 ,1, 申笑涵1, 施奇惠,11
,1, 申笑涵1, 施奇惠,11 Advances in single-cell whole genome sequencing technology and its application in biomedicine
Wang Zhuo,1, Shen Xiaohan1, Shi Qihui,11 通讯作者: 通讯作者: 施奇惠,博士,研究员,研究方向:液态活检与单细胞分析。E-mail:qihuishi@fudan.edu.cn
第一联系人:
收稿日期:2020-12-1
| 基金资助: |
Received:2020-12-1
| Fund supported: |

摘要
随着单细胞基因组测序技术的建立与发展,对细胞基因组特征的分析进入了单细胞水平。单细胞的基因组分辨率不但使研究人员能够在单细胞尺度上分析肿瘤细胞的异质性,也使得传统上难以检测的稀有细胞的基因组研究成为可能。这些稀有细胞往往具有重要的生物学意义或临床价值,如癌症患者血液中循环肿瘤细胞(circulating tumor cell, CTC)的基因组检测或三代试管婴儿植入前胚胎细胞的遗传缺陷诊断与筛查(preimplantation genetic diagnosis/screening, PGD/PGS)。本文总结了近年来发展的各种单细胞基因组扩增技术及其优缺点,并介绍了单细胞基因组测序技术在肿瘤生物学和临床检测中的应用,以期为单细胞基因组测序技术在临床检测中应用开发提供参考。
关键词:
Abstract
Keywords:
PDF (1072KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
王卓, 申笑涵, 施奇惠. 单细胞基因组测序技术新进展及其在生物医学中的应用. 遗传[J], 2021, 43(2): 108-117 doi:10.16288/j.yczz.20-363
Wang Zhuo.
细胞是构成生命体的基本单元,正如物理在原子层面进行研究,化学在分子层面进行研究,生物学基于细胞进行研究。细胞在统一的基因组蓝图和时空特异性调控下,由一个受精卵分化出各种形态、位置、功能不同的细胞,从而构成一个完整的生命体。肿瘤作为一种由基因组异常导致的恶性病变,其细胞之间存在着基因组层面上的异质性,这就使肿瘤成为了一组具有不同基因组特征构成的细胞的集合体。这种异质性的解析对于理解肿瘤的演化规律、耐药机制以及发展有效的治疗方法有着重要的意义。但是,早期的测序技术只能用于包含大量细胞的样本,然后通过计算机重构模拟细胞之间的差异。单细胞测序技术的建立和发展使研究人员能够真正从单细胞水平上研究细胞间的异质性,包括单细胞基因组单碱基突变差异(single-nucleotide variants, SNV)、短序列插入/缺失(insertions and deletions, Indel)和拷贝数变异(copy number variants, CNV)等,以及单细胞基因表达差异和单细胞蛋白修饰差异等[1,2]。除了肿瘤异质性研究以外,单细胞测序所具有的分辨率也使得对具有重要生物学意义或临床价值的稀有细胞的基因组研究成为可能,如循环肿瘤细胞(circulating tumor cell, CTC)或三代试管婴儿植入前的遗传缺陷诊断与筛查(preimplantation genetic diagnosis/screening, PGD/PGS)。单细胞基因组测序主要包括3个部分:单细胞获取、单细胞全基因组扩增和扩增产物测序及分析。本文总结了近年来单细胞基因组扩增技术的发展及优缺点,并介绍了单细胞基因组测序技术在肿瘤生物学和临床检测中的应用。
1 单细胞的获取
单细胞测序首先需要获取感兴趣的单细胞样本。研究中通常采用机械剪切结合酶解方法将组织消化成单细胞悬液,单细胞消化过程为了避免消化不充分从而导致细胞类型不均一,通常采用多种水解酶混合使用,单细胞样本的回收根据实验需要选择合适的技术平台(表1)。使用口吸管[3]或显微操作仪(micromanipulation)[4]回收单细胞,可以借助显微镜直观的观察目标单细胞,可视化、准确、回收成功率高,但对操作人员技术要求较高,单细胞回收通量比较低。口吸管或显微操作技术适用于目标细胞较少且珍贵样本,如循环肿瘤细胞或辅助生殖移植前胚胎细胞。流式细胞分选技术(fluorescence activated cell sorting, FACS)[5]的应用非常广泛,FACS可以高通量、高效的将目标单细胞样本回收到96孔板或384孔板中,与高通量、标准化实验操作兼容。如果借助荧光标记的单克隆抗体可以标记细胞亚群,FACS还可以有选择的回收某一特定类型单细胞。缺点是FACS对细胞有一定损伤,对起始细胞数量有一定要求,细胞数量较少的细胞亚群或珍贵样本不适合使用FACS进行单细胞回收。激光显微切割捕获技术(laser-capture microdissection, LCM)[6]通常被用于分离回收固定染色切片上的目标细胞样本,LCM技术的优点是可以确定单细胞在组织样本中的空间位置,但设备操作者需要对组织样本非常熟悉,因为是从组织原位回收目的细胞,需要操作者分辨出目标细胞和非目标细胞[7],对操作者技术要求也比较高,LCM是唯一能够获取目标细胞空间位置的技术。微流控芯片平台[8,9]用于单细胞回收的优点是通量高,效率高,自动化程度高,能够与下游分子生物学反应集成化,全部在微流控芯片上完成,降低污染,降低人为操作导致的实验偏差,反应体积小,反应效率高,节约试剂使用量,但微流控平台对技术要求比较高,如果自己搭建微流控芯片平台很多普通生物学实验室没有相关技术支持,使用全自动商业化仪器,设备成本较高。Table 1
表1
表1单细胞分离技术优缺点比较
Table 1
| 技术方式 | 分选功能 | 通量 | 费用 | 自动化 | 操作难易 | 效率 |
|---|---|---|---|---|---|---|
| 口吸管或显微操作 | 不可分选 | 低 | 低 | 人工 | 难 | 低 |
| FACS | 可分选 | 高 | 低 | 自动化 | 容易 | 高 |
| LCM | 可分选 | 低 | 高 | 半自动化 | 难 | 低 |
| 微流控芯片 | 不可分选 | 高 | 低 | 自动化 | 容易 | 高 |
新窗口打开|下载CSV
2 单细胞全基因组扩增技术的发展
