 ,, 王浩宇,, 曹悦岩, 朱强, 舒潘寅, 侯婷芸, 王雨婷, 张霁,四川大学华西基础医学与法医学院,成都 610041
,, 王浩宇,, 曹悦岩, 朱强, 舒潘寅, 侯婷芸, 王雨婷, 张霁,四川大学华西基础医学与法医学院,成都 610041Forensic genomics research on microhaplotypes
Xi Li,, Haoyu Wang,, Yueyan Cao, Qiang Zhu, Panyin Shu, Tingyun Hou, Yuting Wang, Ji Zhang,West China School of Basic Medical Sciences & Forensic Medicine, Sichuan University, Chengdu 610041, China通讯作者: 张霁,博士,教授,研究方向:法医遗传学。E-mail:zhangj@scu.edu.cn
编委: 朱波峰
收稿日期:2021-05-26修回日期:2021-07-29
| 基金资助: |
Received:2021-05-26Revised:2021-07-29
| Fund supported: |
作者简介 About authors
李茜,在读硕士研究生,专业方向:法医遗传学。E-mail:
王浩宇,在读硕士研究生,专业方向:法医遗传学。E-mail:

摘要
微单倍型(microhaplotype, MH)是在一定DNA片段范围之内,由至少两个单核苷酸多态性位点组成的遗传标记。MH兼具无stutter伪峰、多态性丰富以及扩增子较小等特点,有望成为法医学上的一种新型遗传标记。为了从全基因组维度上分析MH的特征,进一步发掘其应用潜能,本研究基于千人基因组计划中105个中国南方汉族个体的全基因组测序数据,构建了迄今为止最全面的MH数据集。结果表明,人类基因组中350 bp范围之内的MH位点数量共计9,490,075个,且微单倍型分布密度对染色体变异水平具有提示作用。从多种碱基跨度范围对MH的多态性分析表明,其多态性潜能可达到或者超过常用短串联重复序列位点的水平。此外,本文归纳总结了MH组装灵活等特点,并提出了构建微单倍型数据库的方案。
关键词:
Abstract
Microhaplotype loci (microhaplotype, MHs), defined by two or more closely linked single nucleotide polymorphisms, are a type of molecular marker within a short segment of DNA. As emerging forensic genetic markers, MHs have no stutter artefacts and higher polymorphism, and permit the design of smaller amplicons. In order to identify the markers from a genome wide perspective and explore their potential application further, we constructed the most comprehensive MH dataset to date, based on the whole genome sequencing data of 105 Han individuals in Southern China from 1000 Genomes Project. The results showed that there were 9,490,075 MH loci in the range of 350 bp in the human genome, and the distribution density of microhaplotypes suggests gene variation. Polymorphism analysis of MHs from various base spans showed that the polymorphism of MHs could reach or exceed common short tandem repeat sites. In addition, based on their flexible assembly, a scheme to build the public database of microhaplotypes was proposed.
Keywords:
PDF (2000KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
李茜, 王浩宇, 曹悦岩, 朱强, 舒潘寅, 侯婷芸, 王雨婷, 张霁. 微单倍型遗传标记的法医基因组学研究. 遗传[J], 2021, 43(10): 962-971 doi:10.16288/j.yczz.21-186
Xi Li.
近些年,微单倍型(microhaplotype,MH)逐渐受到法医学领域研究人员的关注。MH由Kidd实验室(美国,耶鲁大学医学院)在2013年首先提出[1],是一种在几百个核苷酸以内,由两个或多个紧密连锁的单核苷酸多态性(single nucleotide polymorphism,SNP)位点组合而成的多等位基因分子标记。MH与其他遗传标记相比具有以下特点:(1)扩增子没有stutter峰。微单倍型没有短串联重复结构,不会出现stutter峰所带来的诸如增加不平衡混合样本分析的复杂性等干扰问题[2,3]。(2)多态性通常高于SNP。MH具有多个等位基因,经过筛选的微单倍型拥有比SNP位点更高的杂合度[4]。(3)MH为序列多态性,其检测依赖于对碱基序列的读取。二代测序技术可以在几百个碱基的DNA单链上进行连续测序,直接对紧密排列的SNP位点进行“定相”(phase),获得真实的单倍型[5]。MH的可检测片段长度随着测序技术的发展一直在增加,由最初定义的200 bp逐渐扩大到300~500 bp[1,6,7]。而其片段长度下限,根据报道可低至20 bp或70 bp[8,9]。MH的这些特点,使其有望成为短串联重复序列(short tandem repeat, STR)位点基因分型的补充方法。
