 ,1, 朱化星,2,*
,1, 朱化星,2,*Optimization of CUT&Tag product recovery and library construction method
Ye Wei1,2, Ke Li2, Daru Lu,1, Huaxing Zhu,2,*通讯作者: 卢大儒,博士,教授,研究方向:遗传学。E-mail:drlu@fudan.edu.cn;朱化星,博士,高级工程师,研究方向:遗传学。E-mail:zhuhuaxing@novoprotein.com.cn
编委: 孟卓贤
收稿日期:2021-01-15修回日期:2021-03-4网络出版日期:2021-04-16
| 基金资助: |
Received:2021-01-15Revised:2021-03-4Online:2021-04-16
| Fund supported: |
作者简介 About authors
韦晔,在读硕士研究生;专业方向:生物工程。E-mail:

摘要
新兴的染色质靶向切割和标签化(clevage under target and tagment, CUT&Tag)技术利用转座酶在目标蛋白结合的DNA附近进行切割并对切割下的DNA片段进行标签化,通过后续的二代测序可以快速鉴定蛋白质- DNA相互作用,极大的简化了染色质免疫共沉淀测序(chromatin immunoprecipitation sequencing, ChIP-seq)的实验过程。CUT&Tag中转座酶完成标签化后需要DNA回收或其他后处理才能进行建库PCR,不同的回收方法对CUT&Tag结果有着显著的影响。通过建立生物素化转座体-链霉亲和素磁珠体系(streptavidin beads recovery CUT&Tag, srCUT&Tag),可以快速便捷地完成CUT&Tag的产物回收。本文在K562细胞中展开H3K4me3、RNA聚合酶II (RNA polymerase II, RNAPII)、转录因子CTCF和HMGA1的CUT&Tag实验,并利用现有的乙醇沉淀、片段分选(solid-phase reversible immobilization, SPRI)磁珠回收和直接PCR法,以及本研究建立的srCUT&Tag方法对产物进行回收。结果表明,从整体上看,SPRI磁珠回收和srCUT&Tag方法着较高的回收效率,而乙醇沉淀法则回收效率低下。在全部4种CUT&Tag产物回收过程中,SPRI磁珠回收均会损失大部分小于150 bp的产物片段。在CTCF和HMGA1 CUT&Tag产物的回收中,直接PCR法则损失了大部分大于300 bp的片段并与其他回收方法的结果有较大的差别。因此,srCUT&Tag能够比其他三种回收方法提供更多更完整的测序信息。综上所述,新建立srCUT&Tag回收方法相比现有的CUT&Tag产物回收方法能提高建库效率并得到更好的数据质量,为表观遗传学研究提供了更好的技术选择。
关键词:
Abstract
The emerging cleavage under target and tagment (CUT&Tag) technology uses Tn5 transposase to cleavage near the DNA binding site of target protein and study the generated DNA fragments by the next-generation sequencing. It can quickly identify protein-DNA interactions, which greatly simplifies the experimental process of ChIP-Seq. After CUT&Tag tagment reaction, DNA recovery or other post-processing is required to perform library construction PCR. Different recovery methods have significant impact respectively. By establishing Streptavidin beads recovery CUT&Tag(srCUT&Tag), we can quickly and conveniently complete the product recovery of CUT&Tag. We carried out CUT&Tag assay of H3K4me3, RNA Polymerase II (RNA polymerase II, RNAPII), transcription factor CTCF and HMGA1 in K562 cells with different recovery methods, including ethanol precipitation, fragment separation magnetic beads (SPRI) Magnetic bead recovery, direct PCR method, as well as our srCUT&Tag recovery method. The results show that among the CUT&Tag results of four different targets, the SPRI magnetic bead recovery and our srCUT&Tag methods have higher recovery efficiency than the direct PCR method and ethanol precipitation method. All CUT&Tag results showed that the recovery of SPRI magnetic beads would lose most of the product fragments less than 150 bp. In the recovery of CTCF and HMGA1, direct PCR lost most of the fragments larger than 300 bp and has significant difference from result of other recovery method. This enables srCUT&Tag to provide more real and higher-resolution information than other recovery method. In summary, the newly established srCUT&Tag recovery method can improve the efficiency of CUT&Tag library construction and obtain better data quality compared with the existing CUT&Tag product recovery method, providing a better technical choice for epigenetics research.
