 ,1,2
,1,2Epigenetic control of transposable elements and cell fate decision
Jiangping He1,2, Jiekai Chen,1,2通讯作者: 陈捷凯,研究员,博士生导师,研究方向:表观遗传与细胞命运决定。E-mail:chen_jiekai@gibh.ac.cn
编委: 陆发隆
收稿日期:2021-03-25修回日期:2021-07-23
| 基金资助: |
Received:2021-03-25Revised:2021-07-23
| Fund supported: |
作者简介 About authors

何江平,博士,研究方向:转座元件的表观遗传调控及功能。E-mail:
2014—2019年就读于中国科学院广州生物医药与健康研究院,在陈捷凯课题组攻读博士学位,目前任生物岛实验室副研究员。博士期间,主要研究转座元件的表观遗传调控机制及其在细胞命运决定中的功能。利用生物信息学方法系统性解析了小鼠胚胎干细胞中所有转座元件的表观遗传修饰图谱,发现不同转座元件存在多种不同的表观遗传修饰调控模式,包括抑制型的H3K9me3和激活型的H3K27ac、H3K4me1等,并进一步筛选到了调控胚胎干细胞往2细胞样细胞重编程的新型调控因子。此外,还研究了转座元件LTR6B在人定型内胚层分化及转座元件RLTR13B2在小鼠上胚层干细胞往胚胎干细胞重编程等细胞命运决定中的功能及机制。博士论文《转座元件表观遗传调控与细胞命运决定》获得2020年中国科学院优秀博士生论文。截止目前,以第一作者(含共同)在Nature、Cell、NatureCellBiology、NatureCommunications和Protein&Cell等杂志发表论文8篇,以其他作者身份发表论文10篇,累积引用超过600次。

摘要
转座元件是哺乳动物基因组内含量最多的元素。尽管转座元件的存在对基因组稳定性具有潜在的危险,但它们同时还是潜在的基因调控序列、蛋白质编码序列和染色质结构序列,并参与物种进化过程。因此,基因组中转座元件的有害性和有益性保持着谨慎的平衡,并且这种平衡主要由表观遗传修饰来调控。本文详细介绍了异染色质类型表观遗传修饰如H3K9me3和DNA甲基化在转座元件沉默中的功能;转座元件作为增强子元件富集激活型表观遗传修饰如H3K4me1和H3K27ac,以及作为转录因子结合靶点、染色质构象锚点等方式参与基因表达调控的模式;从体内胚胎发育到体外细胞命运转变,阐述了转座元件在细胞命运决定中的潜在功能及作用方式;最后,对转座元件领域研究存在的挑战及潜在解决方法提出了见解。总之,本文对转座元件与表观遗传、基因表达调控以及细胞命运决定等方面的研究及存在的问题进行了较全面的综述,旨在为相关领域的研究人员提供参考。
关键词:
Abstract
Transposable elements (TEs) are the most prevalent elements in mammalian genomes. Although potential risks for genome stability, they are a pool of potential regulatory sequences, chromatin control elements, protein-coding genes, and substrates for evolutionary processes. Consequently, a delicate balance is maintained between the potential benefits and deleterious aspects of TEs, and this balance is mediated by the epigenetic regulatory system. In this review, we introduce the role of heterochromatin associated epigentic modifications such as histone 3 lysine 9 trimethylation (H3K9me3) and DNA methylation in the silencing of TEs as well as epigenetic modifications such as histone 3 lysine 4 monomethylation (H3K4me1) and histone 3 lysine 27 acetylation (H3K27ac) in activation of TEs. Further, we elaborate the functions of TEs as binding sites of transcription factors and as anchors of chromosomal conformation in regulation of gene expression. We introduce the impact of TEs on the process of cell fate determination including natural embryonic development in vivo and artificial cell fate transition in vitro. We discuss the main challenges associated with computational TEs analysis and TEs functions exploration, as well as the different experimental and computational strategies in studying these processes. In all, this article provides a comprehensive review of the research advances and existing problems in study of transposable elements in epigenetic regulatory mechanisms, gene transcriptional regulation, and cell fate determination, thereby providing some references for researchers in the fields.
Keywords:
PDF (833KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
何江平, 陈捷凯. 转座元件、表观遗传调控与细胞命运决定. 遗传[J], 2021, 43(9): 822-834 doi:10.16288/j.yczz.21-113
Jiangping He.
转座元件(transposable elements, TEs)是一类在基因组内可以自由移动的DNA,最早由美国遗传学家芭芭拉·麦克林托克(Barbara McClintock)在玉米(Zea mays)基因组中发现,并证明TEs能通过“跳跃”调控玉米粒的颜色[1,2]。继此项开创性的工作之后,研究者们发现TEs几乎在所有真核细胞基因组中都存在,并在生命演化过程中扮演着非常重要的角色。
在人类(Homo sapiens)基因组中,总共含有30亿对碱基,但只有约2%能够编码蛋白质,而接近一半的序列由TEs组成,共有超过300万份不同的拷贝,小鼠(Mus musculus)基因组也基本类似。TEs可以扩增并插入到基因组新的位置,调控基因表达,为物种进化提供原动力,是哺乳动物基因组非常重要的组成部分。而TEs在基因组中“随意”移动则会造成大量遗传突变威胁生命安全,因而基因组进化出了一系列调控机制以限制TEs的活性。本文主要综述了TEs在哺乳动物基因组中的主要类别、TEs在哺乳动物中的表观遗传调控模式及在胚胎发育等细胞命运决定中的功能及作用机制。
1 转座元件