目前已有多个应用于法医DNA分析的MH体系。de la Puente等[10]开发了包含118个MH的复合体系,由于位点的平均长度仅51个核苷酸,对降解的DNA表现出高度的敏感性。MH的等位基因频率在不同大陆群体之间表现出差异,联合使用118个MH可以提供比常用STR体系更低的随机匹配概率。Oldoni等[11]报道的74-MH体系在混合DNA分析方面表现出优势,MH在二代测序平台检测到的等位基因覆盖度(allele coverage, AC)可以一定程度反映混合斑比例,更利于对次要贡献者的等位基因进行拆分。Wu等[12]认为具有较高有效等位基因数(the effective number of alleles, Ae)的MH有利于在混合斑中检测到更多的等位基因,从而减少贡献者之间的等位基因共享,帮助判断贡献者个数。一些研究人员测试了MH体系对法医亲缘关系鉴定的适用性[13,14,15]。结果表明,联合使用30~60个MH在亲子鉴定和全同胞鉴别方面可优于现有STR或SNP体系,但涉及二级或更远的亲缘关系判断仍然需要添加更多的位点。上述研究均强调了开发足够数量的、多等位基因、高多态性MH的重要性。
根据统计,目前约有470个微单倍型被报道[10,12~20],其中多数位点的Ae值在2.0~4.0,Ae达到4.0以上的位点有120个。SNP遗传标记在人类基因组中是广泛存在的,相应的,由多个SNP参与定义的微单倍型的数量也是极为丰富的。相对于MH在全基因组中的广泛分布,目前已开发报道的微单倍型仅是其中很小的一部分。想要进一步了解MH的数目和属性,更好地满足个人识别、混合DNA分析以及亲缘关系鉴定等法医学应用的需求,需要更全面的MH位点信息作为支持。据此,我们从特定群体入手,以期在全基因组维度上对MH的特征进行分析与归纳。本研究使用的是千人基因组计划第三阶段中国南方汉族群体的遗传数据。考虑到法医学领域不同应用目的下的扩增子长度、常用测序平台的阅读长度以及位点侧翼需预留引物设计空间等因素,我们对350 bp范围内的微单倍型进行全面筛查,并统计了多种片段长度限制下MH的多态性,进一步认识和发掘这种新兴法医学遗传标记的应用潜能。
1 材料与方法
1.1 SNP预过滤
本研究使用的全基因组测序数据下载自千人基因组计划第三阶段(GRCh37.p13)的数据库网站[21]。涉及的105个样本均属于中国南方汉族(Southern Han Chinese, CHS)。首先使用VCFtools工具对这些样本的变异检测格式(variant call format, VCF)文件进行预过滤,获取可用于后续组装微单倍型的SNP集合。预过滤的标准如下:(1)染色体定位在1~22号常染色体;(2)排除插入/缺失(insertion or deletion, InDel)变异,即在统计MH的分型和参数时不将InDel纳入考虑;(3) SNP位点在相应群体中的最小等位基因频率(minor allele frequency, MAF)大于0.01;(4)对SNP位点进行Hardy-Weinberg平衡检验,需满足P>0.05。1.2 微单倍型的组装和过滤
本研究对于构建MH的要求是:获取全基因组范围内所有长度在350 bp以内、至少包含2个SNP的潜在微单倍型。通过1.1部分的预过滤,可以在22条常染色体上分别获得SNP物理位置依次递增的预筛选集合。首先,以某一条染色体上第一个SNP (即物理位置最小的SNP)作为潜在MH的“起始SNP”,依次纳入后续相邻的位点。然后,判断当前组合是否为满足要求的潜在微单倍型。每纳入一个SNP,则需判断一次:如果满足要求,则将其输出;如不满足要求,则将“起始SNP”的坐标依次向后移动,循环上述过程。当“起始SNP”的坐标移动至该染色体预筛选集合的最后一个位点时,该染色体的检索结束。最后,对所有常染色体进行检索,并对输出的微单倍型进行编号。
该组装过程可能将规定片段长度范围内的SNP进行多次组合并重复输出。对于目标片段长度范围内存在的n个SNP,至多可输出$\sum\limits_{i=1}^{n-1}{i}$种微单倍型,包含SNP数目最多的MH被称为“最长片段MH”,其余位点被描述为“子集”。例如,在350个碱基跨度内存在5个SNP,则至多可输出10种MH,其中由全部5个SNP定义的微单倍型即是“最长片段MH”,其余由2~4个连续的SNP定义的MH合称为“子集”。在之后的一些分析中,为了减少冗余数据,可能会将子集移除,得到由各目标区域内“最长片段MH”组成的“最长片段集”。“最长片段集”与“子集”合称为“完整集”。需要注意的是,如果研究者关注的片段长度范围发生改变,那么相对应的“完整集”、“最长片段集”以及“子集”中所包含的MH都将发生变化。以上过程均由实验室内部基于Python的脚本实现。
1.3 统计学分析
上述MH的组装和过滤过程,仅对SNP在参考基因组中的物理位置(position)进行输出。之后,对于所有输出的微单倍型,按其构成提取VCF文件中相应SNP的基因分型并组装成MH等位基因,然后计算每个位点在中国南方汉族中的群体遗传学参数,包括杂合度观测值(observed heterozygosity, Ho)、个体识别概率(discrimination power, DP)以及有效等位基因数(Ae)。有效等位基因数(Ae)是一个经典的群体遗传学概念,它的值代表遗传标记所等价的频率相等的等位基因的个数。例如,某遗传标记的Ae值为n,则表示该遗传标记等价于包含n个频率相等的等位基因,即每个等位基因的频率均为1/n。通多该指标可以实现对多等位基因遗传标记的比较和排序。Ae值的计算公式为:${1}/{\sum{p{{i}^{2}}}}\;$,其中pi表示某基因座上等位基因i的频率[22]。2 结果与分析