Keywords:
PDF (2388KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
韦晔, 李科, 卢大儒, 朱化星. CUT&Tag产物回收和建库方法的优化. 遗传[J], 2021, 43(4): 362-374 doi:10.16288/j.yczz.21-016
Ye Wei.
染色质免疫共沉淀测序(chromatin immunoprecipitation sequencing, ChIP-seq)是经典的蛋白质-染色质相互作用研究方法[1,2]。传统的染色质免疫共沉淀操作繁琐,背景较高,需要很大的测序量,实验可控性差。这些限制了其在低起始细胞量上的使用[3]。CUT&Tag是近年兴起的一种新型蛋白质-染色质相互作用研究方法[4,5,6],该方法利用Protein A/Protein G融合的Tn5转座子,通过Protein A/G-抗体相互作用将转座体栓系在靶抗体周围的区域,从而实现目标特异性的片段化反应。相比于传统的ChIP-seq,CUT&Tag操作相对简单,无需甲醛交联,信噪比较高,所需测序量少,且适用于极低起始量和单细胞的应用场景[7,8]。CUT&Tag的主要流程包括:(1)将细胞固定在ConA磁珠上;(2)利用表面活性剂digitonin透化细胞,先后加入一抗和二抗进行孵育;(3)洗去多余的抗体后,加入融合ProteinA/G的转座体使其与靶位点附近的抗体结合;(4)洗去多余的转座体,加入镁离子,在37℃的条件下进行片段化反应;(5)加入SDS终止反应,通过乙醇沉淀或其他方法将酶切产生的DNA片段;(6) PCR建库及二代测序[9]。在CUT&Tag实验中,高效的CUT&Tag产物DNA回收对获取良好的CUT&Tag结果至关重要,本文主要针对CUT&Tag产物DNA提取方法进行讨论。
由于CUT&Tag所使用的转座体需要用SDS失活,体系中存在的SDS则会干扰后续的PCR反应,因此必须进行DNA的提取或利用其他方法消除SDS的干扰。早期的CUT&Tag实验方法中,CUT&Tag产物DNA提取方法主要包括酚氯仿抽提-乙醇沉淀(后文简称乙醇沉淀)或片段分选磁珠(固相可逆固定磁珠,即solid-phase reversible immobilization, SPRI,后文简称SPRI磁珠)磁珠回收。酚氯仿抽提-乙醇沉淀是DNA回收的传统方法,其优点是原理简单,且没有明显的偏好性。缺陷则是操作相对繁琐,给回收操作带来了显著困难。特别是在低细胞起始量的CUT&Tag实验中,很容易造成产物DNA损失,增加了CUT&Tag实验的难度。SPRI磁珠回收相对于乙醇沉淀耗时较短,回收效率高,操作难度低。但是目前市售的SPRI磁珠是为进行二代测序文库构建的片段分选而设计,不适用于回收150 bp以下的小片段。因此,SPRI磁珠回收所得到的文库可能具有偏好性,有损失相应信息的风险。离心柱回收是另一种常见的DNA回收方法并且被广泛用于CUT&Tag的前身CUT&RUN[10]的产物回收中。虽然高质量的离心柱往往成本较高,但是离心柱法可以高效回收微量核酸片段且不具有明显的选择偏好。除此之外,CUT&Tag技术的提出者Henikoff在近期也开发了一种不需要回收直接进行一管式建库的流程:控制SDS终止液的体积,并利用Triton X-100中和SDS后进行直接PCR。这种方法特别适用于以细胞核为底物的CUT&Tag,并且已经被报道其回收效率较传统的提取法更高[11,12]。
为改善CUT&Tag实验中DNA回收效率,本研究建立了一种新的CUT&Tag实验流程(图1),通过生物素化CUT&Tag中所用的pAG-Tn5转座体接头末端,使产生的生物素化的CUT&Tag产物DNA能够被链霉亲和素磁珠所结合并回收。链霉亲和素磁珠仅需要20 min的室温孵育时间就能完成和生物素化CUT&Tag产物的结合,洗涤后可直接PCR制备NGS文库。为比较现有不同CUT&Tag产物回收方法的优劣,本研究选取了甲基化组蛋白H3K4me3,RNAPII,常见转录因子CTCF和HMGA1。用102~105 K562细胞进行CUT&Tag实验。使用相同的起始细胞量,不同的回收方法,比较其回收效率,片段选择性和整体的CUT&Tag数据结果,寻找能够获得最优的CUT&Tag的数据信息的实验流程。
1 材料与方法
1.1 耗材及试剂
胎牛血清FBS、RPMI 1640培养基、磷酸盐缓冲液(PBS)购自Gibco公司;NovoNGS? CUT&Tag 2.0 High-Sensitivity Kit (for Illumina?) (N259)由近岸蛋白质科技有限公司保存;抗体Histone H3K4me3 antibody (pAb) (AB_2615077)购自Active Motif公司;抗体Anti-RNA polymerase II[CTD 4H8] (Ab00832-1.1)购自Absolute Antibody公司;抗体CTCF(D31H2) XP?Rabbit mAb(#3418)购自CST公司;抗体Anti- HMGA1 [EPR7839](ab129153)购自Abcam公司;抗体Rabbit Anti-Mouse IgG H&L (ab6709)购自Abcam公司;酚∶氯仿∶异戊醇=25∶24∶1(PCI)(P1011)购自北京索莱宝科技有限公司;Sreptavidin magnetic beads(S1420S)购自NEB公司;链霉亲和素磁珠购自上海英莱盾生物技术有限公司。图1
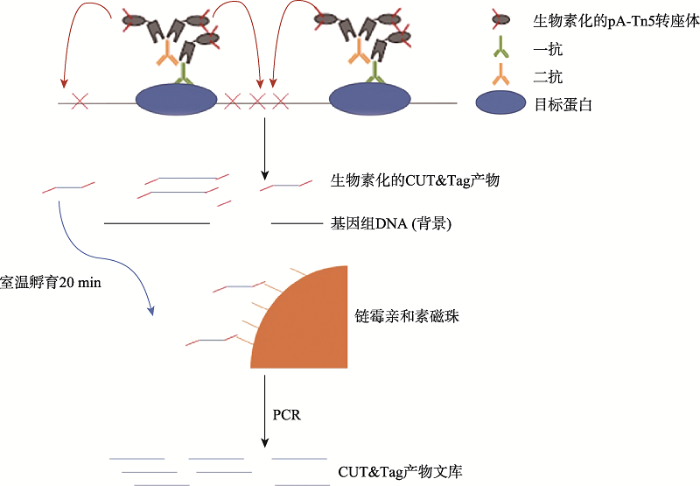 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1srCUT&Tag系统示意图
Fig. 1Overview of srCUT&Tag system
1.2 仪器设备
细胞超净台、细胞培养箱、台式低温离心机购自、PCR仪(Veriti 96)、紫外分光光度计(Nanodrop)为Thermo Fisher公司产品;涡旋振荡仪(Vertex-5)、旋转混合仪(QB-228)购自海门其林贝尔仪器制造有限公司;金属浴加热器(TU-100)及电热恒温培养箱(DHP-9052)购自上海一恒科学仪器公司。1.3 细胞收集
K562细胞复苏后于10 mL含有10% FBS的RPMI 1640培养基中,放入37℃、5%CO2浓度的细胞培养箱中。隔天按照1∶2进行传代培养,细胞浓度稳定在0.5×106~1.0×106/mL。以保持实验细胞状态的稳定。室温下600× g离心收集新鲜细胞并计数,取所需数量的细胞于新的1.5 mL EP管中,室温下600× g离心5 min,小心吸去上清;加入等体积Wash Buffer (Novoprotein, N259)清洗一遍后重悬,根据需要使得每个反应的细胞浓度为102~105/100 μL。1.4 CUT&Tag实验
CUT&Tag实验按照NovoNGS? CUT&Tag 2.0 High-Sensitivity Kit (for Illumina?)(N259)说明书上的标准流程进行。孵育一抗时一抗稀释比为1∶100,孵育二抗时二抗稀释比为1∶200,每个反应转座体用量为1 μL最终的标签化步骤反应体积为100 μL。1.5 DNA的回收和建库
