transgenic Arabidopsis plants, suggesting that the AtHD2A gene product was important in the reproductive development of Arabidopsis thaliana.
DOI:10.1126/science.aai8898URL [本文引用: 1]
PMID:16627744 [本文引用: 1]
Plants and animals activate defenses after perceiving pathogen-associated molecular patterns (PAMPs) such as bacterial flagellin. In Arabidopsis, perception of flagellin increases resistance to the bacterium Pseudomonas syringae, although the molecular mechanisms involved remain elusive. Here, we show that a flagellin-derived peptide induces a plant microRNA (miRNA) that negatively regulates messenger RNAs for the F-box auxin receptors TIR1, AFB2, and AFB3. Repression of auxin signaling restricts P. syringae growth, implicating auxin in disease susceptibility and miRNA-mediated suppression of auxin signaling in resistance.
DOI:10.1073/pnas.0608258103URL [本文引用: 1]
DOI:10.1094/MPMI-05-13-0137-RURL [本文引用: 1]
PMID:2022628 [本文引用: 1]
The effect of O6-alkylation of guanine residues on the enzymic methylation of cytosine has been studied using synthetic oligonucleotides in which all guanines in cytosine-guanine sequences at potentially methylatable sites are replaced by O6-methylguanine. In contrast with the unmodified forms, which showed high acceptance activity for methyl-3H-labeled groups from S-adenosyl-L-[methyl-3H]methionine in the presence of DNA methylase, the modified oligonucleotides were not substrates for the enzyme either in the single-stranded or annealed forms. In view of the importance of cytosine methylation in the down-regulation of certain genes, the potential to affect gene expression by this mechanism may be a contributory factor in the toxic and carcinogenic effects of chemical methylating agents.
[本文引用: 1]
[本文引用: 1]
DOI:10.1104/pp.17.01054URL [本文引用: 1]
PMID:17486309 [本文引用: 1]
Understanding dynamics and inheritance of DNA methylation represents important facets for elucidating epigenetic paradigms in plant development and evolution. Using four sets of sorghum (Sorghum bicolor L.) inter-strain hybrids and their inbred parents, the developmental stability and inheritance of cytosine methylation in two tissues, leaf and endosperm, by MSAP analysis were investigated. It was found that in all lines (inbred and hybrid) studied, endosperm exhibited a markedly reduced level of full methylation of the external cytosine or both cytosines at the CCGG sites relative to leaf, which caused a variable reduction in the estimated total methylation level in endosperm by 6.89-19.69% (11.47% on average). For both tissues, a great majority of cytosine methylation profiles transmitted to F1 hybrids, however, from 1.69 to 3.22% of the profiles showed altered patterns in hybrids. Both inherited and altered methylation profiles can be divided into distinct groups, and their frequencies are variable among the cross-combinations, and between the two tissues. The variations in methylation level and pattern detected in the hybrids were not caused by parental heterozygosity, and they could be either non-random or stochastic among hybrid individuals. Homology analysis of isolated bands that showed endosperm-specific hypomethylation or variation in hybrids indicated that diverse sequences were involved, including known-function cellular genes and mobile elements. RT-PCR analysis of six genes representing endosperm-specific hypomethylation in MSAP profiles indicated that all showed higher expression in endosperm than in leaf, suggesting involvement of methylation state in regulating tissue-specific or tissue-biased expression in sorghum. Analysis on leaf-RNA from 5-azacytidine-treated plants further corroborated this possibility.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.3389/fpls.2018.00013PMID:29441076 [本文引用: 1]
Heterosis refers to a quantitative phenomenon in which F1 hybrid trait values exceed the mean of the parental values in a positive direction. Generally, it is dependent on a high degree of heterozygosity, which is maintained in hybrid breeding by developing parental lines in separate, genetically distinct heterotic groups. The mobility of small RNAs (sRNAs) that mediate epigenetic regulation of gene expression renders them promising candidates for modulating the action of combined diverse genomes in trans-and evidence already indicates their contribution to transgressive phenotypes. By sequencing small RNA libraries of a panel of 21 maize parental inbred lines we found a low overlap of 35% between the sRNA populations from both distinct heterotic groups. Surprisingly, in contrast to genetic or gene expression variation, parental sRNA expression variation is negatively correlated with grain yield (GY) heterosis. Among 0.595 million expressed sRNAs, we identified 9,767, predominantly 22- and 24-nt long sRNAs, which showed an association of their differential expression between parental lines and GY heterosis of the respective hybrids. Of these sRNAs, 3,485 or 6,282 showed an association with high or low GY heterosis, respectively, thus the low heterosis associated group prevailing at 64%. The heterosis associated sRNAs map more frequently to genes that show differential expression between parental lines than reference sets. Together these findings suggest that trans-chromosomal actions of sRNAs in hybrids might add up to a negative contribution in heterosis formation, mediated by unfavorable gene expression regulation. We further revealed an exclusive accumulation of 22-nt sRNAs that are associated with low GY heterosis in pericentromeric genomic regions. That recombinational suppression led to this enrichment is indicated by its close correlation with low recombination rates. The existence of this enrichment, which we hypothesize resulted from the separated breeding of inbred lines within heterotic groups, may have implications for hybrid breeding strategies addressing the recombinational constraints characteristic of complex crop genomes.
