 ,1,2,3
,1,2,3Progress on the mechanistic research of the trinucleotide repeat instabilities underlying human neurodegenerative diseases
Kenao Lv1, Xuefeng Pan,1,2,3通讯作者: 潘学峰,教授,研究方向:分子药理学、分子生物学、药物递送。E-mail:xuefengpancam@aliyun.com
编委: 何淑君
收稿日期:2021-05-24修回日期:2021-07-25
| 基金资助: |
Received:2021-05-24Revised:2021-07-25
| Fund supported: |
作者简介 About authors
吕柯孬,在读硕士研究生,专业方向:分子生物学。E-mail:

摘要
三核苷酸重复DNA序列扩增或缺失不稳定性与50多种人类神经退行性疾病有关。与疾病相关的三核苷酸重复拷贝数的增加或减少,影响了特定基因的表达,或因之产生具有细胞毒性的RNA和蛋白质已成为相关疾病的共有病理机制。现有的研究表明,疾病相关的三核苷酸重复拷贝数的改变有可能起因于相关三核苷酸重复DNA序列的异常DNA复制、修复、重组以及基因转录。有关人类遗传学研究也提示,发生在疾病相关的三核苷酸重复DNA部位的异常DNA复制、修复、重组或基因转录确有可能在三核苷酸重复DNA不稳定过程中发挥着关键作用。本文根据本课题组的研究经验,综述了近年来有关疾病相关三核苷酸重复不稳定性机制的研究进展,包括碱基突变不稳定、重复单元的扩增和缺失不稳定,以助更好地理解疾病相关三核苷酸重复DNA序列不稳定性的分子机制。
关键词:
Abstract
The expansion and deletion instabilities shown by some trinucleotide repeated DNA sequences are associated with more than 50 neurodegenerative diseases in humans. The increase or decrease of the trinucleotide repeat units underlying the diseases are not yet clearly explained using any mechanism, but has been found to affect the expression of specific genes, or produces cytotoxic RNA and protein, which has now become a common pathological mechanism of the diseases. The ongoing studies have shown that the changes in the copy numbers of the disease-related trinucleotide repeats may result from abnormal DNA replication, repair, recombination, and gene transcription. Human genetical studies also suggest that abnormal DNA replication, repair, recombination, or gene transcription that occurred in the disease-related trinucleotide repeat DNA sites may play a key role in the trinucleotide repeat DNA instabilities. Based on the research experiences of our research group, this paper reviews the recent research progress on the mechanisms of the disease-associated trinucleotide repeat DNA instabilities including their base mutation instabilities, the amplification and deletion instabilities of the repeat units, to better understand the molecular mechanism of the disease-associated trinucleotide repeats instabilities.
Keywords:
PDF (826KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
吕柯孬, 潘学峰. 人类神经退行性疾病相关的三核苷酸重复DNA序列不稳定性机制研究进展. 遗传[J], 2021, 43(9): 835-848 doi:10.16288/j.yczz.21-182
Kenao Lv.
自1991年Verkerk等[1]发现位于人类脆性X染色体综合征(fragile X syndrome) FMR-1基因非编码区的简单串联重复DNA序列为CGG三核苷酸重复DNA序列(trinucleotide repeats, TNRs),以及La Spada等[2]发现位于肯尼迪氏病(Kennedy’disease,又名X连锁脊髓和延髓肌萎缩,X-linked spinal and bulbar muscular atrophy)肾上腺受体基因(androgen receptor gene, AR)编码区内的简单串联重复DNA序列为CAG三核苷酸重复DNA序列,迄今已发现50种以上的神经退行性疾病与三核苷酸重复DNA序列扩增有关。与疾病有关的三核苷酸重复DNA序列的不稳定性扩增可能位于基因的非编码区,如内含子或编码序列侧翼的非翻译调控区[如脆性X染色体综合征、弗洛里德共济失调(Friedreich ataxia, FRDA)、强直性肌营养不良(myotonic dystrophy, MD)及脊髓小脑共济失调8型(spinocerebellar ataxia type 8)等]。在这种情况下,三核苷酸重复DNA序列可能因扩增而变得很长,由成千上万个重复单位组成。这些新增的三核苷酸重复DNA通过多种机制对受累基因产生影响,如诱导基因沉默而造成功能丧失(loss of function),或通过其他方式获得新功能(gain of function),如从有义链或反义链产生“毒性”RNA[3,4]。这些毒性RNA干扰mRNA正常加工以及与特定蛋白的结合。位于编码序列内的三核苷酸重复DNA序列的扩增与非编码区域内发生的三核苷酸重复DNA序列扩增相比,其长度往往适中[5,6],而且均为CAG重复序列,编码聚谷氨酰胺(polyglutamine, polyQ)[7]。据此,人们通常把三核苷酸重复DNA序列有关的神经退行疾病分为三类:第一类由CAG重复序列引起,包含亨廷顿舞蹈病(Huntington’s disease, HD)和脊髓小脑共济失调(spinocerebellar ataxia)等在内的16种疾病[8,9,10];第二类是在表型上更加多样化且异常扩增规模较小,由存在于外显子中的三核苷酸重复DNA序列不稳定性扩增引起[11,12];第三类则与位于内含子等非编码区的三核苷酸重复DNA序列不稳定有关,包括脆性X染色体综合征、强直性肌营养不良、脊髓小脑共济失调、青少年肌阵挛性癫痫(juvenile myoclonic epilepsy, JME)和弗里德里希共济失调[13]。除此以外,还可依据病理过程中涉及由扩增的CAG三核苷酸重复DNA序列编码而产生的polyQ的影响,分为polyQ疾病和非polyQ疾病(表1)。其中属于polyQ疾病的病理共性涉及蛋白质聚集、蛋白水解切割、转录失调、自噬损伤和线粒体功能障碍等分子机制[14]。但最近的研究对polyQ长度决定发病率这一观点提出质疑,转而认为polyQ疾病的发病率取决于多个DNA维持基因多态性变异[15]。
尽管对引发多种神经系统疾病的三核苷酸重复DNA序列异常扩增机制和表现出染色体“脆性”(fragility)现象的研究已历时数十年之久,但迄今依然对疾病状态下三核苷酸重复DNA序列异常扩增的分子机制缺乏透彻理解。在相当长的一段时间里,很多人相信疾病相关三核苷酸重复DNA序列在特定情况下通过形成非B型DNA结构(non-B DNA structure)影响DNA复制、基因转录、修复、重组等过程,并由此影响自身在基因组内的稳定性,出现碱基突变、重复单位的扩增或缺失[32,33]。但已报道的研究表明,至少在细菌、酵母(Saccharomyces)、线虫(Caenorhabditis elegans)或人类细胞系(human cell lines)等模式生物中疾病相关的三核苷酸重复DNA序列所能表现的不稳定性多为“变短”(缺失)[34],而不是患者基因中所表现的“变长”(扩增)。更为遗憾的是,很多利用模式生物所得到的研究结果也通常自相或者相互矛盾,因此很难对现有的任何一种机制达成共识,很难真正明确疾病状态下三核苷酸重复DNA序列体内扩增机制及与疾病发生和发展相关的病理机制[35,36]。基于本课题组多年的研究经验,并结合人类基因组内不同位点处三核苷酸重复DNA序列以纯合(pure repeats)和非纯合(interrupted repeats)形式分布所表现出的稳定性差异,本文尝试把疾病相关的三核苷酸重复DNA序列的稳定性分为碱基突变不稳定(base mutation instability)和重复单位扩增和缺失不稳定(repeats expansion instability),并针对疾病状态下这些不稳定性发生的分子机制进行了系统论述。
Table 1
表1
表1与三核苷酸重复扩增相关的疾病信息汇总
Table 1
| 类型 | 疾病名称 | 基因 | 三核苷酸重 复DNA序列 | 基因 结构 | 染色体 定位 | 正常 重复数 | 异常 重复数 | 首次报 道时间 | 参考文献 |
|---|---|---|---|---|---|---|---|---|---|
| PolyQ 三核苷酸扩增性疾病 | 脊髓小脑共济失调 1型 | ATXN1 | CAG | ORF | 6p22.3 | 6~9 | 41~83 | 1993 | [16] |
| 脊髓小脑共济失调 2型 | ATXN2 | CAG | ORF | 12q24.12 | 14~32 | 33~200 | 1996 | [17,18] | |
| 脊髓小脑共济失调 3型 | ATXN3 | CAG | ORF | 14q32.12 | 12~40 | 55~86 | 1994 | [19] | |