(a)对于乙醇沉淀回收的样品,向每份CUT&Tag产物中加入3 μL 10% SDS,10 μL 0.5 mol/L EDTA,和2.5 μL 20 mg/mL Proteinase K,涡旋振荡混匀后,50℃消化1 h;每管样品中加入300 μL酚氯仿混合物,涡旋振荡充分混匀后移至锁相管中,16000× g室温离心5 min。向每管样品中加入100 μL氯仿,涡旋振荡充分混匀后,16000× g室温离心5 min,吸取水相至新的1.5 mL EP管,加入1 mL 100%乙醇,用移液器吹打混匀;将样品管置于-20℃静置1 h, 4℃离心20 min,离心产物小心地倒出液体并在纸巾上沥干,向每管样品中分别加入1 mL 80%乙醇漂洗,4℃下16000× g离心1 min,小心地倒出液体并在纸巾上沥去水珠后自然干燥。干燥后,向每管样品中加入35 μL TE-RA-Buffer,吹打重悬溶解DNA沉淀。(b)对于SPRI磁珠回收的样品,向每份CUT&Tag产物中加入2 μL 10% SDS,55℃加热10 min,然后向每管中加入200 μL在室温平衡后的磁珠,吹打混匀,室温静置5 min,磁珠结合后置于磁力架上静置5 min,移去上清,80%乙醇洗涤2遍,室温晾干。35 μL TE buffer洗脱,回收后的CUT&Tag产物在 -20℃下储存或直接进行下一步PCR扩增。
(c)对于srCUT&Tag回收的样品,向每份CUT&Tag产物中加入2 μL 10% SDS,55℃加热10 min,然后向每管中加入1 μL洗涤后按照1∶1比例复溶的的链霉亲和素磁珠,吹打混匀,室温静置20 min,磁珠结合后置于磁力架上静置2 min,移去上清,1×Wash buffer洗2遍,室温晾干。35 μL TE buffer复溶,回收后的CUT&Tag产物在-20℃下储存或直接进行下一步PCR扩增。
(d)对于直接PCR的样品,在CUT&Tag实验结束后移去片段化溶液上清,利用100 μL TAPs Wash buffer (10 mmol/L TAPs, 0.2 mmol/L EDTA)清洗样品,加入5 μL 0.2%SDS,70℃加热1 h。失活后的体系加入15 μL 0.67% Triton X-100中和SDS。直接进行下一步PCR扩增。
每组回收后的CUT&Tag产物按照NovoNGS? CUT&Tag 2.0 High-Sensitivity Kit (for Illumina?)的流程建库并进行二代测序。
1.6 生物信息学分析
用FastQC和MulitiQC检查文库的质量;用Cutadaptor进行接头序列的去除和数据清洗;用Bowtie2进行比对。用Samtools进行过滤和去重;用Macs2进行识别峰;用IGV进行数据可视化。用Deeptoools绘制插入片段大小图,不同实验组数据相关性图。2 结果与分析
2.1 srCUT&Tag回收体系中链霉亲和素用量的选择
为了确定回收使用链霉亲和素磁珠的用量和回收后直接PCR的可行性,使用细胞数为1×104的K562细胞进行H3K4me3的CUT&Tag实验。不同的平行实验组分别用0.2 μL,0.5 μL,1 μL,2 μL,3 μL,4 μL洗涤后的NEB streptavidin magnetic beads和英莱顿的链霉亲和素磁珠进行回收。产物按照NovoNGS? CUT&Tag 2.0 High-Sensitivity Kit (for Illumina?)建库的流程进行14个循环的PCR,利用琼脂糖凝胶电泳检验回收的片段分布(图2)。对于NEB的磁珠,0.2~4.0 μL均可得到预期结果类似的PCR条带。而对于浓度稍高的英莱盾的磁珠,当链霉亲和素磁珠用量为0.1~2.0 μL时,所得到的的PCR产物大小分布与预期结果类似。当磁珠用量大于2 μL时,PCR条带的扩增水平降低;4 μL磁珠的回收产物则无法扩增出PCR条带。实验结果说明当链霉亲和素磁珠浓度较大时,有可能会抑制后续的PCR扩增,从而对建库过程造成干扰。在本文后续的研究中,选择1 μL的英莱盾链霉亲和素磁珠作为srCUT&Tag所使用的链霉亲和素磁珠用量。图2
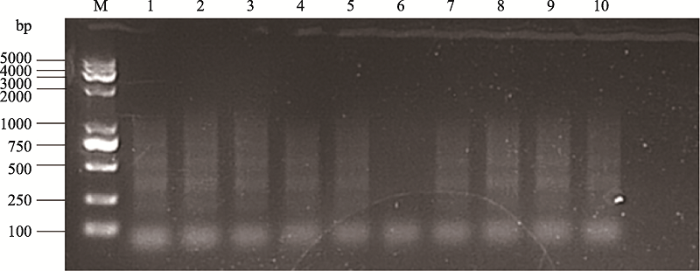 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2不同链霉亲和素磁珠及不同用量回收效果测试
M:DNA ladder 2000 plus;1:4 μL NEB磁珠;2:2 μL NEB磁珠;3:1 μL NEB磁珠;4:0.5 μL NEB磁珠;5:0.2 μL NEB磁珠;6:4 μL 英莱盾磁珠;7:2 μL 英莱盾磁珠;8:1 μL英莱盾磁珠;9:0.5 μL英莱盾磁珠;10:0.2 μL英莱盾磁珠;
Fig. 2Measurement of CUT&Tag PCR products amplified from DNA fragments recovered using different Streptavidin magnetic beads or its volume
2.2 不同回收方法效率的比较
利用生物素化的pAG-Tn5,对于104 K562细胞分别进行H3K4me3、RNAPII、CTCF和HGAM1的CUT&Tag实验。所得到的产物分别用链霉亲和素磁珠(srCUT&Tag)、SPRI磁珠、乙醇沉淀和直接PCR法进行回收或后处理后建库,文库以等体积混匀以后在同一块芯片上进行二代测序,其中H3K4me3和RNAPII进行了三重复平行实验,而CTCF和HMGA1则进行了双重复实验。得到的数据在清洗后进行回贴比对。统计所有实验组产出的unique reads和total mapping reads数量,结果显示,在H3K4me3和RNAPII的CUT&Tag实验结果中,srCUT&Tag在回收unique reads产量和去除重复后的total mapping reads数上仅仅略低于最高的SPRI磁珠回收(图3)。而在CTCF和HGAM1的CUT&Tag结果中,srCUT&Tag回收则产生最多的unique reads和total mapping reads数。直接PCR法和乙醇沉淀法贡献的数据量都相对较低,在4种不同的CUT&Tag结果中直接法PCR得到的total mapping read数相当于产出数据量最高方法的43%~60%,而乙醇沉淀法产出的数据量则相当于产出数据量最高方法的24%~30%。整体而言,srCUT&Tag在4种回收方法中都保持着稳定的高回收效率。而若采用回收效率较低的乙醇沉淀法,则有损失超过2/3原始产物信息的风险。图3
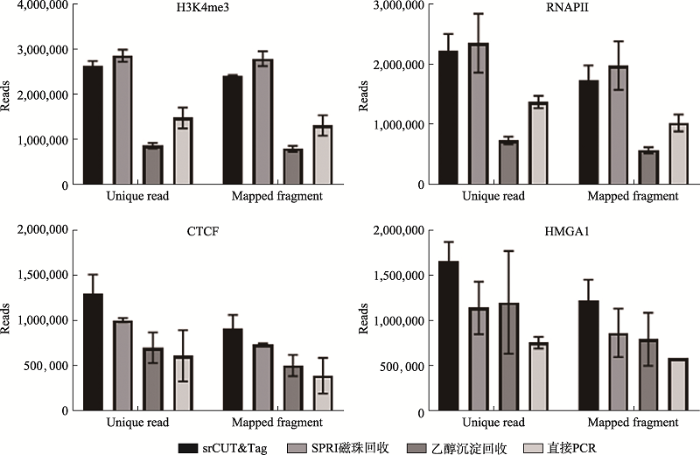 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3不同回收方法文库产出规模比较
Fig. 3Comparison of library output scale of different recovery methods
2.3 不同回收方法产出CUT&Tag文库片段大小和文库信息的差异
