 ,, 张禹, 姜雨,西北农林科技大学动物科技学院,杨凌 712100
,, 张禹, 姜雨,西北农林科技大学动物科技学院,杨凌 712100Pan-genome: setting a new standard for high-quality reference genomes
Peipei Bian,, Yu Zhang, Yu Jiang,College of Animal Science and Technology, Northwest A&F University, Yangling 712100, China通讯作者: 姜雨,博士,教授,研究方向:动物遗传。E-mail:yu.jiang@nwafu.edu.cn
编委: 李海鹏
收稿日期:2021-08-26修回日期:2021-10-28
| 基金资助: |
Received:2021-08-26Revised:2021-10-28
| Fund supported: |
作者简介 About authors
边培培,在读博士研究生,专业方向:动物遗传。E-mail:

摘要
随着三代测序组装的高质量参考基因组的陆续发布,以及大规模重测序和群体遗传学分析的广泛进行,研究人员发现来自单一个体的参考基因组远不能涵盖整个物种的所有遗传序列,大量缺失序列导致群体遗传变异图谱不完整,而构建来自多个个体的泛基因组能很好地解决这一缺陷,其研究内容包括负责基本生物学功能及该物种主要表型特征的核心基因组以及与物种的遗传多样性和个体独特性相关的可变基因组。根据核心和可变基因组所占比例的不同,泛基因组存在开放型和闭合型两种类型。本文主要综述了细菌、真菌和动植物的泛基因组学研究进展,讨论了其在各生物类群中的特征,其中哺乳动物泛基因组是相对闭合的,而目前已知的微生物、被子植物和部分低等动物的泛基因组倾向于开放,通过泛基因组的构建可以完善现有参考基因组并获取整个物种的完整变异信息,将有助于深入研究遗传多样性和表型变异产生的分子机制。
关键词:
Abstract
With the release of high-quality reference genomes assembled by long reads from the third-generation sequencing technology, as well as extensive re-sequencing and population genetic analysis, researchers found that a single reference genome does not represent the diversity within a species. The missing sequences on the reference genome result in an incomplete population genetic polymorphism map. The emergence of pan-genome can well repair the deficiency of single reference genome, which include core genome (responsible for basic biological functions and the main phenotypic characteristics within a species) and the variable genome (related to the genetic diversity or biological characteristics). According to the core and variable genome proportion, the types of pan-genomes can be either open or closed. Here, we review the current exploring of pan-genome for a range of species, to discuss the characteristics of pan-genome in various biological groups. The pan-genome of mammals are more likely closed, while the pan-genomes of microbes, angiosperms, and some invertebrates are likely non-closed. It is possible to complete the reference genome and obtain complete variation information through the pan-genomic study, which will contribute to the study of molecular mechanism for genetic diversity and phenotypic evolution.
Keywords:
PDF (779KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
边培培, 张禹, 姜雨. 泛基因组:高质量参考基因组的新标准. 遗传[J], 2021, 43(11): 1023-1037 doi:10.16288/j.yczz.21-214
Peipei Bian.
随着功能基因组学对基因功能的研究越来越细致,一个物种是否拥有高质量的参考基因组成为了深入解析其遗传与表型关系的重要前提。然而在群体水平上,研究人员发现来自同一物种不同个体的基因组序列并不能完全与该物种的参考基因组一一对应。因此建立一个能够包含这个物种全部基因组序列和变异信息情况的完整集合对基因组学的研究变得极为重要。
2005年,Tettelin等[1]首次在细菌研究中提出泛基因组(pan-genome)的概念,指整个物种基因组序列的非冗余集合,其中包括存在于该物种几乎所有个体中的核心基因组(core genome)和仅在部分个体中存在的可变基因组(accessory/variable/dispensable genome)。相对于细菌来说,真核生物无法频繁的跨物种交换遗传物质,被认为存在相对较少的存在/缺失变异(presence and absence variations, PAVs)[2]。但是随着对动植物个体基因组之间的比较研究,研究者发现高等生物同样具有普遍的跨物种基因交流,也存在相当数量的PAVs,且许多位于功能性区域,承担重要的生物学功能[3,4,5]。泛基因组现已在植物、真菌、动物基因组学研究中被广泛用于更为全面地评估物种内遗传多样性,探究跨物种的基因交流和驯化及改良过程。研究表明利用泛基因组可以获取更为准确全面的变异信息,通过与表型进行关联,筛查出可变基因组中的功能基因或功能序列,这将为物种的遗传改良提供宝贵的遗传资源[6,7,8,9,10,11]。在微生物方面,利用泛基因组还可以对菌种进化、适应性及群体结构进行研究分析[12];同时可应用于菌株重要毒力因子的发现和疫苗的设计[13]。
本文综述了细菌、真菌和动植物的泛基因组学研究进展,讨论了其在各生物类群中的特征,并对其在完善参考基因组以及获取完整变异信息上的应用进行了分析和展望。
1 泛基因组的概念和特点