基因组中TEs的分类非常庞大而复杂。根据转座方式的不同可以分为两大类:DNA转座元件(DNA transposons)和逆转座元件(retrotransposons)。逆转座元件在转座过程中需要先转录成RNA,由RNA逆转录成cDNA再发生转座。逆转座元件转座后原DNA序列仍然保留,通过逆转录的cDNA插入到新的基因组位点,所以这类转座元件是通过“复制-粘贴”的方式完成“跳跃”。哺乳动物基因组中大部分转座元件为逆转座元件,在人和小鼠基因组序列中占比超过40%[3]。与逆转座元件不同,DNA转座元件则不需要RNA的介导,直接通过转座酶将原位点DNA序列切下来插入到新的位点即可发生转座,通过“剪切-粘贴”的方式完成。人类基因组中大约有500,000个DNA转座元件,约占基因组序列3% (图1A)。在灵长类基因组中,大部分DNA转座元件通常已经失去了转座的能力;相反,活跃的逆转座元件几乎在所有的灵长类基因组中都有被发现[4]。逆转座元件根据其两端是否有长末端重复序列(long terminal repeat, LTR)可分为LTR和非LTR转座元件。LTR转座元件因其在序列上与外源性逆转录病毒非常相似,因此又常被称为内源性逆转录病毒(endogenous retrovirus, ERV)。进化学上认为,这主要是在进化过程中外源逆转录病毒感染宿主或其祖先后,这些逆转录病毒的序列保留在宿主基因组中,并获得生殖系传递,所以这些序列一直保留至今。因此,完整的ERVs元件和外源性逆转录病毒一样,主要由两端的LTR以及中间编码的gag、pol以及env编码序列组成(图1B)。但在进化过程中,由于突变等因素,这些ERVs元件大多被截断掉,导致大部分是非完整性的,很多甚至只保留了单独的LTR序列。在人类基因组中,大约有650,000个ERVs元件,但其中只有2000个保留着几乎完整的ERV序列[3]。人类基因组中有超过300万个非LTR转座元件,占基因组含量约30%,而LTR转座元件只占基因组含量约8%[3]。非LTR转座元件中,又可分为长的散在元件(long interspersed nuclear elements, LINEs)和短的散在元件(short interspersed nuclear elements, SINEs)。其中,LINEs包含编码转座所需要的两个蛋白编码框ORF1和ORF2 (图1B),但与ERVs元件一样,大部分LINEs元件在进化过程中被截断进而失去了完整的编码序列。人类基因组中有约950,000个LINEs元件,但其中只有不到1%具有完整的蛋白编码序列[5]。与LINEs不同,SINEs不包含转座所需要蛋白的编码序列(图1B),它们需要借助LINEs编码的蛋白进行转座。所以ERVs、LINEs常被称为自主性转座元件,而SINEs由于不能编码转座所需要的蛋白,又被称为非自主性转座元件。ERVs、LINEs和SINEs根据序列的不同,又可进一步分为1000多种不同的家族,散在分布在基因组每一条染色体上。图1
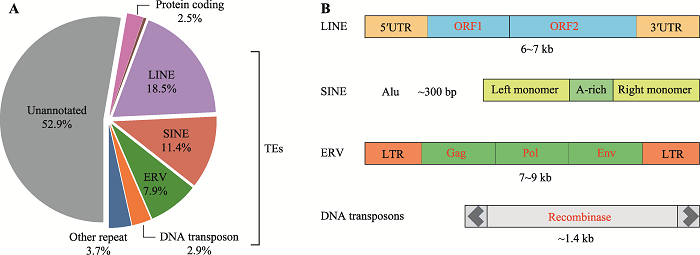 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1小鼠基因组中转座元件分类及含量
A:小鼠基因组中不同序列占基因组的比例。基因注释来源于GENECODE v20,TEs注释来源于RepeatMasker。B:不同种类转座元件结构示意图。
Fig. 1Classification and content of transposable elements within the mouse genome
与小鼠基因组相比,除LINE、SINE、DNA和LTR转座元件外,人类基因组中存在一类特有的被称为SVA(SINE-VNTR-Alu)的转座元件。SVA是类人科(hominoid)物种进化过程中活跃的TEs,大约在2500万年前产生,在人基因组中有约3000个拷贝[6]。经典的SVA全长约2 kb,序列由(CCCTCT)n、Alu样区域、串联重复区域(variable number of tandem repeats, VNTR)、HERV-K10样区域以及Poly-A尾组成。SVA为非自主转座元件,目前的研究证据提示其转座可能依赖于L1的转座机器[6],其中,KRAB锌指蛋白ZNF91/93和SVA/L1在进化过程中互相博弈,抑制进化过程中新生SVA/L1的转座活性[7]。此外,基因组中还存在其他KRAB-ZNF蛋白与转座元件的共进化[8,9]。
TEs是物种进化的主要动力之一。科学家们通过比较鼠-人之间序列的同源性,发现存在大量的TEs序列在真哺乳亚纲形成之前就已经存在[10,11]。并且,在人基因组中发现的数千个保守的调控元件中,其中大部分来源于不同类别的TEs[12]。除存在保守的调控序列来源于TEs外,同时还存在大量物种特异性的调控元件,其中很多也来源于TEs。例如,通过比较有袋类动物(如负鼠)和真哺乳亚纲动物(人、狗、小鼠和大鼠等)基因组,发现至少有16%的真哺乳亚纲动物特异性的保守基因调控元件来源于TEs[13]。同样与小鼠基因组比较,人类基因组中也存在许多特异性的调控元件由TEs构成,反之亦然[10,14]。暗示这些非保守TEs的获得,可能是新物种形成的主要原因之一,并且TEs是不同物种进化的主要痕迹之一。
1.1 转座元件与表观遗传调控
基因的表达主要受表观遗传修饰调控,TEs亦是如此。TEs由于转座特性会导致基因组不稳定性,所以其表观遗传调控一直以来都是科学研究的重要问题之一。基因组中大部分转座元件通常会被表观遗传所沉默,包括转录水平异染色质的修饰以及转录后水平RNA的修饰。DNA甲基化和组蛋白H3K9me3是负责基因表达沉默的两种最主要的表观遗传修饰,通常高富集这些修饰的染色质会处于沉默状态,如形成异染色质。尽管TEs上的表观遗传调控机制还不是非常清楚,但可以确定的是这两种修饰在TEs表达调控上都起着非常重要的主导作用[15,16]。DNA甲基化通常被认为是体细胞中控制转座元件活性的主要因素[11]。然而,在早期胚胎发育过程中,基因组中发生整体DNA去甲基化,因此会导致部分转座元件活化而表达[17]。然而基因组会以其他的表观遗传方式来代偿DNA甲基化以控制TEs的表达,防止转座元件过度活跃而导致基因组不稳定,H3K9me3介导的异染色质化是其中最主要的方式。锌指蛋白(Zinc-finger proteins, ZFPs)是已知可以直接结合到TEs上并招募Trim28,Trim28能够进一步募集组蛋白H3K9me3的甲基转移酶Setdb1使转TEs异染色质化而被沉默[9,18~20]。能够招募Trim28的KRAB-ZFP蛋白还包括Zfp809、Yy1、Zfp819以及多能性因子Zfp42等[18,20~22],这些锌指蛋白都参与了部分TEs的沉默。有研究表明,与H3K9me3修饰相关的其他表观遗传因子如Suv39h1/2、G9a、Daxx/Atrx等都与TEs沉默有关[17,23,24]。不同H3K9me3甲基转移酶调控的TEs也有所不同,其中Setdb1主要调控ERVs元件,而Suv39h1/2主要调控LINEs元件[15,23]。近期研究还发现,具有m6A修饰的TEs来源的RNA及其相关的甲基转移酶Mettl3以及识别因子Ythdc1等在对TEs的沉默起着至关重要的作用[25,26,27] (图2A)。除H3K9me3外,H4K20me3、H4R3me2以及组蛋白变体H3.3都被发现在TEs区富集并参与了转座元件的转录调控[28,29]。此外,许多其他表观遗传因子如Uhrf1、Kdm1a、Rif1、Sumo2、Chaf1a/b、Tet2、胞嘧啶脱氨脱氨酶APOBEC3家族以及RNA结合蛋白TRIM33等在转座元件调控中起着非常重要的作用[22,30-37]。除以上提到的表观遗传修饰外,本课题组的研究还发现不同的TEs之间表观遗传调控方式存在明显差异[38],基因组中TEs的表观遗传调控非常精密而复杂(图2B)。
图2
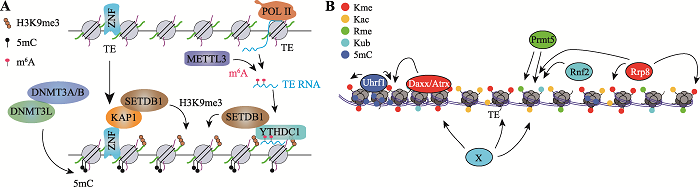 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2小鼠基因组中转座元件主要表观遗传沉默模式