哺乳动物由受精卵发育成完整个体,受精卵基因组遗传自父亲和母亲,分析基因组特定基因碱基序列可以分析家系遗传追踪。肿瘤细胞积累新的基因突变可以形成新的肿瘤细胞克隆亚群。应用基因测序可以分析肿瘤细胞基因组单碱基突变、插入和缺失、拷贝数变异和染色体结构变异。单细胞基因组DNA大约5.6 pg,满足不了测序要求,即使最新的三代测序技术对样本量要求降低很多[10],也需要对单细胞基因组进行预扩增。单细胞培养可以实现核酸在胞内的扩增,但细胞培养只适用于少量细胞类型,如部分干细胞、肿瘤细胞系等,临床检测所分离的稀有细胞原代培养成功率还不高,暂时不具备普遍应用的价值,肿瘤细胞在人为培养下,可能会积累新的基因突变,因此临床上单细胞基因组预扩增主要使用体外试剂扩增技术。单细胞基因组预扩增技术主要有基于聚合酶链式反应(polymerase chain reaction, PCR)扩增、多重置换扩增(multiple displacement amplification, MDA)和体外转录(in vitro transcription technology, IVT)等技术(图1)。
图1
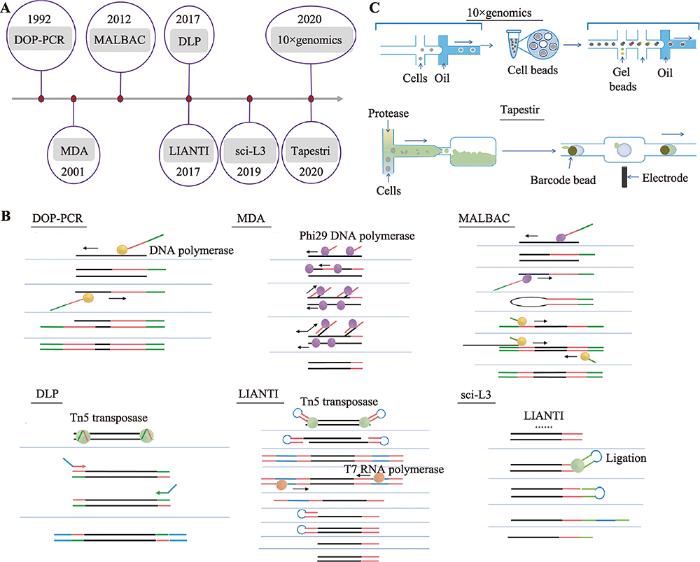 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1单细胞全基因组扩增技术的发展
A:单细胞全基因组扩增技术发展时间轴;B:人工单细胞全基因组扩增技术;C:自动化单细胞基因组扩增技术。
Fig. 1Development of single cell whole genome amplification technologies
2.1 基于PCR的单细胞基因组扩增技术
随机寡核苷酸引物PCR技术(primer-extension preamplification PCR, PEP-PCR)[11,12]或简并引物PCR技术(degenerate oligonucleotide-primed PCR, DOP-PCR)[13,14]首先应用引物随机结合在单细胞基因组上,通过PCR反应随机扩增全基因组序列。而接头介导的PCR技术(ligation-mediated PCR, LM-PCR)[15]则是使用限制性内切酶Mse I在基因组5?-TTAA-3?位置随机切断单细胞基因组DNA,然后在DNA片段末端加上扩增引物,也是通过PCR反应扩增基因组DNA片段。PEP-PCR、DOP-PCR和LM-PCR技术作为单细胞基因组DNA扩增技术的雏形,最先实现了单细胞基因组DNA的扩增,满足了某些研究的需要。但PCR扩增反应具有碱基偏好性导致基因组扩增存在偏向性,全基因组扩增产物覆盖度不足10%[16],PCR扩增反应碱基错配率偏高,此类单细胞基因组扩增技术不适用于检测基因点突变,假阳性率较高。2.2 多重置换扩增技术
Dean等[17,18]创造性的开发了新的单细胞基因组扩增方法—多重置换扩增技术,phi29 DNA聚合酶在恒温条件下扩增单细胞基因组,phi29 DNA聚合酶具有很强DNA合成活性和链置换活性,扩增产物可大于10 kb,以新合成的DNA子链为模板,继续合成新的DNA子链,所以MDA反应后扩增产量高,而且phi29 DNA聚合酶保真性好,与PCR反应使用的DNA聚合酶相比,扩增产物保真度提高1000倍,基因组扩增覆盖度更高,MDA技术比较适合检测单细胞基因组SNV,但MDA指数扩增会导致有些基因组区域偏向性[19],因此不适用于基因组CNV检测,另外,高效的phi29 DNA聚合酶会导致基因融合和等位基因丢失的现象[20]。为了解决MDA指数扩增所导致的偏向性,Fu等[21]将单细胞基因组随机片段化,借助微流控芯片生成的乳滴将DNA片段和预扩增试剂包裹起来,形成小的空间,借助乳滴中扩增反应最小限制因子来终止扩增反应,避免了传统MDA技术基因组扩增时,基因组某些区域无限扩增的偏向性,同时借助乳滴物理隔离DNA片段,降低了单细胞扩增时人为导致的基因融合,但phi29 DNA聚合酶对不完整DNA扩增效率较低,可能会降低扩增产物基因组覆盖度。MDA技术比较适合新鲜样本的单细胞基因组扩增,不适合用于固定后的单细胞样本。2.3 多次退火环状循环扩增技术
多次退火环状循环扩增技术(multiple annealing and looping-based amplification cycles, MALBAC)[22]将PCR和MDA技术联合,同时兼顾了单细胞基因组扩增的保真度和均一性。MALBAC单细胞基因组扩增分为两个过程:前5个循环采用Bst DNA聚合酶完成多重置换扩增,完整的扩增产物两端引入互补序列可以形成loop结构,封闭新扩增的DNA子链,避免新合成的DNA子链被当作扩增反应DNA模板;PCR扩增利用高温变性将完整的扩增子loop结构打开,经过PCR反应完成单细胞基因组扩增。MALBAC技术利用loop结构抑制了指数扩增,尽可能的从单细胞原始基因组模板合成子链DNA,第二步扩增使用PCR方法尽可能的保证扩增产物的均一性。MALBAC技术第一步应用多重置换反应将扩增产物全基因组覆盖度提高到90%以上,在一定程度保证了扩增产物的保真性,适用于SNV的检测,同时loop结构巧妙的应用实现了单细胞基因组准线性扩增,抑制了第一步反应指数扩增,尽可能的从单细胞原始基因组模板合成子链DNA,保证了扩增产物的均一性,可用于基因组拷贝数变异的检测。MALBAC技术可以同时用于新鲜样本和固定后的单细胞样本的单细胞基因组扩增,MALBAC技术的应用促进了临床辅助生殖技术的发展[23]。2.4 单细胞基因组线性扩增技术
Tn5转座酶系统因其可以在基因组DNA上随机打断,在打断位置插入一段已知序列,并且稳定性好的特点,已经成为分子遗传研究和基因诊断应用的常用工具酶[24]。通过转座子介导的单细胞基因组线性扩增技术(linear amplification via transposon insertion, LIANTI)[25]利用Tn5转座酶打断基因组DNA时引入T7启动子,通过体外转录以基因组DNA片段为模板,大量合成mRNA,再通过mRNA合成大量cDNA双链,每次转录合成mRNA都是以最原始的DNA片段为模板,实现了单细胞基因组线性扩增。LIANTI技术使得单细胞基因组测序覆盖提高到97%,同时兼顾了扩增的均一性,而且降低了扩增错误率,等位基因丢失率仅17%。LIANTI技术可同时适用于检测单细胞SNV、Indel和CNV。实验中扩增后的cDNA双链仍然使用传统的高通量测序文库构建方法,仍然是每个单细胞扩增后的DNA片段单独构建文库,所以LIANTI测序通量有限。2.5 高通量单细胞基因组扩增技术