利用deeptool中的collectinsertsizemetrics统计不同实验组插入片段大小分布。比较使用不同回收方法所产生的文库中插入片段的大小分布(图4A),可以发现不同回收方法所保留的底物样本的片段大小分布并不相同。在4种不同的回收方法中,srCUT&Tag和乙醇沉淀法产出的文库片段大小分布相似。对于小于150 bp的片段,SPRI磁珠法的回收率均低于其他几种回收方法。对H3K4me3的CUT&Tag结果进行比较,使用srCUT&Tag、乙醇沉淀和直接PCR法产出的文库中小于150 bp的片段在整体文库中的平均占比分别为23.07%、15.81%和22.16%,而SPRI磁珠所产出的文库中小于150 bp片段的占比仅为10.80%。对转录因子CTCF的CUT&Tag结果进行比较,srCUT&Tag、乙醇沉淀和直接PCR法所产出的文库中小于150 bp的片段在整体文库中的平均占比分别为39.98%,32.40%和62.03%,而SPRI磁珠回收文库中小于150 bp片段的占比则仅为20.43% (图4B)。结合2.2中所展示的不同回收方法的文库产量,在H3K4me3的CUT&Tag结果中,srCUT&Tag产出的小于150 bp片段的reads数为SPRI磁珠回收法的1.75倍;而在CTCF的CUT&Tag结果中,srCUT&Tag产出的小于150 bp片段的reads数则达到SPRI磁珠回收法的2.4倍。由于CTCF和HMGA1文库中含有更多的小于150 bp的片段,一定程度上解释了SPRI磁珠在CTCF和HMGA1样品的回收效率现象。此外,在CTCF和HMGA1的CUT&Tag结果中,直接PCR法产生的大片段含量明显低于使用其他回收方法所产出的文库。其文库中几乎不含有大于300 bp的大片段,且直接PCR法也影响了CTCF和HMGA1文库的数据产量,低于链霉亲和素磁珠回收法产出的文库数据产量。图4
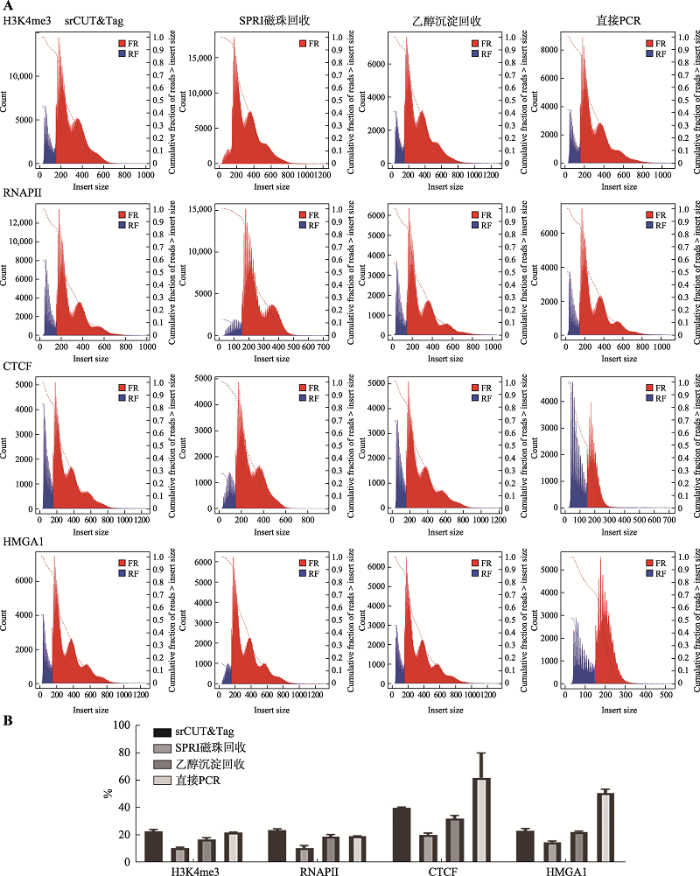 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4不同回收方法文库片段大小分布
A:4种不同回收方法文库片段的大小分布;B:不同回收方法中小于150 bp片段的占比
Fig. 4Size distribution of library fragments by different recovery methods
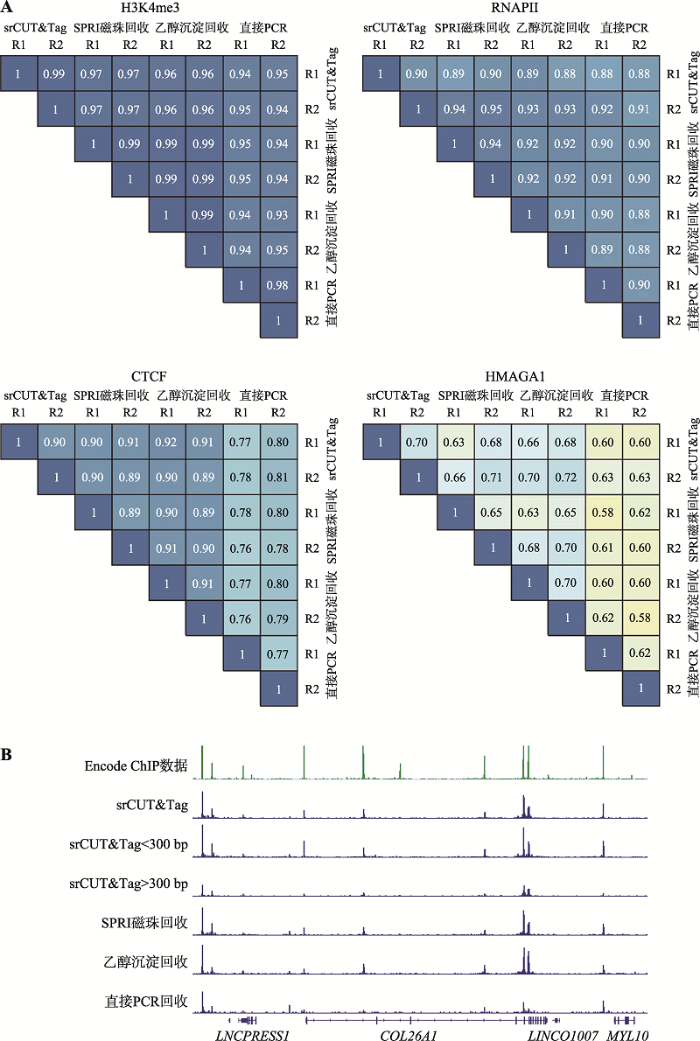
为了探究不同回收方法产生文库的差异,对H3K4me3,RANPII部分平行组数据和CTCF和HMGA1全部的平行组的数据结果进行关联性分析(图5A)。在H3K4me3,RNAPII和CTCF的CUT&Tag实验中,srCUT&Tag,SPRI磁珠和乙醇沉淀法均有较好的相关性(>0.9),而在HMGA1的CUT&Tag结果中,所有回收方法平行组和相互间的相关性都较低,但是srCUT&Tag方法平行组间仍然有0.7的相关性系数,仅次于乙醇沉淀法平行组间的0.71。这证明了srCUT&Tag具有和传统SPRI磁珠和乙醇沉淀回收相当的可重复性。直接PCR法在H3K4me3实验中具有较高的可重复性,并且不同回收方法的信号和信噪比相仿。但是在其余的3组条件CUT&Tag中均展现出较低的平行组内相关性。同时,在H3K4me3以外的其他3类CUT&Tag结果中,直接PCR法与其他3种方法间的相关性系数均较低,这意味着直接PCR法与srCUT&Tag及传统回收方法所得到的的结果均存在明显差异。在相关性系数较低的CTCF的CUT&Tag结果中,可以明显看到直接PCR法产出的信号分布与其他回收方法的结果不同,包括在多个位置的信号强度较弱或完全缺失(图5B)。通过将srCUT&Tag的测序结果拆分为大于300 bp和小于300 bp的部分,可以看到,这些峰高的差异并非是简单的由上文所提到的直接PCR法中大于300 bp片段损失所带来,这暗示了在直接PCR法中TritonX-100对扩增酶的抑制可能了造成更复杂的带有偏好性的扩增,并对CUT&Tag的结果造成了显著的影响。
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5不同回收方法文库的相关性
A:4种不同CUT&Tag产物中不同回收方法产生结果之间的相关性;B:CTCF CUT&Tag结果中,直接PCR法和其他三种方法的结果存在明显的差异。
Fig. 5Correlation of CUT&Tag generated by different recovery methods
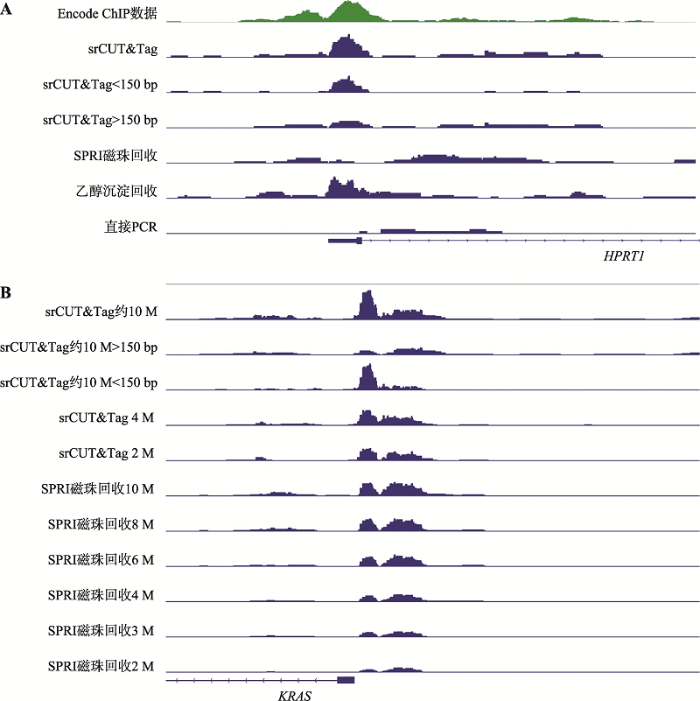
相关性分析显示,SPRI磁珠与srCUT&Tag和乙醇沉淀的结果差别并不大。为了进一步研究小片段损失对CUT&Tag结果可能的影响,选取RNAPII R1实验组的CUT&Tag结果,将srCUT&Tag的测序结果拆分为插入片段小于150 bp和大于150 bp两部分,比较不同大小的插入片段产生的峰的差异(图6)。
图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6片段选择性对CUT&Tag结果的影响
A:小于150bp的片段贡献了RNAPII CUT&Tag中HPRT1启动子位点上的主要部分;B:小于150bp的片段贡献了RNAPII CUT&Tag中KRAS启动子位点上的主要部分,srCUT&Tag和SPRI磁珠的差异在数据量较小时尤其明显。
Fig. 6The effect of fragment selectivity on CUT&Tag results
在次黄嘌呤腺苷酸转移酶基因HPRT1位点上,小于150 bp的插入片段贡献了启动子峰的主要部分。控制不同回收方法投入的起始unique reads数为5 M,在HPRT1位点上,对小于150 bp的片段保留效果较好的链霉亲和素磁珠回收法、乙醇沉淀法产生结果一致的峰图,而对小于150 bp的片段保留效果较差的SPRI磁珠回收法则没有信号(图6A)。同样的现象也可以在KRAS基因位点上观察到。当投入数据量从10 M下降到2 M时,SPRI磁珠文库在KRAS基因启动子上的信号急速衰减,但是链霉亲和素磁珠回收法产生的文库受投入数据量下降的影响较小,在2 M数据量投入时仍有与SPRI磁珠8~10 M回收结果类似的效果。显示了在起始数据量较低的情况下,小于150 bp的片段回收率差距带来的信号差异尤其明显(图6B)。
综上所述,比较使用4种不同的回收方法产出的CUT&Tag结果,基于srCUT&Tag的链霉亲和素磁珠回收法对于不同抗体都有较高的数据产量,而SPRI磁珠法均会损失一部分小于150 bp的小片段,直接PCR法则在转录因子CTCF和HMGA1的回收中损失了大部分大于300 bp的片段,从而导致一部分结合位点信息的损失,乙醇沉淀法的回收效率相对较低。因此,总体上而言,基于srCUT&Tag的链霉亲和素磁珠法是对CUT&Tag结果信息保留效率最高的回收方法。
2.4 srCUT&Tag有利于低起始细胞量的CTCF CUT&Tag