A:小鼠基因组中逆转座元件的表观遗传沉默主要方式。H3K9me3和DNA甲基化占主导作用,RNA的m6A修饰通过其书写器蛋白METTL3、阅读器蛋白YTHDC1招募SETDB1,指导TEs区H3K9me3异染色质化沉默。B:参与沉默TEs表观遗传修饰及各类表观遗传因子。
Fig. 2Mechanism of transposable elements silencing
除大部分TEs被表观遗传修饰沉默外,也存在少部分TEs在特定的细胞类型中被特定的表观遗传修饰所激活。组蛋白修饰H3K27ac、H3K4me1和H3K4me3通常被认为是基因激活的标签,其中H3K27ac和H3K4me1是活性增强子的表观修饰标记,H3K4me3是活跃启动子区表观遗传修饰,这些修饰同样也在部分TEs染色质区域富集从能激活其表达。比如在小鼠滋养外胚层干细胞(trophoblast stem cell, TSCs)中,转座元件RLTR13D5上就显著富集H3K4me1和H3K27ac修饰,并且激活RLTR13D5作为增强子进一步招募TSCs核心转录因子CDX2、EOMES和ELF5的结合[39]。类似的,在人胚胎干细胞中,转座元件LTR77等富集H3K4me1修饰[40];在小鼠胚胎干细胞中,转座元件RLTR9、RLTR13等富集H3K27ac和H3K4me1修饰被激活并调控基因表达[41。
1.2 转座元件与基因转录调控
TEs可以通过多种方式调控基因表达[42,43]。总体而言,大多数TEs在基因组内呈散在分布,但对部分TEs而言,它们的分布并非完全随机。人类基因组中约25%的基因启动子由TEs构成[44],并且基因的表达与其附近TEs的分布及含量有显著联系,其中基因附近SINEs的含量与基因表达呈正相关关系,而LINEs的含量与基因表达呈负相关关系[45]。此外,TEs也可通过作为增强子调控基因表达,并对多种生物学功能如胚胎干细胞多能性维持、胎盘发生、神经发生、免疫反应以及生殖系细胞形成等过程具有重要调控作用[14,46,47]。并且,TEs也可在转录后水平调控基因表达,如影响基因的可变剪接、RNA编辑、细胞内定位等[48,49]。此外,TEs也可为许多转录因子如多能性因子POU5F1、SOX2、NANOG、ZFP42,中内胚层相关转录因子FOXA2、GATA4、SOX17等提供结合位点进而调控基因表达[50,51]。值得一提的是,TEs对基因组三维结构的维持与构建也发挥着重要的作用,如LINE1、B1/Alu (属于SINEs家族)转座元件对三维基因组A/B区隔化起到重要的调控作用[52]。同样,转座元件MERVL和HERVH分别为小鼠早期胚胎和人心肌细胞发育过程中拓扑结构域(Topologically associating domain, TAD)的形成提供锚点,以完成对基因的区块化调控[53,54]。此外,许多TEs还能作为染色质高级结构塑造因子CTCF的结合位点,形成增强子/绝缘子-启动子相互作用套环,实现针对单个基因的精确调控[55,56,57]。同时,TEs还可通过反式作用如通过lncRNA、miRNA等非编码RNA的形式调控基因表达[48,50]。总之,TEs通过影响染色质修饰、转录因子结合、RNA编辑以及染色质高级结构等,对基因的表达起到重要的调控作用。1.3 表观遗传、转座元件与细胞命运决定
从原核生物到真核生物,从单细胞生物到多细胞生物,这是物种进化上两次重大的突破。不同细胞命运状态的形成是多细胞生物形成的基础,细胞命运决定是多细胞生物发育过程中必须经历的生物事件。多细胞生物个体中所有的细胞均来源于同一个受精卵细胞,随着发育的过程,逐渐形成多种命运状态不同的细胞,这一过程究其根本原因是表观遗传修饰导致的遗传信息非均一性展示。表观遗传修饰在细胞命运决定过程中起着根本性调控作用。在发育过程中,不同细胞命运形成过程发生不同细胞命运决定,表观遗传修饰对正常发育、不同细胞谱系细胞的形成起着非常重要的作用。例如,组蛋白修饰H3K9me3甲基转移酶Setdb1敲除的小鼠早期胚胎致死[58],同样DNA甲基转移酶Dnmt3a/b也是小鼠正常发育所必需,敲除后致死[59],其他表观遗传因子如Kdm2a、Kdm2b、Kdm1a等敲除亦是胚胎致死[60,61,62]。这些表观遗传因子敲除致死通常是因为特定谱系细胞命运决定,在发育过程中不能在特定的时间点和特定的组织中精确完成或者维持,在动物表型上,则会进一步导致胚胎发育异常从而致死。作为发育/分化过程相对应的体细胞重编程和转分化,同样也伴随着大量细胞命运决定的过程。研究表明,许多表观遗传修饰参与了体细胞重编程过程中细胞命运决定,如异染色质修饰H3K9me3的擦除是体细胞重编程末期非常关键的节点,许多未能完全重编程的细胞中,其多能性基因如Oct4、Nanog、Sox2以及Dppa5a等位点都存在较高的H3K9me3修饰,并且敲低H3K9me3甲基转移酶Setdb1基因可以显著促进未完全重编程的细胞继续重编程[63]。此外,DNA去甲基化酶Tet1/2基因也能够调控体细胞重编程效率[64,65],其他表观遗传因子如Kdm1a、Kdm2a/b、Kdm5b、Dot1l、Chaf1a/b、Ncor2和Rybp等均能显著影响体细胞重编程过程[66,67,68,69,70,71,72,73]。此外,以上表观遗传因子在TEs活性调控上也起着至关重要的作用[3,38,74],提示表观遗传修饰可能通过调控TEs对细胞命运决定和维持起着及其重要的作用。
作为表观遗传修饰主要锚点的TEs也参与细胞命运决定调控。在早期胚胎发育过程中,小鼠特有转座元件MERVL和LINEs参与早期胚胎发育过程中2-细胞期到囊胚的细胞命运转变的调控[75,76,77]。在神经发育过程中,转座元件L1参与了神经祖细胞向神经元的细胞命运转变[78,79]。在体细胞重编程过程中,部分转座元件在重编程中后期被激活[80]。类似的,人原始态(naïve)胚胎干细胞中也存在部分活化的TEs,如进化过程中较为年轻的人猿科类特有的转座元件SVA、HERVK和HERVH等,并且这些TEs的激活与沉默,通过改变染色质开放状态、组蛋白修饰和DNA甲基化修饰等,改变基因调控网络,直接参与了人naïve胚胎干细胞命运状态的维持和决定[81,82,83]。在人体细胞重编程过程中,转座元件HERVH也会被激活,并且其过度激活将导致获得的诱导多能性干细胞分化缺陷[84]。在免疫系统中,ERVs的激活也直接参与了naïve T细胞向Th2和Th1的细胞命运转变,ERVs的激活直接促进了naïve T细胞向Th1细胞命运的转变,而抑制向Th2细胞命运的转变[85]。
2 转座元件研究领域存在的主要挑战
随着测序技术和系统生物学的发展,全基因组/转录组的深度测序、染色质修饰表征及表达谱分析等生物信息学分析成为驱动转座元件领域研究发展的有力工具,这对理解非编码RNA、TEs和宿主基因之间的关系有重要意义。然而,针对TEs的生物信息学分析和功能研究同样也面临着诸多挑战,如测序数据的匹配。TEs由于其多拷贝重复序列的特性,同一家族不同TEs拷贝间序列高度相似,因此在序列分析上难以确定测序数据来源的准确位点。TEs来源的测序片段通常能够匹配到基因组多个不同的位置,这种序列称为多次匹配序列。通常处理这类多次匹配序列的策略大致可以分为3种:第一种为去除所有多次匹配的序列,只保留单次匹配的数据,常规针对基因表达调控的分析采用的即是这种策略;第二种是保留所有多次匹配的序列,并将所有可匹配位点记录下来用于后续的分析计算;第三种策略与第二种策略类似,保留了所有多次匹配序列,但针对每条序列只保留其中一个最佳可匹配位点,如果存在多个最佳匹配位点时,则随机保留其中一个用于后续的分析计算。这3种策略各有利弊,其中策略一得到的匹配结果最准确,但大部分TEs来源的信息被去除,这对进化上年轻的TEs尤为明显,由于它们突变较少,很难找到可靠的突变位点以区分不同的拷贝;策略二保留了重复序列信息,但会导致部分位点重复序列信号过度放大,产生过多的假阳性;策略三在保留了重复序列信息的同时也没有过度放大重复序列的信号,值得注意的是,虽然这种策略在整个家族层面比较真实的反应了TEs的整体水平,但由于是随机分配,所以这种方法计算的单个拷贝位点信号同样并不是非常准确的[86,87]。