肿瘤细胞基因组进化重建研究往往需要一次性研究大量肿瘤单细胞基因组突变特征,低通量的单细胞测序技术成本高,高人力成本。2017年,加拿大英属哥伦比亚大学的Hansen课题组和美国俄勒冈健康与科学大学的Adey课题组同时发表文章,报道利用Tn5转座酶开发了大规模、低成本的单细胞基因组CNV测序技术[26,27]。直接建库技术(direct library preparation, DLP)[26]借助微流控平台实现单细胞样本的获取,利用Tn5转座酶打断DNA引入接头时并没有添加index,通过11个PCR循环反应引入index和测序接头,将多个单细胞文库混合后进行基因测序。单细胞组合标签测序技术(single-cell combinatorial index sequencing, SCI-seq)[27]使用FACS将单细胞回收到96孔板中,在Tn5转座酶打断DNA引入接头时添加index1,然后将添加index1的DNA片段混匀后重新分组,通过PCR循环反应引入index2,再将文库混匀后进行基因测序。DLP和SCI-seq技术适用于高通量的、低成本单细胞CNV分析,由于Tn5转座酶系统酶切后的DNA片段存在一些大片段,超出了二代测序读长范围,导致基因组覆盖度降低,所以不适用于单细胞SNV和Indel检测,而且DLP和SCI-seq技术在PCR扩增过程中仍然会引入一定偏向性。为了降低PCR扩增DNA片段带来的偏向性,Yin等[28]将sci技术和LIANTI技术结合并进行了优化,开发了一种高通量单细胞基因组线性扩增技术(sci-L3-WGS)。借助sci组合标签技术,sci-L3-WGS大大提高了LIANTI技术的通量。第一轮加标签,Tn5转座酶随机打断单细胞基因组并加上标签1,第二轮加标签2和T7转录启动子序列连接到DNA片段末端,T7转录启动子的引入,可以实现体外转录反应,以DNA片段为模板,线性扩增的方式合成大量mRNA,再将mRNA逆转录合成cDNA,合成cDNA第二条链的时候再引入标签3,最后在DNA片段末端加上测序接头,完成的文库DNA片段含有3种标签组合,因此sci-L3- WGS技术可以实现高通量单细胞基因组线性扩增。sci-L3-WGS技术细胞回收率高达90%,以更少的测序数据达到更高覆盖度,单细胞基因组可覆盖约97,000片段,测序数据高达86%为有效reads,比LIANTI技术61%的有效reads提高了25%。以上高通量技术虽然一定程度上解决了单细胞基因组测序细胞通量问题,自动化平台则可以进一步减少人为操作,大大提高实验效率。Tapestri技术平台最先使用微流控平台实现单细胞多基因突变检测[29],采用油包水的方式高通量的分离单细胞,使用多重PCR特异性扩增多个目标区域靶序列,引入barcode,通过高通量测序实现单细胞靶基因SNV检测,同时利用扩增的目标片段可以低分辨率的分析单细胞CNV,只是扩增区域有限,覆盖度不是很高。单细胞捕获效率有限,需要一次性上机大量单细胞样本,不适合少量单细胞样本的检测。10× genomics是另一款基于微流控自动化、高通量的检测单细胞基因组拷贝数变异的平台[30],10×genomics进行单细胞基因组测序时需要进行两次捕获,第一次将磁球与单细胞包裹在一起,通过磁球上引物随机扩增单细胞基因组片段,第二次将凝胶球与磁球包裹起来,将barcode标记到单细胞DNA片段上,一次操作可以检测数千个肿瘤单细胞基因组拷贝数变异,进而基于肿瘤单细胞CNV特征对肿瘤细胞进行分群,因为需要进行两次捕获,所以捕获率只有15%左右。
3 单细胞基因组测序在生物医学中的应用
3.1 单细胞基因组测序用于肿瘤异质性研究
肿瘤组织由肿瘤祖细胞发展而来,在肿瘤形成过程中,肿瘤细胞不断积累新的基因突变,形成肿瘤内异质性。肿瘤异质性是肿瘤复发、转移和耐药的主要因素之一。研究肿瘤异质性与肿瘤临床诊断和治疗息息相关。肿瘤基因测序通常是提取肿瘤组织中大量细胞基因组DNA,检测肿瘤细胞基因突变,但这样检测的基因突变结果代表的是多种类型细胞平均化的结果。单细胞基因组扩增技术的建立,可以从单细胞水平研究肿瘤细胞基因突变,进而比较肿瘤单细胞之间的基因突变异质性。肿瘤异质性研究早期,单细胞基因组扩增主要基于PCR技术完成, 虽然扩增后的基因组覆盖度有限,仍然可以满足基因组拷贝数变异的分析。Klein等[31]从乳腺癌患者骨髓中分离到稀有肿瘤细胞,扩增肿瘤细胞单细胞基因组,利用比较基因组杂交技术探索肿瘤但细胞CNV突变分析。Martelotto 等[32]基于PCR技术建立了肿瘤组织石蜡切片(formalin-fixed paraffin-embedded, FFPE)样本单细胞CNV 分析方法。应用NGS建立了一种重现性较好、准确度较高的FFPE样本单细胞CNV 分析方法,有助于利用临床肿瘤FFPE样本,研究肿瘤进化机制。美国德州大学MD安德森癌症中心Navin课题组应用单细胞基因组测序技术分析乳腺癌肿瘤单细胞CNV特征,加深了对乳腺癌肿瘤细胞进化机制的理解;应用单细胞基因组测序技术比较了原位肿瘤组织和转移肿瘤组织中肿瘤单细胞CNV变异差异,结果发现转移灶肿瘤细胞与原位灶肿瘤细胞具有类似的CNV变异,通过肿瘤单细胞CNV分析明确了乳腺癌转移确实是由原位肿瘤进展转移到其他器官形成转移病灶的,而不是由完全不同的独立肿瘤细胞克隆形成的[16]。通过乳腺癌肿瘤单细胞CNV分析,确定了乳腺癌肿瘤细胞“爆发式”进化模式,即肿瘤细胞CNV突变在乳腺癌原位肿瘤病灶早期就已经出现,并形成单克隆或多克隆,在一段时间内保持一定的稳定[33]。应用空间单细胞基因组测序技术研究乳腺原位导管癌和侵袭性导管癌肿瘤细胞克隆组成,揭示了乳腺癌多克隆共转移模式。肿瘤原位灶肿瘤细胞多个克隆可以同时转移到新的位置形成转移病灶[34]。
MDA技术的建立大大提高了单细胞SNV和Indel的检测,Xu等[35]应用MDA技术详细的描绘了一位肾癌患者肿瘤单细胞SNV突变谱,从单细胞水平证明了肾癌相关突变基因VHL和PBRM1可能与肿瘤发生没有直接关系,而且此肾癌患者肿瘤组织中并没有鉴定出明显的主克隆亚群,通过单细胞基因突变检测预示着肿瘤异质性要比之前的认识复杂的多。Hou等[36]和Wang等[37]应用单细胞测序分析了骨髓瘤和乳腺癌单细胞SNV突变谱,揭示了肿瘤单细胞之间碱基突变异质性。应用Tapestri高通量技术平台,在急性髓细胞性白血病(acute myeloid leukemia, AML)患者治疗后肿瘤单细胞中检测到携带NRAS、KRAS和FLT3突变的,提供了新的用药靶点[38]。虽然肿瘤细胞系具有均匀的染色体拷贝数变异,但Velazquez-Villarreal等[30]借助10×genomics高通量单细胞DNA拷贝数检测平台,从单细胞水平揭示了肿瘤细胞系单细胞之间拷贝数变异差异,发现了肿瘤细胞系中多个亚克隆,提示肿瘤细胞系较我们之前认识的更复杂。
3.2 单细胞基因组测序用于CTC检测
单细胞测序技术的发展促进了稀有细胞的检测与应用。血液CTC是由肿瘤组织脱落进入血液循环系统的肿瘤细胞,CTC计数可以用于患者预后疗效评估。Jonas等[39]应用单细胞测序分析了非转移性乳腺癌患者骨髓中播散肿瘤细胞(disseminated tumor cell, DTC)和原位肿瘤基因突变特征,确定了DTC的来源。Carlotta等[40]应用单细胞测序分析了脑转移乳腺癌患者CTC基因突变特征,揭示了乳腺癌脑转移相关信号通路基因突变潜在靶点。应用单细胞测序比较肺癌肺静脉血和外周血中CTC和原位肿瘤细胞及转移病灶肿瘤细胞基因突变特征,结果发现CTC携带了一部分原位肿瘤细胞基因突变特征,而与转移病灶肿瘤细胞基因突变特征更相近,明确了CTC是肺癌转移的中间途径[41,42]。Carter 等[43]和Su等[44]在各自研究中,应用单细胞测序检测了小细胞肺癌患者外周血中CTC单细胞CNV 变异特征,基于CTC单细胞CNV突变特征提出了患者接受化疗反应预测模型,用于患者化疗预后评估。吴保军等[45]应用单细胞基因组CNV变异分析,从肺癌患者恶性胸腔积液中鉴定出携带不同CNV变异的肿瘤细胞亚群,从单细胞水平揭示了肺癌患者恶性肿瘤细胞异质性。3.3 单细胞基因组测序用于胚胎移植前基因诊断和筛查
