 ,, 李金环, 吴强上海交通大学系统生物医学研究院比较生物医学研究中心,系统生物医学教育部重点实验室,上海 200240
,, 李金环, 吴强上海交通大学系统生物医学研究院比较生物医学研究中心,系统生物医学教育部重点实验室,上海 200240Combinatorial CRISPR inversions of CTCF sites in HOXD cluster reveal complex insulator function
Xianglong He,, Jinhuan Li, Qiang WuCenter for Comparative Biomedicine, Key laboratory of Systems Biomedicine (Ministry of Education), Institute of Systems Biomedicine, Shanghai Jiao Tong University, Shanghai 200240, China通讯作者: 吴强,博士,教授,研究方向:三维基因组架构与功能。E-mail:qwu123@gmail.com
编委: 谷峰
收稿日期:2021-04-11修回日期:2021-05-12网络出版日期:2021-08-20
| 基金资助: |
Received:2021-04-11Revised:2021-05-12Online:2021-08-20
| Fund supported: |
作者简介 About authors
何象龙,在读硕士研究生,专业方向:生物学。E-mail:

摘要
CTCF (CCCTC-binding factor)是一种重要的染色质架构蛋白,其与绝缘子的方向性结合在哺乳动物基因组三维空间结构形成和维持中起着至关重要的作用。正向-反向相对方向的CTCF结合位点(简称CTCF位点)可以在染色质黏连蛋白(cohesin)的协助下,形成染色质环,介导远距离DNA元件之间的相互作用;而在染色质拓扑结构域边界区域的CTCF位点呈现反向-正向相背方向分布,发挥绝缘子的功能。为进一步研究CTCF介导染色质环的形成与其绝缘功能之间的关系,本研究采用DNA片段编辑方法通过设计成对sgRNA (dual sgRNA)构建了一系列HOXD基因簇区域CTCF位点反转的单细胞克隆。定量高分辨率染色质构象捕获实验显示边界区域CTCF位点反转会改变原有的染色质环方向,通过环挤出模型(loop extrusion)形成新的染色质环,引起染色质拓扑结构域边界漂移至新形成的一对反向-正向CTCF位点处。此外,串联排列的CTCF位点可以通过阻碍反方向渗透的黏连蛋白继续滑动发挥绝缘子的功能。RNA-seq实验发现CTCF位点反转引起的局部基因组空间结构变化会进一步影响基因的表达。上述研究表明相邻两个染色质拓扑结构域边界区域的反向-正向CTCF位点可以通过与各自所在拓扑结构域内相向的CTCF位点形成染色质环,阻碍黏连蛋白滑动,该发现为进一步研究CTCF的绝缘功能和其对基因组拓扑结构的影响提供了参考。
关键词:
Abstract
CTCF (CCCTC-binding factor) is a zinc-finger protein which plays a vital role in the three-dimensional (3D) genome architecture. A pair of forward-reverse convergent CTCF binding sites (CBS elements) mediates long-distance DNA interactions to form chromatin loops with the assistance of the cohesin complex, while CBS elements at the chromatin domain boundaries show reverse-forward divergent patterns and function as insulators to discriminate against DNA interaction between chromatin domains. However, there are still many unresolved problems regarding CTCF-mediated insulation function. In order to study the connections between chromatin loops and the insulation function of CBS elements, we combinatorically inverted CBS elements at the HOXD locus by using CRISPR/Cas9 DNA-fragment editing methods in the HEK293T cell line and obtained five different kinds of single-cell CRISPR clones. By performing quantitative high-resolution chromosome conformation capture copy (QHR-4C) experiments, we found that boundary CBS inversions abolish original chromatin loops and establish new loops from the opposite direction, thus shifting the insulator boundary to the new divergent CTCF sites. Furthermore, tandem CBS elements block cohesin permeated from the opposite orientation to function as insulators. RNA-seq experiments showed that alterations of local three-dimensional genome architecture would further influence gene expression of the HOXD cluster. In conclusion, a pair of divergent CBS elements function as insulators by forming chromatin loops within chromatin domains to block cohesin sliding.
Keywords:
PDF (1974KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
何象龙, 李金环, 吴强. HOXD基因簇内一系列CTCF位点反转揭示绝缘子功能. 遗传[J], 2021, 43(8): 758-774 doi:10.16288/j.yczz.21-131
Xianglong He.
大约2 m长的人类基因组通过多次盘曲折叠,最终被包裹在直径约5 μm的细胞核中,形成了复杂的染色质三维空间结构[1,2,3]。特定的染色质空间结构对于DNA复制[4]、基因转录调控[5,6]和DNA修复[7,8]等生物学过程有着重要作用。早期通过光学显微镜研究发现,有丝分裂间期染色体有规律的分布在细胞核的不同区域,被称为染色质领地(chromosome territories)[9];随着染色质构象捕获(chromosome conformation capture, 3C)[10]、环状染色质构象捕获(circular chromosome conformation capture, 4C)[11]、Hi-C[12]和配对末端标签测序分析染色质相互作用(chromatin interaction analysis by paired-end tag sequencing, ChIA-PET)[13]等技术的发展,A/B隔室(compartment)、拓扑相关结构域(topologically associated domain, TAD)和染色质环(chromatin loop)等概念被相继提出[2,12,14~18],促进人类从多个层次对染色质高级结构的认识。基于Hi-C数据,基因组上平均大小约为1 Mb、内部DNA相互作用紧密的区域,被定义为TAD[13,14]。在人的不同细胞系中,TAD表现出较好的保守性[15]。通常认为在TAD内部,增强子(enhancer)、绝缘子(insulator)与沉默子(silencer)等顺式作用元件可以通过CTCF(CCCTC-binding factor)和YY1等染色质架构蛋白介导的染色质环与基因的启动子(promoter)发生相互作用[5,19~22],来调控基因的表达。