| 脊髓小脑共济失调 6型 | CACNA1A | CAG | ORF | 19p13.13 | 4~18 | 21~33 | 1997 | [20] | |
| 脊髓小脑共济失调 7型 | ATXN7 | CAG | ORF | 3p14.1 | 7~17 | 38~120 | 1997 | [21] | |
| 脊髓小脑共济失调 12型 | PPP2R2B | CAG | 5′UTR | 5q32 | 7~41 | >51 | 1999 | [22] | |
| 脊髓小脑共济失调 17型 | TBP | CAG | ORF | 6q27 | 25~44 | 47~63 | 2001 | [23] | |
| 亨廷顿舞蹈病 | HTT | CAG | ORF | 4p16.3 | 6~35 | 36~250 | 1993 | [24] | |
| 脊髓延髓肌萎缩 | AR | CAG | ORF | Xq12 | 9~36 | 38~62 | 1991 | [25] | |
| 非polyQ 三核苷 酸扩增 性疾病 | 易碎性X综合征 | FMR1 | CGG | 5′UTR | Xq27.3 | 6~53 | >230 | 1991 | [26,27] |
| 易碎性X智力低下(马丁-贝尔综合征) | FMR2 | CGG | 5′UTR | Xq28 | 6~53 | >200 | 1993 | [28] | |
| 弗洛里德共济失调 | FXN | GAA | 内含子 | 9q21.11 | 7~34 | >70 | 1996 | [29] | |
| 强直性肌营养不良 | DMPK | CTG | 3′UTR | 19q13.32 | 5~37 | >50 | 1992 | [30] | |
| 脊髓小脑共济失调 8型 | SCA8 | CTG | ORF | 13q21.33 | 16~37 | 90~250 | 1999 | [31] |
新窗口打开|下载CSV
1 三核苷酸重复DNA序列不稳定诱发的细胞毒性与疾病病理关联
三核苷酸重复DNA序列疾病的致病原因可分为基因功能丧失和所编码产物功能的改变。与基因功能丧失有关的症候多以常染色体隐性或者X连锁隐性方式外显;而起因于基因编码产物功能改变的症候则一般以常染色体显性或者X连锁显性遗传方式外显[37]。三核苷酸重复不稳定性扩增会诱导细胞产生代谢改变,从而损害特定组织的特定功能,其机制主要包括蛋白质功能的丧失、RNA或蛋白质水平上的毒性增加[38]。位于可编码区的重复序列扩增会导致转录缺陷,从而使功能基因失活丧失作用。这种转录缺陷通常表现为改变基因启动子的甲基化模式[39,40]、改变mRNA的剪接模式[41],或者直接改变蛋白表达水平[42]。较长的重复序列会阻碍相应基因的转录,因为它需要更长的转录时间以及资源,会使相应基因产物减少[43]。例如,重复序列可能会形成R-环(RNA-DNA杂交体),这种异常的二级结构会直接影响基因转录。在FRDA中,由于FXN基因中的GAA重复序列形成了R环结构,导致RNA聚合酶无法通过从而阻碍基因的正常转录[29,44,45]。越来越多的证据表明,某些重复序列自身会表现出或强或弱的细胞毒性。转录形成的RNA会参与多价碱基配对,从而导致其凝胶化,最终聚集汇成核灶[46]。这些RNA会螯合某些结合蛋白,通过破坏核孔复合物,损害mRNA的转运[47]。Thomas等[48]发现虽然组蛋白甲基转移酶与富含GC的RNA体外结合能力较弱,但受到体内解旋酶和RNA结合蛋白等蛋白因子的调节会形成高不溶性复合物。蛋白质毒性增加也会导致细胞毒性。例如,CAG重复序列会翻译成多聚谷氨酰胺片段(polyQ片段),polyQ片段错误折叠形成β-折叠结构会表现出异常的极性从而非特异性结合各种调节蛋白,最终在神经元细胞中汇集成毒性包涵体[49]。2 三核苷酸重复DNA序列不稳定性的分子机制
人类基因组中含有大量的串联重复DNA序列[50],主要分布在染色体的着丝粒、端粒和某些基因的调节区域[51]。已知的大多数串联重复DNA序列并不会出现急剧扩增,只有部分三核苷酸重复DNA序列会易于大规模扩增导致疾病(表1)。体外研究发现,与疾病相关的三核苷酸重复DNA序列通常会形成分子内结构影响基因转录、翻译和与某些特定蛋白结合。比如,部分三核苷酸重复DNA序列易形成R环阻碍基因转录,导致编码蛋白表达的降低。而富含GC的三核苷酸重复DNA序列则会发生超甲基化,从而沉默基因(例如FXS、FRAXE)或者采用高度稳定的分子内折叠结合RNA结合蛋白(例如DM1、FXTAS、SCA6、SCA3)[52]。在亨廷顿氏病和肌强直性营养不良中重复序列还会通过复制叉停滞引发三核苷酸重复扩增疾病[53,54]。针对这些现象,研究者们提出了不同的分子机制模型试图解释三核苷酸重复DNA序列的扩增机制。2.1 非B型DNA二级结构介导的扩增机制
除经典的B型构象(B conformation)之外,DNA分子还能在特定条件下形成非B型二级结构(non-B DNA secondary structure)[55],与三核苷酸重复不稳定有关的非B型DNA二级结构见表2。表2列出了三核苷酸重复DNA序列所能形成的“slipped DNA”(滑脱DNA)、H-DNA、G4偶联体和含错配对的发卡结构等主要形式(表2)。这些非B型DNA二级结构一旦出现在DNA复制、修复、重组和转录过程中则有可能介导三核苷酸重复DNA序列的碱基突变不稳定和重复单位扩增和缺失不稳定的发生。Table 2
Table 2Schematic diagram of non-B DNA secondary structures adopted by trinucleotide repeats DNA
| 非B型 DNA二级结构 | Slipped DNA | H-DNA | G4偶联体 | 含错配发卡结构 |
|---|---|---|---|---|
| 示意图 | 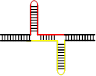 | 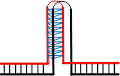 | 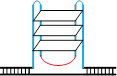 | 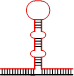 |
| 三核苷酸重复DNA序列 | 所有10种三核苷酸重复 | GAA | CGG | CAG |
| TTC | AGG | CTG | ||
| TGG | CGG | |||
| CCG |
新窗口打开|下载CSV
2.1.1 DNA滑脱复制机制
DNA滑脱复制(slippage DNA replication)模型见图1。此模型的建立是基于疾病相关的三核苷酸重复DNA序列在复制或修复过程中常伴随局部的缺失或插入(deletion/insertion, D/I) (图1)[56,57]。大量的体外研究证据表明,体外复制三核苷酸重复DNA序列时,DNA复制酶会与重复DNA序列模板频繁发生“脱离-识别”,使得新合成链的3′端有机会沿模板链“回缩”,然后再与重复序列中的另外一组重复单位配对,重新进行DNA复制(图1)。这种DNA复制使得三核苷酸重复DNA的某些区段会被更多或者更少的复制[57]。尽管DNA滑脱模型可以通过体外实验得以验证,但该模型无法解释为什么只有部分重复序列会出现急剧不稳定性扩增,而其他序列不容易发生;也无法解释扩增的不稳定性是指数增加而非线性增加。
图1
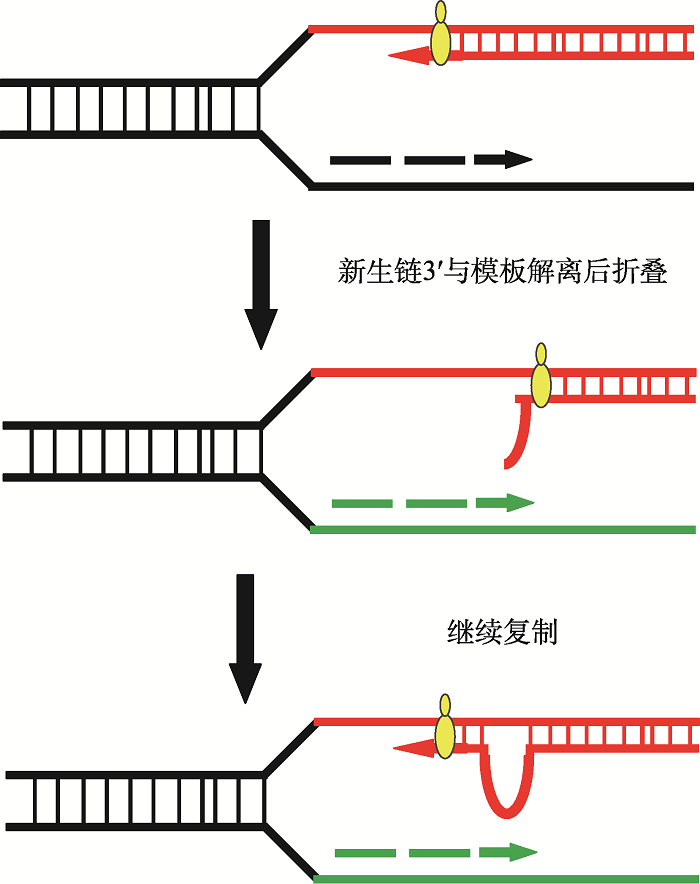 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1三核苷酸重复DNA序列的滑脱复制
复制或修复过程中,新合成链3′段回缩导致DNA局部错位;复制或转录过程继续进行,导致重复序列异常增加或减少。
Fig. 1Slippage DNA replication of the trinucleotide repeats
2.1.2 DNA解链元件
因为DNA滑脱模型无法完美解释观察到的现象,研究者转而试图探究在包括DNA复制、转录、修复和重组等代谢过程中双链DNA被解链呈DNA单链后形成可替代DNA结构(H-DNA、G4偶联体、含错配的发卡等(结构见表2)对三核苷酸重复DNA序列扩增和缺失不稳定性的可能贡献。
2.1.2.1 H-DNA与三核苷酸重复DNA序列不稳定
1987年首次报道了嘌呤-嘧啶镜面重复序列可以形成分子内三螺旋DNA结构,被称为H-DNA (表2)。它们通过分子内的Hoogsteen或反向Hoogsteen碱基对堆叠以及??-??相互作用而稳定[58],一半的DNA链通过折回与对应镜像链结合形成三重螺旋。H-DNA的形成在负超螺旋DNA中是有利的,因为它在拓扑学上等同于DNA的展开。H-DNA结构在真核生物中普遍存在,这暗示它们在基因组功能上有进化保守作用。它的存在可能涉及DNA复制及转录的停止,促进双链断裂,引发相应的修复[59],因此H-DNA的形成可能是部分三核苷酸重复DNA序列不稳定性扩增的关键。
2.1.2.2 G4偶联体与三核苷酸重复DNA序列不稳定
G4偶联体首次发现是在免疫球蛋白转换区,它是四链DNA结构,由多个π-π键相互作用构成。在单价阳离子钠和钾的存在下,G4偶联体可以稳定存在(表2)。G4偶联体具有多种异构体形式,以很高的丰度存在于诸多生物的基因组中,在人等真核生物染色体端粒及一些区域最为常见。能够形成G4偶联体的三核苷酸重复DNA序列是CGG、AGG和TGG,其中CGG与包括脆性X染色体综合征在内的人类多种退行性疾病相关联[60],而TGG则与人群中复发性14q32.2染色体微缺失(recurrent 14q32.2 microdeletion)有关[61,62]。G4偶联体可以干扰转录、影响基因组的完整性,甚至会促进DNA的双链断裂,这预示着G4偶联体在三核苷酸重复DNA序列不稳定性扩增中起着重要作用。
2.1.2.3 含错配的发卡结构与三核苷酸重复DNA序列不稳定
最早由美国McMurray实验室证明的含错配发卡结构(也称作DNA十字形或S-DNA)[63],由其上游序列通过正常的Watson-Crick碱基配对与互补的下游区域退火结合形成(表2)。实验证明三核苷酸重复DNA序列CAG、CTG、CGG和CCG等会形成含错配的发卡结构。DNA十字形和S-DNA在形成动力学上存在差异[64]。DNA十字形初期的形成非常缓慢,它需要形成中心气泡,然后DNA链再退火形成十字形;而S-DNA则需要先解链一段较长的DNA,然后通过单链DNA进行退火配对,因其构象在热力学上是不利的,因而S-DNA无法轻易转化为双链DNA。涉及DNA双链展开的含错配的发卡结构在拓扑异构学上等同于完全解链的DNA,因而在DNA复制、转录以及修复中起重要作用[65]。
2.1.2.4 基于非B型DNA二级结构的其他机制