胚胎移植前PGD主要用于具有家族式遗传病家庭进行试管婴儿移植前胚胎基因诊断,可以确保新生儿不携带相关基因异常而进行的胚胎移植前基因检测;胚胎移植前PGS主要是针对体外受精胚胎进行的染色体倒位等异常的筛查。通常用来做PGD 或PGS的样本为极体或囊胚期单细胞,能够用于检测的DNA含量很少,并且能够检测的靶点比较少。单细胞基因组高通量测序技术的发展大大提高了PGD或PGS检测成功率:如CGH芯片、SNP杂交芯片以及NGS测序。Wells等[46]借助DOP-PCR单细胞扩增技术第一次完成了第一极体单细胞全基因组扩增,并采用CGH芯片成功检测到胚胎染色体异常情况。Daina等[47]使用MDA扩增胚胎活检单细胞样本,第一次完成了Lynch综合征家庭开展单细胞双因子PGD,可以从体外培养的胚胎中挑选既健康又具有整数倍性的胚胎用于胚胎移植。Tobler等[48]回顾性分析了498个胚胎的PGD/PGS结果,比较了SNP 芯片和CGH 杂交芯片用于MDA扩增胚胎单细胞染色体异常检测情况,提示SNP芯片和CGH杂交芯片具有类似的异常检出率。Huang等[49]利用3位孕妇捐献23枚胚胎样本,应用MALBAC技术,比较了MALBAC-scWGS技术、SNP芯片及CGH芯片用于PGD/PGS的准确性,MALBAC-scWGS检测结果与SNP芯片或者CGH芯片结果重复性达到78.26%,其中有8个胚胎使用三种检测技术的检测结果完全一致,验证了MALBAC-scWGS在PGD/ PGS检测中的应用价值。Wang等[50]将此技术应用拓展到线粒体基因异常性疾病的PGD/PGS检测中。单细胞基因组测序技术的发展极大的提高了临床产前诊断技术的应用和准确性。4 结语与展望
单细胞基因组扩增技术和高通量测序技术相结合极大的促进了单细胞基因组测序技术的发展,使得研究细胞之间基因差异达到单细胞水平。应用单细胞基因组测序技术,研究者对肿瘤多样性、肿瘤异质性以及肿瘤克隆进化机制不断加深理解,同时单细胞基因组测序技术促进了CTC检测和PGD/ PGS临床的应用。多种单细胞基因组测序技术各有优缺点,在科研或临床应用时应该根据实际需求选择合适的单细胞基因组测序技术。单细胞基因组测序技术的应用促进了肿瘤异质性研究,但仍然需要发展能够同时兼顾基因组扩增保真性和均一性的技术,提高肿瘤单细胞SNV、CNV、SV等基因突变的检测。随着肿瘤单细胞研究不断深入,人们对单细胞多组学的需求会越来越高。Macaulay等[51]最早将单细胞基因组和转录组测序联合使用,同时检测同一个单细胞样本的基因组和转录组,通过单细胞测序发现HCC38-BL细胞系10%细胞存在11号染色体三倍体,并通过FISH技术做了验证,Tang等[52]应用单细胞基因组和转录组测序技术分析了黑色素瘤患者不同部位肿瘤单细胞的突变负荷和基因表达特征。肿瘤细胞基因突变负荷检测揭示了被遮蔽的部位黑色素细胞突变负荷少于暴露于阳光的部位,间歇性暴露于阳关的背部和四肢的突变负荷多于脸部、脖子等暴露在阳光下的部位。Zhou等[53]联合单细胞基因组和转录组测序技术对结直肠癌肿瘤组织、淋巴结转移组织、癌旁组织、外周血样品以及健康人外周血中非上皮细胞进行了单细胞测序。研究发现人体正常组织中免疫细胞、成纤维细胞以及血管内皮细胞中广泛存在少量正常携带拷贝数变异。转录组测序分析在肿瘤组织成纤维细胞中BGN、RCN3、TAGLN、MYL9和TPM2表达量明显上调,鉴定出肿瘤微环境中成纤维细胞特异性表达的肿瘤标志基因。Hou等[54]又开发了单细胞基因组、甲基化组和转录组测序技术,Zachariadis等[55]应用Tn5转座子系统进一步提高了单细胞基因组和转录组测序的细胞通量。未来单细胞组学技术开发不仅仅考虑提高细胞通量,多组学技术联合开发,同时还需要考虑增加组织空间位置信息。随着技术的发展,单细胞测序技术会展现出更强的应用价值,供研究者选择在科学研究中选择。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1084/jem.192.3.393URLPMID:10934227 [本文引用: 1]

Clonal composition and T cell receptor (TCR) repertoire of CD4(+) and CD8(+) T cells infiltrating actively demyelinating multiple sclerosis (MS) lesions were determined with unprecedented resolution at the level of single cells. Individual CD4(+) or CD8(+) T cells were isolated from frozen sections of lesional tissue by micromanipulation and subjected to single target amplification of TCR-beta gene rearrangements. This strategy allows the assignment of a TCR variable region (V region) sequence to the particular T cell from which it was amplified. Sequence analysis revealed that in both cases investigated, the majority of CD8(+) T cells belonged to few clones. One of these clones accounted for 35% of CD8(+) T cells in case 1. V region sequence comparison revealed signs of selection for common peptide specificities for some of the CD8(+) T cells in case 1. In both cases, the CD4(+) T cell population was more heterogeneous. Most CD4(+) and CD8(+) clones were represented in perivascular infiltrates as well as among parenchymal T cells. In case 2, two of the CD8(+) clones identified in brain tissue were also detected in peripheral blood. Investigation of the antigenic specificities of expanded clones may help to elucidate their functional properties.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:16642997 [本文引用: 1]