CTCF蛋白是一种参与染色质折叠的重要架构蛋白。CTCF蛋白最早发现于对鸡(Gallus gallus) c-myc基因的研究,由于其与DNA结合的序列含有高度保守的CCCTC序列,因而被称为CCCTC结合因子[23]。CTCF蛋白通过11个串联的锌指结构与人类基因组上成千上万的绝缘子调控元件结合[19,24~27]。在哺乳动物中,CTCF结合位点(CTCF-binding site, CBS)是最主要的绝缘子调控元件,包含4个模块(module),即module 1~4,其具有方向性,module 1~4的方向是正向,module 4~1的方向则为反向[5]。CTCF在黏连蛋白的协助下,通过环挤出模型介导远距离的DNA相互作用[16,18]。在TAD边界处分布着大量反向-正向的CBS,通过阻断cohesin的滑动,起到绝缘子的功能[2,5,28~30]。删除CBS导致CTCF无法结合,会破坏原有DNA之间的染色质环,引起局部基因组构象的改变[28,31~33]。反之,插入异位CBS会形成新的染色质环,同时作为绝缘子阻碍原有染色质环的形成[34,35]。在Pchdα基因簇的增强子和启动子之间插入正向或者反向的CBS,都可以起到绝缘子的作用,显示绝缘子没有方向性[34]。在原钙粘蛋白(protocadherin, Pcdh)、免疫球蛋白 (immunoglobulin, Ig)、HoxD等基因簇存在着成串同向的CBS,这些CBS均有机会形成染色质环[5,36,37],显示正向-反向CBS对cohesin的阻碍作用是有渗透性的,这可能是由于CTCF与DNA的动态结合[34,38]。然而,当前对于成串CBS绝缘子功能的研究主要侧重于启动子与增强子的相互作用,在染色质拓扑结构域边界处的成串CBS的绝缘子功能还缺少系统性的研究。
HoxD基因簇是控制四肢发育时空顺序的重要基因簇,被广泛应用于对基因组三维空间结构的研究[19,39]。HoxD基因簇区域存在一系列的CBS,与两侧调控区域内的CBS通过环挤压模型分别形成两个TAD[19,39~42]。HoxD基因有序的表达受到两侧TAD内增强子簇的调控,其表达分为两个阶段;在四肢发育早期,3′端粒侧TAD (telomeric domain, T-DOM)内由CS38-41 (conserved sequence elements 38-41)和CS65 (conserved sequence element 65)组成的下游增强子簇或超级增强子(downstream enhancer cluster or super-enhancer)共同调节Hoxd1-11的表达,并在四肢近端维持这种表达模式;在肢端发育后期,5′中心粒侧TAD (centromeric domain, C-DOM)内由Island 1-5、GCR和Prox组成的上游增强子簇或超级增强子(downstream enhancer cluster or super-enhancer)协同调节四肢远端Hoxd9-13的表达[19,42,43]。HoxD基因簇区域大范围的片段反转破坏原来的基因组三维结构,影响HoxD基因簇的正常表达[39,41,44],但这些大范围的片段反转主要针对增强子调控元件,针对绝缘子CTCF位点的DNA片段反转研究较少。
成簇规律间隔短回文重复系统(clustered regularly interspaced short palindromic repeats/CRISPR-associated nuclease 9, CRISPR/Cas9)源于古菌和细菌中的一种适应性免疫系统,近年来被广泛应用于DNA片段反转、DNA片段删除和染色体异位等编辑实验[22,45~52]。HEK293T细胞系是一种常用的模式细胞系,常被用于对染色质三维空间结构的研究[5,53,54]。本实验室之前通过对HEK293T细胞系以及HEC-1B等细胞系中CBS位点反转的研究发现,CBS方向性是哺乳动物细胞中普遍存在的规律[5]。本研究利用CRISPR DNA片段编辑技术在HEK293T细胞系中对HOXD基因簇所在区域中串联排列的CBS元件进行一系列组合性的反转,利用定量高分辨率染色质构象捕获(quantitative high-resolution chromosome conformation capture copy, QHR-4C)技术[34]研究CBS反转后染色质空间构象的变化,发现CBS反转会改变该CBS原有的远距离DNA相互作用,引起染色质拓扑结构域边界漂移至新形成的一对反向-正向的CBS处;同时,相邻两个染色质拓扑结构域边界区域的反向-正向CBS可以通过与各自所在拓扑结构域内的相向CBS形成染色质环,发挥绝缘子功能,这种绝缘子的作用有助于稳定基因组三维空间构象。
1 材料与方法
1.1 实验材料
人胚肾细胞系HEK293T细胞从中国科学院细胞库购买;pcDNA3.1-Cas9-WT质粒由北京大学席建忠教授惠赠;pGL3-U6-sgRNA质粒由上海科技大学黄行许教授惠赠;DMEM培养基从美国Hyclone公司购买;胎牛血清购自上海ExCell公司;Bsa Ⅰ内切酶、Dpn Ⅱ内切酶、T4 DNA连接酶、蛋白酶K和ChIP-Seq建库试剂盒均购自美国NEB公司;DNA凝胶回收试剂盒和质粒DNA小量试剂盒购自美国AXYGEN公司;嘌呤霉素、NP-40、Triton X-100、DMSO、Tris base、SDS、醋酸钠购自美国SIGMA公司;Lipofectamine 3000、RNaseA、糖原、16%甲醛溶液购自美国Thermo Fisher Scientific公司;胰酶和青链霉素购自美国Gibco公司;Streptavidin Magnetic Beads购自美国Invitrogen公司;RNA-seq试剂盒和Phanta酶购自南京诺维赞生物科技有限公司;TRIzol购自美国Ambion公司;AMPure XP beads购自美国Beckman Coulter公司;CTCF抗体从英国Abcam公司购买;三氯甲烷、硼酸和乙醇购自上海沪试实验室器材股份有限公司;异丙醇和氯化钠购自上海凌峰化学试剂有限公司;引物均由上海生物工程股份有限公司和北京擎科新业生物技术有限公司提供。1.2 HOXD基因簇TAD分析
从SRA数据库下载HEK293细胞的Hi-C数据[54]:SRR710075、SRR710076、SRR710077,以hg19为参考基因组,利用HiC-Pro[55]软件进行分析,获得标准化矩阵;截取chr2:175,760,000~178,200,000范围的矩阵,利用TADtool绘制热图[56]。1.3 CTCF结合序列分析
分析CTCF蛋白ChIP-seq数据,确定HOXD基因簇区域CBS位点分布情况;利用MEME[57]软件分析获得CTCF的结合基序(motif),以此基序作为参考,使用FIMO[57]软件查找HOXD基因簇区域CTCF结合序列,并确定CBS方向。1.4 细胞培养
HEK293T细胞用89% DMEM、10%胎牛血清和1%青链霉素双抗配制的完全培养基培养,在5% CO2浓度、37℃恒温培养箱中培养。1.5 构建sgRNA质粒
使用Bsa I酶将pGL3-U6-sgRNA质粒线性化,并用琼脂糖凝胶进行分离纯化。将购买的sgRNA正反脱氧核苷酸单链溶解并稀释到20 μmol/L (表1),配制引物退火溶液,体系如下:正反单链sgRNA各2 μL,2 μL NEB buffer2,14 μL ddH2O,并置于水浴锅中95℃ 5 min,然后自然冷却至室温。带有悬挂端的双链sgRNA与获得的线性化载体连接,然后用感受态细胞转化摇菌。利用质粒小量试剂盒提取质粒,并用桑格测序(Sanger sequencing)确认质粒是否正确连接。Table 1
表1
表1引物序列
Table 1
| 类型 | 引物名称 | 序列(5′→3′) |
|---|---|---|
| sgRNA | a-sgRNA1F | accgCGAAGAGTGCGGGAGAACGG |
| a-sgRNA1R | aaacCCGTTCTCCCGCACTCTTCG | |
| b-sgRNA1F | accgGGCGCATCAGGAATGTAAG | |
| b-sgRNA1R | aaacCTTACATTCCTGATGCGCC | |
| b-sgRNA2F | accgTTCCAGAGATTATGAGCCAG | |
| b-sgRNA2R | aaacCTGGCTCATAATCTCTGGAA | |
| c-sgRNA2F | accgAGTAAAACTGGGTGTTGCC | |
| c-sgRNA2R | aaacGGCAACACCCAGTTTTACT | |
| e-sgRNA1F | accgAACTGTGCTCAAACGCTCTC | |
| e-sgRNA1R | aaacGAGAGCGTTTGAGCACAGTT | |
| e-sgRNA2F | accgGAGGCGCAAACAGCTGTTGT | |
| e-sgRNA2R | aaacACAACAGCTGTTTGCGCCTC | |
| PCR | a-1F | TTCCAGCACCTCGGCTTTGTC |
| a-1R | CCCACTTTCCACCTCTGTCCTG | |
| b-1F | GTCCGCCCGTGAGCTTCTGAA | |
| b-1R | GCGTTGGCAGGGCTGGACTC | |
| b-1F1 | TTCCCTGACCCTCCAAGCAC | |
| b-1R1 | CTCACAGCAGCCGAAACCG | |