2.1 人类基因组中MH位点的数量
对千人基因组计划数据进行初步筛选之后,在22条人类常染色体上共得到5,977,655个SNP位点。按照1.2所述策略进行无差别组装,获取350 bp范围之内所有可能的MH (“完整集”)共计9,490,075个。过滤子集之后,仍保留30.47%的位点(2,891,927个),其中2号染色体的MH最多,22号染色体的MH最少,分别为235,330和40,808 (表1)。平均每百万个碱基对(Mb)检索到大约1000 (2,891,927/3000 Mb)个微单倍型。Table 1
表1
表1SNP及MH在不同染色体上的数量统计
Table 1
| 染色体 | #SNPs a | #MHs≤350 bp | #MHs≤150 bp | #MHs≤100 bp | #MHs≤50 bp | ||||
|---|---|---|---|---|---|---|---|---|---|
| A | B | A | B | A | B | A | B | ||
| 1 | 463,261 | 684,624 | 224,137 | 307,923 | 160,070 | 211,609 | 127,357 | 112,562 | 80,220 |
| 2 | 485,172 | 697,551 | 235,330 | 312,548 | 167,213 | 213,273 | 132,583 | 113,234 | 82,973 |
| 3 | 429,260 | 648,976 | 208,260 | 289,892 | 149,992 | 197,993 | 119,687 | 104,404 | 75,230 |
| 4 | 444,134 | 719,321 | 214,966 | 322,127 | 158,179 | 220,652 | 127,643 | 116,157 | 81,220 |
| 5 | 369,559 | 536,677 | 179,135 | 240,372 | 127,576 | 164,475 | 101,232 | 87,636 | 63,535 |
| 6 | 406,810 | 855,491 | 197,195 | 380,915 | 145,792 | 258,670 | 118,720 | 134,514 | 77,942 |
| 7 | 357,021 | 560,129 | 172,793 | 252,329 | 125,618 | 172,901 | 100,917 | 91,761 | 64,047 |
| 8 | 323,908 | 538,902 | 157,309 | 239,578 | 116,180 | 163,404 | 93,911 | 85,902 | 60,124 |
| 9 | 264,750 | 408,354 | 128,051 | 183,076 | 94,273 | 125,067 | 75,556 | 65,695 | 47,502 |
| 10 | 310,578 | 493,463 | 150,444 | 221,083 | 109,703 | 151,560 | 88,309 | 80,883 | 56,820 |
| 11 | 290,938 | 445,661 | 140,840 | 199,579 | 102,589 | 136,071 | 81,970 | 71,787 | 52,025 |
| 12 | 287,513 | 430,602 | 139,153 | 194,229 | 100,173 | 133,241 | 79,814 | 70,791 | 50,312 |
| 13 | 217,352 | 335,132 | 105,264 | 150,042 | 76,429 | 102,775 | 61,026 | 54,319 | 38,801 |
| 14 | 194,482 | 293,076 | 94,194 | 131,568 | 67,706 | 90,024 | 53,957 | 47,739 | 34,160 |
| 15 | 176,222 | 274,166 | 84,933 | 123,892 | 61,570 | 85,058 | 49,433 | 45,461 | 31,845 |
| 16 | 187,593 | 331,113 | 90,597 | 148,132 | 68,613 | 101,035 | 56,023 | 53,349 | 36,743 |
| 17 | 155,592 | 237,431 | 74,611 | 108,076 | 54,056 | 74,898 | 43,371 | 40,510 | 27,684 |
| 18 | 172,512 | 267,593 | 83,271 | 121,279 | 60,482 | 83,014 | 48,506 | 43,974 | 30,786 |
| 19 | 142,771 | 254,075 | 67,813 | 117,069 | 51,542 | 81,348 | 42,076 | 44,002 | 27,689 |
| 20 | 127,126 | 186,986 | 61,427 | 84,197 | 44,252 | 57,717 | 35,181 | 30,649 | 22,301 |
| 21 | 86,049 | 140,790 | 41,396 | 63,550 | 31,033 | 43,662 | 25,070 | 22,955 | 16,084 |
| 22 | 85,052 | 149,962 | 40,808 | 68,111 | 30,586 | 47,028 | 24,982 | 25,065 | 16,427 |