无论何种分析方法,都无法准确衡量TEs单个拷贝在测序数据中的信号程度。解决这一问题的根本办法或许还得在测序技术上获得突破,使得测序技术能够更长更准确的读取碱基序列。另一个主要的挑战是TEs在基因组中的注释问题。TEs在基因组中含量有数百万条不同的拷贝,如何准确识别每一个拷贝在基因组中的具体位置对TEs调控基因表达、染色质高级结构、TEs来源的嵌合转录本等研究非常重要。目前不同基因组版本中TEs位置信息变动较大,TEs的位置也是染色体组装的主要困难之一[86]。其次,不同个体之间TEs的多态性对TEs的分析也会存在较大的影响,例如不同品系小鼠中TEs在基因组中的位置信息就存在很大差异[88]。个体之间的表观遗传变异性是科学家们需要解决的一个重要问题,TEs在不同个体之间插入的多态性也许能够部分地解释这种变异性,因此TEs各拷贝在基因组中位置的准确注释就显得及其重要。这一问题的解决同样很大程度上依赖于测序技术的改进。如何准确鉴定细胞命运决定相关的TEs还存在一定的挑战。无论是发育还是疾病等一系列细胞命运决定过程中,细胞都存在非常大的异质性。单细胞技术是解析细胞命运决定过程中最有利的技术,通过单细胞测序技术解析TEs的动态变化将对鉴定一系列潜在细胞命运决定相关TEs提供极大的帮助[89,90]。同时,实验上针对TEs的功能验证也存在巨大挑战。尽管基因编辑技术的发展给基因功能的研究带来了极大的便利,但由于TEs多拷贝的特性,这些技术一般难以针对TEs进行编辑。尽管近年来也有研究报道利用改进的基因编辑技术可以针对TEs进行整体调控[77,91,92],但一般仅适用于非常保守的TEs元件,并且存在效率低以及高脱靶性的问题。针对TEs的基因编辑主要存在两个难点:其一,由于其在基因组中数目非常大,难以同时靶向所有/大部分拷贝位点,只靶向个别/部分位点时,对整体水平影响不大,或其他位点可能存在补偿效应;其二,TEs在基因组中散在分布,作为染色质的骨架,多个位点同时编辑时会影响染色质整体结构,从而导致细胞死亡。总之,针对TEs的生物信息学分析以及生物学功能实验验证目前在技术上还存在一定的挑战。
3 结语与展望
TEs是基因组中含量最多的元素。尽管TEs的转座特性会给基因组完整性带来潜在的危险,但它们同时又是基因表达调控序列,并且是物种进化的主要动力和产物。基因组中TEs的有害性和有益性保持在严格的平衡状态,这种平衡主要由表观遗传系统控制,特别是染色质层面上的表观遗传修饰。异染色质的形成是抑制转座元件表达的主要机制,本文重点总结了近年来关于H3K9me3修饰及其相关机制介导的异染色质在TEs沉默中的关键作用,介绍了基因组中部分处于“激活”状态的TEs在基因表达调控与细胞命运决定过程中的功能。尽管前人的研究已经付出了很多的努力,但仍存在许多悬而未决的问题,比如:不同TEs是如何产生的,物种特异性的TEs进化在进化过程中的作用是什么?不同TEs间的具体调控机制是什么?不同细胞命运状态下是如何精确区分并激活/抑制特异的TEs?这些TEs在细胞命运转变过程中具有什么功能?个体间TEs插入位点的多态性和表观遗传变异性的关系是什么?越来越多的TEs被检测到存在多样的表观遗传修饰和特定的表达模式,这背后的生物学意义是什么?直至目前,这些问题都尚不清楚,对细胞命运转变过程中TEs的生理功能及调控机制的了解仍然十分匮乏。尽管人们还不能全面认识TEs的精细调控机制及在细胞命运转变过程中的规律和功能,但随着测序技术、单细胞技术、生物信息分析方法和基因编辑技术的不断发展,相信未来该领域会取得新的突破。同时,研究TEs在细胞命运转变过程中的变化情况及分子调控机制,将为细胞命运转换过程揭示一个全新的、重要的遗传因素。从新的角度来定义并研究细胞状态与命运决定,也许将为揭示影响细胞命运决定因素的研究提供新的突破口和原创性的发现,并且有助于人们对细胞命运转变更深层次的理解,以及将加深人们对TEs在再生医学与人类疾病中的认识。
(责任编委: 陆发隆)
中国科学院广州生物医药与健康研究院陈捷凯课题组简介
中国科学院广州生物医药与健康研究院陈捷凯课题组成立于2013年,课题组长为陈捷凯研究员。课题组专注于细胞命运决定机理的研究,以干细胞为模型(如类器官、重编程等),主要研究表观遗传调控的特异性机制,及其与信号转导、转录因子、非编码遗传信息之间的联系。除了干细胞实验平台和生物化学平台之外,课题组擅长通过计算生物学在表观遗传组和单细胞转录组等大数据中挖掘重要生物学机理,并根据生物学问题开发新算法和新软件,在转座元件和细胞谱系方面开发了一系列工具。课题组近年来取得了一系列研究成果,在Nature、Cell、Cell Stem Cell、Nature Cell Biology、Nature Communications、National Science Review和Cell Reports等国际知名期刊发表论文数十篇。与此同时,课题组还承担了科技部、国家自然科学基金委及中国科学院等一系列重大课题。课题组网站:
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
PMID:14942727 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 4]
DOI:10.1101/gr.5826307URL [本文引用: 1]
DOI:10.1146/annurev-genom-082509-141802URL [本文引用: 1]
DOI:10.1038/nrg2640URL [本文引用: 2]
DOI:10.1038/nature13760URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/nature21683URL [本文引用: 2]
DOI:10.1017/S0016672303006268URL [本文引用: 2]
DOI:10.1146/annurev-cellbio-100814-125514PMID:26393776 [本文引用: 2]

Transposable elements (TEs) account for at least 50% of the human genome. They constitute essential motors of evolution through their ability to modify genomic architecture, mutate genes and regulate gene expression. Accordingly, TEs are subject to tight epigenetic control during the earliest phases of embryonic development via histone and DNA methylation. Key to this process is recognition by sequence-specific RNA- and protein-based repressors. Collectively, these mediators are responsible for silencing a very broad range of TEs in an evolutionarily dynamic fashion. As a consequence, mobile elements and their controllers exert a marked influence on transcriptional networks in embryonic stem cells and a variety of adult tissues. The emerging picture is not that of a simple arms race but rather of a massive and sophisticated enterprise of TE domestication for the evolutionary benefit of the host.