广义的泛基因组是一个捕获了物种全部遗传信息的集合。对于包含一定数量个体基因组信息的泛基因组来说,整个基因或序列集合可以被分为核心基因组和可变基因组(图1A),核心基因组(core,一般认为存在于超过95%的个体基因组中);可变基因组又可以被进一步分为壳基因组(shell,在所有个体基因组中存在比例大约为5%~95%)和云基因组(cloud,仅存在约少于5%的个体基因组中),shell和cloud作为可变基因组的子集,一般与生物对特定环境的适应或生物学特性有关。上述分类能够弥补在实际定义不同基因组类别时所面临的不确定性,核心基因组为95%以上而不是100%的存在比例,可以避免某个个体的低质量基因组序列或者是基因组缺陷而造成的分类错误,确保真实的核心基因组在注释和分类过程中不被遗漏;而cloud则可能是个别个体基因组意外获得的外源基因,或者是来自于该个体基因组异常装配或者是外源污染[14]。具体的分类比例并不固定,研究人员可以根据实际物种研究情况,进行合理定义。一些研究证明了泛基因组中基因频率呈不对称的“U”型分布(图1B),这说明大部分基因或以核基因组的方式存在于绝大多数个体中,或以云基因组的方式存在于个别个体中[4,14~16]。根据泛基因组中核心基因组的比例,将泛基因组分为开放型和闭合型两种,具体状态取决于所分析的物种特征,如物种整合外源DNA的能力,以及物种的生活方式和环境[17]。与具有开放程度较小泛基因组的物种相比,具有大型开放泛基因组的物种可能占据更多样的生态位和具有更复杂的群落[18]以及更大的有效群体规模,多态性水平更高。一般认为完全闭合的泛基因组是不存在的,在构建泛基因组的时候随着个体数量的增加,无论是开放型还是闭合型的泛基因组,整个泛基因组的大小都是逐步增加的,而核心基因组的大小都是逐渐减少的(图1:C, D)。对于一个既定的物种来说,除去云序列(仅存在于物种极少数个体中)以外的核心基因组和可变基因组是一个定值。对于闭合型的泛基因组,有限数量个体的增加,可以使核心基因组和整个泛基因组含量迅速到达平台期,趋近于真实的水平。而开放程度高的泛基因组需要大量的个体才能获取这个真实值,在逐个增加研究个体时,到达平台期获得这个值的速度是缓慢的。基于以上差异,在进行闭合型泛基因组研究时,通过汇总有限数量个体的基因组序列,人们可以获取这个物种几乎全部的遗传信息。哺乳动物的泛基因组是比较典型的闭合型,其基因数量以及结构相对稳定,可变基因数量有限[5,19~20],保证了高度复杂化的基因调节网络的稳定。而开放型泛基因组意味着,随着人们不断加入研究个体,其总是会有一定数量的新基因或者新序列的增长,也就是说通过一定数量的研究对象获取物种内全部遗传信息是不现实的,但是这种开放的模式为物种提供了丰富的遗传资源库,增加其功能多样性和复杂性,提高了其对动态环境的适应性。细菌、真菌和被子植物表现出开放型的特征,许多物种的核心基因比例小于80%[2,21]。
图1
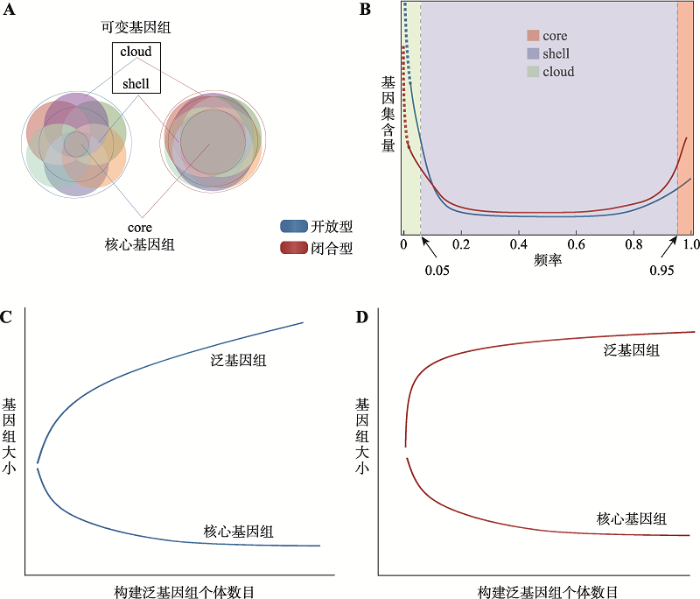 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1开放程度不同的泛基因组特征
A:泛基因组的组成。B:泛基因组中基因频率的不规则“U”型分布。C:开放型泛基因组:随着构建泛基因组个体的增加,整个泛基因组以及核心基因组大小的增长趋势。D:闭合型泛基因组:随着构建泛基因组个体的增加,整个泛基因组以及核心基因组大小的增长趋势。
Fig. 1Pan-genome features with varying degrees of openness
2 群体中可变基因组的来源
当前泛基因组的研究主要是强调物种内部完整基因组序列的获取,所以更关注可变基因组,也就是在物种内部个体基因组之间一致性低的多态序列或者是产生了PAVs的序列集合。广义的泛基因组应该能够捕获该物种的全部遗传变异信息,但是当前的研究所构建的泛基因组大多体现不了那些小的插入缺失(insertions and deletions, indels)和单核苷酸多态性(single-nucleotide polymorphisms, SNPs),以及不改变序列组成的易位(translocation)和倒位(inversion)变异等,因此这种泛基因组可以被认为是狭义的泛基因组。最初应用泛基因组概念的细菌,通常具有较小的基因组,其基因占据基因组序列的大部分,几乎没有基因间序列,而且数量差异很大,所以蛋白编码基因的含量是细菌等原核生物泛基因组研究的主要内容。原核基因组以不断变化的状态存在,通过水平基因转移,基因复制甚至可能以从头出现的方式而扩张,并通过基因丢失而收缩。在细菌中广泛的基因损失和水平基因转移(转化、接合和转导)是导致可变基因产生的两个主要进化过程[22]。不同模式真菌物种的泛基因组的研究表明真菌是通过菌株水平的创新来进化的,而不是大规模的水平基因转移。此外被子植物可通过全基因组复制(whole genome duplications, WGDs)、局部串联重复、转座因子(transposable elements, TEs)介导的重复、片段重复、近缘物种渗入、水平基因转移和从头基因诞生(de novo gene birth)获取新基因,同时也能通过染色体内重组和假基因化介导基因和序列的丢失[21]。虽然当前在动物上泛基因组的研究有限,但是众多的基因组学研究已经证明了在动物基因组上存在渗入、水平基因转移以及各种重复事件[23]。综上所述,正是通过序列的重复、近缘物种渗入、基因从头诞生或水平基因转移,以及后续的序列分歧/丢失或基因分裂/融合等多种过程,才产生了物种内广泛的PAVs,形成了泛基因组。但是重复以及从头诞生的新基因一般很难在短时间内与原序列产生足够的分歧,因此在狭义泛基因组中难以被捕获。所以通常认为从狭义上来说,可变基因组的主要来源是基因和序列的丢失,渗入和水平基因转移(图2)。
图2
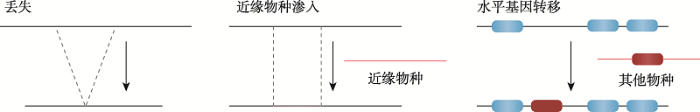 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2可变基因组的主要来源
Fig. 2Origin of variable genome
3 泛基因组的构建与呈现
目前构建泛基因组主要有基于迭代组装和基于从头组装两种方法(表1)。首先出现的是基于从头组装基因组构建泛基因组的方法[1]。这种方法分别对多个个体进行从头组装并注释,然后通过同一物种不同个体基因组间的相互比较,确定出核心基因组序列和可变基因组序列,最后将这些序列去冗余合并后构成一个包含该物种所有个体基因组序列的泛基因组[5]。这种方法的优势在于它能够检测到更多的结构变异(structural variations, SVs),但对计算资源和样品的测序深度有较高的要求,不适用于基因组较大的物种和大规模群体的分析。迭代组装构建泛基因组方法的出现弥补了这些不足,其方式是由参考基因组起始,将每个样本的测序数据映射到参考基因组,提取未比对成功的序列进行组装,然后使用非冗余序列直接更新参考基因组,获得最终的扩展参考基因组即为该物种的泛基因组或者是对个体进行初步组装,从与参考基因组未比对上的contigs中移除冗余序列来构建代表性的非参考序列,结合参考基因组和代表性非参考基因组序列构建泛基因组。这种构建策略可以利用大规模的重测序数据,对测序深度要求很低,同时,因为只对未成功比对到参考基因组上的序列进行了组装,这种方法相对节省了计算资源,已在基因组较大的物种如小麦[24]以及大规模测序物种如水稻[10]中被应用。这种方法会在最终的泛基因组中产生大量的序列片段,并且无法检测每个个体的拷贝数变异(copy number variations, CNVs),但对于基因的PAVs检测非常有效[25]。
Table 1
表1
表1泛基因组构建方法比较
Table 1
| 构建方法 | 基于重测序unmapping 序列的迭代组装 | 基于多个从头组装 基因组的比较 |
|---|---|---|
| (1)计算资源需求 | 低 | 高 |
| (2)测序深度要求 | 低 | 高 |
| (3)研究个体规模 | 大 | 小 |
| (4)结构变异检测能力 | 低 | 高 |
| (5)存在缺失变异 检测能力 | 高 | 较低 |
新窗口打开|下载CSV
这两种方法各有优缺点,目前均已被广泛应用于构建各种物种的泛基因组,研究人员通过将新发现的序列直接加入参考基因组的呈现形式产生了一系列的线性泛基因组,极大地丰富了人们对现有物种基因组的认识。然而,这种展示方式也带来了一些问题如:源于不同个体的变异信息被丢失,也几乎没有相应的程序和算法可以处理这种方式提供的变异信息。