| 总计 | 5,977,655 | 9,490,075 | 2,891,927 | 4,259,567 | 2,103,627 | 2,915,475 | 1,687,324 | 1,543,349 | 1,074,470 |
新窗口打开|下载CSV
图1以密度图的形式展示了每条染色体上MH“最长片段集”的分布情况。一些分布特征与人类已知的变异模式相匹配:例如,在6号染色体主要组织相容性复合体(the major histocompatibility complex, MHC)周围观察到了极大数量的MH;在8q21.2周期性新着丝粒(neocentromere)的附近[23],也发现MH高密度分布区。此外,16号染色体短臂或长臂近端粒处(16q23)的“亮黄色”区域可能提示MH数量高于平均水平。其余MH的分布相对均匀。
图1
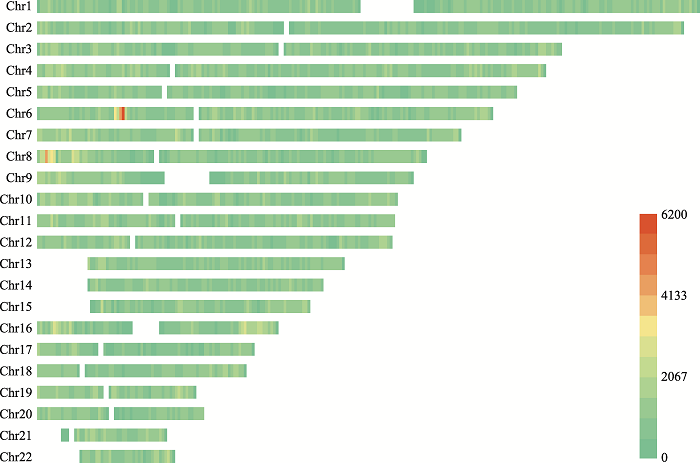 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1人类基因组中微单倍型遗传标记的密度分布图
使用350 bp范围内、移除子集的MH数据绘制。色阶代表每Mb染色体序列的MH总数。性染色体数据未收集。
Fig. 1Density plots of microhaplotypes identified in the human genome
2.2 350 bp范围内MH的统计学参数
如前所述,微单倍型标记的组装过程会将一定范围内的SNP进行重复组合和输出。为了减少冗余数据,此部分的分析只针对350 bp范围内、移除子集的MH集合(“最长片段集”)。2.2.1 总体特征
用于定义微单倍型的SNP数量在2~51之间,其中由两个SNP构成的标记数量最多,占比45.42%。观察到至少3个等位基因的遗传标记共计2,494,157个,约占86.25%;等位基因数超过10的位点多达14,133个。有50%的微单倍型长度范围集超过263 bp,所有位点的平均长度是239 bp。
根据千人基因组计划数据库中发布的“确定相位”(phased)的基因分型数据,估计微单倍型的等位基因频率信息。总的来说,微单倍型在中国南方汉族群体中,具有非常可观的遗传多态性。Ho值超过0.8的MH共计11,712个;DP值超过0.9的MH多达21,355个。之前一项研究提出了Ae值的阈值(Ae = 3)[22],超过这一阈值的微单体型被认为具有较高的法医学应用价值。本部分共涉及2,891,927个微单倍型,Ae值在1.02~66.62之间。Ae值高于3的标记共计199,176个,高于5的标记共计6935个;387个MH的Ae值在10~20之间(不包括10),41个MH的Ae值大于20。表2给出了Ae值位于前10的微单倍型位点信息,参与构成这些MH的SNP互不重复,且MHC周围的位点没有纳入。
Table 2
表2
表2Ae值前10的微单倍型位点信息
Table 2
| MH_ID | bp | Ae | Ho | DP | #SNPs | #Alleles | Position (GRCh37) |
|---|---|---|---|---|---|---|---|
| mh04zj0146583 | 346 | 66.6163 | 0.9905 | 0.9899 | 41 | 134 | Chr4:30279658~30280003 |
| mh01zj0675568 | 248 | 43.2353 | 0.9714 | 0.9905 | 15 | 88 | Chr1:247032193~247032440 |
| mh20zj0185187 | 347 | 32.6183 | 0.9999 | 0.9892 | 9 | 68 | Chr20:62308266~62308612 |
| mh09zj0366544 | 239 | 28.5622 | 0.9524 | 0.9883 | 12 | 60 | Chr9:129479455~129479693 |
| mh07zj0103025 | 323 | 28.3055 | 0.9714 | 0.9892 | 26 | 77 | Chr7:18772264~18772586 |
| mh04zj0352614 | 346 | 27.1218 | 0.9810 | 0.9859 | 22 | 105 | Chr4:88537078~88537423 |
| mh03zj0068937 | 350 | 24.2308 | 0.8762 | 0.9858 | 20 | 75 | Chr3:11955851~11956200 |
| mh02zj0082461 | 320 | 23.7864 | 0.9810 | 0.9874 | 12 | 56 | Chr2:20701112~20701431 |
| mh01zj0508420 | 337 | 21.7028 | 0.9619 | 0.9872 | 37 | 63 | Chr1:200785797~200786133 |
| mh04zj0474307 | 348 | 20.5307 | 0.9714 | 0.9870 | 11 | 51 | Chr4:129682428~129682775 |
新窗口打开|下载CSV
2.2.2 特征参数之间的关系
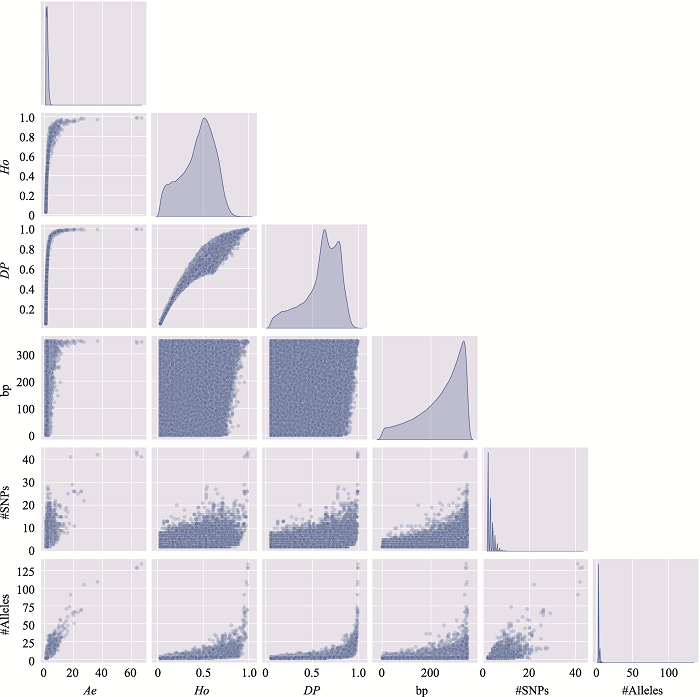
为了探究微单倍型遗传标记Ae值、Ho值、DP值、bp、构成MH的SNP数以及等位基因数之间的关系,研究者分别对每条染色体上的MH绘制这六个特征参数的散点图矩阵。以位点数量居中的9号染色体为例展示了MH特征参数之间的相关性(图2,其余染色体的散点图矩阵见附图1~21)。对角线处分别为各参数的核密度估计图,其余位置为任意两参数之间的散点图。核密度估计是一种从数据样本本身出发研究数据分布特征的方法,曲线下方的面积和等于1;当存在多个波峰时,所有波峰下方的面积之和为1。某区间所对应的曲线下面积越大,代表样本在该区间分布的概率越大。散点图直观的反映了这六个特征参数之间的关系。首先,Ae值、DP值、Ho值三者之间具有较强的相关关系。其次,随着等位基因数的增加,Ae值的最低值逐渐升高,二者存在一定的相关性。其余参数之间的相关程度均较差。