三核苷酸重复DNA序列形成的非B型DNA二级结构可以帮助疾病相关的三核苷酸重复DNA序列逃避细胞内忠实的DNA复制模式,从而造成包括重复序列扩增不稳定在内的多种影响,这些影响有可能转而加剧三核苷酸序列的扩增不稳定。与此有关的研究包括CAG重复扩增产生的siRNA可以通过RNAi在体外有效的杀死癌症细胞,这至少表明癌细胞对三核苷酸不稳定性扩增衍生的siRNA特别敏感[66];而利用锌指蛋白转录因子(ZFP-TFs)或者CRISPR/Cas9靶向CAG致病重复序列选择性降低突变蛋白表达的治疗策略在小鼠脑中表现较好的耐受性,并使小鼠脑在分子生物学、组织病理学、电生理学等多方面得到改进[67,68,69,70]。除此之外,重复相关的非AUG翻译中发现RNA二级结构和启动子保真度是调节FMR1 RNA翻译的关键因素[27]。此类实验结果暗示有必要进一步探究基于非B型DNA二级结构影响的更为复杂的三核苷酸重复DNA序列扩增机制。
2.2 三核苷酸重复DNA不稳定性扩增的其他机制
2.2.1 “ori-shift/switch”模型已有的研究数据表明,疾病相关的三核苷酸重复DNA序列不稳定性可以表现出对DNA复制方向的依赖性。有人据此提出了“ori-shift/switch”模型(图2)。该模型假定重复序列复制时三核苷酸重复DNA序列与复制起始位点ori之间发生了DNA序列插入(图2A),或位于三核苷酸重复DNA序列一侧的复制起始位点失活,重复序列转而利用其相反一侧的DNA复制起点进行复制(图2 B)。这种复制方向的改变会使得特定三核苷酸重复DNA序列的稳定性发生变化,从而表现出模型实验或临床实践中常见的三核苷酸重复DNA序列的扩增和缺失[71]。这个模型强调了三核苷酸重复DNA序列的稳定性可能很大程度上取决于它与最近复制起始位点的距离(图2A)或复制起始位点的使用(图2B)。具有扩增潜力的三核苷酸重复DNA序列本身有可能会影响邻近DNA复制起始位点的使用,因此促发DNA复制起始位点的转换。这种模型可以帮助理解疾病相关的三核苷酸重复DNA序列不稳定性的改变可表现出“阈值”的现象,即当重复序列拷贝数达到一定数目之后,相应的三核苷酸重复DNA序列会愈发不稳定。
图2
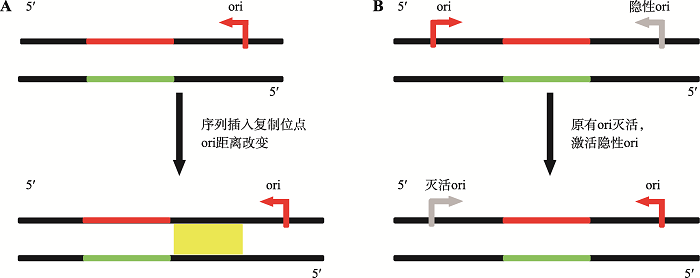 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2ori-shift/switch机制
A:插入因改变三核苷酸重复DNA与ori的距离从而引发三核苷酸重复DNA序列扩增或缺失;B:重复序列一侧的复制起点失活,激活位于相反一侧的隐性复制起始位点,从而使重复序列发生扩增或缺失。
Fig. 2ori-shift/switch mechanism
2.2.2 R-环介导的扩增机制
以上所有机制模型都无法完美的解释三核苷酸重复DNA序列的不稳定性扩增,基于多年的研究积累本课题组在2006年首次提出了R-环介导的扩增机制[72,73]。已有的实验数据证明,重复序列越长给细胞生长造成的压力越大,而当重复数增加达到相应阈值后会造成细胞无法继续生长。有****认为这可能是达到特定阈值的三核苷酸重复DNA序列会首先影响细胞内的DNA复制,而本课题组一步的研究表明,造成这一现象的真正原因是在转录过程中出现了障碍。通过抑制转录过程,可以使原本无法正常生长的细胞恢复生长,这表明复制障碍在其中的作用不大(待发表)。通过对这些三核苷酸重复DNA序列片段的进一步分析发现,带有特定碱基突变或序列缺失的长三核苷酸重复DNA序列可逃逸基因转录压力,从而避免对细胞生长繁殖的影响,而未能获得碱基突变或序列缺失的长三核苷酸重复DNA序列依然能够在基因转录过程中出现问题,从而影响细胞的正常繁殖。这一现象暗示了,在转录过程中,达到阈值的重复序列会产生特定的二级结构,这一构象会阻碍转录的正常进行,进而引发一系列的修复反应,但精确修复过后的细胞仍然面临相同的问题,无法越过转录障碍陷入修复循环,导致细胞无法生长繁殖最终死亡。更直接的证据是在我们选用了不忠实的DNA复制酶后,原本无法正常生长繁殖的带有长重复序列的细胞可以恢复生长。通过分析DNA提取物,我发现长重复序列形成了特殊的二级结构,结合之前的研究结果,我们进一步完善了R-环介导的扩增机制[71,72,73,74,75,76]。
基因转录需要在RNA聚合酶的催化下,以双链 DNA中的一条链为模板合成RNA。正常情形下,基因转录产生的RNA分子必须在转录泡后方与DNA模板分开,以容许两条被打开呈单链状态的DNA分子重新恢复成双链DNA。但包括本课题组在内的一些实验室的研究表明,发生在GAA•TTC、CGG•CCG和CAG•CTG等疾病相关的三核苷酸重复DNA序列部位的基因转录所合成的RNA分子通常难以与其模板DNA分开,或很容易重新与模板DNA复性形成RNA:DNA杂交体。这种RNA:DNA杂交体与非模板链一起被称为R-环结构[45]。R-环结构的形成不仅会阻碍转录复合体的正常通过,使转录暂停并能引发修复反应。当R-环在模板链上形成时,它会留下互补的部分单链,从而诱发非B DNA结构的形成。因此,一方面,R-环在调节基因表达和转录终止过程中起重要作用。另一方面,它们还可以阻止复制和转录,造成复制-转录冲突,并通过引发修复造成DNA链断裂(DSB),造成基因组不稳定。这一机制过程可以完美的解释目前实验中观察到的现象。在实验过程中本课题还发现,当CTG重复序列位于前导链时的抑制程度要比含相同重复单位的CAG重复序列位于前导链更加严重。通过对转录产物进行热力学稳定性分析发现CTG形成的二级结构要比CAG形成的二级结构更加稳定(待发表)。这种情况解释了转录复合体以CAG重复序列为模板进行转录比以CTG重复序列为模板进行转录更具优势的原因。
2.2.3 不忠实同源重组促发三核苷酸重复DNA序列不稳定
人类基因组中以纯合和非纯合形式分布着10种类型的三核苷酸重复DNA序列(图3)[77],其中,尚未明确与任何人类疾病有关的TAA、CAA三核苷酸重复DNA序列多以含有8个重复单位及以上的纯合形式存在,ATG、GGA和TGG则表现为单位重复数多于或等于100的非纯合形式明显多于纯合形式(图3)。而与疾病密切相关的三核苷酸重复DNA序列中的CAG•CTG、CGG•CCG则多以重复单位数小于或等于8的纯合形式存在,鲜见重复单位数大于100非纯合形式,而GAA•TTC则表现为重复单位数多于或等于100的非纯合形式多于含重复单位数为8及以上的纯合形式(图3)。
图3
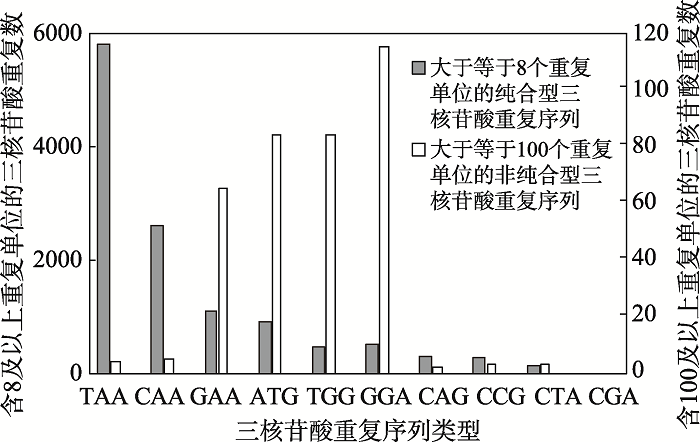 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3人类基因组中三核苷酸重复DNA序列的分布
参照文献[77]等呈现的数据信息绘制。
Fig. 3Distribution of trinucleotide repeats in human genome
本课题组在研究中发现,利用克隆手段构建与疾病相关的长三核苷酸重复DNA序列过程中存在着一个DNA突变阈值,即当所构建的重复序列的重复单位数达到一定值后,所得到的重复序列多为非纯合重复序列,很少或几乎得不到不含碱基突变和缺失的纯合形式的三核苷酸重复DNA序列。显然这种DNA突变阈值有别于三核苷酸重复DNA序列体内所能表现出的扩增阈值。当前这种情况已被多个实验室确证[78,79]。据此,我们推测DNA碱基突变阈值的出现可能与三核苷酸重复以如图4所示的同源复制机制有关(图4)。当三核苷酸序列重复数目达到一定值后,序列本身会对复制或转录造成阻碍。可预见的情形包括DNA聚合酶转换(DNA polymerase switching),即由原本使用具有校对活性的DNA聚合酶转而使用不具校对活性的DNA聚合酶复制处于DNA突变阈值的三核苷酸重复DNA序列。在这个过程中,包括碱基错配对修复蛋白MutS (原核)和MSH2-MSH6(真核)等会参与其中,这种情况反而带来了更大困难和更高的出错率。为了验证这种可能性,本课题组曾把DNA聚合酶的校对活性灭活或者高表达缺乏校对活性的不忠实DNA聚合酶DinB (又称DNA聚合酶IV,与人类细胞内的DNA聚合酶Pol κ相当,是三界生物共有的一种不具3′→5′校对活性的非忠实性DNA复制酶)反而会大大降低重复序列出错的几率。之所以会出现这种DNA复制忠实性的悖论,我们认为这是由于已处于DNA突变阈值的三核苷酸重复DNA序列本身不容易被复制和转录,特别当碱基错配修复蛋白试图修复DNA复制或转录过程中的问题模板时反而会进一步增加相关三核苷酸重复DNA序列区的DNA复制和基因转录的难度,此时,有可能进一步转化出包括DNA链断裂(如Mre11等结构特异性核酸酶的内切)在内的DNA损伤,迫使细胞调用包括同源重组依赖的DNA复制机制参与三核苷酸重复DNA序列的复制。此时一旦发生不忠实性同源重组(如图5所示)就有可能使处于DNA突变阈值的三核苷酸重复DNA序列进入重复单位数扩增阈值。这种策略是生物体为了降低复制和转录时的出错率进化出来的;当只有不忠实酶时会更容易跨越障碍,降低出错率。这一策略对本身有复制转录障碍的重复序列而言是可行的,但是对生物体复制转录其他正常序列时则是一个灾难。目前本课题组已经利用大肠杆菌的DNA聚合酶IV (DinB)不忠实复制系统初步证实了这一猜测。简言之,重复序列本身存在的复制和转录困难,会造成双链断裂,同源重组时为了精准修复带来了更高的出错率,这可能是三核苷酸重复DNA序列碱基突变不稳定和重复单位数扩增或缺失不稳定性发生的一种综合机制。
图4
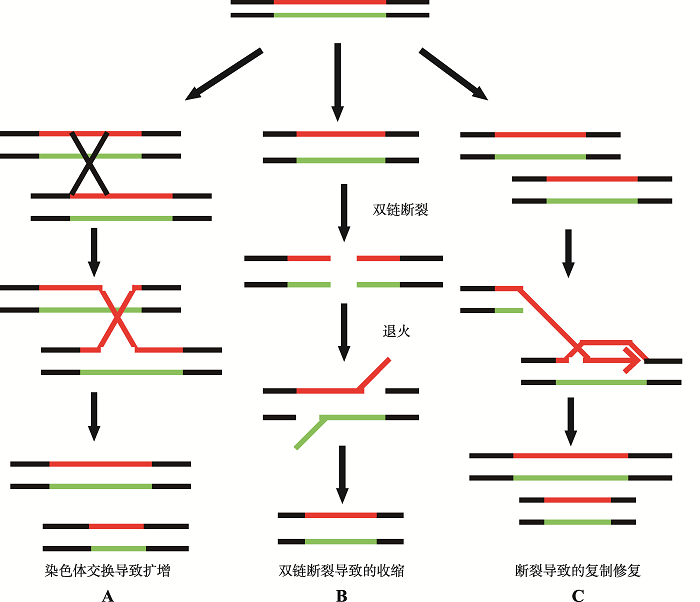 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4三核苷酸重复DNA序列同源复制机制
A:同源染色体在重复序列发生交换,一条染色体获得收缩的重复序列,而另一条染色体获得扩增的重复序列;B:重复序列发生双链断裂,单链重复末端退火连接,多余末端切除导致重复序列收缩;C:重复序列处双链断裂,引发修复机制,导致重复序列的异常扩增。
Fig. 4The homologous replication mechanism of TNRs
图5
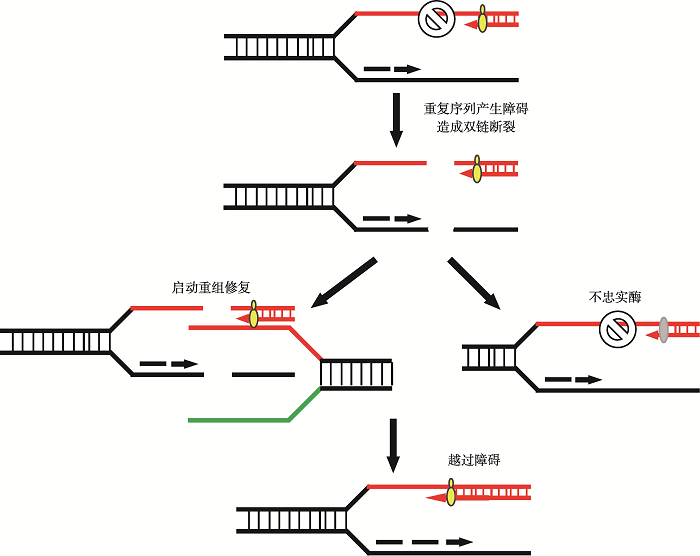 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5同源重组不忠实性机制
重复序列阻碍复制正常进行,导致双链断裂,诱发修复机制,在忠实修复失败后,以出错率更高的不忠实酶进行修复越过重复序列障碍。
Fig. 5Mechanism of unfaithful homologous recombination
3 结语与展望