获取可变基因组的序列组成和位置信息是展示和应用泛基因组的关键。但是线性泛基因组方式只呈现了可变基因组的序列组成,丢失了重要的染色体位置信息,因此在构建泛基因组的过程中,为防止重要信息的丢失,有两种方法:要么在线性泛基因组中标注序列位置信息,要么构建图结构的泛基因组。和线性基因组不同的是,图结构泛基因组是一个二维序列图谱,它以参考基因组为框架,以单个碱基作为图的节点,碱基间的前后关系作为图的边,存在序列差异的地方会自然形成不同的分支,呈现出一个图结构。这个图结构基因组可以依据新序列的加入不断扩展变化,最终它将会成为一个符合全物种的泛基因组图谱[26]。这种展示形式可以包含变异的嵌套,将同一位置的变异整合而不是单独占据一个区域,从而达到将所有变异精确纳入图谱的效果。这使得物种内大量复杂的变异可以紧凑的形式呈现。目前已有大量软件被开发用于这种图结构泛基因组的分析[27],如vg[28]、minigraph[26]、GraphType2[29]等,并且已在动植物基因组学研究中得到了初步应用[19,26,30~32]。
随着测序技术以及生物信息学工具的进步,包含全部序列变异信息的图结构泛基因组出现,尽管它受限于计算和存储当前只能应用于部分个体,但仍旧是向着广义泛基因组研究迈进的重要一步。未来技术的发展会让构建一个包含物种内全部遗传信息的泛基因组成为可能,实现精确处理大量基因组中的序列和变异信息,那时的基因组学研究才是真正在利用一个“参考”基因组。
4 泛基因组在不同物种中的研究进展
由于微生物基因组的可塑性和多样性,泛基因组的研究对其十分重要,同时,近年来测序和基因组组装成本的降低,研究人员在真核生物物种中发现了大规模的基因组变异,促使了泛基因组研究在真核生物中的扩展(图3,表2)。图3
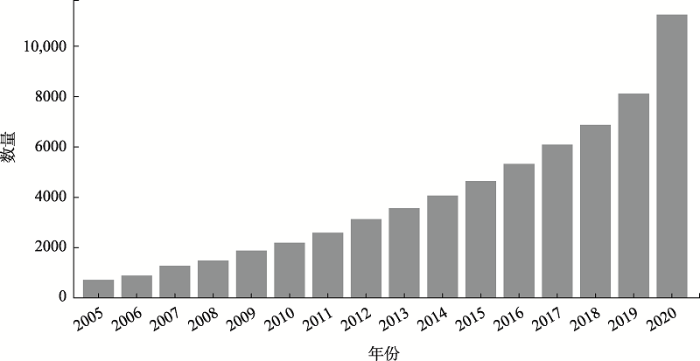 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3泛基因组相关研究数量的增长
泛基因组的概念在2005年被首次提出之后,关键词“pangenome”或者“pan-genome”在Europe Pubmed Central (
Fig. 3The growth of pan-genome publications
Table 2
表2
表2泛基因组代表性研究
Table 2
| 年份 | 物种 | 基因组大小 | 个体数目 | 构建策略 | 主要新进展 | 参考文献 |
|---|---|---|---|---|---|---|
| 2005 | 无乳链球菌 (Streptococcus agalactiae) | ~2 Mb | 8 | 基于多个从头组装的基因组的比较 | 泛基因组概念的引入 | [1] |
| 2007 | N/A | N/A | N/A | N/A | 综述文章,第一次在植物中应用泛基因组这个术语 | [45] |
| 2010 | 人(Homo sapiens) | ~3.2 Gb | 3 | 基于多个从头组装的基因组的比较 | 估计一个完整的泛基因组可能包含19~ 40 Mb在当前参考基因组中不存在的新序列,鉴定了额外86个新基因 | [5] |
| 2014 | 大豆(Glycine soya) | ~0.9 Gb | 7 | 基于多个从头组装的基因组的比较 | 第一个植物泛基因组文章,测序和重新组装了野生大豆个体的基因组,将注释基因聚类到基因家族,核心基因簇的比例为49% | [46] |
| 玉米(Zea mays) | ~2.4 Gb | 503 | 基于多个从头组装的转录组的比较 | 获得了约8600个有代表性的在参考基因组中不存在的转录本,其中的16.4%在所有品系中表达,82.7%在部分品系中表达 | [50] | |
| 2016 | 甘蓝(Brassica oleracea ) | ~650 Mb | 10 | reads映射到参考基因组;unmapping reads的组装;通过新组装的contigs更新旧序列来产生新的参考序列。(将从每个基因组获得的reads映射到不断增长的泛基因组) | 核心基因簇比例占泛基因组总数的81%,近20%的基因受到存在/缺失变异的影响 | [53] |
| 2018 | 水稻(Oryza sativa) | ~400 Mb | 3010 | 对个体测序数据进行组装,通过从与参考基因组unaligned的contigs中移除冗余序列来构建具有代表性的非参考序列,结合参考基因组构建泛基因组 | 鉴定了超过10,000个新的全长蛋白编码基因和大量的存在-缺失变异,核心基因簇比例占泛基因组总数的54%~62% | [10] |
| 2019 | 人(Homo sapiens) | ~3.2Gb | 910 | reads映射到参考基因组,组装unmapping的reads,保留新组装的长度大于1 kb的非参考序列的contigs用于构建泛基因组 | 利用非洲血统的人类群体基因组构建泛基因组,获取了参考基因组中296 Mb不存在的序列 | [20] |
| 番茄(Solanum lycopersicum) | ~810Mb | 725 | 对个体测序数据进行组装,通过从与参考基因组unaligned的contigs中移除冗余序列来构建具有代表性的非参考序列,结合参考基因组构建泛基因组 | 鉴定出一个约 4 kb 与风味相关的基因TomLoxC的启动子的存在缺失变异,表明泛基因组研究可以帮助物种恢复驯化或者改良过程中丢失的理想性状 | [4] | |
| 猪(Sus scrofa) | ~2.7 Gb | 12 | 基于从头组装的基因组之间的相互比较 | 第一个家养动物的线性泛基因组,获得了额外的72.5 Mb序列 | [3] | |
| 山羊(Capra hircus) | ~2.9 Gb | 10 | 基于从头比较来自近缘物种的基因组 | 第一个跨物种比对的泛基因组,从参考基因组中寻找缺失序列的有效且可靠的策略,获得了38.3 Mb 序列 | [70] | |
| 2020 | 大豆 (Glycine soja和 Glycine max) | ~1 Gb | 29 | 基于从头组装的基因组之间的相互比较,图结构泛基因组 | 鉴定了大的结构变异和基因融合事件,将结构变异与基因表达和农艺性状联系起来 | [30] |
| 牛(Bos taurus) | ~2.6 Gb | 300 | 集成了线性参考基因组坐标和预先选择的变异(<50 bp),图结构泛基因组 | 第一个家养动物的图结构泛基因组,在人类以外的大基因组动物上对图结构泛基因组的首个尝试 | [32] | |
| 贻贝 (Mytilus galloprovincialis) | ~1.28 Gb | 16 | 测序reads被映射到贻贝参考基因组上,收集未映射的reads从头组装。新组装的contigs被添加到参考基因组中,构建了一个贻贝泛基因组(将从每个基因组获得的reads映射到不断增长的泛基因组)。 | 开放型的动物泛基因组,高比例的可变基因组(45%),展示了动物泛基因组的潜能 | [71] | |
| 2021 | 水稻(Oryza sativa和Oryza glaberrima) | ~400 Mb | 33 | 基于从头组装的基因组之间的相互比较,图结构泛基因组 | 共鉴定了171,072个SVs和25,549个gCNVs,可以用于全基因组关联研究 | [31] |
| 牛(Bos taurus) | ~2.6 Gb | 6 | 基于从头组装的基因组之间的相互比较,图结构泛基因组 | 70 Mb的非参考基因组等位序列,提供了一个构建图结构泛基因组的框架,适合于多种物种 | [19] |
新窗口打开|下载CSV
4.1 细菌泛基因组
首个细菌泛基因组由无乳链球菌(Streptococcus agalactiae)构建,每个菌种的核心基因组约占任何单个基因组的80%[1],这说明有一定数量的可变基因组仅在部分或者个别菌种中存在,很明显单个基因组序列不能反映细菌物种内的整个遗传变异性。细菌栖息在千差万别的生态位中,并具有大量相应的调节机制,以适应多变的环境[33],核心基因的比例可以从5%至98%。除了使基因组垂直向下传给后代外,细菌还具有通过水平转移从环境中获取遗传物质的能力[34],在获得基因的同时,为了维持细菌基因组小而紧凑的结构特征,基因还经常复制或丢失[35]。垂直传播和水平转移的混合作用使细菌基因组的系统发育分析复杂化[22]。在同一种细菌内,在基因组水平上也可能存在很大程度的个体差异。如在大肠杆菌(Escherichia coli)泛基因组中,任何一种大肠杆菌的基因组核心基因的比例都少于泛基因总数的10%,即使在转录因子水平上,大肠杆菌基因组之间也存在巨大差异[36]。考虑到这种高水平的遗传变异,重建细菌的系统发育和种群历史,泛基因组研究是有必要的,并且可以作为细菌分类的重要依据[37]。Freschi等[38]基于1311个铜绿假单胞菌的高质量基因组进行了泛基因组分析,研究了水平基因转移在人类病原体铜绿假单胞菌的抗菌素耐药性和毒力机制中的贡献,基于核心基因组的系统发育为其种群结构提供了强有力的证据。同样分枝杆菌泛基因组学研究证明了水平基因转移在进化过程中对其适应新环境和宿主中有重要作用[39]。随着测序成本降低以及数据库中可用细菌基因组的快速增加促进了泛基因组软件工具的开发[40],一些在线软件例如PanX[41]等,只要遵循特定步骤,即可生成泛基因组分析结果,加速了细菌泛基因组的研究进展。在细菌泛基因组研究中发现一些可变基因在不断变化的环境中具有适应性优势[42],另一些则和菌株的致病性和耐药性相关[18,43]。细菌泛基因组的研究在临床微生物学中有许多应用。它可以揭示细菌的致病潜力和抵抗抗菌素的能力,鉴定特定序列并预测抗原表位,从而可以设计分子或血清学检测方法和疫苗[40]。