图2
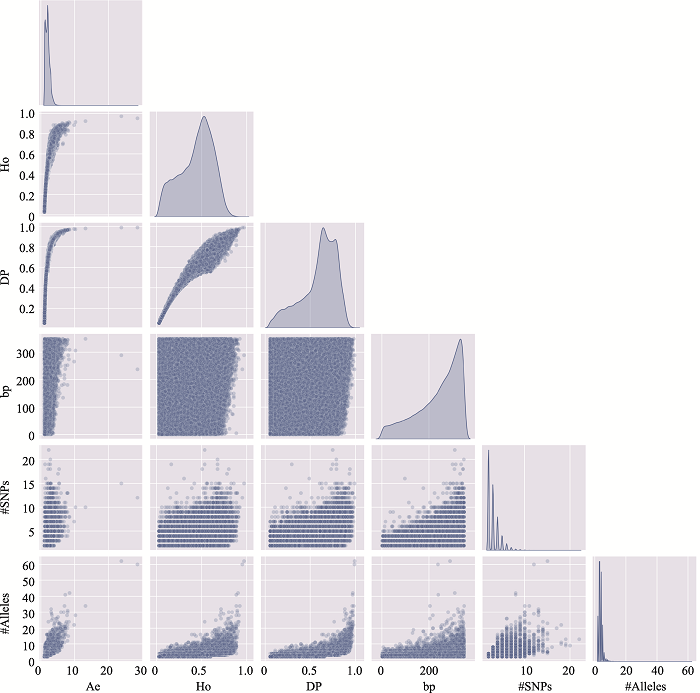 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2微单倍型遗传标记特征参数之间的关系
使用位于9号染色体、350 bp范围内、移除子集的MH数据绘制(共计128,051个)。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 2Relationship among characteristic parameters of microhaplotypes
综合22条常染色体的MH数据,计算这些参数之间的成对Pearson相关系数(r)并绘制热图(图3)。DP值和Ho值的相关系数最高(r=0.97);Ae值与DP值和Ho值的相关系数分别为0.85和0.88;等位基因数与Ae值和构成MH的SNP数呈中等程度相关;其余参数之间的相关系数均小于等于0.4。
图3
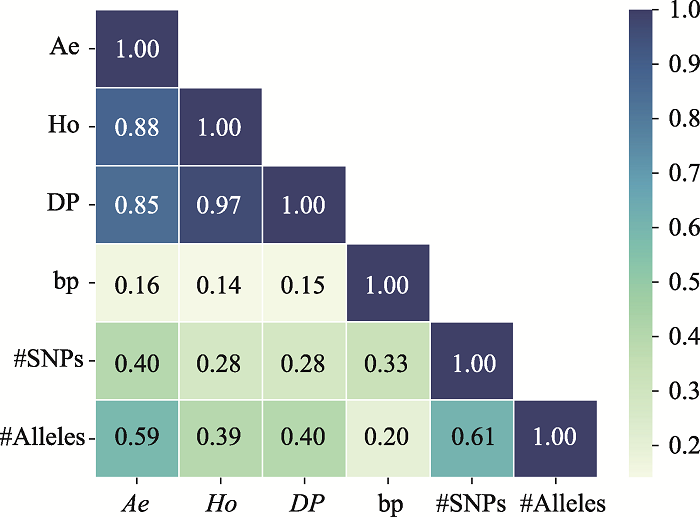 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3微单倍型遗传标记特征参数之间的成对相关系数
使用人类基因组350 bp范围内、移除子集的MH数据绘制(共计2,891,927个)。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 3Pairwise correlation coefficient between characteristic parameters of microhaplotypes
2.3 不同片段长度范围内的MH的数量及Ae值分布情况
如前所述,350 bp范围之内所有可能的MH (即“完整集”)共计9,490,075个;过滤子集之后,仍保留2,891,927个位点(即“最长片段集”,占比30.47%)。当将片段长度的上限分别设置为150 bp、100 bp和50 bp时,相对应的“完整集”中MH的数量分别为4,259,567、2,915,475和1,543,349 (表1);移除子集之后潜在位点的数量分别减少了50.61%、42.13%和30.38% (图4A)。目标区域的碱基跨度越大,可能纳入的SNP数目就会越多,从而产生更多的组合形式,“子集”占比也随之增高。图4
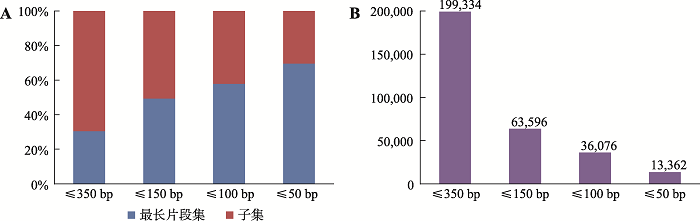 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4不同片段长度范围内的微单倍型遗传标记
A:350 bp、150 bp、100 bp、50 bp内MH“子集”与“最长片段集”的百分比堆积柱形图;B:不同片段长度范围内Ae值大于等于3的MH数量统计,使用“最长片段集”的MH数据绘制。
Fig. 4Microhaplotypes within different lengths of base pairs
Table 3
表3
表3不同片段长度范围内微单倍型Ae值的分布
Table 3
| Ae | #MHs≤350 bp | #MHs≤150 bp | #MHs≤100 bp | #MHs≤50 bp |
|---|---|---|---|---|
| 1~ | 1,634,742 | 1,345,361 | 1,116,786 | 741,819 |
| 2~ | 1,057,851 | 694,670 | 534,462 | 319,289 |
| 3~ | 171,637 | 56,715 | 32,419 | 12,211 |
| 4~ | 20,759 | 4941 | 2683 | 937 |
| 5~ | 6510 | 1875 | 957 | 213 |
| 10~ | 307 | 52 | 11 | 0 |
| 15~ | 80 | 8 | 5 | 1 |
| 20~ | 26 | 0 | 1 | 0 |
| 25~ | 15 | 5 | 0 | 0 |
新窗口打开|下载CSV
本研究对不同片段长度范围内的“最长片段集”微单倍型的Ae值分布情况进行了统计(表3)。在加强碱基长度的限制之后,具有高多态性的微单倍型仍然十分丰富:在150 bp和100 bp范围内,Ae值大于等于3.0的MH数量分别是199,334和63,596;长度降低至50个碱基之内时,仍有13,362个位点的Ae值大于等于3.0 (图4B)。
3 讨论
本研究使用千人基因组计划中国南方汉族群体的基因分型数据,构建了350 bp范围内的微单倍型标记库,展示了迄今为止最全面的人类MH集合,并对MH的特征和应用潜能有了更深刻的认识。第一,微单倍型在人类基因组中的数量极为丰富。为了尽可能不高估MH的数量,本研究仅从“最长片段集”水平考虑,在22条常染色上共检索到2,891,927个位点。法医遗传学****所熟知的STR基因座在人类基因组中的分布密度约100个/Mb[24],相较而言微单倍型遗传标记的数量更为丰富,平均每Mb碱基序列检索到1000个MH位点(2,891,927/ 3000 Mb)。
从微单倍型密度分布图(图1)可以观察MH在基因组测序数据缺失序列(gap)之外的分布情况。MH的高密度分布区与人类基因组中一些已知的高变异区域相匹配,说明MH的分布密度可以一定程度体现人类基因组的变异水平。MH的高密度分布本质上来源于SNP的高密度分布,这提示了微单倍型多态性来源于历史性基因突变的可能性,而MH多态性水平与基因重组的关系则需要在家系中进一步探究。我们建议在解决亲缘关系鉴定的问题时,对于MH位点的选择和使用需要慎重考虑。