三核苷酸重复不稳定相关疾病的发病年龄和疾病进展由两种不同机制共同控制,以等位基因存在的三核苷酸重复的拷贝数的多少并不直接表现出明确的细胞毒性。但是,这些重复序列会在某些组织的体细胞中扩增,当重复序列的重复单位数目超过疾病所需阈值时,则会通过快速出现的细胞毒性引发疾病。随后,症候进程主要决定于扩增所能产生的细胞毒性大小。这解释了杂合性和纯合性患者的发病年龄对遗传重复序列数目的依赖性。大量的研究表明,三核苷酸重复DNA序列通常在复制、转录或者修复时形成非B型DNA二级结构,然后诱发多种类型的修复。将DNA二级结构切除或转而衍生出包括DNA双链断裂在内的DNA损伤,对于这种损伤的重组修复可能导致了三核苷酸重复DNA的扩增或缺失。显然,上述过程受制于三核苷酸重复DNA序列的种类、在染色体中的位置,以及所处细胞的类型等因素。尽管目前已在包括人类细胞系在内的多种模式生物中试图重现疾病状态下的三核苷酸重复不稳定性扩增过程,但由于技术限制,有关三核苷酸重复是否在体内形成非B型DNA构象,以及形成何种类型的非B型DNA构象很难直接呈现。临床状态下,三核苷酸重复DNA序列可能形成的非B型DNA二级结构如何通过影响DNA的复制、转录发生扩增需深入研究。简言之,尽管研究使用的模式生物拥有完整的复制、转录、修复机制,但它们在疾病发生发展过程中的实际作用依然难以确认。特别是疾病相关的三核苷酸重复DNA序列所在细胞内的蛋白组差异(功能不同的细胞,其蛋白组不同)、所在染色体上的位置差异是否会直接影响非B型DNA二级结构的形成以及非B型DNA二级结构形成后细胞对它们的修复也需进一步研究。到目前为止,虽然三核苷酸重复相关的绝大多数人类疾病尚缺乏有效的治疗手段,但随着对疾病相关的三核苷酸重复DNA序列扩增机制的深入了解,相信终将会获得有效治疗的方法。比如,利用类似于CRISPR/Cas9的基因编辑技术删除扩增后的三核苷酸重复DNA序列,避免这些三核苷酸重复DNA序列处在拷贝数扩增阈值之上,通过降低非B型二级DNA结构形成的能力,可能会是一种很有前途的治疗的策略。(责任编委: 何淑君)
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
PMID:1710175 [本文引用: 1]

Fragile X syndrome is the most frequent form of inherited mental retardation and is associated with a fragile site at Xq27.3. We identified human YAC clones that span fragile X site-induced translocation breakpoints coincident with the fragile X site. A gene (FMR-1) was identified within a four cosmid contig of YAC DNA that expresses a 4.8 kb message in human brain. Within a 7.4 kb EcoRI genomic fragment, containing FMR-1 exonic sequences distal to a CpG island previously shown to be hypermethylated in fragile X patients, is a fragile X site-induced breakpoint cluster region that exhibits length variation in fragile X chromosomes. This fragment contains a lengthy CGG repeat that is 250 bp distal of the CpG island and maps within a FMR-1 exon. Localization of the brain-expressed FMR-1 gene to this EcoRI fragment suggests the involvement of this gene in the phenotypic expression of the fragile X syndrome.
DOI:10.1038/352077a0URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.omtn.2019.07.004URL [本文引用: 1]
DOI:10.1001/jamaneurol.2019.2368 [本文引用: 1]
IMPORTANCE In Huntington disease (HD), mutation severity is defined by the length of the CAG trinucleotide sequence, a well-known predictor of clinical onset age. The association with disease trajectory is less well characterized. Quantifiable summary measures of trajectory applicable over decades of early disease progression are lacking. An accurate model of the age-CAG association with early progression is critical to clinical trial design, informing both sample size and intervention timing. OBJECTIVE To succinctly capture the decades-long early progression of HD and its dependence on CAG repeat length. DESIGN, SETTING, AND PARTICIPANTS Prospective study at 4 academic HD treatment and research centers. Participants were the combined sample from the TRACK-HD and Track-On HD studies consisting of 290 gene carriers (presymptomatic to stage II), recruited from research registries at participating centers, and 153 nonbiologically related controls, generally spouses or friends. Recruitment was targeted to match a balanced, prespecified spectrum of age, CAG repeat length, and diagnostic status. In the TRACK-HD and Track-On HD studies, 13 and 5 potential participants, respectively, failed study screening. Follow-up ranged from 0 to 6 years. The study dates were January 2008 to November 2014. These analyses were performed between December 2015 and January 2019. MAIN OUTCOMES AND MEASURES The outcome measures were principal component summary scores of motor-cognitive function and of brain volumes. The main outcome was the association of these scores with age and CAG repeat length. RESULTS We analyzed 2065 visits from 443 participants (247 female [55.8%]; mean [SD] age, 44.4 [10.3] years). Motor-cognitive measures were highly correlated and had similar CAG repeat length-dependent associations with age. A composite summary score accounted for 67.6% of their combined variance. This score was well approximated by a score combining 3 items (total motor score, Symbol Digit Modalities Test, and Stroop word reading) from the Unified Huntington's Disease Rating Scale. For either score, initial progression age and then acceleration rate were highly CAG repeat length dependent. The acceleration continues through at least stage II disease. In contrast, 3 distinct patterns emerged among brain measures (basal ganglia, gray matter, and a combination of whole-brain, ventricular, and white matter volumes). The basal ganglia pattern showed considerable change in even the youngest participants but demonstrated minimal acceleration of loss with aging. Each clinical and brain summary score was strongly associated with the onset and rate of decline in total functional capacity. CONCLUSIONS AND RELEVANCE Results of this study suggest that succinct summary measures of function and brain loss characterize HD progression across a wide disease span. CAG repeat length strongly predicts their decline rate. This work aids our understanding of the age and CAG repeat length-dependent association between changes in the brain and clinical manifestations of HD.