[本文引用: 1]
DOI:10.1038/nbt.1561URLPMID:19668243 [本文引用: 1]
Recent advances in high-throughput DNA sequencing technologies have enabled order-of-magnitude improvements in both cost and throughput. Here we report the use of single-molecule methods to sequence an individual human genome. We aligned billions of 24- to 70-bp reads (32 bp average) to approximately 90% of the National Center for Biotechnology Information (NCBI) reference genome, with 28x average coverage. Our results were obtained on one sequencing instrument by a single operator with four data collection runs. Single-molecule sequencing enabled analysis of human genomic information without the need for cloning, amplification or ligation. We determined approximately 2.8 million single nucleotide polymorphisms (SNPs) with a false-positive rate of less than 1% as validated by Sanger sequencing and 99.8% concordance with SNP genotyping arrays. We identified 752 regions of copy number variation by analyzing coverage depth alone and validated 27 of these using digital PCR. This milestone should allow widespread application of genome sequencing to many aspects of genetics and human health, including personal genomics.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
loss of heterozygosity
and DNA sequence analysis of single cells
[本文引用: 1]
DOI:10.1038/nature09807URLPMID:21399628 [本文引用: 2]
Genomic analysis provides insights into the role of copy number variation in disease, but most methods are not designed to resolve mixed populations of cells. In tumours, where genetic heterogeneity is common, very important information may be lost that would be useful for reconstructing evolutionary history. Here we show that with flow-sorted nuclei, whole genome amplification and next generation sequencing we can accurately quantify genomic copy number within an individual nucleus. We apply single-nucleus sequencing to investigate tumour population structure and evolution in two human breast cancer cases. Analysis of 100 single cells from a polygenomic tumour revealed three distinct clonal subpopulations that probably represent sequential clonal expansions. Additional analysis of 100 single cells from a monogenomic primary tumour and its liver metastasis indicated that a single clonal expansion formed the primary tumour and seeded the metastasis. In both primary tumours, we also identified an unexpectedly abundant subpopulation of genetically diverse 'pseudodiploid' cells that do not travel to the metastatic site. In contrast to gradual models of tumour progression, our data indicate that tumours grow by punctuated clonal expansions with few persistent intermediates.