考虑到片段选择性在低数据量水平上差异更加显著,推测对于极低的起始细胞量,且当目的位为丰度相对较低的转录因子时,不同回收方法贡献的数据质量可能有更大的差异。利用生物素化的pAG-Tn5,分别在200、500、1000个K562细胞水平上进行CTCF的CUT&Tag实验,产物利用srCUT&Tag、SPRI磁珠、乙醇沉淀酚氯仿抽提和直接PCR回收后建库测序。与104细胞的回收结果类似,在200~1000细胞水平上,产生的文库从大到小排列为:srCUT&Tag>SPRI磁珠法>直接PCR法>乙醇沉淀法(表1)。其中链霉亲和素磁珠回收文库在去除重复后的mapping reads数比SPI磁珠文库高15%~23%,比乙醇沉淀文库高108%~211%,比直接PCR文库高118%~128%。在链霉亲和素磁珠法产出的CUT&Tag结果中,macs2软件在200,500,1000细胞水平上分别识别到8301、13389和16269个峰,多于在相同的细胞数量上,SPRI磁珠法产出的峰值数(7630、11875和14145),乙醇沉淀法产出的峰值数(2949、5749和9752)和直接PCR产出的峰值数(5992、9780和11028)。Table 1
表1
表1低细胞数量时不同回收方法的回收效率比较
Table 1
| 回收方法 | 细胞数量 | 起始unique read | 去重后的mapping reads | 峰值数 |
|---|---|---|---|---|
| srCUT&Tag | 200.00 | 6384518 | 2436658 | 8301 |
| srCUT&Tag | 500.00 | 15961295 | 4628092 | 13389 |
| srCUT&Tag | 1000.00 | 29650679 | 6318990 | 16269 |
| SPRI 磁珠回收 | 200.00 | 5407687 | 2100856 | 7630 |
| SPRI 磁珠回收 | 500.00 | 13168069 | 3751782 | 11875 |
| SPRI 磁珠回收 | 1000.00 | 23021799 | 5238500 | 14145 |
| 乙醇沉淀回收 | 200.00 | 1580168 | 783550 | 2497 |
| 乙醇沉淀回收 | 500.00 | 4243044 | 1707974 | 5749 |
| 乙醇沉淀回收 | 1000.00 | 7608218 | 3012370 | 9752 |
| 直接PCR | 200.00 | 2809188 | 1116464 | 5992 |
| 直接PCR | 500.00 | 7461906 | 2027234 | 9870 |
| 直接PCR | 1000.00 | 13058335 | 3446298 | 11028 |
新窗口打开|下载CSV
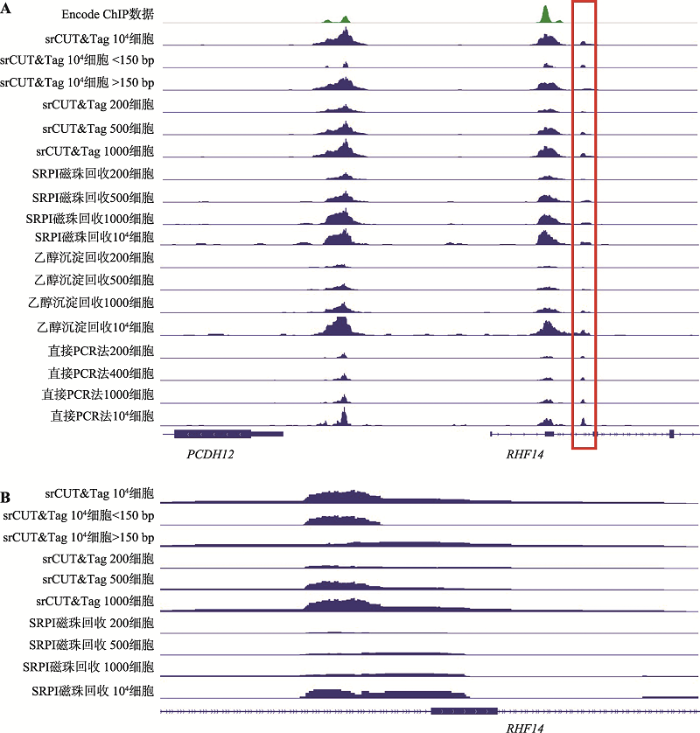
在基因组上选取一段具有代表性的区域,通过IGV的可视化获取不同回收方法得到的覆盖度图(图7A)。在500细胞和1000细胞水平上,srCUT&Tag和SPRI磁珠法均能产生与105细胞水平相类似的CUT&Tag产物峰图。而直接PCR法和乙醇沉淀法在500细胞产生的覆盖图谱峰高则远低于105细胞的结果,1000细胞的覆盖图谱整体上同样弱于105细胞产生的图谱结果。另一方面,与前文所述的结果类似,在RNF14基因主峰侧翼的较弱信号位点上,低细胞起始量状态下的SPRI磁珠回收几乎损失了全部的信号。与104细胞的SPRI磁珠回收结果相比,可以看出SPRI磁珠法损失小于150 bp片段的问题在细胞数量更少的情况下更加显著,从而相比于srCUT&Tag提供的结果显现出明显的劣势(图7B)。总体来看srCUT&Tag在起始细胞量较少的情况下可以产出更完整和丰富的CUT&Tag文库信息,适用于极低起始细胞量的CTCF CUT&Tag实验。
图7
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7不同回收方法在少量细胞上的对比
A:200~1000细胞水平上的不同CTCF回收结果;B:在RNF14基因侧翼的弱结合位点,SRPI磁珠回收相比于srCUT&Tag出现了明显的信号缺失。
Fig. 7Comparison of different recovery methods in few cells condition
Table 2
表2
表2srCUT&Tag与其他CUT&Tag产物回收方法的比较
Table 2
| 回收方法 | 耗时 | 偏好性 | 单个反应成本(¥) | 回收效率 |
|---|---|---|---|---|
| 酚氯仿抽提-乙醇沉淀 | 2 h~1 d | 无明显选择性 | - | 较低 |
| SPRI磁珠回收 | 0.5 h | 损失<150bp片段 | 10 | 高 |
| srCUT&Tag | 0.5 h | 无明显选择性 | 0.5~2.5 | 高 |
| 直接PCR法 | 1 h | 部分情况下损失>300bp片段,并造成信号损失 | - | 中等 |
| 离心柱回收 | 1 h | 无明显选择性 | >30 | 高 |
新窗口打开|下载CSV
3 讨论
Henikoff在其早期关于CUT&Tag方法的文献报道中主要使用2×SPRI磁珠回收,随后将酚氯仿抽提乙醇沉淀作为CUT&Tag产物的标准回收方法加以推广,而在近期的文献中,则主要推荐其基于Triton中和的PCR直接扩增法[9]。针对现有的CUT&Tag回收方法各自存在的缺陷,本研究提出了一种新的srCUT&Tag体系:利用含有生物素化接头的pAG-Tn5转座酶进行DNA片段化和生物素化,产物DNA通过链霉亲和素磁珠回收。在K562细胞中使用4种不同抗体开展CUT&Tag实验,srCUT&Tag的链霉亲和素回收法仅对H3K4me3和RNAPII的回收效率略低于传统的SPRI磁珠回收效率,对CTCF和HMGA1的回收效率则高于SPRI磁珠回收法,对于4种抗体其回收效率都远高于乙醇沉淀回收法和直接PCR法。并且srCUT&Tag回收的DNA片段大小分布中与乙醇沉淀回收法相似,不存在2XSPRI磁珠丢失小片段或者直接PCR扩增法丢失部分大片段的问题,更有利于CUT&Tag产物完整信息的保留(表2)。本研究发现,使用不同的抗体所产生的CUT&Tag文库有着不同的DNA片段大小分布,因此可能导致不同回收方法搭配不同抗体产出的CUT&Tag文库有不同的表现。以链霉亲和素磁珠回收法产出的文库作为标准,H3K4me3和RNAPII的CUT&Tag文库中,小片段含量本身并不高,特别是在H3K4me3的文库中,不同建库方法产生的片段差异并不大。150 bp左右的长度是单个核小体DNA的大小,而在CTCF和HMGA1文库中,由于转录因子结合区域本身有着比组蛋白周围更广阔的核小体耗尽区域,因此转录因子的CUT&Tag文库中,含有更多小于单个核小体大小的150 bp以下片段。更多小片段的产出不仅凸显了SPRI磁珠在片段选择性上的差异,也造成直接PCR法的扩增偏好。在加入了Triton X-100后,扩增酶的活性在一定程度上受到了影响,从而使得PCR扩增大片段能力下降,使得整体的文库大小向小片段的方向偏移。而在CTCF文库中,直接PCR法与srCUT&Tag回收法的差异不仅仅来源于300 bp以上大片段回收效率的低下;将文库中小于300 bp的片段分离后,在相同文库投入数据量的情况下,直接PCR法仍然存在许多不同大小片段的缺失。