DOI:10.1146/annurev-pathmechdis-012418-012857PMID:30089230 [本文引用: 1]
Among the age-dependent protein aggregation disorders, nine neurodegenerative diseases are caused by expansions of CAG repeats encoding polyglutamine (polyQ) tracts. We review the clinical, pathological, and biological features of these inherited disorders. We discuss insights into pathogenesis gleaned from studies of model systems and patients, highlighting work that informs efforts to develop effective therapies. An important conclusion from these analyses is that expanded CAG/polyQ domains are the primary drivers of neurodegeneration, with the biology of carrier proteins influencing disease-specific manifestations. Additionally, it has become apparent that CAG/polyQ repeat expansions produce neurodegeneration via multiple downstream mechanisms, involving both gain- and loss-of-function effects. This conclusion indicates that the likelihood of developing effective therapies targeting single nodes is reduced. The evaluation of treatments for premanifest disease will likely require new investigational approaches. We highlight the opportunities and challenges underlying ongoing work and provide recommendations related to the development of symptomatic and disease-modifying therapies and biomarkers that could inform future research.
PMID:8896555 [本文引用: 1]
The gene for spinocerebellar ataxia type 2 (SCA2) has been mapped to 12q24.1. A 1.1-megabase contig in the candidate region was assembled in P1 artificial chromosome and bacterial artificial chromosome clones. Using this contig, we identified a CAG trinucleotide repeat with CAA interruptions that was expanded in patients with SCA2. In contrast to other unstable trinucleotide repeats, this CAG repeat was not highly polymorphic in normal individuals. In SCA2 patients, the repeat was perfect and expanded to 36-52 repeats. The most common disease allele contained (CAG)37, one of the shortest expansions seen in a CAG expansion syndrome. The repeat occurs in the 5'-coding region of SCA2 which is a member of a novel gene family.
DOI:10.1242/dmm.031930URL [本文引用: 1]
DOI:10.1007/978-3-319-71779-1_1PMID:29427096 [本文引用: 1]
Huntington's disease (HD) is the most common monogenic neurodegenerative disease and the commonest genetic dementia in the developed world. With autosomal dominant inheritance, typically mid-life onset, and unrelenting progressive motor, cognitive and psychiatric symptoms over 15-20 years, its impact on patients and their families is devastating. The causative genetic mutation is an expanded CAG trinucleotide repeat in the gene encoding the Huntingtin protein, which leads to a prolonged polyglutamine stretch at the N-terminus of the protein. Since the discovery of the gene over 20 years ago much progress has been made in HD research, and although there are currently no disease-modifying treatments available, there are a number of exciting potential therapeutic developments in the pipeline. In this chapter we discuss the epidemiology, genetics and pathogenesis of HD as well as the clinical presentation and management of HD, which is currently focused on symptomatic treatment. The principles of genetic testing for HD are also explained. Recent developments in therapeutics research, including gene silencing and targeted small molecule approaches are also discussed, as well as the search for HD biomarkers that will assist the validation of these potentially new treatments.
DOI:10.1074/jbc.R118.003237URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.cell.2019.06.036URL [本文引用: 1]
PMID:8358429
Spinocerebellar ataxia type 1 (SCA1) is an autosomal dominant disorder characterized by neurodegeneration of the cerebellum, spinal cord and brainstem. A 1.2-Megabase stretch of DNA from the short arm of chromosome 6 containing the SCA1 locus was isolated in a yeast artificial chromosome contig and subcloned into cosmids. A highly polymorphic CAG repeat was identified in this region and was found to be unstable and expanded in individuals with SCA1. There is a direct correlation between the size of the (CAG)n repeat expansion and the age-of-onset of SCA1, with larger alleles occurring in juvenile cases. We also show that the repeat is present in a 10 kilobase mRNA transcript. SCA1 is therefore the fifth genetic disorder to display a mutational mechanism involving an unstable trinucleotide repeat.