4.2 植物泛基因组
从不同植物中获得的数据向人们展示了植物基因组的可塑性[44],单个基因组已无法表征全部的遗传多样性,促使在基因组学研究中引入了植物泛基因组的概念[45],这有助于深入了解植物产生遗传多样性和表型变异的过程。首个植物泛基因组在2014年被报道,其基于对7份代表性野生大豆(Glycine soja)全基因组的组装比较,发现了与生物抗性、种子组成、开花和成熟时间等重要农艺性状有关的可变基因[46]。泛基因组分析使人们能够追踪驯化和育种过程中基因的保留和丢失,开发将基因重新引入现代品种的潜力,恢复物种失去的遗传多样性。Gao等[4]使用了具有广泛品种和地理代表性的725个番茄(Solanum lycopersicum)个体,揭示了参考基因组中不存在的4873个基因,PAVs分析表明,在驯化和改良过程中有大量的基因丢失以及基因和启动子的负选择,并且丢失或者受到负选择的基因具有重要功能,尤其是与抗病性相关。此外,该研究还鉴定出在驯化阶段受选择的TomLoxC启动子上与番茄风味有关的稀有等位基因,利用其杂合子优势,可直接应用于生产中的性状改良。目前对泛基因组的研究并不局限于基因本身,基因以外的基因组区域也解释了作物表型变异的很大一部分,许多重要的农艺性状可能是由基因调控的变化而不是基因的存在/缺失变异决定的[21]。由于SVs的大小能够造成更多的核苷酸序列差异,因此可能会表现出不成比例的大表型效应[47],已被确定为许多罕见和常见疾病的致病因素,并且通常被认为它们是通过影响基因的表达来起作用的。多个植物泛基因组研究也发现,SVs导致基因组变异的同时,能够引起表型变异[48]。2020年对番茄PanSV基因组的深入研究揭示了这一点,几乎一半的SVs与基因或调控序列重叠,并且半数影响编码序列的SVs与基因差异表达有关[49]。
泛基因组对于揭示物种内完整的遗传变异信息至关重要,尤其是近年来图结构泛基因组的发展,其构建及应用策略越来越稳定和完善,包含的功能元素和序列空间越来越充足,能够作为分析其他个体的参考,提高了研究人员对许多个体和物种基因组复杂性的理解。2020年,有研究将26个大豆株系从头组装的基因组和3个先前报道的基因组构建了一个基于图形的泛基因组,结合2898个不同株系的重测序数据,揭示了众多仅用单个参考基因组无法检测到的变异,为大豆的进化和功能基因组学研究提供了更加完整的基因组图谱,并且通过对全基因组复制区域及SVs的研究,表明基因组复制是SVs进化的重要驱动力[30]。同样基于多个参考基因组水平的高质量组装基因组,2021年,Qin等[31]构建了高质量的水稻(Oryza sativa和Oryza glaberrima)图结构泛基因组。其研究提供了水稻基因组变异和驯化的遗传资源,并推断了整个水稻种群中SVs的派生状态,分析了SVs的分布并评估了SVs形成的机制以及SVs对基因表达的影响。此研究提供了SVs和基因的拷贝数变异(gene copy number variations, gCNVs)如何直接影响环境适应性和农艺性状的示例,展示了高质量基因组组装和图结构泛基因组在植物基因组学和功能基因组学中的重要作用。
迄今为止,已经有10余种植物建立了泛基因组包括玉米(Zea mays)[50]、大豆[30,46]、二穗短柄草(Brachypodium distachyon)[14]、辣椒(Capsicum)[51]、小麦(Triticum spp.)[24,52]、甘蓝(Brassica oleracea)[53]、水稻[10,31,54]、番茄[4,49]、狗尾草(Setaria viridis)[55]、向日葵(Helianthus annuus L.)[56]、大麦(Hordeum vulgare ssp.)[57,58]、桃子(Prunus persica)[6],高粱(Sorghum bicolor)[59,60]等,除了重要农作物还包括驯化作物的野生和杂草近缘种,在每个被研究的物种中都有一个可观的可变基因含量(10%~60%)。可变基因经常被注释为与生物和非生物胁迫耐受性相关,同时这些基因相较于核心基因具有较低的进化约束和表达水平。通过泛基因组研究可以获取更多准确和大片段的SVs,其中一些涉及改变基因剂量和表达水平的SVs影响了许多重要的农艺性状,包括水果的味道、大小和产量。这些发现强调了泛基因组研究在作物改良中的重要性和效用。
4.3 真菌泛基因组
研究人员使用长reads组装了驯化酵母及其野生近缘种的12个端到端的基因组,核基因组的大小从11.73到12.14 Mb不等,通过多个参考质量的基因组序列的比较,在驯化和野生个体之间观察到的许多差异可能反映了人类活动对基因组结构进化的影响[8]。接着通过对1011个酿酒酵母分离株的泛基因组构建,结合表型分析工作,提供了酿酒酵母变异的详细信息,为其全基因组关联分析(genome- wide association study, GWAS)奠定了基础,并为基因型-表型关系提供了新的见解,在规模上提供了与其他模式生物体相匹配的群体基因组资源[61]。2019年报道了四种模式真菌的泛基因组:酿酒酵母(Saccharomyces cerevisiae)、白色念珠菌(Candida albicans)、新型隐球菌(Cryptococcus neoformans)和烟曲霉(Aspergillus fumigatus)。研究发现,在这些物种中,每个菌株的所有基因中80%~90%属于核心基因[62],其余的可变基因可能与发病机制和抗菌素耐药有关。对物种祖先核心基因组和可变基因组的分析表明:基因复制等过程可能是影响真菌全基因组进化的主要因素,水平基因转移的作用有限。真菌病原体反复击败农作物抗性,变得对农药耐受,威胁着全球粮食生产,种群内的遗传变异多样性常常助长了这种进化过程[63]。小麦叶枯菌(Zymoseptoria tritici)会导致小麦枯萎病,2019年其泛基因组的研究仅鉴定出了58%的核心基因,其余的可变基因为其适应性进化提供了基础[64]。此外,有研究人员组装了来自六大洲的小麦叶枯菌的19个完整基因组,构建了小麦真菌病原体的高质量泛基因组,表明了染色体重排是广泛的基因存在/缺失变异的基础,同时发现可变基因组中富含与发病机制相关的功能基因[65]。与细菌相似,真菌生物在基因含量上也显示出种内变异性。真菌泛基因组可用于获取大量菌株完整的变异信息,有助于真菌的驯化以及基因型-表型的关联研究。同时研究表明可变基因通常在致病性中起重要作用,通过泛基因组研究可以追踪确定参与感染和宿主反应的新基因的来源,也将有助于解决与作物-病原体共同进化相关的问题。
4.4 动物泛基因组