DOI:10.1073/pnas.0604768103URL [本文引用: 1]
DOI:10.1038/nature05805URL [本文引用: 1]
[本文引用: 2]
DOI:10.1007/s00018-017-2454-8URL [本文引用: 2]
DOI:10.1016/j.stem.2011.04.004URL [本文引用: 1]
DOI:10.1016/j.virol.2010.12.007URL [本文引用: 2]
DOI:10.1016/j.scr.2013.07.006URL [本文引用: 2]
DOI:10.1038/nature08674URL
DOI:10.1038/nature07844URL [本文引用: 2]
DOI:10.3389/fonc.2014.00014PMID:24567914
About half of the mammalian genome is occupied by DNA sequences that originate from transposable elements. Retrotransposons can modulate gene expression in different ways and, particularly retrotransposon-derived long terminal repeats, profoundly shape expression of both surrounding and distant genomic loci. This is especially important in pre-implantation development, during which extensive reprograming of the genome takes place and cells pass through totipotent and pluripotent states. At this stage, the main mechanism responsible for retrotransposon silencing, i.e., DNA methylation, is inoperative. A particular retrotransposon called muERV-L/MERVL is expressed during pre-implantation stages and contributes to the plasticity of mouse embryonic stem cells. This review will focus on the role of MERVL-derived sequences as controlling elements of gene expression specific for pre-implantation development, two-cell stage-specific gene expression, and stem cell pluripotency, the epigenetic mechanisms that control their expression, and the contributions of the pluripotency marker REX1 and the related Yin Yang 1 family of transcription factors to this regulation process.
DOI:10.1016/j.cell.2015.08.037PMID:26365490 [本文引用: 2]
Embryonic stem cells (ESCs) repress the expression of exogenous proviruses and endogenous retroviruses (ERVs). Here, we systematically dissected the cellular factors involved in provirus repression in embryonic carcinomas (ECs) and ESCs by a genome-wide siRNA screen. Histone chaperones (Chaf1a/b), sumoylation factors (Sumo2/Ube2i/Sae1/Uba2/Senp6), and chromatin modifiers (Trim28/Eset/Atf7ip) are key determinants that establish provirus silencing. RNA-seq analysis uncovered the roles of Chaf1a/b and sumoylation modifiers in the repression of ERVs. ChIP-seq analysis demonstrates direct recruitment of Chaf1a and Sumo2 to ERVs. Chaf1a reinforces transcriptional repression via its interaction with members of the NuRD complex (Kdm1a, Hdac1/2) and Eset, while Sumo2 orchestrates the provirus repressive function of the canonical Zfp809/Trim28/Eset machinery by sumoylation of Trim28. Our study reports a genome-wide atlas of functional nodes that mediate proviral silencing in ESCs and illuminates the comprehensive, interconnected, and multi-layered genetic and epigenetic mechanisms by which ESCs repress retroviruses within the genome. Copyright © 2015 Elsevier Inc. All rights reserved.
DOI:10.1016/j.molcel.2014.05.029PMID:24981170 [本文引用: 2]
Heterochromatin is required to restrict aberrant expression of retrotransposons, but it remains poorly defined due to the underlying repeat-rich sequences. We dissected Suv39h-dependent histone H3 lysine 9 trimethylation (H3K9me3) by genome-wide ChIP sequencing in mouse embryonic stem cells (ESCs). Refined bioinformatic analyses of repeat subfamilies indicated selective accumulation of Suv39h-dependent H3K9me3 at interspersed repetitive elements that cover ~5% of the ESC epigenome. The majority of the ~8,150 intact long interspersed nuclear elements (LINEs) and endogenous retroviruses (ERVs), but only a minor fraction of the >1.8 million degenerate and truncated LINEs/ERVs, are enriched for Suv39h-dependent H3K9me3. Transcriptional repression of intact LINEs and ERVs is differentially regulated by Suv39h and other chromatin modifiers in ESCs but governed by DNA methylation in committed cells. These data provide a function for Suv39h-dependent H3K9me3 chromatin to specifically repress intact LINE elements in the ESC epigenome.Copyright © 2014 Elsevier Inc. All rights reserved.
DOI:10.1016/j.stem.2015.07.022URL [本文引用: 1]
DOI:10.1038/s41586-021-03210-1URL [本文引用: 1]
DOI:10.1038/s41586-020-03135-1URL [本文引用: 1]
DOI:10.1126/science.aay6018URL [本文引用: 1]
DOI:10.1038/nature14345URL [本文引用: 1]
DOI:10.1016/j.molcel.2014.10.003URL [本文引用: 1]
DOI:10.1038/nature03238URL [本文引用: 1]
DOI:10.7554/eLife.02008URL
PMID:16537839
We demonstrated previously that the cytosine deaminase APOBEC3G inhibits retrotransposition of two active murine endogenous retroviruses, namely intracisternal A-particles (IAP) and MusD, in an ex vivo assay where retrotransposition was monitored by selection of neo-marked elements. Sequencing of the transposed copies further disclosed extensive editing, resulting in a high load of G-to-A mutations. Here, we asked whether this G-to-A editing was associated with an impact of APOBEC3G on viral cDNA yields. To this end, we used a specially designed quantitative PCR method to selectively measure the copy number of transposed retroelements, in the absence of G418 selection. We show that human APOBEC3G severely reduces the number of MusD and IAP transposed cDNA copies, with no effect on the level of the intermediate RNA transcripts. The magnitude of the decrease closely parallels that observed when transposed copies are assayed by selection of G418-resistant cells. Moreover, sequencing of transposed elements recovered by PCR without prior selection of the cells reveals high-level editing. Using this direct method with a series of cytosine deaminases, we further demonstrate a similar dual effect of African green monkey APOBE3G, human APOBEC3F and murine APOBEC3 on MusD retrotransposition, with a distinct extent and site specificity for each editing activity. Altogether the data demonstrate that cytosine deaminases have a protective effect against endogenous retroviruses both by reducing viral cDNA levels and by introducing mutations in the transposed copies, thus inactivating them for subsequent rounds of retrotransposition. This dual, two-step effect likely participates in the efficient defense of the cell genome against invading endogenous retroelements.