PMID:8896557 [本文引用: 1]
Two forms of the neurodegenerative disorder spinocerebellar ataxia are known to be caused by the expansion of a CAG (polyglutamine) trinucleotide repeat. By screening cDNA expression libraries, using an antibody specific for polyglutamine repeats, we identified six novel genes containing CAG stretches. One of them is mutated in patients with spinocerebellar ataxia linked to chromosome 12q (SCA2). This gene shows ubiquitous expression and encodes a protein of unknown function. Normal SCA2 alleles (17 to 29 CAG repeats) contain one to three CAAs in the repeat. Mutated alleles (37 to 50 repeats) appear particularly unstable, upon both paternal and maternal transmissions. The sequence of three of them revealed pure CAG stretches. The steep inverse correlation between age of onset and CAG number suggests a higher sensitivity to polyglutamine length than in the other polyglutamine expansion diseases.
DOI:10.1016/j.neurobiolaging.2018.03.019URL [本文引用: 1]
PMID:7874163 [本文引用: 1]
We have identified a novel gene containing CAG repeats and mapped it to chromosome 14q32.1, the genetic locus for Machado-Joseph disease (MJD). In normal individuals the gene contains between 13 and 36 CAG repeats, whereas most of the clinically diagnosed patients and all of the affected members of a family with the clinical and pathological diagnosis of MJD show expansion of the repeat-number (from 68-79). Southern blot analyses and genomic cloning demonstrates the existence of related genes. These results raise the possibility that similar abnormalities in related genes may give rise to diseases similar to MJD.
PMID:8988170 [本文引用: 1]
A polymorphic CAG repeat was identified in the human alpha 1A voltage-dependent calcium channel subunit. To test the hypothesis that expansion of this CAG repeat could be the cause of an inherited progressive ataxia, we genotyped a large number of unrelated controls and ataxia patients. Eight unrelated patients with late onset ataxia had alleles with larger repeat numbers (21-27) compared to the number of repeats (4-16) in 475 non-ataxia individuals. Analysis of the repeat length in families of the affected individuals revealed that the expansion segregated with the phenotype in every patient. We identified six isoforms of the human alpha 1A calcium channel subunit. The CAG repeat is within the open reading frame and is predicted to encode glutamine in three of the isoforms. We conclude that a small polyglutamine expansion in the human alpha 1A calcium channel is most likely the cause of a newly classified autosomal dominant spinocerebellar ataxia, SCA6.
PMID:9288099 [本文引用: 1]
The gene for spinocerebellar ataxia 7 (SCA7) has been mapped to chromosome 3p12-13. By positional cloning, we have identified a new gene of unknown function containing a CAG repeat that is expanded in SCA7 patients. On mutated alleles, CAG repeat size is highly variable, ranging from 38 to 130 repeats, whereas on normal alleles it ranges from 7 to 17 repeats. Gonadal instability in SCA7 is greater than that observed in any of the seven known neuro-degenerative diseases caused by translated CAG repeat expansions, and is markedly associated with paternal transmissions. SCA7 is the first such disorder in which the degenerative process also affects the retina.
PMID:10581021 [本文引用: 1]
PMID:11448935 [本文引用: 1]
Genetic etiologies of at least 20% of autosomal dominant cerebellar ataxias (ADCAs) have yet to be clarified. We identified a novel spinocerebellar ataxia (SCA) form in four Japanese pedigrees which is caused by an abnormal CAG expansion in the TATA-binding protein (TBP) gene, a general transcription initiation factor. Consequently, it has been added to the group of polyglutamine diseases. This abnormal expansion of glutamine tracts in TBP bears 47--55 repeats, whereas the normal repeat number ranges from 29 to 42. Immunocytochemical examination of a postmortem brain which carried 48 CAG repeats detected neuronal intranuclear inclusion bodies that stained with anti-ubiquitin antibody, anti-TBP antibody and with the 1C2 antibody that recognizes specifically expanded pathological polyglutamine tracts. We therefore propose that this new disease be called SCA17 (TBP disease).
DOI:10.1093/hmg/ddz138 [本文引用: 1]
Triplet repeat diseases (TRDs) are caused by pathogenic expansions of trinucleotide sequence repeats within coding and non-coding regions of different genes. They are typically progressive, very disabling and frequently involve the nervous system. Currently available symptomatic therapies provide modest benefit at best. The development of interventions that interfere with the natural history of these diseases is a priority. A common pathogenic process shared by most TRDs is the presence of toxicity from the messenger RNA or protein encoded by the gene harboring the abnormal expansion. Strategies to interfere with the expression of these genes using different molecular approaches are being pursued and have reached the clinical stage. This review will summarize the significant progress made in this field in the last few years, focusing on three main areas: the discovery of biomarkers of disease progression and target engagement, advances in preclinical studies for the polyglutamine ataxias and the initial clinical application in myotonic dystrophy type 1 and Huntington's disease.
DOI:10.1038/352077a0URL [本文引用: 1]
PMID:1675488 [本文引用: 1]
The sequence of a Pst I restriction fragment was determined that demonstrate instability in fragile X syndrome pedigrees. The region of instability was localized to a trinucleotide repeat p(CCG)n. The sequence flanking this repeat were identical in normal and affected individuals. The breakpoints in two somatic cell hybrids constructed to break at the fragile site also mapped to this repeat sequence. The repeat exhibits instability both when cloned in a nonhomologous host and after amplification by the polymerase chain reaction. These results suggest variation in the trinucleotide repeat copy number as the molecular basis for the instability and possibly the fragile site. This would account for the observed properties of this region in vivo and in vitro.
[本文引用: 2]
PMID:8334699 [本文引用: 1]
We have cloned the fragile site FRAXE and demonstrate that individuals with this fragile site possess amplifications of a GCC repeat adjacent to a CpG island in Xq28 of the human X chromosome. Normal individuals have 6-25 copies of the GCC repeat, whereas mentally retarded, FRAXE-positive individuals have > 200 copies and also have methylation at the CpG island. This situation is similar to that seen at the FRAXA locus and is another example in which a trinucleotide repeat expansion is associated with a human genetic disorder. In contrast with the fragile X syndrome, the GCC repeat can expand or contract and is equally unstable when passed through the male or female line. These results also have implications for the understanding of chromosome fragility.
PMID:8596916 [本文引用: 2]
Friedreich's ataxia (FRDA) is an autosomal recessive, degenerative disease that involves the central and peripheral nervous systems and the heart. A gene, X25, was identified in the critical region for the FRDA locus on chromosome 9q13. This gene encodes a 210-amino acid protein, frataxin, that has homologs in distant species such as Caenorhabditis elegans and yeast. A few FRDA patients were found to have point mutations in X25, but the majority were homozygous for an unstable GAA trinucleotide expansion in the first X25 intron.
PMID:1349364 [本文引用: 1]
A variable DNA sequence has been detected in patients with myotonic dystrophy. We set out to determine whether identification of this specific molecular defect would improve clinical management of patients and families with myotonic dystrophy. 127 affected patients who were studied had an expanded DNA fragment not seen in 73 normal controls. The increase in length of the fragment correlated broadly with disease severity, and we noted expansion of the sequence in successive generations of the same family. Progressive expansion of the affected gene provides a molecular explanation for an apparently earlier onset in successive generations (anticipation) in myotonic dystrophy and supports the role of an unstable repeat sequence as the basis of the defect. The specificity of this finding will assist in accurate diagnosis of myotonic dystrophy and genetic counselling of affected families.
PMID:10192387 [本文引用: 1]
Myotonic dystrophy (DM) is the only disease reported to be caused by a CTG expansion. We now report that a non-coding CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). This expansion, located on chromosome 13q21, was isolated directly from the genomic DNA of an ataxia patient by RAPID cloning. SCA8 patients have expansions similar in size (107-127 CTG repeats) to those found among adult-onset DM patients. SCA8 is the first example of a dominant SCA not caused by a CAG expansion translated as a polyglutamine tract.
DOI:10.1186/1752-0509-4-29URL [本文引用: 1]
DOI:10.3390/molecules24112078URL [本文引用: 1]
DOI:10.1016/j.celrep.2019.02.008URL [本文引用: 1]
DOI:10.1038/s41598-018-37102-8PMID:30631090 [本文引用: 1]
Antagonist pleiotropy, where a gene exerts a beneficial effect at early stages and a deleterious effect later on in an animal's life, may explain the evolutionary persistence of devastating genetic diseases such as Huntington's disease (HD). To date, however, there is little direct experimental evidence to support this theory. Here, we studied a transgenic mouse carrying the HD mutation with a repeat of 50 CAGs (R6/2_50) that is within the pathological range of repeats causing adult-onset disease in humans. R6/2_50 mice develop characteristic HD brain aggregate pathology, with aggregates appearing predominantly in the striatum and cortex. However, they show few signs of disease in their lifetime. On the contrary, R6/2_50 mice appear to benefit from carrying the mutation. They have extended lifespans compared to wildtype (WT) mice, and male mice show enhanced fecundity. Furthermore, R6/2_50 mice outperform WT mice on the rotarod and show equal or better performance in the two choice discrimination task than WT mice. This novel mouse line provides direct experimental evidence that, although the HD mutation causes a fatal neurodegenerative disorder, there may be premorbid benefits of carrying the mutation.