目前,相对于微生物和植物来说,动物泛基因组的研究范围还很有限,主要集中在人类(Homo sapiens)和家养动物。2010年,Li等[5]整合了亚洲人和非洲人新组装的基因组以及当时的人类参考基因组,构建了人类的第一个泛基因组。该研究在每个新组装基因组中获取了~5 Mb在参考基因组中不存在的新序列,推断完整的人类泛基因组将包含现有参考基因组中不存在的19~40 Mb新序列。跨物种保守性分析表明这些新序列中包含的某些基因在哺乳动物基因组之间是保守的,很可能具有生物学功能。此研究证实了单个人类基因组序列中存在大量未证明的遗传区域,并且可以通过非常深的测序和从头组装来鉴定。对来自冰岛的15,219个人进行测序,仅关注非重复,非参考基因组序列,该研究共发现了3719个约0.33 Mb的新序列[66]。2019年构建的汉人泛基因组发现了~29.5 Mb的新序列,还鉴定了188个新的蛋白质编码基因[67],而对1000个瑞典基因组的分析发现了~46 Mb的新序列,大部分为重复序列(56%)[68]。Sherman等[20]利用910个非洲后裔个体组成的深度测序数据集,构建的泛基因组比当前参考基因组多近300Mb的新序列,这是迄今为止报道的找到最多新序列的人类泛基因组。这些研究说明,单一参考基因组不足以进行基于群体的人类遗传学研究,更好的方法可能是为不同的人类群体创建参考基因组。猪、牛和羊在畜牧业中都占据重要地位,猪也是重要的生物医学模型[69],构建猪、牛和羊的泛基因组对优质种质资源的保护和利用,解析人类驯化动物的历史及作为模式动物探究生命奥秘有重要意义。Tian等[3]使用了来自欧亚大陆的12个基因组构建了猪的泛基因组,相较于参考基因组(Sscrofa11.1)共获取了72.5 Mb的非冗余的新序列,且发现了脂肪分解的必要调节基因TIG3在猪群中显示为PAVs,并且可能导致不同猪种之间的生理差异。山羊泛基因组研究利用其他9个从头组装的Caprini物种基因组共鉴定出了38.3 Mb山羊参考基因组上不存在的新序列,通过山羊全基因组重测序和转录组数据进一步验证了它们在山羊中的存在,证明了对亲缘关系近的物种基因组进行比较是一种基于参考基因组寻找缺失序列的有效且可靠的策略,这种方法也可能适用于其他物种[70]。这两项研究都表明使用泛基因组作为参考可产生更高质量的变异集合和更准确的基因表达量化,改善广泛的基因组分析。2020年,研究人员使用来自约300头牛的变异信息(<50 bp)构建了家养动物的第一个图结构泛基因组[32],提高了序列比对和基因分型的准确性,这是在人类以外的大基因组动物上对图结构泛基因组的首个尝试,为其他动物的研究提供了重要参考。稍后研究人员利用6只牛的基因组构建了图结构泛基因组,发现了参考基因组中缺失的功能序列[19],其中包括参与免疫反应和免疫调节的基因,此研究提供了用于建立和利用更多样化的参考基因组的方法和框架。
除了上述哺乳动物以外,研究人员还报道了地中海贻贝的开放型泛基因组[71]。贻贝是具有生态和经济意义的食用双壳类生物,对生物和非生物应激源具有高度的侵袭性和复原力,其泛基因组具有15,000个可变基因,占全部泛基因组数量的25%,平均出现的时间晚,表达水平低并且容易受到PAVs的影响,开放阅读框较短,基因结构复杂性低,并且参与了与防御和生存相关的功能,对生物适应性具有重要价值。此外,泛基因组也在昆虫基因组学的研究中得到了应用。蜱虫(Acari: Ixodidae)是传播最多样化的人类和动物病原体,对其泛基因组的研究揭示了不同蜱种的遗传结构和病原体组成主要受生态和地理因素的影响,并进一步确定了与不同宿主范围、生命周期和分布相关的物种特异性决定因素[72],这也将为蜱虫生物学、病媒-病原体相互作用、疾病传播和控制策略的研究开辟新途径。熊蜂(Hymenoptera: Apidae)的泛基因组研究表明在系统发育框架中对多个基因组进行比较分析,大大提高了进化分析的精度和灵敏度,并可以提供识别基因组稳定和动态特征的可靠结果[73]。此研究也将助力于功能基因定位和克隆,以及重测序和群体基因组学研究,为熊蜂在农业中的使用提供基础的遗传信息。
上述研究表明,目前的动物单一参考基因组对于具有高适应能力,高杂合度,高水平重复元素以及复杂群体历史的物种还远远不够完整,并且强调了参考基因组缺失的基因对于临床和农业应用的潜在影响。后续研究应集中于动物高质量泛基因组的构建,获取完整的泛基因组序列,以及构建可用的图结构泛基因组,寻找更多可应用于经济动物选育和改良的遗传信息。
5 结语与展望
基因组时代的前期,研究人员采取的主要策略就是为目标物种提供一个单一的“参考”基因组,该基因组成为各种遗传分析(包括研究物种内部和物种之间的变异)的基础[25]。随着测序新技术的发展,测序质量进一步提高,同时成本大大降低,成千上万的新基因组被测序,物种间大量变异被获取,人们开始意识到单一参考基因组不足以代表一个物种全部的遗传信息。泛基因组分析提供了一个平台,可通过收集物种的整个基因组信息库来获取其全部的遗传多样性,在细菌、真菌以及动植物中已经得到了广泛的应用。在目前泛基因组的研究中仍存在一些问题亟待解决:各种生物的基因组组装还不完整,尽管长reads测序被证明已经能够解析基因组中一些具有挑战性的区域,检测以前无法获取的SVs[74,75,76],但是为物种中每一个个体实现完整、无间隙的装配是不现实的;此外,基因组的测序、组装,泛基因组的构建策略,序列注释,判断PAVs等一系列方法并没有标准化的流程,导致不同研究获取的泛基因组序列不能直接比较,汇集所有数据建立一个完整的泛基因组将是一个巨大的挑战。
微生物和被子植物相比于哺乳动物,基因组可塑性更高,物种内的遗传多样性更为丰富,因而有了相对广泛的研究。哺乳动物基因组相对保守,通常只有基因间或片段化的基因区域参与基因组序列的增减,但是这并不意味着动物泛基因组的重要性降低。从对贻贝的研究[71]中可以看到动物泛基因组的潜力,随着泛基因组研究扩展到更多的物种,才能真正准确地评估一个生物类群的多样性水平。近年来泛基因组学研究为植物多样性研究和改良提供了新的思路[21,44],但在除人类以外的动物中泛基因组学研究有限,在其他动物泛基因组的研究上还需要努力,以期为动物遗传相关研究打下坚实基础。
当前泛基因组研究的核心是用更丰富的数据结构取代传统的线性参考基因组[27],相对于传统的单一线性参考基因组,泛基因组作为参考基因组能更加全面地呈现群体基因组信息,同时更有益于变异信息的获取和利用。随着图结构泛基因组的构建方式和分析策略的逐步完善,利用泛基因组将会更加高效地辅助解决功能基因组学研究的难题,从而彻底改变基因组学的研究。
责任编委: 李海鹏
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1073/pnas.0506758102URL [本文引用: 4]
DOI:10.1016/j.tig.2019.11.006URL [本文引用: 2]
DOI:10.1007/s11427-019-9551-7URL [本文引用: 3]
DOI:10.1038/s41588-019-0410-2URL [本文引用: 5]
DOI:10.1038/nbt.1596URL [本文引用: 5]
[本文引用: 2]
DOI:10.1186/s13059-018-1528-8PMID:30241487 [本文引用: 1]

Understanding how crop plants evolved from their wild relatives and spread around the world can inform about the origins of agriculture. Here, we review how the rapid development of genomic resources and tools has made it possible to conduct genetic mapping and population genetic studies to unravel the molecular underpinnings of domestication and crop evolution in diverse crop species. We propose three future avenues for the study of crop evolution: establishment of high-quality reference genomes for crops and their wild relatives; genomic characterization of germplasm collections; and the adoption of novel methodologies such as archaeogenetics, epigenomics, and genome editing.