综上所述,与传统建库方法相比较,srCUT&Tag回收法兼顾了现有回收方法中酚氯仿抽提乙醇沉淀回收法没有回收偏好性,SPRI磁珠回收效率高和直接PCR法一管式操作便捷的优势,提高了实验的便捷性,避免了更繁琐的实验流程。基于srCUT&Tag系统的链霉亲和素磁珠在104~200细胞水平上都有较高的回收效率,并且在细胞量较低的情况下仍能够提供较好的回收效果,其CUT&Tag文库产量和数据信息优于现有的酚氯仿抽提乙醇沉淀回收、SPRI磁珠回收和直接PCR法。本研究建立了基于srCUT&Tag的链霉亲和素磁珠回收系统,并比较了该回收方法与现有的主流回收方法,对各自的优缺点进行了系统的分析,在保证数据完整性的基础上进一步优化了CUT&Tag的易用性。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1038/ni.2117URLPMID:21934668 [本文引用: 1]

Chromatin immunoprecipitation followed by next-generation sequencing analysis (ChIP-Seq) is a powerful method with which to investigate the genome-wide distribution of chromatin-binding proteins and histone modifications in any genome with a known sequence. The application of this technique to a variety of developmental and differentiation systems has provided global views of the cis-regulatory elements, transcription factor function and epigenetic processes involved in the control of gene transcription. Here we describe several technical aspects of the ChIP-Seq assay that diminish bias and background noise and allow the consistent generation of high-quality data.
URLPMID:29028882 [本文引用: 1]
DOI:10.1242/dev.170217URLPMID:31249006 [本文引用: 1]
Understanding chromatin regulation holds enormous promise for controlling gene regulation, predicting cellular identity, and developing diagnostics and cellular therapies. However, the dynamic nature of chromatin, together with cell-to-cell heterogeneity in its structure, limits our ability to extract its governing principles. Single cell mapping of chromatin modifications, in conjunction with expression measurements, could help overcome these limitations. Here, we review recent advances in single cell-based measurements of chromatin modifications, including optimization to reduce DNA loss, improved DNA sequencing, barcoding, and antibody engineering. We also highlight several applications of these techniques that have provided insights into cell-type classification, mapping modification co-occurrence and heterogeneity, and monitoring chromatin dynamics.
DOI:10.1038/s41467-019-09982-5URLPMID:31036827 [本文引用: 1]
Many chromatin features play critical roles in regulating gene expression. A complete understanding of gene regulation will require the mapping of specific chromatin features in small samples of cells at high resolution. Here we describe Cleavage Under Targets and Tagmentation (CUT&Tag), an enzyme-tethering strategy that provides efficient high-resolution sequencing libraries for profiling diverse chromatin components. In CUT&Tag, a chromatin protein is bound in situ by a specific antibody, which then tethers a protein A-Tn5 transposase fusion protein. Activation of the transposase efficiently generates fragment libraries with high resolution and exceptionally low background. All steps from live cells to sequencing-ready libraries can be performed in a single tube on the benchtop or a microwell in a high-throughput pipeline, and the entire procedure can be performed in one day. We demonstrate the utility of CUT&Tag by profiling histone modifications, RNA Polymerase II and transcription factors on low cell numbers and single cells.
DOI:10.1038/s41467-020-18761-6URLPMID:33009411 [本文引用: 1]