第二,MH多态性不仅优于SNP,而且可达到甚至超过常用的STR基因座。MH拥有比SNP位点更高的杂合度,这一观点基本被法医遗传学家所公认。其与STR基因座之间的比较,Oldoni等[11,25]认为后者更具优势。本研究虽然没有考虑引物设计、位点序列与基因组对齐(BLAST)结果等因素对最终能够用于构建实验体系的MH位点数量的影响,但从理论上对MH的多态性潜能做出了评估。基于105个CHS样本的数据统计, Ho值超过0.8、DP值超过0.9的MH数量分别为11,712和21,355;Ae达到4.0的位点数量也由已报到的120个[10,12~20],增加至27,697个。更有14,133个MH的等位基因数超过10,870个MH的等位基因数超过50,这完全超出了研究人员对于MH以往的印象。因此我们认为,通过筛选可以得到等位基因数和多态性都优于STR的微单倍型,而这样的MH有望在DNA混合物的分析中,特别是在混合斑的确认以及贡献者数量的推断方面发挥巨大优势。
第三,MH的Ae值与DP值和Ho值之间均具有较强的线性相关关系。三者分别由不同的参数计算得到(等位基因频率、表型频率、杂合子频率),其中Ae值与Ho值是表征遗传标记本身多态性的指标,而DP值是评价遗传标记识别不同个体效能大小的指标,三者无法直接由公式推导而进行转换。作者通过对数百万个MH位点的Ae值、DP值和Ho值进行成对相关性分析,观察到Ae值与DP值、Ho值之间具有较强的相关性(r分别为0.85、0.88)。这再次印证了当筛选MH应用于法医学领域时,以Ae值(而不计算DP值、Ho值)作为主要筛选标准具有一定的合理性。此外,Ae值与位点的等位基因数之间存在一定的相关性(r=0.59),提示一些研究以等位基因数作为MH筛选标准具有理论依据。Ae值与片段长度、构成MH的SNP数之间的相关系数不超过0.4。这表明,虽然随着片段长度范围的增加、可纳入SNP数量的增多可能会丰富微单倍型位点的基因多样性,但提升效果非常有限。在评价MH效能之时,不能仅以片段长度或构成MH的SNP数作为标准。
第四,MH包含大量的“子集”,这使MH的组装既灵活又复杂。如在一段目标碱基序列上存在n个SNP,至多可组装$\sum\limits_{i=1}^{n-1}{i}$种微单倍型,其中$\sum\limits_{i=1}^{n-1}{i-1}$种均属于“子集”。根据本课题的统计结果,MH的片段跨度越广,包含的子集数量就越多。当片段长度的上限由50 bp增加至350 bp时,相应子集占比从30.38%增加至69.53%。以上情况设定了SNP位点在特定群体中MAF,如若MAF或目标群体发生变化,靶序列可输出的MH“子集”将会变得更加复杂。也正是因为“子集”的存在,使得MH的拼装具有极大的灵活性。理论上,任何定义微单倍型的SNP只要被检测到,就可为后续个人识别或亲缘鉴定等法医学分析提供有价值的遗传信息,即使是不完整的MH位点(即MH的“子集”)也可被充分利用,这与传统的STR遗传标记是截然不同的。MH受靶片段完整性的限制更小,将这一特性与单引物延伸技术相结合,可以为降解DNA样本的分析提供新思路。
与此同时,由于组装“灵活性”而产生的大量子集也给MH数据库构建以及遗传标记频率信息共享带来挑战。随着MH的研究与应用越来越广泛,各科研团队由于研究目的不同,采用的位点组装标准(例如群体、MAF、片段长度等)也会有所差异。那么同一段靶序列可能会记录多种MH,或者多个MH中包含有相同的SNP。这会导致数据记录缺乏兼容性,不利于数据库的整合与共享。因此我们提议,除了将MH作为整体进行一系列信息的记录和储存之外,参与定义MH的SNP基因分型,尤其是“确定相位”(phased)的基因分型结果也应被记录在数据库之中。这样的数据储存方式,具有良好的“向后兼容性”,可以使任何公开发表的MH信息与之后的研究人员充分共享。
综上所述,本研究提供了一套详尽的微单倍型组装方案,证明了MH在人类全基因组中数量丰富,同时在不同的碱基范围尺度上揭示了MH多态性水平。对MH的特征进行了更全面的展示,并结合其特点提出构建微单倍型数据库的方案,为未来群体遗传学和法医遗传学的研究与应用提供支持。
(责任编委: 朱波峰)
附录:
附加材料详见文章电子版图1附
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1附1号染色体上微单倍型遗传标记特征参数之间的关系
使用1号染色体、350 bp范围内、移除子集的MH数据绘制。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 1 SupplementaryRelationship among characteristic parameters of microhaplotypes on chromosome 1
图2附
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2附2号染色体上微单倍型遗传标记特征参数之间的关系
使用2号染色体、350 bp范围内、移除子集的MH数据绘制。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 2 SupplementaryRelationship among characteristic parameters of microhaplotypes on chromosome 2
图3附
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3附3号染色体上微单倍型遗传标记特征参数之间的关系
使用3号染色体、350 bp范围内、移除子集的MH数据绘制。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 3 SupplementaryRelationship among characteristic parameters of microhaplotypes on chromosome 3