| b-2F | GACACCTTGGTTCAGGCTCG | |
| b-2R | CAGAGGCTGCATCAAACCAC | |
| c-1F | TGATGCAGCCTCTGTGACCG | |
| c-1R | AGTTTTCCCGTGGCGTCTGA | |
| c-2F | GGACAACCCCTCACCTTGAA | |
| c-2R | TATGGTGCCCAAAGTCCCAC | |
| e-1F | TTCCCTGTCCCAGCTTGATTTC | |
| e-1R | TCAACAGTGAAGGGCGGTGC | |
| e-2F | CAAGCCACTCTCCCGCCACTA | |
| e-2R | TCGCTCTCGTCCTCTCTTGGG | |
| bio-primer | upstream-a-bioprimer | 5′biotin-GGACGAGGGACATAGAGAGTTC |
| downstream-e-bioprimer | 5′biotin- AGGAGCCCAGGCATAGAGAC | |
| CS38-bioprimer | 5′biotin-CCGAATTAAATCCCCGTGAC | |
| Island5-bioprimer | 5′biotin-AAACACAAATGCATCAACCTG | |
| P5 primer | upstream-a-P5 | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTCTGTCTAGAGATTCAAGTATTTCCCA |
| P5 primer | downstream-e-P5 | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTACGCGATACTCAAGGGAATACAAGCC |
| CS38-P5 | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTAGGTTACAGTGTCATTTTCCCTG | |
| Island5-P5 | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTATGCATCTCATGAAGCTGGCATCT | |
| P7 index | P7-index-1 | CAAGCAGAAGACGGCATACGAGATCGAGTAATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| P7-index-2 | CAAGCAGAAGACGGCATACGAGATTCTCCGGAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-3 | CAAGCAGAAGACGGCATACGAGATAATGAGCGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-4 | CAAGCAGAAGACGGCATACGAGATGGAATCTCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-5 | CAAGCAGAAGACGGCATACGAGATTTCTGAATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-6 | CAAGCAGAAGACGGCATACGAGATACGAATTCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-7 | CAAGCAGAAGACGGCATACGAGATAGCTTCAGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-8 | CAAGCAGAAGACGGCATACGAGATGCGCATTAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-9 | CAAGCAGAAGACGGCATACGAGATCATAGCCGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-10 | CAAGCAGAAGACGGCATACGAGATTTCGCGGAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-11 | CAAGCAGAAGACGGCATACGAGATGCGCGAGAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-12 | CAAGCAGAAGACGGCATACGAGATCTATCGCTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-13 | CAAGCAGAAGACGGCATACGAGATAGAGTACTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-14 | CAAGCAGAAGACGGCATACGAGATGCTCCGTAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-15 | CAAGCAGAAGACGGCATACGAGATCATGAGAGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-16 | CAAGCAGAAGACGGCATACGAGATTGAATCGCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-17 | CAAGCAGAAGACGGCATACGAGATGTCTGACTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-18 | CAAGCAGAAGACGGCATACGAGATCTGAATGCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-19 | CAAGCAGAAGACGGCATACGAGATCGCTTCTGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-20 | CAAGCAGAAGACGGCATACGAGATTCGCATGAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-21 | CAAGCAGAAGACGGCATACGAGATAATAGCAGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-22 | CAAGCAGAAGACGGCATACGAGATGTCGCGTAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-23 | CAAGCAGAAGACGGCATACGAGATACGCGATAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7 index | P7-index-24 | CAAGCAGAAGACGGCATACGAGATTGATCGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| P7-index-25 | CAAGCAGAAGACGGCATACGAGATCCGCATGAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-26 | CAAGCAGAAGACGGCATACGAGATCCACAATCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-27 | CAAGCAGAAGACGGCATACGAGATGATGTTCGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-28 | CAAGCAGAAGACGGCATACGAGATGAATACGTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-29 | CAAGCAGAAGACGGCATACGAGATTAGATACCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| P7-index-30 | CAAGCAGAAGACGGCATACGAGATTATGGTCGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| adapter | adapter-upper | GACGTGTGCTCTTCCGATCTGNNNNNN-3`NH2 C6 |
| adapter-lower | 5′P-CAGATCGGAAGAGCACACGTC-3`NH2 C6 |
新窗口打开|下载CSV
1.6 筛选CTCF位点反转的单细胞克隆