The HUSH complex represses retroviruses, transposons and genes to maintain the integrity of vertebrate genomes. HUSH regulates deposition of the epigenetic mark H3K9me3, but how its three core subunits - TASOR, MPP8 and Periphilin - contribute to assembly and targeting of the complex remains unknown. Here, we define the biochemical basis of HUSH assembly and find that its modular architecture resembles the yeast RNA-induced transcriptional silencing complex. TASOR, the central HUSH subunit, associates with RNA processing components. TASOR is required for H3K9me3 deposition over LINE-1 repeats and repetitive exons in transcribed genes. In the context of previous studies, this suggests that an RNA intermediate is important for HUSH activity. We dissect the TASOR and MPP8 domains necessary for transgene repression. Structure-function analyses reveal TASOR bears a catalytically-inactive PARP domain necessary for targeted H3K9me3 deposition. We conclude that TASOR is a multifunctional pseudo-PARP that directs HUSH assembly and epigenetic regulation of repetitive genomic targets.
DOI:10.3389/fgene.2020.00338URLPMID:32318100 [本文引用: 1]
ZNF143, a human homolog of the transcriptional activator Staf, is a C2H2-type protein consisting of seven zinc finger domains. As a transcription factor (TF), ZNF143 is sequence specifically binding to chromatin and activates the expression of protein-coding and non-coding genes on a genome scale. Although it is ubiquitous expressed, its expression in cancer cells and tissues is usually higher than that in normal cells and tissues. Therefore, abnormal expression of ZNF143 is related to cancer cell survival, proliferation, differentiation, migration, and invasion, suggesting that new small molecules can be designed by targeting ZNF143 as it may be a good potential biomarker and therapeutic target for related cancers. However, the mechanism on how ZNF143 regulates its targeting gene remains unclear. Recently, with the development of chromatin conformation capture (3C) and its derivatives, and high-throughput sequencing technology, new findings have been obtained in the study of ZNF143. Pioneering studies have showed that ZNF143 binds directly to promoters and contributes to chromatin interactions connecting promoters to distal regulatory elements, such as enhancers. Further, it has proved that ZNF143 is involved in CCCTC-binding factor (CTCF) in establishing the conserved chromatin loops by cooperating with cohesin and other partners. These results indicate that ZNF143 is a key loop formation factor. In addition, we report ZNF143 is dynamically bound to chromatin during the cell cycle demonstrated that it is a potential mitotic bookmarking factor. It may be associated with CTCF for mitosis-to-G1 phase transition and chromatin loop re-establishment in early G1 phase. In the future, researchers could further clarify the fine mechanism of ZNF143 in mediating chromatin loops with the help of CUT&RUN (CUT&Tag) and Cut-C technology. Thus, in this review, we summarize the research progress of TF ZNF143 in detail and also predict the potential functions of ZNF143 in cell fate and identity based on our recent discoveries.