[本文引用: 1]
[本文引用: 1]
DOI:10.1371/journal.pone.0105585URLPMID:25136831 [本文引用: 1]
Single-cell sequencing is emerging as an important tool for studies of genomic heterogeneity. Whole genome amplification (WGA) is a key step in single-cell sequencing workflows and a multitude of methods have been introduced. Here, we compare three state-of-the-art methods on both bulk and single-cell samples of E. coli DNA: Multiple Displacement Amplification (MDA), Multiple Annealing and Looping Based Amplification Cycles (MALBAC), and the PicoPLEX single-cell WGA kit (NEB-WGA). We considered the effects of reaction gain on coverage uniformity, error rates and the level of background contamination. We compared the suitability of the different WGA methods for the detection of copy-number variations, for the detection of single-nucleotide polymorphisms and for de-novo genome assembly. No single method performed best across all criteria and significant differences in characteristics were observed; the choice of which amplifier to use will depend strongly on the details of the type of question being asked in any given experiment.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1101/gr.177881.114URL [本文引用: 1]
Massively parallel DNA sequencing of thousands of samples in a single machine-run is now possible, but the preparation of the individual sequencing libraries is expensive and time-consuming. Tagmentation-based library construction, using the Tn5 transposase, is efficient for generating sequencing libraries but currently relies on undisclosed reagents, which severely limits development of novel applications and the execution of large-scale projects. Here, we present simple and robust procedures for Tn5 transposase production and optimized reaction conditions for tagmentation-based sequencing library construction. We further show how molecular crowding agents both modulate library lengths and enable efficient tagmentation from subpicogram amounts of cDNA. The comparison of single-cell RNA-sequencing libraries generated using produced and commercial Tn5 demonstrated equal performances in terms of gene detection and library characteristics. Finally, because naked Tn5 can be annealed to any oligonucleotide of choice, for example, molecular barcodes in single-cell assays or methylated oligonucleotides for bisulfite sequencing, custom Tn5 production and tagmentation enable innovation in sequencing-based applications.
URLPMID:28408603 [本文引用: 1]
URLPMID:28068316 [本文引用: 2]
[本文引用: 2]
URLPMID:31495564 [本文引用: 1]
URLPMID:30087104 [本文引用: 1]
URLPMID:32587328 [本文引用: 2]
[本文引用: 1]
DOI:10.1038/nm.4279URLPMID:28165479 [本文引用: 1]
A substantial proportion of tumors consist of genotypically distinct subpopulations of cancer cells. This intratumor genetic heterogeneity poses a substantial challenge for the implementation of precision medicine. Single-cell genomics constitutes a powerful approach to resolve complex mixtures of cancer cells by tracing cell lineages and discovering cryptic genetic variations that would otherwise be obscured in tumor bulk analyses. Because of the chemical alterations that result from formalin fixation, single-cell genomic approaches have largely remained limited to fresh or rapidly frozen specimens. Here we describe the development and validation of a robust and accurate methodology to perform whole-genome copy-number profiling of single nuclei obtained from formalin-fixed paraffin-embedded clinical tumor samples. We applied the single-cell sequencing approach described here to study the progression from in situ to invasive breast cancer, which revealed that ductal carcinomas in situ show intratumor genetic heterogeneity at diagnosis and that these lesions may progress to invasive breast cancer through a variety of evolutionary processes.
DOI:10.1038/ng.3641URLPMID:27526321 [本文引用: 1]
Aneuploidy is a hallmark of breast cancer; however, knowledge of how these complex genomic rearrangements evolve during tumorigenesis is limited. In this study, we developed a highly multiplexed single-nucleus sequencing method to investigate copy number evolution in patients with triple-negative breast cancer. We sequenced 1,000 single cells from tumors in 12 patients and identified 1-3 major clonal subpopulations in each tumor that shared a common evolutionary lineage. For each tumor, we also identified a minor subpopulation of non-clonal cells that were classified as metastable, pseudodiploid or chromazemic. Phylogenetic analysis and mathematical modeling suggest that these data are unlikely to be explained by the gradual accumulation of copy number events over time. In contrast, our data challenge the paradigm of gradual evolution, showing that the majority of copy number aberrations are acquired at the earliest stages of tumor evolution, in short punctuated bursts, followed by stable clonal expansions that form the tumor mass.