图4附
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4附4号染色体上微单倍型遗传标记特征参数之间的关系
使用4号染色体、350 bp范围内、移除子集的MH数据绘制。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 4 SupplementaryRelationship among characteristic parameters of microhaplotypes on chromosome 4
图5附
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5附5号染色体上微单倍型遗传标记特征参数之间的关系
使用5号染色体、350 bp范围内、移除子集的MH数据绘制。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. SupplementaryRelationship among characteristic parameters of microhaplotypes on chromosome 5
图6附
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6附6号染色体上微单倍型遗传标记特征参数之间的关系
使用6号染色体、350 bp范围内、移除子集的MH数据绘制。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 6 SupplementaryRelationship among characteristic parameters of microhaplotypes on chromosome 6
图7附
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7附7号染色体上微单倍型遗传标记特征参数之间的关系
使用7号染色体、350 bp范围内、移除子集的MH数据绘制。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 7 SupplementaryRelationship among characteristic parameters of microhaplotypes on chromosome 7
图8附
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8附8号染色体上微单倍型遗传标记特征参数之间的关系
使用8号染色体、350 bp范围内、移除子集的MH数据绘制。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 8 SupplementaryRelationship among characteristic parameters of microhaplotypes on chromosome 8
图9附
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图9附10号染色体上微单倍型遗传标记特征参数之间的关系
使用10号染色体、350 bp范围内、移除子集的MH数据绘制。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 9 SupplementaryRelationship among characteristic parameters of microhaplotypes on chromosome 10
图10附
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图10附11号染色体上微单倍型遗传标记特征参数之间的关系
使用11号染色体、350 bp范围内、移除子集的MH数据绘制。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 10 SupplementaryRelationship among characteristic parameters of microhaplotypes on chromosome 11
图11附
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图11附12号染色体上微单倍型遗传标记特征参数之间的关系
使用12号染色体、350 bp范围内、移除子集的MH数据绘制。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 11 SupplementaryRelationship among characteristic parameters of microhaplotypes on chromosome 12
图12附
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图12附13号染色体上微单倍型遗传标记特征参数之间的关系
使用13号染色体、350 bp范围内、移除子集的MH数据绘制。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 12 SupplementaryRelationship among characteristic parameters of microhaplotypes on chromosome 13
图13附
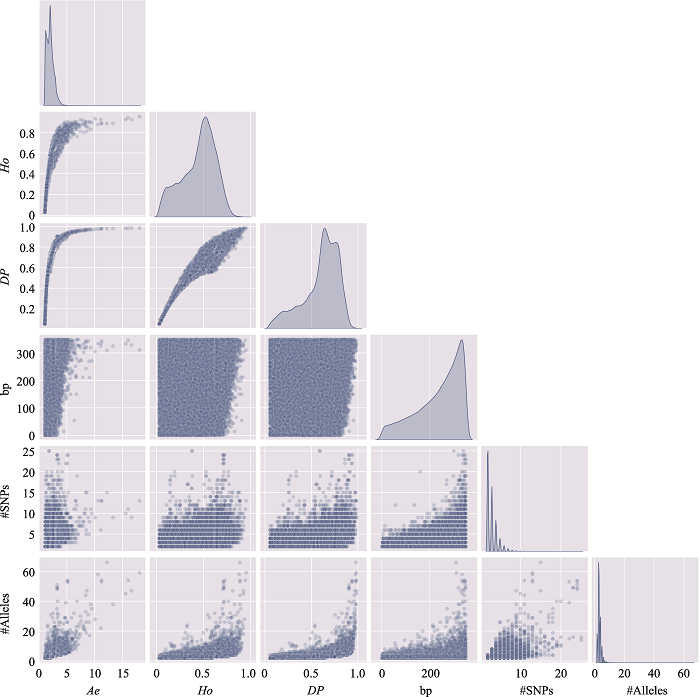 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图13附14号染色体上微单倍型遗传标记特征参数之间的关系