DOI:10.1101/2021.03.29.437516URLPMID:33821279 [本文引用: 1]
There is a persistent male bias in the prevalence and severity of COVID-19 disease. Underlying mechanisms accounting for this sex difference remain incompletely understood. Interferon responses have been implicated as a modulator of disease in adults, and play a key role in the placental anti-viral response. Moreover, the interferon response has been shown to alter Fc-receptor expression, and therefore may impact placental antibody transfer. Here we examined the intersection of viral-induced placental interferon responses, maternal-fetal antibody transfer, and fetal sex. Placental interferon stimulated genes (ISGs), Fc-receptor expression, and SARS-CoV-2 antibody transfer were interrogated in 68 pregnancies. Sexually dimorphic placental expression of ISGs, interleukin-10, and Fc receptors was observed following maternal SARS-CoV-2 infection, with upregulation in males. Reduced maternal SARS-CoV-2-specific antibody titers and impaired placental antibody transfer were noted in pregnancies with a male fetus. These results demonstrate fetal sex-specific maternal and placental adaptive and innate immune responses to SARS-CoV-2.
DOI:10.7554/eLife.63274URLPMID:33191916 [本文引用: 1]
Chromatin accessibility mapping is a powerful approach to identify potential regulatory elements. A popular example is ATAC-seq, whereby Tn5 transposase inserts sequencing adapters into accessible DNA ('tagmentation'). CUT&Tag is a tagmentation-based epigenomic profiling method in which antibody tethering of Tn5 to a chromatin epitope of interest profiles specific chromatin features in small samples and single cells. Here, we show that by simply modifying the tagmentation conditions for histone H3K4me2 or H3K4me3 CUT&Tag, antibody-tethered tagmentation of accessible DNA sites is redirected to produce chromatin accessibility maps that are indistinguishable from the best ATAC-seq maps. Thus, chromatin accessibility maps can be produced in parallel with CUT&Tag maps of other epitopes with all steps from nuclei to amplified sequencing-ready libraries performed in single PCR tubes in the laboratory or on a home workbench. As H3K4 methylation is produced by transcription at promoters and enhancers, our method identifies transcription-coupled accessible regulatory sites.
DOI:10.1038/s41596-020-0373-xURLPMID:32913232 [本文引用: 2]
We recently introduced Cleavage Under Targets & Tagmentation (CUT&Tag), an epigenomic profiling strategy in which antibodies are bound to chromatin proteins in situ in permeabilized nuclei. These antibodies are then used to tether the cut-and-paste transposase Tn5. Activation of the transposase simultaneously cleaves DNA and adds adapters ('tagmentation') for paired-end DNA sequencing. Here, we introduce a streamlined CUT&Tag protocol that suppresses DNA accessibility artefacts to ensure high-fidelity mapping of the antibody-targeted protein and improves the signal-to-noise ratio over current chromatin profiling methods. Streamlined CUT&Tag can be performed in a single PCR tube, from cells to amplified libraries, providing low-cost genome-wide chromatin maps. By simplifying library preparation CUT&Tag requires less than a day at the bench, from live cells to sequencing-ready barcoded libraries. As a result of low background levels, barcoded and pooled CUT&Tag libraries can be sequenced for as little as $25 per sample. This enables routine genome-wide profiling of chromatin proteins and modifications and requires no special skills or equipment.
DOI:10.1038/nprot.2018.015URLPMID:29651053 [本文引用: 1]
Cleavage under targets and release using nuclease (CUT&RUN) is an epigenomic profiling strategy in which antibody-targeted controlled cleavage by micrococcal nuclease releases specific protein-DNA complexes into the supernatant for paired-end DNA sequencing. As only the targeted fragments enter into solution, and the vast majority of DNA is left behind, CUT&RUN has exceptionally low background levels. CUT&RUN outperforms the most widely used chromatin immunoprecipitation (ChIP) protocols in resolution, signal-to-noise ratio and depth of sequencing required. In contrast to ChIP, CUT&RUN is free of solubility and DNA accessibility artifacts and has been used to profile insoluble chromatin and to detect long-range 3D contacts without cross-linking. Here, we present an improved CUT&RUN protocol that does not require isolation of nuclei and provides high-quality data when starting with only 100 cells for a histone modification and 1,000 cells for a transcription factor. From cells to purified DNA, CUT&RUN requires less than a day at the laboratory bench and requires no specialized skills.
DOI:10.1101/2021.03.29.437516URLPMID:33821279 [本文引用: 1]
There is a persistent male bias in the prevalence and severity of COVID-19 disease. Underlying mechanisms accounting for this sex difference remain incompletely understood. Interferon responses have been implicated as a modulator of disease in adults, and play a key role in the placental anti-viral response. Moreover, the interferon response has been shown to alter Fc-receptor expression, and therefore may impact placental antibody transfer. Here we examined the intersection of viral-induced placental interferon responses, maternal-fetal antibody transfer, and fetal sex. Placental interferon stimulated genes (ISGs), Fc-receptor expression, and SARS-CoV-2 antibody transfer were interrogated in 68 pregnancies. Sexually dimorphic placental expression of ISGs, interleukin-10, and Fc receptors was observed following maternal SARS-CoV-2 infection, with upregulation in males. Reduced maternal SARS-CoV-2-specific antibody titers and impaired placental antibody transfer were noted in pregnancies with a male fetus. These results demonstrate fetal sex-specific maternal and placental adaptive and innate immune responses to SARS-CoV-2.
DOI:10.1186/s13007-020-00664-8URLPMID:32884577 [本文引用: 1]
Background: In 2019, Kaya-Okur et al. reported on the cleavage under targets and tagmentation (CUT&Tag) technology for efficient profiling of epigenetically modified DNA fragments. It was used mainly for cultured cell lines and was especially effective for small samples and single cells. This strategy generated high-resolution and low-background-noise chromatin profiling data for epigenomic analysis. CUT&Tag is well suited to be used in plant cells, especially in tissues from which small samples are taken, such as ovules, anthers, and fibers. Results: Here, we present a CUT&Tag protocol step by step using plant nuclei. In this protocol, we quantified the nuclei that can be used in each CUT&Tag reaction, and compared the efficiency of CUT&Tag with chromatin immunoprecipitation with sequencing (ChIP-seq) in the leaves of cotton. A general workflow for the bioinformatic analysis of CUT&Tag is also provided. Results indicated that, compared with ChIP-seq, the CUT&Tag procedure was faster and showed a higher-resolution, lower-background signal than did ChIP. Conclusion: A CUT&Tag protocol has been refined for plant cells using intact nuclei that have been isolated.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}