DOI:10.1038/ng.3847URL [本文引用: 2]
DOI:10.1186/1471-2164-13-577URL [本文引用: 1]
DOI:10.1038/s41586-018-0063-9URL [本文引用: 4]
DOI:10.1038/s41477-019-0577-7URL [本文引用: 1]
DOI:10.1038/s41587-018-0008-8URL [本文引用: 1]
DOI:10.1186/s12859-019-2713-9URL [本文引用: 1]
DOI:10.1038/s41467-017-02292-8PMID:29259172 [本文引用: 3]
While prokaryotic pan-genomes have been shown to contain many more genes than any individual organism, the prevalence and functional significance of differentially present genes in eukaryotes remains poorly understood. Whole-genome de novo assembly and annotation of 54 lines of the grass Brachypodium distachyon yield a pan-genome containing nearly twice the number of genes found in any individual genome. Genes present in all lines are enriched for essential biological functions, while genes present in only some lines are enriched for conditionally beneficial functions (e.g., defense and development), display faster evolutionary rates, lie closer to transposable elements and are less likely to be syntenic with orthologous genes in other grasses. Our data suggest that differentially present genes contribute substantially to phenotypic variation within a eukaryote species, these genes have a major influence in population genetics, and transposable elements play a key role in pan-genome evolution.
DOI:10.1093/gbe/evx270URL
[本文引用: 1]
DOI:10.1093/gbe/evq048PMID:20688752 [本文引用: 1]
Defining bacterial species and understanding the relative cohesiveness of different components of their genomes remains a fundamental problem in microbiology. Bacterial species tend to be comprised of both a set of core and dispensable genes, with the sum of these two components forming the species pan-genome. The role of the core and dispensable genes in defining bacterial species and the question of whether pan-genomes are finite or infinite remain unclear. Here we demonstrate, through the analysis of 96 genome sequences derived from two closely related sympatric sister species of pathogenic bacteria (Campylobacter coli and C. jejuni), that their pan-genome is indeed finite and that there are unique and cohesive features to each of their genomes defining their genomic identity. The two species have a similar pan-genome size; however, C. coli has acquired a larger core genome and each species has evolved a number of species-specific core genes, possibly reflecting different adaptive strategies. Genome-wide assessment of the level of lateral gene transfer within and between the two sister species, as well as within the core and non-core genes, demonstrates a resistance to interspecies recombination in the core genome of the two species and therefore provides persuasive support for the core genome hypothesis for bacterial species.
DOI:10.1016/j.nmni.2015.06.005URL [本文引用: 2]
[本文引用: 3]
DOI:10.1038/s41588-018-0273-yPMID:30455414 [本文引用: 3]
We used a deeply sequenced dataset of 910 individuals, all of African descent, to construct a set of DNA sequences that is present in these individuals but missing from the reference human genome. We aligned 1.19 trillion reads from the 910 individuals to the reference genome (GRCh38), collected all reads that failed to align, and assembled these reads into contiguous sequences (contigs). We then compared all contigs to one another to identify a set of unique sequences representing regions of the African pan-genome missing from the reference genome. Our analysis revealed 296,485,284?bp in 125,715 distinct contigs present in the populations of African descent, demonstrating that the African pan-genome contains ~10% more DNA than the current human reference genome. Although the functional significance of nearly all of this sequence is unknown, 387 of the novel contigs fall within 315 distinct protein-coding genes, and the rest appear to be intergenic.
DOI:10.1038/s41477-020-0733-0URL [本文引用: 4]
DOI:10.1186/s12915-014-0066-4URL [本文引用: 2]
[本文引用: 1]
DOI:10.1111/tpj.2017.90.issue-5URL [本文引用: 2]
DOI:10.1038/s41576-020-0210-7PMID:32034321 [本文引用: 2]
Since the early days of the genome era, the scientific community has relied on a single 'reference' genome for each species, which is used as the basis for a wide range of genetic analyses, including studies of variation within and across species. As sequencing costs have dropped, thousands of new genomes have been sequenced, and scientists have come to realize that a single reference genome is inadequate for many purposes. By sampling a diverse set of individuals, one can begin to assemble a pan-genome: a collection of all the DNA sequences that occur in a species. Here we review efforts to create pan-genomes for a range of species, from bacteria to humans, and we further consider the computational methods that have been proposed in order to capture, interpret and compare pan-genome data. As scientists continue to survey and catalogue the genomic variation across human populations and begin to assemble a human pan-genome, these efforts will increase our power to connect variation to human diversity, disease and beyond.
DOI:10.1186/s13059-020-02168-zURL [本文引用: 3]
DOI:10.1146/genom.2020.21.issue-1URL [本文引用: 2]
DOI:10.1038/nbt.4227PMID:30125266 [本文引用: 1]
Reference genomes guide our interpretation of DNA sequence data. However, conventional linear references represent only one version of each locus, ignoring variation in the population. Poor representation of an individual's genome sequence impacts read mapping and introduces bias. Variation graphs are bidirected DNA sequence graphs that compactly represent genetic variation across a population, including large-scale structural variation such as inversions and duplications. Previous graph genome software implementations have been limited by scalability or topological constraints. Here we present vg, a toolkit of computational methods for creating, manipulating, and using these structures as references at the scale of the human genome. vg provides an efficient approach to mapping reads onto arbitrary variation graphs using generalized compressed suffix arrays, with improved accuracy over alignment to a linear reference, and effectively removing reference bias. These capabilities make using variation graphs as references for DNA sequencing practical at a gigabase scale, or at the topological complexity of de novo assemblies.
DOI:10.1038/s41467-019-13341-9PMID:31776332 [本文引用: 1]
Analysis of sequence diversity in the human genome is fundamental for genetic studies. Structural variants (SVs) are frequently omitted in sequence analysis studies, although each has a relatively large impact on the genome. Here, we present GraphTyper2, which uses pangenome graphs to genotype SVs and small variants using short-reads. Comparison to the syndip benchmark dataset shows that our SV genotyping is sensitive and variant segregation in families demonstrates the accuracy of our approach. We demonstrate that incorporating public assembly data into our pipeline greatly improves sensitivity, particularly for large insertions. We validate 6,812 SVs on average per genome using long-read data of 41 Icelanders. We show that GraphTyper2 can simultaneously genotype tens of thousands of whole-genomes by characterizing 60 million small variants and half a million SVs in 49,962 Icelanders, including 80 thousand SVs with high-confidence.
DOI:10.1016/j.cell.2020.05.023URL [本文引用: 4]
DOI:10.1016/j.cell.2021.04.046URL [本文引用: 3]
DOI:10.1186/s13059-020-02105-0PMID:32718320 [本文引用: 3]
The current bovine genomic reference sequence was assembled from a Hereford cow. The resulting linear assembly lacks diversity because it does not contain allelic variation, a drawback of linear references that causes reference allele bias. High nucleotide diversity and the separation of individuals by hundreds of breeds make cattle ideally suited to investigate the optimal composition of variation-aware references.We augment the bovine linear reference sequence (ARS-UCD1.2) with variants filtered for allele frequency in dairy (Brown Swiss, Holstein) and dual-purpose (Fleckvieh, Original Braunvieh) cattle breeds to construct either breed-specific or pan-genome reference graphs using the vg toolkit. We find that read mapping is more accurate to variation-aware than linear references if pre-selected variants are used to construct the genome graphs. Graphs that contain random variants do not improve read mapping over the linear reference sequence. Breed-specific augmented and pan-genome graphs enable almost similar mapping accuracy improvements over the linear reference. We construct a whole-genome graph that contains the Hereford-based reference sequence and 14 million alleles that have alternate allele frequency greater than 0.03 in the Brown Swiss cattle breed. Our novel variation-aware reference facilitates accurate read mapping and unbiased sequence variant genotyping for SNPs and Indels.We develop the first variation-aware reference graph for an agricultural animal ( https://doi.org/10.5281/zenodo.3759712 ). Our novel reference structure improves sequence read mapping and variant genotyping over the linear reference. Our work is a first step towards the transition from linear to variation-aware reference structures in species with high genetic diversity and many sub-populations.