[本文引用: 1]
DOI:10.1016/j.cell.2012.02.025URL [本文引用: 1]
Clear cell renal cell carcinoma (ccRCC) is the most common kidney cancer and has very few mutations that are shared between different patients. To better understand the intratumoral genetics underlying mutations of ccRCC, we carried out single-cell exome sequencing on a ccRCC tumor and its adjacent kidney tissue. Our data indicate that this tumor was unlikely to have resulted from mutations in VHL and PBRM1. Quantitative population genetic analysis indicates that the tumor did not contain any significant clonal subpopulations and also showed that mutations that had different allele frequencies within the population also had different mutation spectrums. Analyses of these data allowed us to delineate a detailed intratumoral genetic landscape at a single-cell level. Our pilot study demonstrates that ccRCC may be more genetically complex than previously thought and provides information that can lead to new ways to investigate individual tumors, with the aim of developing more effective cellular targeted therapies.
DOI:10.1016/j.cell.2012.02.028URL [本文引用: 1]
Tumor heterogeneity presents a challenge for inferring clonal evolution and driver gene identification. Here, we describe a method for analyzing the cancer genome at a single-cell nucleotide level. To perform our analyses, we first devised and validated a high-throughput whole-genome single-cell sequencing method using two lymphoblastoid cell line single cells. We then carried out whole-exome single-cell sequencing of 90 cells from a JAK2-negative myeloproliferative neoplasm patient. The sequencing data from 58 cells passed our quality control criteria, and these data indicated that this neoplasm represented a monoclonal evolution. We further identified essential thrombocythemia (ET)-related candidate mutations such as SESN2 and NTRK1, which may be involved in neoplasm progression. This pilot study allowed the initial characterization of the disease-related genetic architecture at the single-cell nucleotide level. Further, we established a single-cell sequencing method that opens the way for detailed analyses of a variety of tumor types, including those with high genetic complex between patients.
DOI:10.1038/nature13600URL [本文引用: 1]
[本文引用: 1]
DOI:10.1186/s13059-016-1109-7URLPMID:27931250 [本文引用: 1]
BACKGROUND: Single-cell micro-metastases of solid tumors often occur in the bone marrow. These disseminated tumor cells (DTCs) may resist therapy and lay dormant or progress to cause overt bone and visceral metastases. The molecular nature of DTCs remains elusive, as well as when and from where in the tumor they originate. Here, we apply single-cell sequencing to identify and trace the origin of DTCs in breast cancer. RESULTS: We sequence the genomes of 63 single cells isolated from six non-metastatic breast cancer patients. By comparing the cells' DNA copy number aberration (CNA) landscapes with those of the primary tumors and lymph node metastasis, we establish that 53% of the single cells morphologically classified as tumor cells are DTCs disseminating from the observed tumor. The remaining cells represent either non-aberrant
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/nm.4239URLPMID:27869802 [本文引用: 1]
In most patients with small-cell lung cancer (SCLC)-a metastatic, aggressive disease-the condition is initially chemosensitive but then relapses with acquired chemoresistance. In a minority of patients, however, relapse occurs within 3 months of initial treatment; in these cases, disease is defined as chemorefractory. The molecular mechanisms that differentiate chemosensitive from chemorefractory disease are currently unknown. To identify genetic features that distinguish chemosensitive from chemorefractory disease, we examined copy-number aberrations (CNAs) in circulating tumor cells (CTCs) from pretreatment SCLC blood samples. After analysis of 88 CTCs isolated from 13 patients (training set), we generated a CNA-based classifier that we validated in 18 additional patients (testing set, 112 CTC samples) and in six SCLC patient-derived CTC explant tumors. The classifier correctly assigned 83.3% of the cases as chemorefractory or chemosensitive. Furthermore, a significant difference was observed in progression-free survival (PFS) (Kaplan-Meier P value = 0.0166) between patients designated as chemorefractory or chemosensitive by using the baseline CNA classifier. Notably, CTC CNA profiles obtained at relapse from five patients with initially chemosensitive disease did not switch to a chemorefractory CNA profile, which suggests that the genetic basis for initial chemoresistance differs from that underlying acquired chemoresistance.
DOI:10.1158/1078-0432.CCR-18-3571URLPMID:31113842 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/s0015-0282(02)03271-5URLPMID:12215331 [本文引用: 1]
OBJECTIVE: To develop a preimplantation genetic diagnosis (PGD) protocol that allows any form of chromosome imbalance to be detected. DESIGN: Case report employing a method based on whole-genome amplification and comparative genomic hybridization (CGH). SETTING: Clinical IVF laboratory. PATIENT(S): A 40-year-old IVF patient. INTERVENTION(S): Polar body and blastomere biopsy. MAIN OUTCOME MEASURE(S): Detection of aneuploidy. RESULT(S): Chromosome imbalance was detected in 9 of 10 polar bodies. A variety of chromosomes were aneuploid, but chromosomal size was found to be an important predisposing factor. In three cases, the resulting embryos could be tested using fluorescence in situ hybridization, and in each case the CGH diagnosis was confirmed. A single embryo could be recommended for transfer on the basis of the CGH data, but no pregnancy ensued. CONCLUSION(S): Evidence suggests that preferential transfer of chromosomally normal embryos can improve IVF outcomes. However, current PGD protocols do not allow analysis of every chromosome, and therefore a proportion of abnormal embryos remains undetected. We describe a method that allows every chromosome to be assessed in polar bodies and oocytes. The technique was accurate and allowed identification of aneuploid embryos that would have been diagnosed as normal by standard PGD techniques. As well as comprehensive cytogenetic analysis, this protocol permits simultaneous testing for multiple single-gene disorders.