待到12孔板中HEK293T细胞长到约60%时,利用Lipofectamine 3000试剂将Cas9质粒、sgRNA质粒一起转染到细胞中。2天后,用含有2 ng/μL嘌呤霉素的DMEM培养基培养4天。换用普通DMEM完全培养基培养2天后,进行单克隆铺板。约10天后,用胰酶消化单克隆细胞,取部分细胞使用碱裂法裂解获得基因组。使用特异性引物进行PCR鉴定,鉴定各单细胞克隆类型,鉴定引物如表1所示。PCR产物经TA克隆后,进行桑格测序,确定单细胞克隆基因型。
1.7 定量高分辨率染色质构象捕获实验QHR-4C
QHR-4C实验方法参考本实验室之前的研究[34],步骤如下:培养鉴定好的单细胞克隆,收取处于对数生长期的细胞500万个左右,用4C裂解液(配方为:50 mmol/L Tris-HCl, 150 mmol/L NaCl, 5 mmol/L EDTA, 0.5% NP-40, 1% Triton X-100)裂解细胞两次。获得的细胞核沉淀经Dpn Ⅱ内切酶过夜酶切后,用T4 DNA连接酶在16℃过夜连接,之后利用超声将DNA片段打断为200~1000 bp长度范围。超声后的DNA片段利用酚氯仿进行纯化,后使用带有biotin的引物进行线性扩增。扩增产物加上接头后进行PCR扩增和产物纯化,获得QHR-4C文库。文库用Illumina HiSeq测序获得原始数据,根据index和barcode拆分数据。用fastuniq去重,通过bowtie2比对到hg19参考基因组。比对文件经过samtools排序,用r3Cseq进行计数和标准化,获得每个酶切片段的RPKM值。每个单细胞克隆株至少做两个重复实验,共计99个QHR-4C文库,每个文库测序约800万条序列。所用index见表1。1.8 RNA-seq实验
待细胞长到12孔板约80%~90%时,使用TRIzol处理细胞,并提取细胞总RNA。随后用诺维赞RNA-seq建库试剂盒进行RNA-seq文库构建。步骤如下:用beads捕获mRNA,热打断至150~200 bp,使用随机引物逆转录获得cDNA,加A和加接头,最后进行PCR扩增。文库用Illumina HiSeq进行测序,数据经index拆分后,利用trim_galore去除低质量reads,使用STAR比对到hg19参考基因组,利用cufflinks对转录本进行计数和标准化,得到每个基因的FPKM值。每个单细胞克隆株做三个重复实验,共计33个RNA-seq文库,每个文库测序约1000万条序列。1.9 ChIP-seq实验
待细胞汇合度达到10 cm培养皿约80%~90%时收取细胞,用甲醛交联固定,用超声将基因组DNA打断为100~1000 bp,孵育protein G凝胶珠。4℃过夜孵育一抗后,再次加入protein G凝胶珠孵育,经洗脱DNA和用酚氯仿纯化DNA后,利用DNA建库试剂盒进行建库,包括末端修复、末端加A、加接头和PCR扩增等步骤,获得ChIP-seq文库。文库用Illumina HiSeq进行测序,测序数据经过index拆分获得双端测序序列,使用trim_galore进行数据过滤,bowtie2比对到hg19参考基因组。之后用samtools对比对结果进行排序,使用macs2成峰。实验做两个重复,每个重复测序约2000万条序列。1.10 高通量测序数据信息
该研究的相关结果数据已收录在国家基因库生命大数据平台(CNGBdb)[58]的国家基因库序列归档系统(CNSA)[59],项目编号:CNP0001772。2 结果与分析
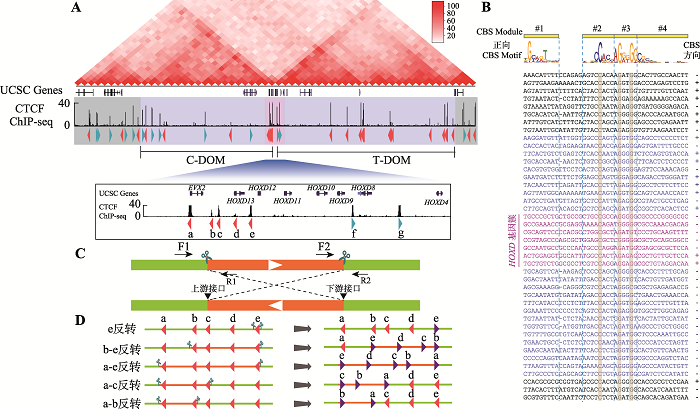
2.1 HOXD基因簇区域CTCF位点序列分析
为了研究TAD边界处成串CBS的绝缘子作用,本研究选定HOXD基因簇为研究对象。Hi-C数据显示HOXD基因簇位于两个TAD边界处,CTCF ChIP-seq数据证实HOXD基因簇以及两侧的C-DOM和T-DOM分布着大量CTCF结合位点(图1A)。为了判断CBS的方向性,对每一个CTCF结合峰进行序列分析(图1B)。这些CBS基序共包括4个module,其中module 2~4包含最为保守的20 bp核心基序,少部分在核心基序上游含有module 1,正向CBS方向为module 1到module 4 (图1B)。在module 2~3中最为保守的C20、G25和G28是判断CBS存在的重要参考,而module 1和位于24位的碱基是否为A则可以用于判断CBS方向性;这些CBS序列特性也与之前发表文章相符[2,5]。在HEK293T细胞中,HOXD基因簇分布着5个反向CBS和2个正向CBS,分别命名为CBS a-e和f-g (图1A)。在C-DOM中大部分是正向CBS,只有3个反向CBS (图1B);T-DOM中CBS呈反向串联排布(图1B)。2.2 构建HOXD基因簇一系列CTCF位点反转的单细胞克隆株
利用CRISPR/Cas9 DNA片段编辑技术,针对预反转的DNA片段设计一对sgRNA,将Cas9和sgRNA质粒转染到HEK293T细胞中,Cas9核酸酶在成对sgRNA的介导下对目的DNA片段进行切割。断裂的双链DNA在细胞DNA修复系统的作用下,可以产生DNA片段原位反转,反转后的DNA片段具有两个不同的接口(上游接口和下游接口)(图1C)。利用此策略,我们对HOXD基因簇内CBS a-e进行组合反转(图1D)。共获得5种CBS反转单细胞克隆株,分别为e反转、b-e反转、a-e反转、a-c反转和a-b反转(图1D),由于不同sgRNA具有不同的编辑效率,单克隆筛选数相差较大(图2A),其中a-b反转共筛选1307个单细胞克隆只获得了一个预期反转克隆。最后,e反转获得5个纯合子克隆株,b-e反转、a-e反转、a-c各获得2个纯合子克隆株,a-b反转获得1个纯合子克隆株(图2A)。全部单细胞克隆株通过PCR进行鉴定,DNA片段反转的接口还进行TA克隆和桑格测序确定其具体的基因型,大部分接口为两种基因型,除了在PAM位点上游3 bp处精确切割后的连接产物外,单碱基插入占据相当的比例(图2,B~J),推测Cas9核酸酶在PAM位点上游4 bp处切割导致[48,60]。在b-e#127单细胞克隆株的一个等位基因下游接口存在518 bp的碱基删除,但删除区域不包括CBS核心基序(图2G);在a-c#81单细胞克隆株的一个等位基因上游接口存在258 bp的碱基插入,经比对发现插入序列为质粒序列(图2I),因此这两个细胞株仍是纯合的CBS反转单细胞克隆株。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1人类HOXD基因簇区域CTCF位点序列分析
A:HOXD基因簇区域基因组三维空间结构示意图。HOXD基因簇位于C-DOM和T-DOM两个TAD边界处,位于HOXD基因簇附近的CBS a-e与C-DOM中的正向CBS形成染色质环,CBS f-g与T-DOM中的成串反向CBS形成染色质环。B:HOXD基因簇区域CBS序列比对图。正向CBS的方向为module 1~4,其中module 1与module 2之间相隔5 bp或者6 bp。DNA序列中的“-”表示该CBS的module 1与module 2间隔为5 bp,故用“-”补齐。右侧CBS方向列,“+”表示CBS为正向,“-”表示该CBS为反向且显示DNA序列是反向互补序列。C:利用CRISPR/Cas9系统进行DNA片段反转示意图。针对预反转片段设计一对sgRNA进行片段编辑,会产生上游接口和下游接口;在切割点附近设计引物,用引物F1+F2、R1+R2进行PCR鉴定,若出现目的条带且F1+R1、F2+R2和F1+R2无目的条带,则为纯合反转单细胞克隆。D:HOXD基因簇内CBS组合性反转示意图,共构建五种反转单细胞克隆株。
Fig. 1Sequence analyses of CTCF sites of the human HOXD cluster
图2
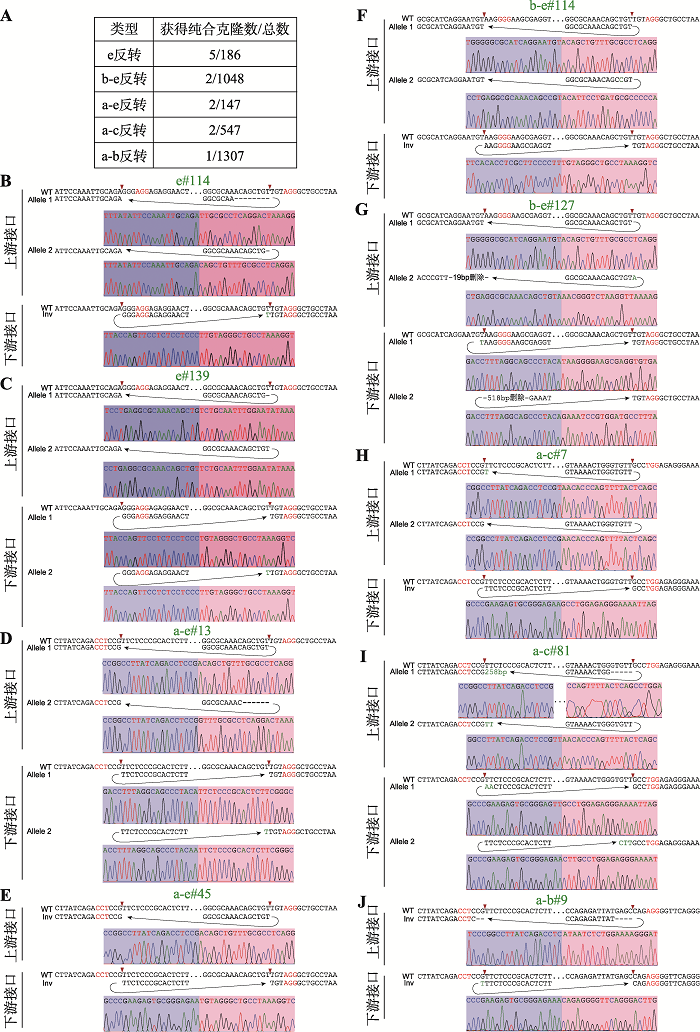 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2反转单细胞克隆株基因型鉴定
A:获得纯合CBS反转单细胞克隆株个数以及筛选克隆总数目。B~J:每一个纯合反转单细胞克隆株的基因型鉴定(TA克隆桑格测序图)。序列中红色表示PAM所在位置,红色箭头指示PAM上游-3位。序列中“-”表示存在碱基删除,绿色表示存在碱基增加。
Fig. 2Genotyping of single-cell clones of combinatorial CBS inversions
2.3 CTCF位点反转引起染色质拓扑结构域边界漂移