DOI:10.1016/j.tim.2013.01.002PMID:23419217 [本文引用: 1]
Bacteria inhabit enormously diverse niches and have a correspondingly large array of regulatory mechanisms to adapt to often inhospitable and variable environments. The stringent response (SR) allows bacteria to quickly reprogram transcription in response to changes in nutrient availability. Although the proteins controlling this response are conserved in almost all bacterial species, recent work has illuminated considerable diversity in the starvation cues and regulatory mechanisms that activate stringent signaling proteins in bacteria from different environments. In this review, we describe the signals and genetic circuitries that control the stringent signaling systems of a copiotroph, a bacteriovore, an oligotroph, and a mammalian pathogen -Escherichia coli, Myxococcus xanthus, Caulobacter crescentus, and Mycobacterium tuberculosis, respectively - and discuss how control of the SR in these species is adapted to their particular lifestyles.Copyright © 2013 Elsevier Ltd. All rights reserved.
DOI:10.1038/nrg3962URL [本文引用: 1]
DOI:10.1186/gb-2007-8-5-r71URL [本文引用: 1]
DOI:10.1111/emi.2013.15.issue-12URL [本文引用: 1]
DOI:10.1186/s12864-015-1968-4URL [本文引用: 1]
DOI:10.1093/gbe/evy259PMID:30496396 [本文引用: 1]
The huge increase in the availability of bacterial genomes led us to a point in which we can investigate and query pan-genomes, for example, the full set of genes of a given bacterial species or clade. Here, we used a data set of 1,311 high-quality genomes from the human pathogen Pseudomonas aeruginosa, 619 of which were newly sequenced, to show that a pan-genomic approach can greatly refine the population structure of bacterial species, provide new insights to define species boundaries, and generate hypotheses on the evolution of pathogenicity. The 665-gene P. aeruginosa core genome presented here, which constitutes only 1% of the entire pan-genome, is the first to be in the same order of magnitude as the minimal bacterial genome and represents a conservative estimate of the actual core genome. Moreover, the phylogeny based on this core genome provides strong evidence for a five-group population structure that includes two previously undescribed groups of isolates. Comparative genomics focusing on antimicrobial resistance and virulence genes showed that variation among isolates was partly linked to this population structure. Finally, we hypothesized that horizontal gene transfer had an important role in this respect, and found a total of 3,010 putative complete and fragmented plasmids, 5% and 12% of which contained resistance or virulence genes, respectively. This work provides data and strategies to study the evolutionary trajectories of resistance and virulence in P. aeruginosa.
DOI:10.1093/gbe/evw001URL [本文引用: 1]
DOI:10.1016/j.micpath.2020.104275URL [本文引用: 2]
[本文引用: 1]
DOI:10.1016/j.mib.2014.11.016PMID:25483351 [本文引用: 1]
Next generation sequencing technologies have engendered a genome sequence data deluge in public databases. Genome analyses have transitioned from single or few genomes to hundreds to thousands of genomes. Pan-genome analyses provide a framework for estimating the genomic diversity of the dataset at hand and predicting the number of additional whole genomes sequences that would be necessary to fully characterize that diversity. We review recent implementations of the pan-genome approach, its impact and limits, and we propose possible extensions, including analyses at the whole genome multiple sequence alignment level. Copyright © 2014 Elsevier Ltd. All rights reserved.
[本文引用: 1]
[本文引用: 1]
DOI:10.1111/pbi.12499URL [本文引用: 2]
PMID:17300983 [本文引用: 2]
The comparative sequencing of several grass genomes has revealed that transposable elements are largely responsible for extensive variation in both intergenic and local genic content, not only between closely related species but also among individuals within a species. These observations indicate that a single genome sequence might not reflect the entire genomic complement of a species, and prompted us to introduce the concept of the plant pan-genome, which includes core genomic features that are common to all individuals and a dispensable genome composed of partially shared and/or non-shared DNA sequence elements. Uncovering the intriguing nature of the dispensable genome, namely its composition, origin and function, represents a step forward towards an understanding of the processes that generate genetic diversity and phenotypic variation. The developing view of transcriptional regulation as a complex and modular system, in which long-range interactions and the involvement of transposable elements are frequently observed, lends support to the possibility of an important functional role for the dispensable genome and could make it less dispensable than previously thought.
DOI:10.1038/nbt.2979 [本文引用: 3]
Wild relatives of crops are an important source of genetic diversity for agriculture, but their gene repertoire remains largely unexplored. We report the establishment and analysis of a pan-genome of Glycine soja, the wild relative of cultivated soybean Glycine max, by sequencing and de novo assembly of seven phylogenetically and geographically representative accessions. Intergenomic comparisons identified lineage-specific genes and genes with copy number variation or large-effect mutations, some of which show evidence of positive selection and may contribute to variation of agronomic traits such as biotic resistance, seed composition, flowering and maturity time, organ size and final biomass. Approximately 80% of the pan-genome was present in all seven accessions (core), whereas the rest was dispensable and exhibited greater variation than the core genome, perhaps reflecting a role in adaptation to diverse environments. This work will facilitate the harnessing of untapped genetic diversity from wild soybean for enhancement of elite cultivars.
DOI:10.1038/nature08516URL [本文引用: 1]
DOI:10.1007/s11427-020-1808-0URL [本文引用: 1]
DOI:10.1016/j.cell.2020.05.021URL [本文引用: 2]
DOI:10.1105/tpc.113.119982URL [本文引用: 2]
DOI:10.1111/nph.15413URL [本文引用: 1]
DOI:10.1038/s41586-020-2961-xURL [本文引用: 1]
DOI:10.1038/ncomms13390URL [本文引用: 2]
DOI:10.1038/s41588-018-0041-zURL [本文引用: 1]
DOI:10.1038/s41587-020-0681-2PMID:33020633 [本文引用: 1]
Wild and weedy relatives of domesticated crops harbor genetic variants that can advance agricultural biotechnology. Here we provide a genome resource for the wild plant green millet (Setaria viridis), a model species for studies of C grasses, and use the resource to probe domestication genes in the close crop relative foxtail millet (Setaria italica). We produced a platinum-quality genome assembly of S. viridis and de novo assemblies for 598 wild accessions and exploited these assemblies to identify loci underlying three traits: response to climate, a 'loss of shattering' trait that permits mechanical harvest and leaf angle, a predictor of yield in many grass crops. With CRISPR-Cas9 genome editing, we validated Less Shattering1 (SvLes1) as a gene whose product controls seed shattering. In S. italica, this gene was rendered nonfunctional by a retrotransposon insertion in the domesticated loss-of-shattering allele SiLes1-TE (transposable element). This resource will enhance the utility of S. viridis for dissection of complex traits and biotechnological improvement of panicoid crops.
DOI:10.1038/s41477-018-0329-0URL [本文引用: 1]
DOI:10.1186/s12864-018-5357-7URL [本文引用: 1]
DOI:10.1038/s41586-020-2947-8URL [本文引用: 1]
DOI:10.1038/s41477-021-00925-xURL [本文引用: 1]
[本文引用: 1]
DOI:10.1038/s41586-018-0030-5URL [本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.pbi.2020.04.009URL [本文引用: 1]
DOI:10.1186/s12915-017-0457-4PMID:29325559 [本文引用: 1]
Background: Structural variation contributes substantially to polymorphism within species. Chromosomal rearrangements that impact genes can lead to functional variation among individuals and influence the expression of phenotypic traits. Genomes of fungal pathogens show substantial chromosomal polymorphism that can drive virulence evolution on host plants. Assessing the adaptive significance of structural variation is challenging, because most studies rely on inferences based on a single reference genome sequence.Results: We constructed and analyzed the pangenome of Zymoseptoria tritici, a major pathogen of wheat that evolved host specialization by chromosomal rearrangements and gene deletions. We used single-molecule real-time sequencing and high-density genetic maps to assemble multiple genomes. We annotated the gene space based on transcriptomics data that covered the infection life cycle of each strain. Based on a total of five telomere-to-telomere genomes, we constructed a pangenome for the species and identified a core set of 9149 genes. However, an additional 6600 genes were exclusive to a subset of the isolates. The substantial accessory genome encoded on average fewer expressed genes but a larger fraction of the candidate effector genes that may interact with the host during infection. We expanded our analyses of the pangenome to a worldwide collection of 123 isolates of the same species. We confirmed that accessory genes were indeed more likely to show deletion polymorphisms and loss-of-function mutations compared to core genes.Conclusions: The pangenome construction of a highly polymorphic eukaryotic pathogen showed that a single reference genome significantly underestimates the gene space of a species. The substantial accessory genome provides a cradle for adaptive evolution.