使用14号染色体、350 bp范围内、移除子集的MH数据绘制。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 13 SupplementaryRelationship among characteristic parameters of microhaplotypes on chromosome 14
图14附
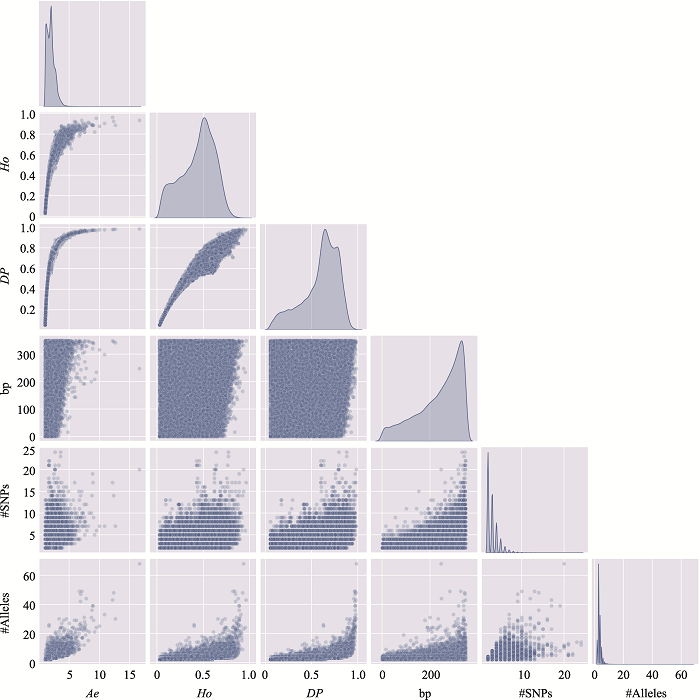 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图14附15号染色体上微单倍型遗传标记特征参数之间的关系
使用15号染色体、350 bp范围内、移除子集的MH数据绘制。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 14 SupplementaryRelationship among characteristic parameters of microhaplotypes on chromosome 15
图15附
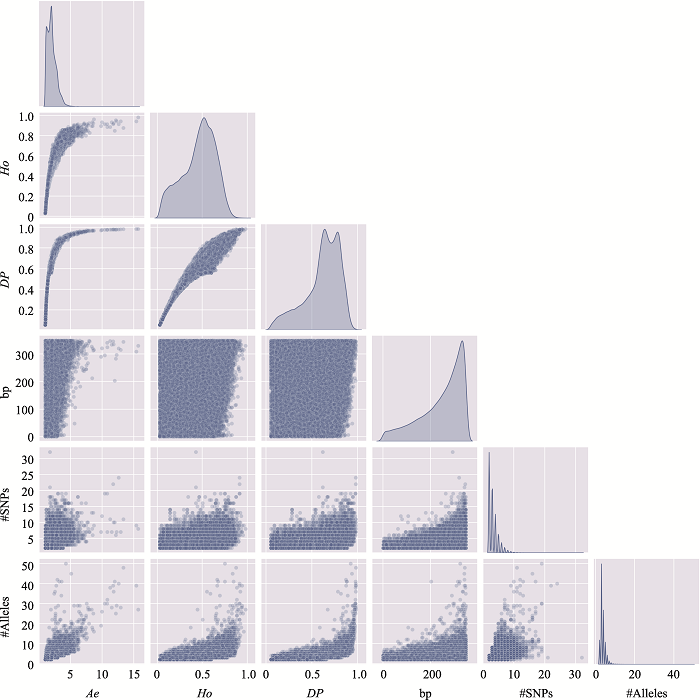 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图15附16号染色体上微单倍型遗传标记特征参数之间的关系
使用16号染色体、350 bp范围内、移除子集的MH数据绘制。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 15 SupplementaryRelationship among characteristic parameters of microhaplotypes on chromosome 16
图16附
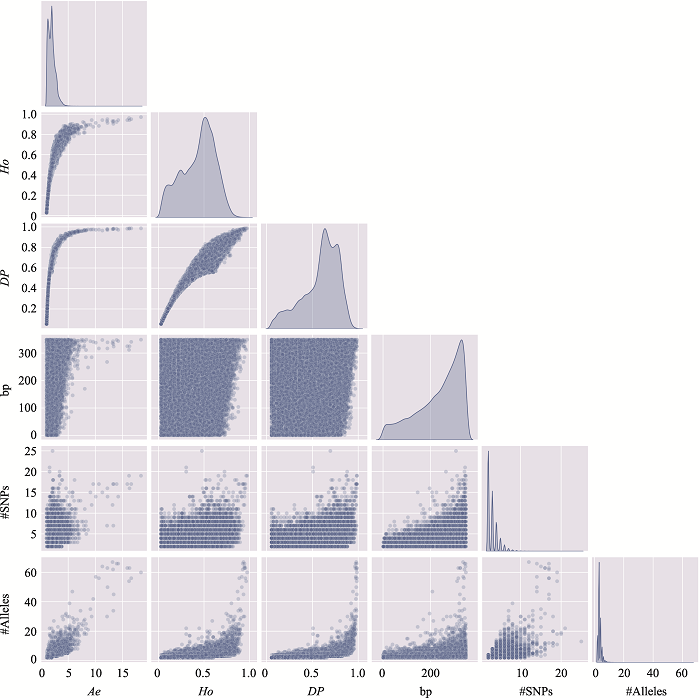 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图16附17号染色体上微单倍型遗传标记特征参数之间的关系
使用17号染色体、350 bp范围内、移除子集的MH数据绘制。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 16 SupplementaryRelationship among characteristic parameters of microhaplotypes on chromosome 17
图17附
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图17附18号染色体上微单倍型遗传标记特征参数之间的关系
使用18号染色体、350 bp范围内、移除子集的MH数据绘制。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 17 SupplementaryRelationship among characteristic parameters of microhaplotypes on chromosome 18
图18附
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图18附19号染色体上微单倍型遗传标记特征参数之间的关系
使用19号染色体、350 bp范围内、移除子集的MH数据绘制。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 18 SupplementaryRelationship among characteristic parameters of microhaplotypes on chromosome 19
图19附
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图19附20号染色体上微单倍型遗传标记特征参数之间的关系
使用20号染色体、350 bp范围内、移除子集的MH数据绘制。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 19 SupplementaryRelationship among characteristic parameters of microhaplotypes on chromosome 20
图20附
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图20附21号染色体上微单倍型遗传标记特征参数之间的关系
使用21号染色体、350 bp范围内、移除子集的MH数据绘制。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 20 SupplementaryRelationship among characteristic parameters of microhaplotypes on chromosome 21
图21附
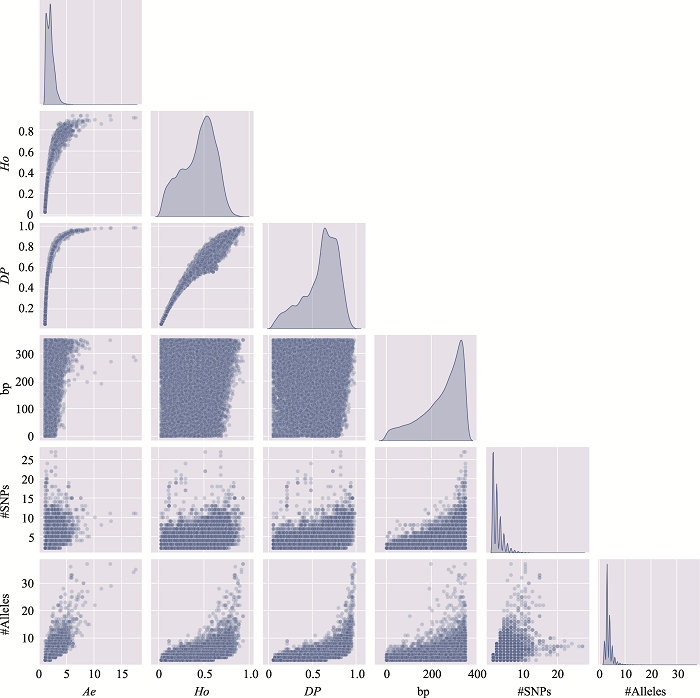 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图21附22号染色体上微单倍型遗传标记特征参数之间的关系
使用22号染色体、350 bp范围内、移除子集的MH数据绘制。#SNPs:构成MH的SNP数;#Alleles:等位基因数。
Fig. 21 SupplementaryRelationship among characteristic parameters of microhaplotypes on chromosome 22
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 3]
[本文引用: 2]
[本文引用: 3]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}