DOI:10.1016/S1474-4422(16)30350-7URL [本文引用: 1]
DOI:10.1002/ajmg.a.v179.7URL [本文引用: 1]
DOI:10.1016/j.biochi.2019.05.001URL [本文引用: 1]
DOI:10.1016/j.bbadis.2017.12.037URL [本文引用: 1]
DOI:10.3390/brainsci9050096URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1002/ana.25488PMID:30973967 [本文引用: 1]
X-linked dystonia parkinsonism (XDP) is a neurodegenerative movement disorder caused by a single mutation: SINE-VNTR-Alu (SVA) retrotransposon insertion in TAF1. Recently, a (CCCTCT) repeat within the SVA insertion has been reported as an age-at-onset (AAO) modifier in XDP. Here we investigate the role of this hexanucleotide repeat in modifying expressivity of XDP.We genotyped the hexanucleotide repeat in 355 XDP patients and correlated the repeat number (RN) with AAO (n = 295), initial clinical manifestation (n = 294), site of dystonia onset (n = 238), disease severity (n = 28), and cognitive function (n = 15). Furthermore, we investigated i) repeat instability by segregation analysis and Southern blotting using postmortem brain samples from two affected individuals and ii) relative TAF1 expression in blood RNA from 31 XDP patients.RN showed significant inverse correlations with AAO and with TAF1 expression and a positive correlation with disease severity and cognitive dysfunction. Importantly, AAO (and not RN) was directly associated with whether dystonia or parkinsonism will manifest at onset. RN was lower in patients affected by mouth/tongue dystonia compared with blepharospasm. RN was unstable across germline transmissions with an overall tendency to increase in length and exhibited somatic mosaicism in brain.The hexanucleotide repeat within the SVA insertion acts as a genetic modifier of disease expressivity in XDP. RN-dependent TAF1 repression and subsequent differences in TAF1 mRNA levels in patients may be potentiated in the brain through somatic variability leading to the neurological phenotype. ANN NEUROL 2019;85:812-822.© 2019 American Neurological Association.
PMID:9603975 [本文引用: 1]
Friedreich ataxia (FRDA) is associated with the expansion of a GAA. TTC triplet repeat in the first intron of the frataxin gene, resulting in reduced levels of frataxin mRNA and protein. To investigate the mechanisms by which the intronic expansion produces its effect, GAA.TTC repeats of various lengths (9 to 270 triplets) were cloned in both orientations in the intron of a reporter gene. Plasmids containing these repeats were transiently transfected into COS-7 cells. A length- and orientation-dependent inhibition of reporter gene expression was observed. RNase protection and Northern blot analyses showed very low levels of mature mRNA when longer GAA repeats were transcribed, with no accumulation of primary transcript. Replication of plasmids carrying long GAA.TTC tracts (approximately 250 triplets) was greatly inhibited in COS-7 cells compared with plasmids carrying (GAA.TTC)9 and (GAA.TTC)90. Replication inhibition was five times greater for the plasmid whose transcript contains (GAA)230 than for the plasmid whose transcript contains (UUC)270. Our in vivo investigation revealed that expanded GAA.TTC repeats from intron I of the FRDA gene inhibit transcription rather than post-transcriptional RNA processing and also interfere with replication. The molecular basis for these effects may be the formation of non-B DNA structures.
PMID:17693431 [本文引用: 2]
Expansion of an unstable GAA.TTC repeat in the first intron of the FXN gene causes Friedreich ataxia by reducing frataxin expression. Deficiency of frataxin, an essential mitochondrial protein, leads to progressive neurodegeneration and cardiomyopathy. The degree of frataxin reduction correlates with GAA.TTC tract length, but the mechanism of reduction remains controversial. Here we show that transcription causes extensive RNA.DNA hybrid formation on GAA.TTC templates in bacteria as well as in defined transcription reactions using T7 RNA polymerase in vitro. RNA.DNA hybrids can also form to a lesser extent on smaller, so-called 'pre-mutation' size GAA.TTC repeats, that do not cause disease, but are prone to expansion. During in vitro transcription of longer repeats, T7 RNA polymerase arrests in the promoter distal end of the GAA.TTC tract and an extensive RNA.DNA hybrid is tightly linked to this arrest. RNA.DNA hybrid formation appears to be an intrinsic property of transcription through long GAA.TTC tracts. RNA.DNA hybrids have a potential role in GAA.TTC tract instability and in the mechanism underlying reduced frataxin mRNA levels in Friedreich Ataxia.
DOI:10.1038/nature22386URL [本文引用: 1]
DOI:10.7554/eLife.08881URL [本文引用: 1]
DOI:10.1261/rna.071191.119URL [本文引用: 1]
DOI:10.1073/pnas.91.12.5355URL [本文引用: 1]
DOI:10.1038/nrg3117PMID:22124482 [本文引用: 1]
Repetitive DNA sequences are abundant in a broad range of species, from bacteria to mammals, and they cover nearly half of the human genome. Repeats have always presented technical challenges for sequence alignment and assembly programs. Next-generation sequencing projects, with their short read lengths and high data volumes, have made these challenges more difficult. From a computational perspective, repeats create ambiguities in alignment and assembly, which, in turn, can produce biases and errors when interpreting results. Simply ignoring repeats is not an option, as this creates problems of its own and may mean that important biological phenomena are missed. We discuss the computational problems surrounding repeats and describe strategies used by current bioinformatics systems to solve them.
DOI:10.1146/annurev-genet-072610-155046PMID:20809801 [本文引用: 1]
Genotype-to-phenotype mapping commonly focuses on two major classes of mutations: single nucleotide polymorphisms (SNPs) and copy number variation (CNV). Here, we discuss an underestimated third class of genotypic variation: changes in microsatellite and minisatellite repeats. Such tandem repeats (TRs) are ubiquitous, unstable genomic elements that have historically been designated as nonfunctional "junk DNA" and are therefore mostly ignored in comparative genomics. However, as many as 10% to 20% of eukaryotic genes and promoters contain an unstable repeat tract. Mutations in these repeats often have fascinating phenotypic consequences. For example, changes in unstable repeats located in or near human genes can lead to neurodegenerative diseases such as Huntington disease. Apart from their role in disease, variable repeats also confer useful phenotypic variability, including cell surface variability, plasticity in skeletal morphology, and tuning of the circadian rhythm. As such, TRs combine characteristics of genetic and epigenetic changes that may facilitate organismal evolvability.
DOI:10.3390/ijms20133365URL [本文引用: 1]
DOI:10.1093/nar/gky1195URL [本文引用: 1]
DOI:10.1093/hmg/ddaa139URL [本文引用: 1]
[本文引用: 1]
DOI:10.1038/365207a0URL [本文引用: 1]
DOI:10.1007/s00294-018-0865-1URL [本文引用: 2]
DOI:10.1146/annurev.bi.64.070195.000433URL [本文引用: 1]
DOI:10.1128/MCB.20.3.990-1000.2000PMID:10629056 [本文引用: 1]
The ability to stimulate recombination in a site-specific manner in mammalian cells may provide a useful tool for gene knockout and a valuable strategy for gene therapy. We previously demonstrated that psoralen adducts targeted by triple-helix-forming oligonucleotides (TFOs) could induce recombination between tandem repeats of a supF reporter gene in a simian virus 40 vector in monkey COS cells. Based on work showing that triple helices, even in the absence of associated psoralen adducts, are able to provoke DNA repair and cause mutations, we asked whether intermolecular triplexes could stimulate recombination. Here, we report that triple-helix formation itself is capable of promoting recombination and that this effect is dependent on a functional nucleotide excision repair (NER) pathway. Transfection of COS cells carrying the dual supF vector with a purine-rich TFO, AG30, designed to bind as a third strand to a region between the two mutant supF genes yielded recombinants at a frequency of 0.37%, fivefold above background, whereas a scrambled sequence control oligomer was ineffective. In human cells deficient in the NER factor XPA, the ability of AG30 to induce recombination was eliminated, but it was restored in a corrected subline expressing the XPA cDNA. In comparison, the ability of triplex-directed psoralen cross-links to induce recombination was only partially reduced in XPA-deficient cells, suggesting that NER is not the only pathway that can metabolize targeted psoralen photoadducts into recombinagenic intermediates. Interestingly, the triplex-induced recombination was unaffected in cells deficient in DNA mismatch repair, challenging our previous model of a heteroduplex intermediate and supporting a model based on end joining. This work demonstrates that oligonucleotide-mediated triplex formation can be recombinagenic, providing the basis for a potential strategy to direct genome modification by using high-affinity DNA binding ligands.