为了研究CBS的绝缘子作用,对获得的CBS反转单细胞克隆株进行QHR-4C实验。在野生型细胞中,位于T-DOM中的增强子CS38主要与正向CBS f-g有较强的DNA相互作用(图3A),而位于C-DOM中的增强子Island5主要与反向的CBS a-e具有DNA相互作用(图3B),与Hi-C结果一致(图1A)。CBS e反转后,CBS e与CS38的远距离DNA相互作用增强(图3A),显示反转后CBS e可以作为绝缘子阻碍CBS f-g渗透的cohesin继续滑动;与之相反,CBS e与Island5的远距离DNA相互作用减弱(图3B)。CBS d和反转后的CBS e呈反向-正向分布,形成了一个新的染色质拓扑结构域边界。CBS b-e反转后,CBS a与CBS e形成一对反向-正向的CBS (图1D),CBS b-e与Island5的远距离DNA相互作用下降(图3B),与CS38形成新的远距离DNA相互作用(图3A),阻碍从CBS f-g方向渗透的cohesin继续滑动。CBS a和反转后的CBS e形成了新的染色质拓扑结构域边界,与CBS e反转结果相似,即边界CBS反转引起染色质拓扑结构域边界的漂移。CBS a-e全部反转后,CBS a-e与Island5的DNA相互作用明显减弱(图3B),与CS38形成新的DNA相互作用(图3A),由于缺乏成对反向-正向CBS,破坏了HOXD区域的染色质拓扑边界。
图3
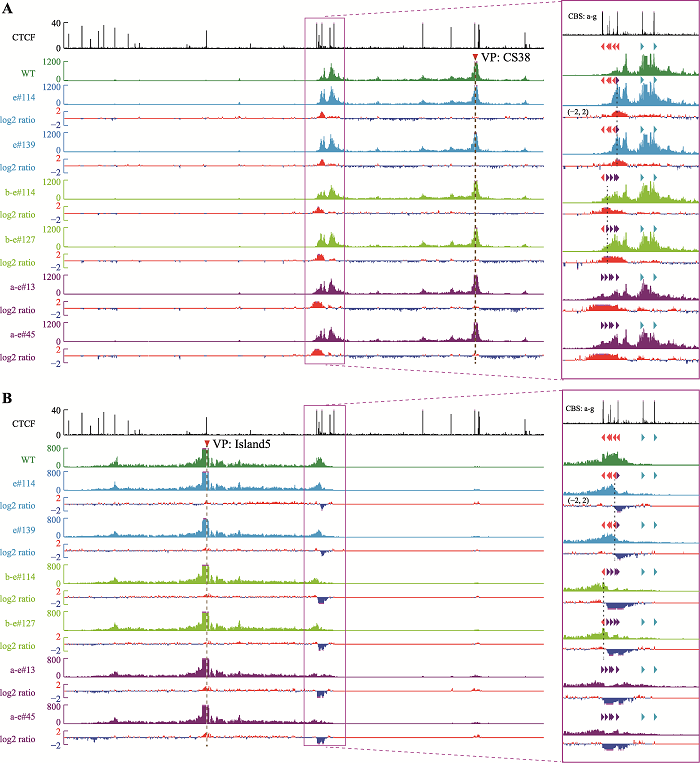 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3从两侧增强子观察CBS反转后局部三维基因组构象变化
A:以CS38为观测点(viewpoint, VP)显示,CBS反转后与CS38的染色质相互作用显著增强。CBS反转单细胞克隆株数据分别与野生型(WT)进行log2转化,获得两者差异数据。B:以Island5为观测点显示,CBS反转后与Island5的原有染色质相互作用减弱。
Fig. 3Alterations of the local 3D chromatin structure observed from viewpoints of flanking enhancers in CBS inversion clones
为了进一步确定这些染色质空间构象的改变,选用CBS a和CBS e附近的DNA片段作为观测点(upstream-a和downstream-e,upstream-a观测点位于CBS a的上游,downstream-e观测点位于CBS e的下游),为了保证观测点不随CBS反转而改变,观测点的位置不在反转片段内。在CBS e反转的细胞中,CS38与CBS a的远距离DNA相互作用没有变化(图4A);而CBS b-e反转后,CS38与CBS a的远距离DNA相互作用有轻微增强(图4A);反转CBS a-e后CBS a主要与CS38形成较强的DNA相互作用(图4A)。从downstream-e为观测点来看,所有反转克隆均与CS38有较强的DNA相互作用(图4B),与上述结果相同。这些结果说明,反转CBS后,该CBS原有的远距离DNA相互作用被破坏,即CBS方向可以决定染色质环的形成,反转的CBS可以阻碍反方向渗透的cohesin继续滑动,发挥绝缘子的功能;同时,边界CBS元件反转会引起染色质拓扑结构域边界的漂移。
图4
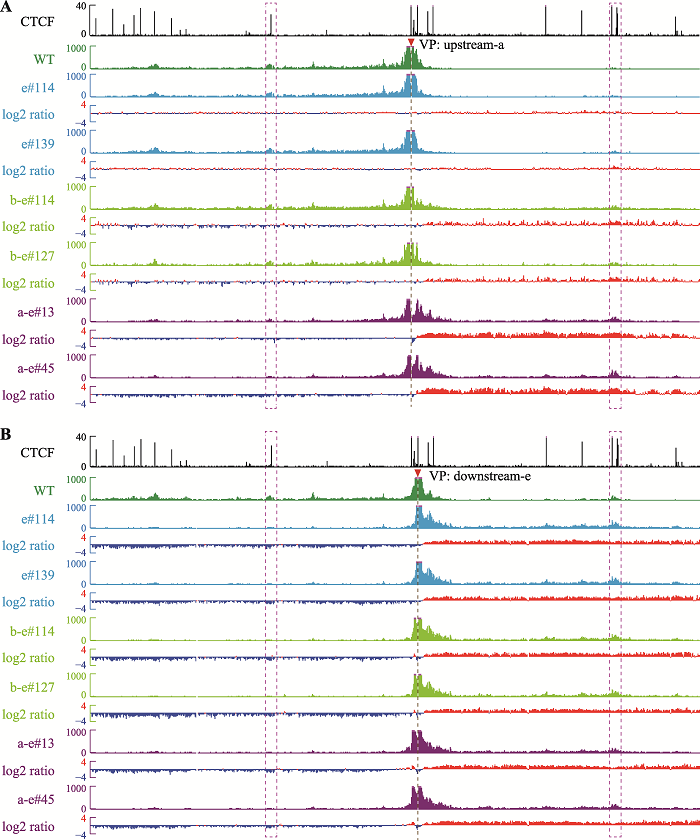 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4从反转位点附近观察CBS反转后局部三维基因组构象变化
A:以upstream-a为观测点,在CBS e反转细胞中观测点与两侧远距离DNA相互作用无明显变化,在CBS b-e反转细胞中观测点与CS38的远距离DNA相互作用增强,在CBS a-e反转细胞中观测点与CS38相互作用明显增强,伴随着与Island5的远距离DNA相互作用减弱。B:以downstream-e为观测点,在三种包含CBS e反转的细胞株中,观测点与CS38的染色质相互作用均增强,伴随着与Island5染色质相互作用减弱。
Fig. 4Alterations of the local 3D chromatin structure observed from viewpoints near the inverted CBS
2.4 非边界CTCF位点也发挥绝缘子功能
本实验室之前的研究发现,位于Pcdhα基因簇启动子区域的正向CBS可以与位于远端的增强子HS5-1形成染色质环,阻碍更远处的启动子CBS与增强子HS5-1的远距离DNA相互作用,具有绝缘子功能[34]。为了研究HOXD基因簇区域CBS是否同样具有绝缘子功能,本研究进一步反转了CBS a-b和CBS a-c (图1D)。以Island5为观测点显示,反转CBS a-b或CBS a-c后,Island5与CBS e的远距离DNA相互作用增强(图5A),而以CS38为观测点显示CS38与CBS a-e的远距离DNA相互作用无明显变化(图5B)。相应地,upstream-a与上游的Island5的远距离DNA相互作用减弱(图5C),downstream-e与Island5的远距离DNA相互作用增强(图5D),与Island5为观测点的结果相同。但是,染色质拓扑结构域边界并没有发生变化。因此,CBS a-b或a-c可以作为绝缘子,通过与Island5形成染色质环,分别阻碍CBS c-e或d-e与Island5的远距离DNA相互作用。反转细胞株的RNA-seq实验显示HOXD基因簇区域局部染色质构象变化后,基因表达水平会发生改变(图6,A~D)。其中,在所有的CBS反转细胞株中,HOXD13的表达水平均显著下降;除CBS b-e反转细胞株外,HOXD3与HOXD10的表达水平均下降;有趣的是,在CBS e反转和CBS a-e反转的细胞中,HOXD9的表达水平显著上升(图6,A~D)。图6
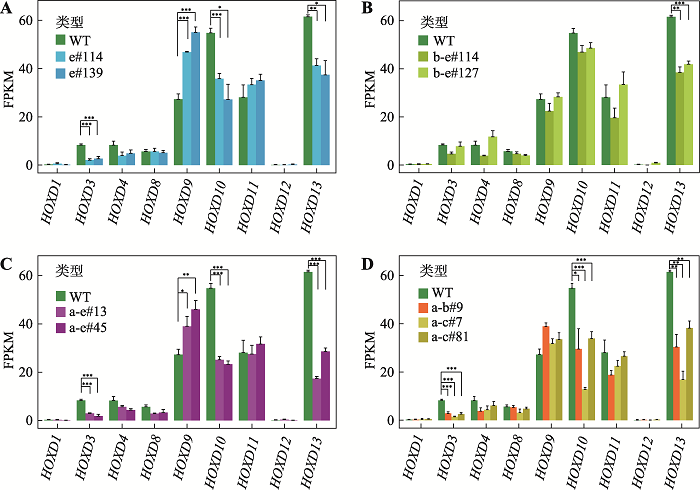 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6CTCF位点反转改变HOXD基因表达
A:CBS e反转后,HOXD3、HOXD10和HOXD13的表达水平明显降低,HOXD9的表达水平升高;*:P<0.05,**:P<0.01,***:P < 0.001;FPKM,fragments per kilobase of transcript per million mapped reads。B:CBS b-e反转后,HOXD13表达水平显著降低。C:CBS a-e反转后,HOXD3、HOXD10和HOXD13的表达水平降低,而HOXD9的表达水平上升。D:CBS a-b或CBS a-c反转后,HOXD3、HOXD10和HOXD13的表达水平降低。
Fig. 6CBS inversions alter expression levels of HOXD genes
3 讨论
CTCF通过阻碍cohesin蛋白在染色质上滑动形成染色质环,介导远距离的DNA相互作用,对于基因组三维空间结构的形成与维持有重要作用[29,61,62]。多个位点的CBS反转和插入实验显示染色质环形成依赖于成对正向-反向CBS[5,28,29],而CBS的绝缘子功能是不依赖方向的[34]。研究CBS的方向性有助于理解CBS在染色质高级拓扑结构中的功能,但是在成串CBS中,理解CBS的功能则更加复杂。原钙粘蛋白基因簇的CBS研究显示,成串CBS可能通过环挤出模型,呈现类似于“葫芦”结构的平衡分布模式[34]。这显示成对正向-反向CBS对于cohesin的阻碍不是完全的,存在cohesin渗透出正向-反向CBS对(CBS pair)限制的染色质范围的情况,可能是由于CTCF与DNA结合是动态的[38],因而,在成串CBS中,CBS呈“葫芦”状分布[34]。但是,位于染色质拓扑结构域边界的成串CBS如何发挥绝缘子功能,目前仍缺少系统研究。图7
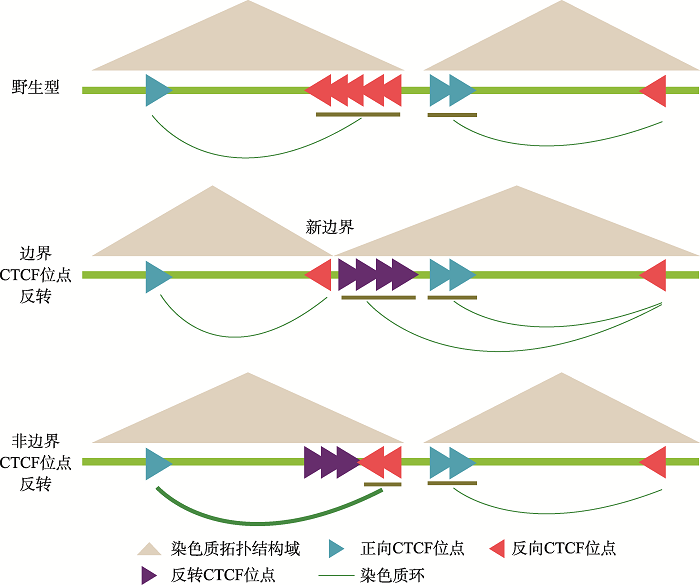 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7染色质拓扑结构域边界成串CTCF位点发挥绝缘子功能的模式图
A:在野生型细胞中,位于染色质拓扑结构域边界成串反向-正向排列的CBS分别与两侧拓扑结构域内的CBS形成染色质环。边界CBS反转后,可以阻碍反方向渗透的cohesin滑动,形成染色质环;染色质结构域边界也漂移至形成的一对反向-正向CBS处,而非边界CBS反转不引起染色质结构域边界漂移。非边界CBS反转后边界CBS与拓扑结构域内的相向CBS的远距离DNA相互作用增强,显示非边界CBS也能发挥绝缘子功能。
Fig. 7A model for insulator function of tandem boundary CBS sites
本研究通过对HOXD基因簇区域的CBS进行CRISPR组合反转,获得了五种纯合反转单细胞克隆株。QHR-4C实验显示,在CBS e、CBS b-e、CBS a-e反转细胞中,由于包含了TAD边界的CBS元件,反转的CBS引起了远距离DNA互作的巨大变化,证实了边界单个CBS反转可以改变染色质环形成方向[29]。在这三种反转细胞中,多个观测点的QHR-4C数据显示,反转后CBS可以阻碍渗透的cohesin继续滑动,发挥绝缘子功能,染色质拓扑结构域边界也漂移至新形成的一对反向-正向CBS处(图7)。其次,CBS a-b和CBS a-c反转结果显示,CBS a-b 或a-c可以作为绝缘子,分别阻碍CBS c-e或d-e与增强子Island5的远距离DNA相互作用,证实在成串CBS a-e中,非边界的CBS也可以作为绝缘子阻碍边界CBS与增强子的染色质环的形成;反转非边界CBS不改变染色质拓扑结构域边界(图7)。CBS反转引起的局部染色质构象变化会进一步影响HOXD基因簇的表达,但是对于不同的基因,引起的表达水平变化不尽相同,显示出HOXD基因簇存在复杂的表达调控机制。综上所述,本研究验证了环挤出模型,同时进一步完善了对CTCF绝缘功能的理解。