DOI:10.1371/journal.pgen.1005693URL
DOI:10.7554/eLife.08851URL
DOI:10.1038/nsmb.3066PMID:26237512
Cellular plasticity is essential for early embryonic cells. Unlike pluripotent cells, which form embryonic tissues, totipotent cells can generate a complete organism including embryonic and extraembryonic tissues. Cells resembling 2-cell-stage embryos (2C-like cells) arise at very low frequency in embryonic stem (ES) cell cultures. Although induced reprogramming to pluripotency is well established, totipotent cells remain poorly characterized, and whether reprogramming to totipotency is possible is unknown. We show that mouse 2C-like cells can be induced in vitro through downregulation of the chromatin-assembly activity of CAF-1. Endogenous retroviruses and genes specific to 2-cell embryos are the highest-upregulated genes upon CAF-1 knockdown. Emerging 2C-like cells exhibit molecular characteristics of 2-cell embryos and higher reprogrammability than ES cells upon nuclear transfer. Our results suggest that early embryonic-like cells can be induced by modulating chromatin assembly and that atypical histone deposition may trigger the emergence of totipotent cells.
DOI:10.1093/nar/gkx884URL
DOI:10.1038/s41588-018-0060-9PMID:29483655 [本文引用: 1]
Ten-eleven translocation (TET) proteins play key roles in the regulation of DNA-methylation status by oxidizing 5-methylcytosine (5mC) to generate 5-hydroxymethylcytosine (5hmC), which can both serve as a stable epigenetic mark and participate in active demethylation. Unlike the other members of the TET family, TET2 does not contain a DNA-binding domain, and it remains unclear how it is recruited to chromatin. Here we show that TET2 is recruited by the RNA-binding protein Paraspeckle component 1 (PSPC1) through transcriptionally active loci, including endogenous retroviruses (ERVs) whose long terminal repeats (LTRs) have been co-opted by mammalian genomes as stage- and tissue-specific transcriptional regulatory modules. We found that PSPC1 and TET2 contribute to ERVL and ERVL-associated gene regulation by both transcriptional repression via histone deacetylases and post-transcriptional destabilization of RNAs through 5hmC modification. Our findings provide evidence for a functional role of transcriptionally active ERVs as specific docking sites for RNA epigenetic modulation and gene regulation.
DOI:10.1038/s41467-018-08006-yURL [本文引用: 2]
DOI:10.1038/ng.2553URL [本文引用: 1]
DOI:10.1038/ng.2649URL [本文引用: 1]
DOI:10.1038/ncomms14550PMID:28348391
Cis-regulatory modules contain multiple transcription factor (TF)-binding sites and integrate the effects of each TF to control gene expression in specific cellular contexts. Transposable elements (TEs) are uniquely equipped to deposit their regulatory sequences across a genome, which could also contain cis-regulatory modules that coordinate the control of multiple genes with the same regulatory logic. We provide the first evidence of mouse-specific TEs that encode a module of TF-binding sites in mouse embryonic stem cells (ESCs). The majority (77%) of the individual TEs tested exhibited enhancer activity in mouse ESCs. By mutating individual TF-binding sites within the TE, we identified a module of TF-binding motifs that cooperatively enhanced gene expression. Interestingly, we also observed the same motif module in the in silico constructed ancestral TE that also acted cooperatively to enhance gene expression. Our results suggest that ancestral TE insertions might have brought in cis-regulatory modules into the mouse genome.
DOI:10.1186/s13059-018-1577-zURL [本文引用: 1]
DOI:10.1007/s11427-015-4993-2URL [本文引用: 1]
PMID:12547512 [本文引用: 1]
Transposable elements (TEs) are abundant in mammalian genomes and have potentially contributed to their hosts' evolution by providing novel regulatory or coding sequences. We surveyed different classes of regulatory region in the human genome to assess systematically the potential contribution of TEs to gene regulation. Almost 25% of the analyzed promoter regions contain TE-derived sequences, including many experimentally characterized cis-regulatory elements. Scaffold/matrix attachment regions (S/MARs) and locus control regions (LCRs) that are involved in the simultaneous regulation of multiple genes also contain numerous TE-derived sequences. Thus, TEs have probably contributed substantially to the evolution of both gene-specific and global patterns of human gene regulation.
DOI:10.1093/gbe/evr015PMID:21362639 [本文引用: 1]
Independent lines of investigation have documented effects of both transposable elements (TEs) and gene length (GL) on gene expression. However, TE gene fractions are highly correlated with GL, suggesting that they cannot be considered independently. We evaluated the TE environment of human genes and GL jointly in an attempt to tease apart their relative effects. TE gene fractions and GL were compared with the overall level of gene expression and the breadth of expression across tissues. GL is strongly correlated with overall expression level but weakly correlated with the breadth of expression, confirming the selection hypothesis that attributes the compactness of highly expressed genes to selection for economy of transcription. However, TE gene fractions overall, and for the L1 family in particular, show stronger anticorrelations with expression level than GL, indicating that GL may not be the most important target of selection for transcriptional economy. These results suggest a specific mechanism, removal of TEs, by which highly expressed genes are selectively tuned for efficiency. MIR elements are the only family of TEs with gene fractions that show a positive correlation with tissue-specific expression, suggesting that they may provide regulatory sequences that help to control human gene expression. Consistent with this notion, MIR fractions are relatively enriched close to transcription start sites and associated with coexpression in specific sets of related tissues. Our results confirm the overall relevance of the TE environment to gene expression and point to distinct mechanisms by which different TE families may contribute to gene regulation.
DOI:10.1126/science.aad5497URL [本文引用: 1]
DOI:10.1038/ng.600PMID:20526341 [本文引用: 1]
Detection of new genomic control elements is critical in understanding transcriptional regulatory networks in their entirety. We studied the genome-wide binding locations of three key regulatory proteins (POU5F1, also known as OCT4; NANOG; and CTCF) in human and mouse embryonic stem cells. In contrast to CTCF, we found that the binding profiles of OCT4 and NANOG are markedly different, with only approximately 5% of the regions being homologously occupied. We show that transposable elements contributed up to 25% of the bound sites in humans and mice and have wired new genes into the core regulatory network of embryonic stem cells. These data indicate that species-specific transposable elements have substantially altered the transcriptional circuitry of pluripotent stem cells.
DOI:10.1126/science.aac7247URL [本文引用: 2]
DOI:10.1038/emboj.2008.94URL [本文引用: 1]
DOI:10.1038/nrg2337PMID:18368054 [本文引用: 2]
The control and coordination of eukaryotic gene expression rely on transcriptional and post-transcriptional regulatory networks. Although progress has been made in mapping the components and deciphering the function of these networks, the mechanisms by which such intricate circuits originate and evolve remain poorly understood. Here I revisit and expand earlier models and propose that genomic repeats, and in particular transposable elements, have been a rich source of material for the assembly and tinkering of eukaryotic gene regulatory systems.