DOI:S0002-9297(19)30200-9PMID:31178126 [本文引用: 1]
Neuronal intranuclear inclusion disease (NIID) is a slowly progressing neurodegenerative disease characterized by eosinophilic intranuclear inclusions in the nervous system and multiple visceral organs. The clinical manifestation of NIID varies widely, and both familial and sporadic cases have been reported. Here we have performed genetic linkage analysis and mapped the disease locus to 1p13.3-q23.1; however, whole-exome sequencing revealed no potential disease-causing mutations. We then performed long-read genome sequencing and identified a large GGC repeat expansion within human-specific NOTCH2NLC. Expanded GGC repeats as the cause of NIID was further confirmed in an additional three NIID-affected families as well as five sporadic NIID-affected case subjects. Moreover, given the clinical heterogeneity of NIID, we examined the size of the GGC repeat among 456 families with a variety of neurological conditions with the known pathogenic genes excluded. Surprisingly, GGC repeat expansion was observed in two Alzheimer disease (AD)-affected families and three parkinsonism-affected families, implicating that the GGC repeat expansions in NOTCH2NLC could also contribute to the pathogenesis of both AD and PD. Therefore, we suggest defining a term NIID-related disorders (NIIDRD), which will include NIID and other related neurodegenerative diseases caused by the expanded GGC repeat within human-specific NOTCH2NLC.Copyright © 2019 The Author(s). Published by Elsevier Inc. All rights reserved.
DOI:10.1093/hmg/ddq075URL [本文引用: 1]
PMID:10931934 [本文引用: 1]
A 24 triplet TGG.CCA repeat array shows length- and orientation-dependent propagation when present in the plasmid pUC18. When TGG(24) is present as template for leading-strand synthesis, plasmid recovery is normal in all strains tested. However, when it acts as template for lagging-strand synthesis, plasmid propagation is seriously compromised. Plasmids carrying deletions in the 5' side of this sequence can be isolated and products carrying 15 TGG triplets do not significantly interfere with plasmid propagation. Mutations in sbcCD, mutS and recA significantly improve the recovery of plasmids with TGG(24) on the lagging-strand template. These findings suggest that TGG(24) can fold into a structure that can interfere with DNA replication in vivo but that TGG(15) cannot. Furthermore, since the presence of the MutS and SbcCD proteins are required for propagation interference, it is likely that stabilisation of mismatched base pairs and secondary structure cleavage are implicated. In contrast, there is no correlation of triplet repeat expansion and deletion instability with predicted DNA folding. These results argue for a dissociation of the factors affecting DNA fragility from those affecting trinucleotide repeat expansion-contraction instability.
PMID:7758107 [本文引用: 1]
We show that repeating units from all reported disease genes are capable of forming hairpins of common structure and threshold stability. The threshold stability is roughly -50 kcal per hairpin and is influenced by the flanking sequence of the gene. Hairpin stability has two components, sequence and length; only DNA of select sequences and the correct length can form hairpins of threshold energy. There is a correlation among the ability to form hairpins of threshold stability, the sequence selectivity of expansion, and the length dependence of expansion. Additionally, hairpin formation provides a potential structural basis for the constancy of the CCG region of the Huntington's disease gene in individuals and explains the stabilizing effects of AGG interruptions in FMR1 alleles.
[本文引用: 1]
PMID:3697079 [本文引用: 1]
There are two alternative pathways by which inverted repeat sequences in supercoiled DNA molecules may extrude cruciform structures, called C-type and S-type. S-type cruciforms, which form the great majority, are characterised by absolute requirement for cations to promote extrusion, which then proceeds at higher temperatures and with lower activation parameters than for C-type cruciforms. The mechanism proposed for S-type extrusion involves an initial opening of basepairs limited to the centre of the inverted repeat, formation of intra-strand basepairing and a four-way junction, and finally branch migration to the fully extruded cruciform. The model predicts that central sequence changes will be more kinetically significant than those removed from the centre. We have studied the kinetics of cruciform extrusion by a series of inverted repeats related to that of pIRbke8 by either one or two mutations in the symmetric unit. We find that mutations in the central 8 to 10 nucleotides may profoundly affect extrusion rates--the fastest being 2000-fold faster than the slowest, whereas mutations further from the centre affect rates to a much smaller extent, typically up to ten-fold. These data support the proposed mechanism for extrusion via central opening.
DOI:S2405-8033(18)30191-2PMID:30292352 [本文引用: 1]
Many neurodegenerative diseases are caused by unstable trinucleotide repeat (TNR) expansions located in disease-associated genes. siRNAs based on CAG repeat expansions effectively kill cancer cell lines in vitro through RNAi. They also cause significant reduction in tumor growth in a human ovarian cancer mouse model with no toxicity to the treated mice. This suggests that cancer cells are particularly sensitive to CAG TNR-derived siRNAs, and explains a reported inverse correlation between the length of CAG TNRs and reduced global cancer incidences in some CAG TNR diseases. This review discusses both mutant proteins and mutant RNAs as a cause of TNR diseases, with a focus on RNAi and its role in contributing to disease pathology and in suppressing cancer.Copyright © 2018 Elsevier Inc. All rights reserved.
DOI:10.1038/s41591-019-0478-3PMID:31263285 [本文引用: 1]
Huntington's disease (HD) is a dominantly inherited neurodegenerative disorder caused by a CAG trinucleotide expansion in the huntingtin gene (HTT), which codes for the pathologic mutant HTT (mHTT) protein. Since normal HTT is thought to be important for brain function, we engineered zinc finger protein transcription factors (ZFP-TFs) to target the pathogenic CAG repeat and selectively lower mHTT as a therapeutic strategy. Using patient-derived fibroblasts and neurons, we demonstrate that ZFP-TFs selectively repress?>99% of HD-causing alleles over a wide dose range while preserving expression of?>86% of normal alleles. Other CAG-containing genes are minimally affected, and virally delivered ZFP-TFs are active and well tolerated in HD neurons beyond 100?days in culture and for at least nine months in the mouse brain. Using three HD mouse models, we demonstrate improvements in a range of molecular, histopathological, electrophysiological and functional endpoints. Our findings support the continued development of an allele-selective ZFP-TF for the treatment of HD.
DOI:10.3390/ijms20153689URL [本文引用: 1]
DOI:10.3389/fnins.2018.00075URL [本文引用: 1]
DOI:10.1093/nar/gky548URL [本文引用: 1]
DOI:10.1038/ng0502-5URL [本文引用: 2]
DOI:10.1016/S0379-4172(06)60001-2URL [本文引用: 2]
[本文引用: 2]
[本文引用: 2]
DOI:10.1016/j.dnarep.2009.08.001URL [本文引用: 1]
DOI:10.1371/journal.pone.0014271URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1093/nar/gkq127PMID:20215431 [本文引用: 2]
Trinucleotide repeats (TNRs) are of interest in genetics because they are used as markers for tracing genotype-phenotype relations and because they are directly involved in numerous human genetic diseases. In this study, we searched the human genome reference sequence and annotated exons (exome) for the presence of uninterrupted triplet repeat tracts composed of six or more repeated units. A list of 32 448 TNRs and 878 TNR-containing genes was generated and is provided herein. We found that some triplet repeats, specifically CNG, are overrepresented, while CTT, ATC, AAC and AAT are underrepresented in exons. This observation suggests that the occurrence of TNRs in exons is not random, but undergoes positive or negative selective pressure. Additionally, TNR types strongly determine their localization in mRNA sections (ORF, UTRs). Most genes containing exon-overrepresented TNRs are associated with gene ontology-defined functions. Surprisingly, many groups of genes that contain TNR types coding for different homo-amino acid tracts associate with the same transcription-related GO categories. We propose that TNRs have potential to be functional genetic elements and that their variation may be involved in the regulation of many common phenotypes; as such, TNR polymorphisms should be considered a priority in association studies.
DOI:S0002-9297(19)30153-3PMID:31104771 [本文引用: 1]
Huntington disease (HD) is caused by a CAG repeat expansion in the huntingtin (HTT) gene. Although the length of this repeat is inversely correlated with age of onset (AOO), it does not fully explain the variability in AOO. We assessed the sequence downstream of the CAG repeat in HTT [reference: (CAG)n-CAA-CAG], since variants within this region have been previously described, but no study of AOO has been performed. These analyses identified a variant that results in complete loss of interrupting (LOI) adenine nucleotides in this region [(CAG)n-CAG-CAG]. Analysis of multiple HD pedigrees showed that this LOI variant is associated with dramatically earlier AOO (average of 25 years) despite the same polyglutamine length as in individuals with the interrupting penultimate CAA codon. This LOI allele is particularly frequent in persons with reduced penetrance alleles who manifest with HD and increases the likelihood of presenting clinically with HD with a CAG of 36-39 repeats. Further, we show that the LOI variant is associated with increased somatic repeat instability, highlighting this as a significant driver of this effect. These findings indicate that the number of uninterrupted CAG repeats, which is lengthened by the LOI, is the most significant contributor to AOO of HD and is more significant than polyglutamine length, which is not altered in these individuals. In addition, we identified another variant in this region, where the CAA-CAG sequence is duplicated, which was associated with later AOO. Identification of these cis-acting modifiers have potentially important implications for genetic counselling in HD-affected families.Copyright © 2019 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
DOI:10.1212/NXG.0000000000000283URL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}