(责任编委: 谷峰)
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1007/s12038-019-9968-1URL [本文引用: 1]
DOI:10.1016/j.jgg.2020.06.008URL [本文引用: 4]
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/s41580-019-0162-yURL [本文引用: 1]
DOI:10.1016/j.cell.2015.07.038PMID:26276636 [本文引用: 9]

CTCF and the associated cohesin complex play a central role in insulator function and higher-order chromatin organization of mammalian genomes. Recent studies identified a correlation between the orientation of CTCF-binding sites (CBSs) and chromatin loops. To test the functional significance of this observation, we combined CRISPR/Cas9-based genomic-DNA-fragment editing with chromosome-conformation-capture experiments to show that the location and relative orientations of CBSs determine the specificity of long-range chromatin looping in mammalian genomes, using protocadherin (Pcdh) and β-globin as model genes. Inversion of CBS elements within the Pcdh enhancer reconfigures the topology of chromatin loops between the distal enhancer and target promoters and alters gene-expression patterns. Thus, although enhancers can function in an orientation-independent manner in reporter assays, in the native chromosome context, the orientation of at least some enhancers carrying CBSs can determine both the architecture of topological chromatin domains and enhancer/promoter specificity. These findings reveal how 3D chromosome architecture can be encoded by linear genome sequences. Copyright © 2015 Elsevier Inc. All rights reserved.
DOI:10.1038/nature12753URL [本文引用: 1]
DOI:10.1038/s41467-020-20047-wPMID:33268790 [本文引用: 1]
The three-dimensional structure of chromosomes plays an important role in gene expression regulation and also influences the repair of radiation-induced DNA damage. Genomic aberrations that disrupt chromosome spatial domains can lead to diseases including cancer, but how the 3D genome structure responds to DNA damage is poorly understood. Here, we investigate the impact of DNA damage response and repair on 3D genome folding using Hi-C experiments on wild type cells and ataxia telangiectasia mutated (ATM) patient cells. We irradiate fibroblasts, lymphoblasts, and ATM-deficient fibroblasts with 5?Gy X-rays and perform Hi-C at 30?minutes, 24?hours, or 5 days after irradiation. We observe that 3D genome changes after irradiation are cell type-specific, with lymphoblastoid cells generally showing more contact changes than irradiated fibroblasts. However, all tested repair-proficient cell types exhibit an increased segregation of topologically associating domains (TADs). This TAD boundary strengthening after irradiation is not observed in ATM deficient fibroblasts and may indicate the presence of a mechanism to protect 3D genome structure integrity during DNA damage repair.
DOI:10.1038/s41586-021-03193-zURL [本文引用: 1]
[本文引用: 1]
PMID:11847345 [本文引用: 1]
We describe an approach to detect the frequency of interaction between any two genomic loci. Generation of a matrix of interaction frequencies between sites on the same or different chromosomes reveals their relative spatial disposition and provides information about the physical properties of the chromatin fiber. This methodology can be applied to the spatial organization of entire genomes in organisms from bacteria to human. Using the yeast Saccharomyces cerevisiae, we could confirm known qualitative features of chromosome organization within the nucleus and dynamic changes in that organization during meiosis. We also analyzed yeast chromosome III at the G1 stage of the cell cycle. We found that chromatin is highly flexible throughout. Furthermore, functionally distinct AT- and GC-rich domains were found to exhibit different conformations, and a population-average 3D model of chromosome III could be determined. Chromosome III emerges as a contorted ring.