DOI:10.1186/s12915-020-0744-3URL [本文引用: 1]
DOI:10.1038/ng.3801 [本文引用: 1]
Genomes usually contain some non-repetitive sequences that are missing from the reference genome and occur only in a population subset. Such non-repetitive, non-reference (NRNR) sequences have remained largely unexplored in terms of their characterization and downstream analyses. Here we describe 3,791 breakpoint-resolved NRNR sequence variants called using PopIns from whole-genome sequence data of 15,219 Icelanders. We found that over 95% of the 244 NRNR sequences that are 200 bp or longer are present in chimpanzees, indicating that they are ancestral. Furthermore, 149 variant loci are in linkage disequilibrium (r(2) > 0.8) with a genome-wide association study (GWAS) catalog marker, suggesting disease relevance. Additionally, we report an association (P = 3.8 x 10(-8), odds ratio (OR) = 0.92) with myocardial infarction (23,360 cases, 300,771 controls) for a 766-bp NRNR sequence variant. Our results underline the importance of including variation of all complexity levels when searching for variants that associate with disease.
DOI:10.1186/s13059-019-1751-yURL [本文引用: 1]
DOI:10.1093/molbev/msz176PMID:31560401 [本文引用: 1]
Novel sequences (NSs), not present in the human reference genome, are abundant and remain largely unexplored. Here, we utilize de novo assembly to study NS in 1,000 Swedish individuals first sequenced as part of the SweGen project revealing a total of 46 Mb in 61,044 distinct contigs of sequences not present in GRCh38. The contigs were aligned to recently published catalogs of Icelandic and Pan-African NSs, as well as the chimpanzee genome, revealing a great diversity of shared sequences. Analyzing the positioning of NS across the chimpanzee genome, we find that 2,807 NS align confidently within 143 chimpanzee orthologs of human genes. Aligning the whole genome sequencing data to the chimpanzee genome, we discover ancestral NS common throughout the Swedish population. The NSs were searched for repeats and repeat elements: revealing a majority of repetitive sequence (56%), and enrichment of simple repeats (28%) and satellites (15%). Lastly, we align the unmappable reads of a subset of the thousand genomes data to our collection of NS, as well as the previously published Pan-African NS: revealing that both the Swedish and Pan-African NS are widespread, and that the Swedish NSs are largely a subset of the Pan-African NS. Overall, these results highlight the importance of creating a more diverse reference genome and illustrate that significant amounts of the NS may be of ancestral origin.© The Author(s) 2019. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
PMID:17384736 [本文引用: 1]
This review is a short update on the diversity of swine biomedical models and the importance of genomics in their continued development. The swine has been used as a major mammalian model for human studies because of the similarity in size and physiology, and in organ development and disease progression. The pig model allows for deliberately timed studies, imaging of internal vessels and organs using standard human technologies, and collection of repeated peripheral samples and, at kill, detailed mucosal tissues. The ability to use pigs from the same litter, or cloned or transgenic pigs, facilitates comparative analyses and genetic mapping. The availability of numerous well defined cell lines, representing a broad range of tissues, further facilitates testing of gene expression, drug susceptibility, etc. Thus the pig is an excellent biomedical model for humans. For genomic applications it is an asset that the pig genome has high sequence and chromosome structure homology with humans. With the swine genome sequence now well advanced there are improving genetic and proteomic tools for these comparative analyses. The review will discuss some of the genomic approaches used to probe these models. The review will highlight genomic studies of melanoma and of infectious disease resistance, discussing issues to consider in designing such studies. It will end with a short discussion of the potential for genomic approaches to develop new alternatives for control of the most economically important disease of pigs, porcine reproductive and respiratory syndrome (PRRS), and the potential for applying knowledge gained with this virus for human viral infectious disease studies.
DOI:10.3389/fgene.2019.01169URL [本文引用: 2]
DOI:10.1186/s13059-020-02180-3PMID:33168033 [本文引用: 3]
The Mediterranean mussel Mytilus galloprovincialis is an ecologically and economically relevant edible marine bivalve, highly invasive and resilient to biotic and abiotic stressors causing recurrent massive mortalities in other bivalves. Although these traits have been recently linked with the maintenance of a high genetic variation within natural populations, the factors underlying the evolutionary success of this species remain unclear.Here, after the assembly of a 1.28-Gb reference genome and the resequencing of 14 individuals from two independent populations, we reveal a complex pan-genomic architecture in M. galloprovincialis, with a core set of 45,000 genes plus a strikingly high number of dispensable genes (20,000) subject to presence-absence variation, which may be entirely missing in several individuals. We show that dispensable genes are associated with hemizygous genomic regions affected by structural variants, which overall account for nearly 580?Mb of DNA sequence not included in the reference genome assembly. As such, this is the first study to report the widespread occurrence of gene presence-absence variation at a whole-genome scale in the animal kingdom.Dispensable genes usually belong to young and recently expanded gene families enriched in survival functions, which might be the key to explain the resilience and invasiveness of this species. This unique pan-genome architecture is characterized by dispensable genes in accessory genomic regions that exceed by orders of magnitude those observed in other metazoans, including humans, and closely mirror the open pan-genomes found in prokaryotes and in a few non-metazoan eukaryotes.
DOI:S0092-8674(20)30931-4PMID:32814014 [本文引用: 1]
Among arthropod vectors, ticks transmit the most diverse human and animal pathogens, leading to an increasing number of new challenges worldwide. Here we sequenced and assembled high-quality genomes of six ixodid tick species and further resequenced 678 tick specimens to understand three key aspects of ticks: genetic diversity, population structure, and pathogen distribution. We explored the genetic basis common to ticks, including heme and hemoglobin digestion, iron metabolism, and reactive oxygen species, and unveiled for the first time that genetic structure and pathogen composition in different tick species are mainly shaped by ecological and geographic factors. We further identified species-specific determinants associated with different host ranges, life cycles, and distributions. The findings of this study are an invaluable resource for research and control of ticks and tick-borne diseases.Copyright © 2020 Elsevier Inc. All rights reserved.
DOI:10.1093/molbev/msaa240URL [本文引用: 1]
DOI:10.1038/s41576-018-0003-4PMID:29599501 [本文引用: 1]
Several new genomics technologies have become available that offer long-read sequencing or long-range mapping with higher throughput and higher resolution analysis than ever before. These long-range technologies are rapidly advancing the field with improved reference genomes, more comprehensive variant identification and more complete views of transcriptomes and epigenomes. However, they also require new bioinformatics approaches to take full advantage of their unique characteristics while overcoming their complex errors and modalities. Here, we discuss several of the most important applications of the new technologies, focusing on both the currently available bioinformatics tools and opportunities for future research.
[本文引用: 1]
DOI:10.1038/nrg2958PMID:21358748 [本文引用: 1]
Comparisons of human genomes show that more base pairs are altered as a result of structural variation - including copy number variation - than as a result of point mutations. Here we review advances and challenges in the discovery and genotyping of structural variation. The recent application of massively parallel sequencing methods has complemented microarray-based methods and has led to an exponential increase in the discovery of smaller structural-variation events. Some global discovery biases remain, but the integration of experimental and computational approaches is proving fruitful for accurate characterization of the copy, content and structure of variable regions. We argue that the long-term goal should be routine, cost-effective and high quality de novo assembly of human genomes to comprehensively assess all classes of structural variation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}