DOI:10.1371/journal.pgen.1006883URL [本文引用: 1]
DOI:10.1038/s41422-020-00466-6URL [本文引用: 1]
[本文引用: 1]
DOI:10.1038/s41588-019-0479-7URL [本文引用: 1]
DOI:10.1038/s41467-020-15520-5PMID:32286261 [本文引用: 1]
Chromatin looping is important for gene regulation, and studies of 3D chromatin structure across species and cell types have improved our understanding of the principles governing chromatin looping. However, 3D genome evolution and its relationship with natural selection remains largely unexplored. In mammals, the CTCF protein defines the boundaries of most chromatin loops, and variations in CTCF occupancy are associated with looping divergence. While many CTCF binding sites fall within transposable elements (TEs), their contribution to 3D chromatin structural evolution is unknown. Here we report the relative contributions of TE-driven CTCF binding site expansions to conserved and divergent chromatin looping in human and mouse. We demonstrate that TE-derived CTCF binding divergence may explain a large fraction of variable loops. These variable loops contribute significantly to corresponding gene expression variability across cells and species, possibly by refining sub-TAD-scale loop contacts responsible for cell-type-specific enhancer-promoter interactions.
DOI:10.1101/gr.235747.118URL [本文引用: 1]
DOI:10.1016/j.cell.2011.11.058URL [本文引用: 1]
DOI:10.1128/MCB.24.6.2478-2486.2004URL [本文引用: 1]
PMID:10555141 [本文引用: 1]
The establishment of DNA methylation patterns requires de novo methylation that occurs predominantly during early development and gametogenesis in mice. Here we demonstrate that two recently identified DNA methyltransferases, Dnmt3a and Dnmt3b, are essential for de novo methylation and for mouse development. Inactivation of both genes by gene targeting blocks de novo methylation in ES cells and early embryos, but it has no effect on maintenance of imprinted methylation patterns. Dnmt3a and Dnmt3b also exhibit nonoverlapping functions in development, with Dnmt3b specifically required for methylation of centromeric minor satellite repeats. Mutations of human DNMT3B are found in ICF syndrome, a developmental defect characterized by hypomethylation of pericentromeric repeats. Our results indicate that both Dnmt3a and Dnmt3b function as de novo methyltransferases that play important roles in normal development and disease.
DOI:10.1172/JCI84014PMID:26808549 [本文引用: 1]
The development of the hematopoietic system is a dynamic process that is controlled by the interplay between transcriptional and epigenetic networks to determine cellular identity. These networks are critical for lineage specification and are frequently dysregulated in leukemias. Here, we identified histone demethylase KDM2B as a critical regulator of definitive hematopoiesis and lineage commitment of murine hematopoietic stem and progenitor cells (HSPCs). RNA sequencing of Kdm2b-null HSPCs and genome-wide ChIP studies in human leukemias revealed that KDM2B cooperates with polycomb and trithorax complexes to regulate differentiation, lineage choice, cytokine signaling, and cell cycle. Furthermore, we demonstrated that KDM2B exhibits a dichotomous role in hematopoietic malignancies. Specifically, we determined that KDM2B maintains lymphoid leukemias, but restrains RAS-driven myeloid transformation. Our study reveals that KDM2B is an important mediator of hematopoietic cell development and has opposing roles in tumor progression that are dependent on cellular context.
DOI:10.1016/j.mod.2014.10.001URL [本文引用: 1]
DOI:10.1038/ng.268URL [本文引用: 1]
DOI:10.1038/ng.2491URL [本文引用: 1]
DOI:10.1038/ng.2807URL [本文引用: 1]
DOI:S1934-5909(18)30401-6PMID:30220521 [本文引用: 1]
Here, we report DNA methylation and hydroxymethylation dynamics at nucleotide resolution using C/EBPα-enhanced reprogramming of B cells into induced pluripotent cells (iPSCs). We observed successive waves of hydroxymethylation at enhancers, concomitant with a decrease in DNA methylation, suggesting active demethylation. Consistent with this finding, ablation of the DNA demethylase Tet2 almost completely abolishes reprogramming. C/EBPα, Klf4, and Tfcp2l1 each interact with Tet2 and recruit the enzyme to specific DNA sites. During reprogramming, some of these sites maintain high levels of 5hmC, and enhancers and promoters of key pluripotency factors become demethylated as early as 1 day after Yamanaka factor induction. Surprisingly, methylation changes precede chromatin opening in distinct chromatin regions, including Klf4 bound sites, revealing a pioneer factor activity associated with alternation in DNA methylation. Rapid changes in hydroxymethylation similar to those in B cells were also observed during compound-accelerated reprogramming of fibroblasts into iPSCs, highlighting the generality of our observations.Copyright © 2018 Elsevier Inc. All rights reserved.
DOI:10.1038/nature15749URL [本文引用: 1]
DOI:10.1038/s41556-018-0047-xPMID:29531310 [本文引用: 1]
Somatic cell reprogramming by exogenous factors requires cooperation with transcriptional co-activators and co-repressors to effectively remodel the epigenetic environment. How this interplay is regulated remains poorly understood. Here, we demonstrate that NCoR/SMRT co-repressors bind to pluripotency loci to create a barrier to reprogramming with the four Yamanaka factors (OCT4, SOX2, KLF4 and c-MYC), and consequently, suppressing NCoR/SMRT significantly enhances reprogramming efficiency and kinetics. The core epigenetic subunit of the NCoR/SMRT complex, histone deacetylase 3 (HDAC3), contributes to the effects of NCoR/SMRT by inducing histone deacetylation at pluripotency loci. Among the Yamanaka factors, recruitment of NCoR/SMRT-HDAC3 to genomic loci is mostly facilitated by c-MYC. Hence, we describe how c-MYC is beneficial for the early phase of reprogramming but deleterious later. Overall, we uncover a role for NCoR/SMRT co-repressors in reprogramming and propose a dual function for c-MYC in this process.
DOI:10.1038/nature10953URL [本文引用: 1]
DOI:10.1016/j.stem.2016.12.002URL [本文引用: 1]
DOI:10.1016/j.celrep.2017.10.091URL [本文引用: 1]
DOI:10.1016/j.stem.2011.10.005PMID:22100412 [本文引用: 1]
Reprogramming of somatic cells into induced pluripotent stem cells (iPSCs) resets the epigenome to an embryonic-like state. Vitamin C enhances the reprogramming process, but the underlying mechanisms are unclear. Here we show that the histone demethylases Jhdm1a/1b are key effectors of somatic cell reprogramming downstream of vitamin C. We first observed that vitamin C induces H3K36me2/3 demethylation in mouse embryonic fibroblasts in culture and during reprogramming. We then identified Jhdm1a/1b, two known vitamin-C-dependent H3K36 demethylases, as potent regulators of reprogramming through gain- and loss-of-function approaches. Furthermore, we found that Jhdm1b accelerates cell cycle progression and suppresses cell senescence during reprogramming by repressing the Ink4/Arf locus. Jhdm1b also cooperates with Oct4 to activate the microRNA cluster 302/367, an integral component of the pluripotency machinery. Our results therefore reveal a role for H3K36me2/3 in cell fate determination and establish a link between histone demethylases and vitamin-C-induced reprogramming.Copyright © 2011 Elsevier Inc. All rights reserved.