DOI:10.1038/ng1891URL [本文引用: 1]
DOI:10.1126/science.1181369PMID:19815776 [本文引用: 2]
We describe Hi-C, a method that probes the three-dimensional architecture of whole genomes by coupling proximity-based ligation with massively parallel sequencing. We constructed spatial proximity maps of the human genome with Hi-C at a resolution of 1 megabase. These maps confirm the presence of chromosome territories and the spatial proximity of small, gene-rich chromosomes. We identified an additional level of genome organization that is characterized by the spatial segregation of open and closed chromatin to form two genome-wide compartments. At the megabase scale, the chromatin conformation is consistent with a fractal globule, a knot-free, polymer conformation that enables maximally dense packing while preserving the ability to easily fold and unfold any genomic locus. The fractal globule is distinct from the more commonly used globular equilibrium model. Our results demonstrate the power of Hi-C to map the dynamic conformations of whole genomes.
DOI:10.1038/nature08497URL [本文引用: 2]
DOI:10.1038/nature11049URL [本文引用: 2]
DOI:10.1038/nature11082URL [本文引用: 1]
DOI:10.1016/j.cell.2014.11.021URL [本文引用: 1]
DOI:10.1073/pnas.1219280110URL
DOI:10.1073/pnas.1518552112URL [本文引用: 2]
DOI:10.1126/science.1234167URL [本文引用: 5]
DOI:10.1016/j.molcel.2016.05.018URL
DOI:S0092-8674(17)31317-XPMID:29224777
There is considerable evidence that chromosome structure plays important roles in gene control, but we have limited understanding of the proteins that contribute to structural interactions between gene promoters and their enhancer elements. Large DNA loops that encompass genes and their regulatory elements depend on CTCF-CTCF interactions, but most enhancer-promoter interactions do not employ this structural protein. Here, we show that the ubiquitously expressed transcription factor Yin Yang 1 (YY1) contributes to enhancer-promoter structural interactions in a manner analogous to DNA interactions mediated by CTCF. YY1 binds to active enhancers and promoter-proximal elements and forms dimers that facilitate the interaction of these DNA elements. Deletion of YY1 binding sites or depletion of YY1 protein disrupts enhancer-promoter looping and gene expression. We propose that YY1-mediated enhancer-promoter interactions are a general feature of mammalian gene control.Copyright © 2017 Elsevier Inc. All rights reserved.
[本文引用: 2]
[本文引用: 2]
PMID:2284094 [本文引用: 1]
The chicken c-myc 5'-flanking sequence has previously been shown to bind multiple proteins present in undifferentiated and differentiated red blood cells. In this report the protein binding to one specific region within a hypersensitive site approximately 200 base pairs upstream of the start of transcription has been analysed in detail. Using a combination of a modified agarose gel retardation assay with O-phenanthroline-copper footprinting in situ, missing contact point and methylation interference techniques, two proteins were found to bind to overlapping sequences within 180-230 bp upstream of the start of transcription. One protein resembles the transcription factor Sp1, the other is a protein which binds to three regularly spaced repeats of the core sequence CCCTC. This CCCTC-binding factor was termed CTCF. It requires additional sequences outside the three recognition motifs for tight binding. CTCF was purified to near homogeneity by sequence-specific DNA chromatography. The approximate molecular weight of the CTCF was estimated to be 130,000. Removal of 110 bp sequence binding both CTCF and Sp1-like proteins leads to a 4 to 8-fold increase in transcription of stably transfected c-myc fusion constructs in chicken embryonic fibroblasts, suggesting that the CTCF is likely to be one of multiple nuclear factors involved in the transcriptional regulation of the chicken c-myc gene.
DOI:10.1038/cr.2017.131URL [本文引用: 1]
DOI:10.1186/s13059-020-02108-xPMID:32782014
Topologically associating domains (TADs) are genomic regions of self-interaction. Additionally, it is known that TAD boundaries are enriched in CTCF binding sites. In turn, CTCF sites are known to be asymmetric, whereby the convergent configuration of a pair of CTCF sites leads to the formation of a chromatin loop in vivo. However, to date, it has been unclear how to reconcile TAD structure with CTCF-based chromatin loops.We approach this problem by analysing CTCF binding site strengths and classifying clusters of CTCF sites along the genome on the basis of their relative orientation. Analysis of CTCF site orientation classes as a function of their spatial distribution along the human genome reveals that convergent CTCF site clusters are depleted while divergent CTCF clusters are enriched in the 5- to 100-kb range. We then analyse the distribution of CTCF binding sites as a function of TAD boundary conservation across seven primary human blood cell types. This reveals divergent CTCF site enrichment at TAD boundaries. Furthermore, convergent arrays of CTCF sites separate the left and right sections of TADs that harbour internal CTCF sites, resulting in unequal TAD 'halves'.The orientation-based CTCF binding site cluster classification that we present reconciles TAD boundaries and CTCF site clusters in a mechanistically elegant fashion. This model suggests that the emergent structure of nuclear chromatin in the form of TADs relies on the obligate alternation of divergent and convergent CTCF site clusters that occur at different length scales along the genome.
DOI:10.1101/gad.277863.116URL
DOI:10.1016/j.cell.2009.06.001URL [本文引用: 1]
DOI:10.1016/j.molcel.2015.09.023URL [本文引用: 3]
DOI:10.1007/s13238-019-00656-yURL [本文引用: 3]
DOI:10.1007/s12264-020-00578-4URL [本文引用: 1]
DOI:10.1126/science.1262088PMID:25722416 [本文引用: 1]
Polycomb and Trithorax group proteins encode the epigenetic memory of cellular positional identity by establishing inheritable domains of repressive and active chromatin within the Hox clusters. Here we demonstrate that the CCCTC-binding factor (CTCF) functions to insulate these adjacent yet antagonistic chromatin domains during embryonic stem cell differentiation into cervical motor neurons. Deletion of CTCF binding sites within the Hox clusters results in the expansion of active chromatin into the repressive domain. CTCF functions as an insulator by organizing Hox clusters into spatially disjoint domains. Ablation of CTCF binding disrupts topological boundaries such that caudal Hox genes leave the repressed domain and become subject to transcriptional activation. Hence, CTCF is required to insulate facultative heterochromatin from impinging euchromatin to produce discrete positional identities. Copyright © 2015, American Association for the Advancement of Science.
DOI:10.1016/j.molcel.2017.08.026URL
DOI:10.1073/pnas.1505463112URL [本文引用: 1]
DOI:10.1186/s13059-020-01984-7URL [本文引用: 9]
DOI:10.1038/s41588-020-0680-8URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/s41586-019-1723-0URL [本文引用: 1]
DOI:10.1021/acs.jpclett.8b01440URL [本文引用: 2]
DOI:10.7554/eLife.52962URL [本文引用: 3]
DOI:10.1016/j.celrep.2019.07.013URL
DOI:10.1186/s13059-017-1278-zURL [本文引用: 1]
DOI:10.1126/science.1207194PMID:21998387 [本文引用: 2]
The spatial and temporal control of Hox gene transcription is essential for patterning the vertebrate body axis. Although this process involves changes in histone posttranslational modifications, the existence of particular three-dimensional (3D) architectures remained to be assessed in vivo. Using high-resolution chromatin conformation capture methodology, we examined the spatial configuration of Hox clusters in embryonic mouse tissues where different Hox genes are active. When the cluster is transcriptionally inactive, Hox genes associate into a single 3D structure delimited from flanking regions. Once transcription starts, Hox clusters switch to a bimodal 3D organization where newly activated genes progressively cluster into a transcriptionally active compartment. This transition in spatial configurations coincides with the dynamics of chromatin marks, which label the progression of the gene clusters from a negative to a positive transcription status. This spatial compartmentalization may be key to process the colinear activation of these compact gene clusters.
DOI:10.1016/j.cell.2011.10.023PMID:22118467 [本文引用: 1]
The evolution of digits was an essential step in the success of tetrapods. Among the key players, Hoxd genes are coordinately regulated in developing digits, where they help organize growth and patterns. We identified the distal regulatory sites associated with these genes by probing the three-dimensional architecture of this regulatory unit in developing limbs. This approach, combined with in vivo deletions of distinct regulatory regions, revealed that the active part of the gene cluster contacts several enhancer-like sequences. These elements are dispersed throughout the nearby gene desert, and each contributes either quantitatively or qualitatively to Hox gene transcription in presumptive digits. We propose that this genetic system, which we call a "regulatory archipelago," provides an inherent flexibility that may partly underlie the diversity in number and morphology of digits across tetrapods, as well as their resilience to drastic variations.Copyright © 2011 Elsevier Inc. All rights reserved.