DOI:10.1016/j.tplants.2009.12.003URL [本文引用: 1]
DOI:10.1073/pnas.1019217108URL [本文引用: 1]
DOI:10.1073/pnas.1202073109URL [本文引用: 1]
DOI:10.1016/j.pbi.2012.01.007URL [本文引用: 1]
DOI:10.1105/tpc.109.072041URL [本文引用: 2]
DOI:10.1073/pnas.1201043109URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/nature07523URL [本文引用: 1]
DOI:10.1534/genetics.106.056580URL [本文引用: 1]
DOI:10.1111/tpj.2011.67.issue-4URL [本文引用: 1]
DOI:10.1360/972013-1103URL [本文引用: 1]
DOI:10.1360/972013-1103URL [本文引用: 1]
DOI:10.1186/s13059-017-1263-6PMID:28655328 [本文引用: 1]
Plants frequently have to weather both biotic and abiotic stressors, and have evolved sophisticated adaptation and defense mechanisms. In recent years, chromatin modifications, nucleosome positioning, and DNA methylation have been recognized as important components in these adaptations. Given their potential epigenetic nature, such modifications may provide a mechanistic basis for a stress memory, enabling plants to respond more efficiently to recurring stress or even to prepare their offspring for potential future assaults. In this review, we discuss both the involvement of chromatin in stress responses and the current evidence on somatic, intergenerational, and transgenerational stress memory.
DOI:10.1016/j.cell.2007.07.007URL [本文引用: 1]
DOI:10.1073/pnas.0510213103URL [本文引用: 1]
DOI:10.1038/nrg.2017.45PMID:28669983 [本文引用: 1]
Plant breeding has traditionally relied on combining the genetic diversity present within a species to develop combinations of alleles that provide desired traits. Epigenetic diversity may provide additional sources of variation within a species that could be captured or created for crop improvement. It will be important to understand the sources of epigenetic variation and the stability of newly formed epigenetic variants over generations to fully use the potential of epigenetic variation to improve crops. The development and application of methods for widespread epigenome profiling and engineering may generate new avenues for using the full potential of epigenetics in crop improvement.
DOI:10.1126/science.15739260URL [本文引用: 1]
DOI:10.1038/ng.3117URL [本文引用: 2]
DOI:10.1038/nature11119URL [本文引用: 1]
DOI:10.1038/srep09192URL [本文引用: 2]
DOI:10.1038/nrg3097PMID:22207165 [本文引用: 1]
The completion of reference genome sequences for many important crops and the ability to perform high-throughput resequencing are providing opportunities for improving our understanding of the history of plant domestication and to accelerate crop improvement. Crop plant comparative genomics is being transformed by these data and a new generation of experimental and computational approaches. The future of crop improvement will be centred on comparisons of individual plant genomes, and some of the best opportunities may lie in using combinations of new genetic mapping strategies and evolutionary analyses to direct and optimize the discovery and use of genetic variation. Here we review such strategies and insights that are emerging.
DOI:10.1038/nature08498URL [本文引用: 1]
DOI:10.1073/pnas.0901942106URL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}