[本文引用: 1]
DOI:S1934-5909(17)30074-7PMID:28366589 [本文引用: 1]
Vertebrate eggs can induce the nuclear reprogramming of somatic cells to enable production of cloned animals. Nuclear reprogramming is relatively inefficient, and the development of the resultant embryos is frequently compromised, in part due to the inappropriate expression of genes previously active in the donor nucleus. Here, we identify H3K4 methylation as a major epigenetic roadblock that limits transcriptional reprogramming and efficient nuclear transfer (NT). Widespread expression of donor-cell-specific genes was observed in inappropriate cell types in NT embryos, limiting their developmental capacity. The expression of these genes in reprogrammed embryos arises from epigenetic memories of a previously active transcriptional state in donor cells that is characterized by high H3K4 methylation. Reducing H3K4 methylation had little effect on gene expression in donor cells, but it substantially improved transcriptional reprogramming and development of NT embryos. These results show that H3K4 methylation imposes a barrier to efficient nuclear reprogramming and suggest approaches for improving reprogramming strategies.Copyright © 2017 The Authors. Published by Elsevier Inc. All rights reserved.
[本文引用: 1]
DOI:S0092-8674(18)30655-XPMID:29937225 [本文引用: 1]
Transposable elements represent nearly half of mammalian genomes and are generally described as parasites, or "junk DNA." The LINE1 retrotransposon is the most abundant class and is thought to be deleterious for cells, yet it is paradoxically highly expressed during early development. Here, we report that LINE1 plays essential roles in mouse embryonic stem cells (ESCs) and pre-implantation embryos. In ESCs, LINE1 acts as a nuclear RNA scaffold that recruits Nucleolin and Kap1/Trim28 to repress Dux, the master activator of a transcriptional program specific to the 2-cell embryo. In parallel, LINE1 RNA mediates binding of Nucleolin and Kap1 to rDNA, promoting rRNA synthesis and ESC self-renewal. In embryos, LINE1 RNA is required for Dux silencing, synthesis of rRNA, and exit from the 2-cell stage. The results reveal an essential partnership between LINE1 RNA, Nucleolin, Kap1, and peri-nucleolar chromatin in the regulation of transcription, developmental potency, and ESC self-renewal.Copyright © 2018 Elsevier Inc. All rights reserved.
DOI:10.1038/nature11244URL [本文引用: 1]
DOI:10.1038/ng.3945PMID:28846101 [本文引用: 2]
After fertilization, to initiate development, gametes are reprogramed to become totipotent. Approximately half of the mammalian genome consists of repetitive elements, including retrotransposons, some of which are transcribed after fertilization. Retrotransposon activation is generally assumed to be a side effect of the extensive chromatin remodeling underlying the epigenetic reprogramming of gametes. Here, we used a targeted epigenomic approach to address whether specific retrotransposon families play a direct role in chromatin organization and developmental progression. We demonstrate that premature silencing of LINE-1 elements decreases chromatin accessibility, whereas prolonged activation prevents the gradual chromatin compaction that occurs naturally in developmental progression. Preventing LINE-1 activation and interfering with its silencing decreases developmental rates independently of the coding nature of the LINE-1 transcript, thus suggesting that LINE-1 functions primarily at the chromatin level. Our data suggest that activation of LINE-1 regulates global chromatin accessibility at the beginning of development and indicate that retrotransposon activation is integral to the developmental program.
DOI:10.1038/nature03663URL [本文引用: 1]
DOI:10.1038/nature08248URL [本文引用: 1]
DOI:10.1101/gr.172809.114PMID:24879558 [本文引用: 1]
Endogenous retroelements (EREs) account for about half of the mouse or human genome, and their potential as insertional mutagens and transcriptional perturbators is suppressed by early embryonic epigenetic silencing. Here, we asked how ERE control is maintained during the generation of induced pluripotent stem cells (iPSCs), as this procedure involves profound epigenetic remodeling. We found that all EREs tested were markedly up-regulated during the reprogramming of either mouse embryonic fibroblasts, human CD34(+) cells, or human primary hepatocytes. At the iPSC stage, EREs of some classes were repressed, whereas others remained highly expressed, yielding a pattern somewhat reminiscent of that recorded in embryonic stem cells. However, variability persisted between individual iPSC clones in the control of specific ERE integrants. Both during reprogramming and in iPS cells, the up-regulation of specific EREs significantly impacted on the transcription of nearby cellular genes. While transcription triggered by specific ERE integrants at highly precise developmental stages may be an essential step toward obtaining pluripotent cells, the broad and unspecific unleashing of the repetitive genome observed here may contribute to the inefficiency of the reprogramming process and to the phenotypic heterogeneity of iPSCs. © 2014 Friedli et al.; Published by Cold Spring Harbor Laboratory Press.
DOI:S1934-5909(16)30161-8PMID:27424783 [本文引用: 1]
Recent studies have aimed to convert cultured human pluripotent cells to a naive state, but it remains unclear to what extent the resulting cells recapitulate in vivo naive pluripotency. Here we propose a set of molecular criteria for evaluating the naive human pluripotent state by comparing it to the human embryo. We show that transcription of transposable elements provides a sensitive measure of the concordance between pluripotent stem cells and early human development. We also show that induction of the naive state is accompanied by genome-wide DNA hypomethylation, which is reversible except at imprinted genes, and that the X chromosome status resembles that of the human preimplantation embryo. However, we did not see efficient incorporation of naive human cells into mouse embryos. Overall, the different naive conditions we tested showed varied relationships to human embryonic states based on molecular criteria, providing a backdrop for future analysis of naive human pluripotency.Copyright © 2016 The Authors. Published by Elsevier Inc. All rights reserved.
DOI:10.1038/nature14308URL [本文引用: 1]
DOI:10.1038/nature13804URL [本文引用: 1]
DOI:10.1073/pnas.1413299111URL [本文引用: 1]
DOI:10.1016/j.immuni.2019.01.003URL [本文引用: 1]
DOI:10.1038/s41576-018-0050-xPMID:30232369 [本文引用: 2]
A substantial proportion of the genome of many species is derived from transposable elements (TEs). Moreover, through various self-copying mechanisms, TEs continue to proliferate in the genomes of most species. TEs have contributed numerous regulatory, transcript and protein innovations and have also been linked to disease. However, notwithstanding their demonstrated impact, many genomic studies still exclude them because their repetitive nature results in various analytical complexities. Fortunately, a growing array of methods and software tools are being developed to cater for them. This Review presents a summary of computational resources for TEs and highlights some of the challenges and remaining gaps to perform comprehensive genomic analyses that do not simply 'mask' repeats.
DOI:10.1038/nrg3117PMID:22124482 [本文引用: 1]
Repetitive DNA sequences are abundant in a broad range of species, from bacteria to mammals, and they cover nearly half of the human genome. Repeats have always presented technical challenges for sequence alignment and assembly programs. Next-generation sequencing projects, with their short read lengths and high data volumes, have made these challenges more difficult. From a computational perspective, repeats create ambiguities in alignment and assembly, which, in turn, can produce biases and errors when interpreting results. Simply ignoring repeats is not an option, as this creates problems of its own and may mean that important biological phenomena are missed. We discuss the computational problems surrounding repeats and describe strategies used by current bioinformatics systems to solve them.
DOI:10.1186/gb-2012-13-6-r45URL [本文引用: 1]
URL [本文引用: 1]
DOI:10.1101/gr.265173.120URL [本文引用: 1]
DOI:10.1126/science.aan4187URL [本文引用: 1]
DOI:10.7554/eLife.35989URL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}