DOI:10.1073/pnas.2015083117URL [本文引用: 1]
DOI:10.1093/jmcb/mjv016URL [本文引用: 1]
DOI:S1097-2765(18)30466-0PMID:30033371 [本文引用: 1]
Chromosomal rearrangements including large DNA-fragment inversions, deletions, and duplications by Cas9 with paired sgRNAs are important to investigate genome structural variations and developmental gene regulation, but little is known about the underlying mechanisms. Here, we report that disrupting CtIP or FANCD2, which have roles in alternative non-homologous end joining, enhances precise DNA-fragment deletion. By analyzing the inserted nucleotides at the junctions of DNA-fragment editing of deletions, inversions, and duplications and characterizing the cleaved products, we find that Cas9 endonucleolytically cleaves the noncomplementary strand with a flexible scissile profile upstream of the -3 position of the PAM site in vivo and in vitro, generating double-strand break ends with 5' overhangs of 1-3 nucleotides. Moreover, we find that engineered Cas9 nucleases have distinct cleavage profiles. Finally, Cas9-mediated nucleotide insertions are nonrandom and are equal to the combined sequences upstream of both PAM sites with predicted frequencies. Thus, precise and predictable DNA-fragment editing could be achieved by perturbing DNA repair genes and using appropriate PAM configurations.Copyright © 2018 Elsevier Inc. All rights reserved.
DOI:10.1126/science.1231143PMID:23287718
Functional elucidation of causal genetic variants and elements requires precise genome editing technologies. The type II prokaryotic CRISPR (clustered regularly interspaced short palindromic repeats)/Cas adaptive immune system has been shown to facilitate RNA-guided site-specific DNA cleavage. We engineered two different type II CRISPR/Cas systems and demonstrate that Cas9 nucleases can be directed by short RNAs to induce precise cleavage at endogenous genomic loci in human and mouse cells. Cas9 can also be converted into a nicking enzyme to facilitate homology-directed repair with minimal mutagenic activity. Lastly, multiple guide sequences can be encoded into a single CRISPR array to enable simultaneous editing of several sites within the mammalian genome, demonstrating easy programmability and wide applicability of the RNA-guided nuclease technology.
DOI:10.1093/jmcb/mjaa060URL
DOI:10.16288/j.yczz.20-184PMID:32952112 [本文引用: 1]
Gene expression and three-dimensional (3D) genome organization are thought to be closely related. The protocadherin (Pcdh) clusters are central for neuronal self-avoidance in brain development and have been used as model genes to explore the role of 3D genome structures in gene regulation. Transcription factor RFX5 (regulatory factor x 5) is a member of the winged-helix subfamily of the helix-turn-helix superfamily proteins. The RFX5 protein contains four domains: oligomerization, DNA-binding, helical, and transactivation domains. RFX5 plays an important role in regulating expression of the major histocompatibility complex class II (MHC II) gene complex. Here we report a role of RFX5 in the regulation of clustered Pcdh genes. We first knocked out the RFX5 gene by using CRISPR/Cas9 DNA-fragment editing in HEC-1-B cell line. By RNA-seq experiments, we found that RFX5 deletion results in a significant increase of expression levels of the Pcdhα6, Pcdhα12 and Pcdhαc2 genes. By ChIP-nexus experiments, we found that RFX5 deletion leads to the increased binding of CTCF and RAD21 in the Pcdhα cluster. Finally, through quantitative high-resolution chromosome conformation capture copy (QHR-4C) experiments, we found that RFX5 deletion results in a significant increase of long-distance chromatin interactions between the HS5-1 enhancer and its target promoters in the Pcdhα cluster. In conclusion, RFX5 regulates gene expression of the Pcdhα cluster through higher-order chromatin structure.
DOI:10.16288/j.yczz.20-184PMID:32952112 [本文引用: 1]
Gene expression and three-dimensional (3D) genome organization are thought to be closely related. The protocadherin (Pcdh) clusters are central for neuronal self-avoidance in brain development and have been used as model genes to explore the role of 3D genome structures in gene regulation. Transcription factor RFX5 (regulatory factor x 5) is a member of the winged-helix subfamily of the helix-turn-helix superfamily proteins. The RFX5 protein contains four domains: oligomerization, DNA-binding, helical, and transactivation domains. RFX5 plays an important role in regulating expression of the major histocompatibility complex class II (MHC II) gene complex. Here we report a role of RFX5 in the regulation of clustered Pcdh genes. We first knocked out the RFX5 gene by using CRISPR/Cas9 DNA-fragment editing in HEC-1-B cell line. By RNA-seq experiments, we found that RFX5 deletion results in a significant increase of expression levels of the Pcdhα6, Pcdhα12 and Pcdhαc2 genes. By ChIP-nexus experiments, we found that RFX5 deletion leads to the increased binding of CTCF and RAD21 in the Pcdhα cluster. Finally, through quantitative high-resolution chromosome conformation capture copy (QHR-4C) experiments, we found that RFX5 deletion results in a significant increase of long-distance chromatin interactions between the HS5-1 enhancer and its target promoters in the Pcdhα cluster. In conclusion, RFX5 regulates gene expression of the Pcdhα cluster through higher-order chromatin structure.
DOI:10.1007/s11427-019-1598-4URL [本文引用: 1]
DOI:10.1073/pnas.1317788111URL [本文引用: 2]
DOI:10.1186/s13059-015-0831-xURL [本文引用: 1]
PMID:27318199 [本文引用: 1]
Eukaryotic genomes are hierarchically organized into topologically associating domains (TADs). The computational identification of these domains and their associated properties critically depends on the choice of suitable parameters of TAD-calling algorithms. To reduce the element of trial-and-error in parameter selection, we have developed TADtool: an interactive plot to find robust TAD-calling parameters with immediate visual feedback. TADtool allows the direct export of TADs called with a chosen set of parameters for two of the most common TAD calling algorithms: directionality and insulation index. It can be used as an intuitive, standalone application or as a Python package for maximum flexibility.TADtool is available as a Python package from GitHub (https://github.com/vaquerizaslab/tadtool) or can be installed directly via PyPI, the Python package index (tadtool).kai.kruse@mpi-muenster.mpg.de, jmv@mpi-muenster.mpg.deSupplementary information: Supplementary data are available at Bioinformatics online.© The Author 2016. Published by Oxford University Press.
DOI:10.1093/nar/gkv416URL [本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/s41421-019-0120-zURL [本文引用: 1]
DOI:10.1126/science.1256917PMID:25414305 [本文引用: 1]
Through their association with a kleisin subunit (Scc1), cohesin's Smc1 and Smc3 subunits are thought to form tripartite rings that mediate sister chromatid cohesion. Unlike the structure of Smc1/Smc3 and Smc1/Scc1 interfaces, that of Smc3/Scc1 is not known. Disconnection of this interface is thought to release cohesin from chromosomes in a process regulated by acetylation. We show here that the N-terminal domain of yeast Scc1 contains two α helices, forming a four-helix bundle with the coiled coil emerging from Smc3's adenosine triphosphatase head. Mutations affecting this interaction compromise cohesin's association with chromosomes. The interface is far from Smc3 residues, whose acetylation prevents cohesin's dissociation from chromosomes. Cohesin complexes holding chromatids together in vivo do indeed have the configuration of hetero-trimeric rings, and sister DNAs are entrapped within these. Copyright © 2014, American Association for the Advancement of Science.
DOI:S0955-0674(18)30166-2PMID:30544080 [本文引用: 1]
Cohesin, one of structural maintenance of chromosomes (SMC) complexes, forms a ring-shaped protein complex, and mediates sister chromatid cohesion for accurate chromosome segregation and precise genome inheritance. The cohesin ring entraps one or two DNA molecules to achieve cohesion, which is further regulated by cohesin-binding proteins and modification enzymes in a cell cycle-dependent manner. Recent significant advancements in Hi-C technologies have revealed numerous cohesin-dependent higher-order chromatin structures. Simultaneously, single-molecule imaging has also unveiled the detailed dynamics of cohesin on DNA and/or chromatin. Thus, those studies are providing novel visions for the authentic chromatin structure regulated by cohesin.Copyright © 2018. Published by Elsevier Ltd.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}