[本文引用: 1]
DOI:10.1007/s10815-014-0230-3URLPMID:24771116 [本文引用: 1]
PURPOSE: To compare single nucleotide polymorphism (SNP) and comparative genomic hybridization (aCGH) microarray platforms to evaluate embryos for parental translocation imbalances and aneuploidy. METHODS: A retrospective review of preimplantation genetic diagnosis and screening (PGD/PGS) results of 498 embryos from 63 couples undergoing 75 in vitro fertilization cycles due to parental carriers of a reciprocal translocation. RESULTS: There was no significant difference between SNP and aCGH microarrays when comparing the prevalence of embryos that were euploid with no translocation imbalance, euploidy with a parental translocation imbalance or aneuploid with or without the parental chromosome imbalance. The clinical pregnancy rates were also equivalent for SNP (60 %) versus aCGH (65 %) microarrays. Of 498 diagnosed embryos, 45 % (226/498) were chromosomally normal without translocation errors or aneuploidy, 22 % (112/498) were euploid but had a parentally derived unbalanced chromosomal segregant, 8 % (42/498) harbored both a translocation imbalance and aneuploidy and 24 % (118/498) of embryos were genetically balanced for the parental reciprocal translocation but were aneuploid for other chromosomes. The overall clinical pregnancy rate per IVF cycle following SNP or aCGH microarray analysis was 61 % and was higher if the biopsy was done on blastocysts (65 %) versus cleavage stage embryos (59 %), although not statistically significant. CONCLUSIONS: SNP or aCGH microarray technologies demonstrate equivalent clinical findings that maximize the pregnancy potential in patients with known parental reciprocal chromosomal translocations.
Design: The 24-chromosome aneuploidy analyses of the blastomeres included comparative genomic hybridization (CGH), single nucleotide polymorphism (SNP), and MALBAC sequencing.
Setting: University-affiliated IVF center.
Patient(s): Three couples who delivered babies from the same IVF cycle, which included 23 donated, frozen cleavage embryos.
Intervention(s): None.
Main Outcome Measure(s): Three blastomeres were selected from each single embryo and subject to CGH, SNP, and MALBAC sequencing for 24-chromosome aneuploidy, respectively. The results of MALBAC sequencing were compared with the results of CGH and SNP. The chromosomal status and occurrence of the abnormal chromosomes were investigated. The relationship between the embryos' morphology and the euploid state was analyzed.
Result(s): Among the 23 donated embryos, the MALBAC sequencing results of 18 (78.26%) embryos were identical to those of CGH or SNP, including 8 embryos that had identical results by all three techniques. In terms of euploidy, only 6 of these 23 embryos (26.09%) were diploid. Blastomere abnormality was observed in all autosomes and sex chromosomes. In addition, the frequency of abnormality was different for certain chromosomes.
Conclusion(s): With sequencing at 0.04 x genome depth, MALBAC sequencing has been validated as a satisfactory method for 24-chromosome aneuploidy screening. The proportion of abnormal chromosomes was high in cleavage-stage embryos, and any chromosome could be abnormal. (C) 2014 by American Society for Reproductive Medicine.
DOI:10.1016/j.fertnstert.2014.08.015URL [本文引用: 1]
DOI:10.1016/j.rbmo.2017.10.110URLPMID:29203383 [本文引用: 1]
Single cell whole genome sequencing helps to decipher the genome heterogeneity within a cell population and facilitates the analysis of trace amounts of genetic material, such as is found in human embryos. The mitochondrial genome, although an important part of the genetic composition of eukaryotic cells, is often neglected in single cell genome analysis. A recently developed single cell whole genome amplification method was used, known as multiple annealing and looping based amplification cycles (MALBAC-NGS), for simultaneous analysis of chromosomal and mitochondrial genomes at the single cell level. The platform was validated by a series of technical and biological replicates and used for chromosomal and mitochondrial copy number analysis in 399 in-vitro fertilized embryos from 81 couples. A positive correlation of maternal age with increased mitochondria quantity (beta = 0.176, P = 0.001) was observed after adjusting for the impact of cell type. Lower numbers of mitochondria were detected in successfully implanted embryos, although the difference was not significant. It is proposed that MALBAC-NGS could potentially be used for an advanced pre-implantation genetic screening procedure with both chromosomal constitution and mitochondrial copy number being evaluated.
DOI:10.1038/nmeth.3370URLPMID:25915121 [本文引用: 1]
The simultaneous sequencing of a single cell's genome and transcriptome offers a powerful means to dissect genetic variation and its effect on gene expression. Here we describe G&T-seq, a method for separating and sequencing genomic DNA and full-length mRNA from single cells. By applying G&T-seq to over 220 single cells from mice and humans, we discovered cellular properties that could not be inferred from DNA or RNA sequencing alone.
DOI:10.1038/s41586-020-2785-8URLPMID:33029006 [本文引用: 1]
Every cell in the human body has a unique set of somatic mutations, but it remains difficult to comprehensively genotype an individual cell(1). Here we describe ways to overcome this obstacle in the context of normal human skin, thus offering a glimpse into the genomic landscapes of individual melanocytes from human skin. As expected, sun-shielded melanocytes had fewer mutations than sun-exposed melanocytes. However, melanocytes from chronically sun-exposed skin (for example, the face) had a lower mutation burden than melanocytes from intermittently sun-exposed skin (for example, the back). Melanocytes located adjacent to a skin cancer had higher mutation burdens than melanocytes from donors without skin cancer, implying that the mutation burden of normal skin can be used to measure cumulative sun damage and risk of skin cancer. Moreover, melanocytes from healthy skin commonly contained pathogenic mutations, although these mutations tended to be weakly oncogenic, probably explaining why they did not give rise to discernible lesions. Phylogenetic analyses identified groups of related melanocytes, suggesting that melanocytes spread throughout skin as fields of clonally related cells that are invisible to the naked eye. Overall, our results uncover the genomic landscapes of individual melanocytes, providing key insights into the causes and origins of melanoma.
URLPMID:33096021 [本文引用: 1]
DOI:10.1038/cr.2016.23URLPMID:26902283 [本文引用: 1]
Single-cell genome, DNA methylome, and transcriptome sequencing methods have been separately developed. However, to accurately analyze the mechanism by which transcriptome, genome and DNA methylome regulate each other, these omic methods need to be performed in the same single cell. Here we demonstrate a single-cell triple omics sequencing technique, scTrio-seq, that can be used to simultaneously analyze the genomic copy-number variations (CNVs), DNA methylome, and transcriptome of an individual mammalian cell. We show that large-scale CNVs cause proportional changes in RNA expression of genes within the gained or lost genomic regions, whereas these CNVs generally do not affect DNA methylation in these regions. Furthermore, we applied scTrio-seq to 25 single cancer cells derived from a human hepatocellular carcinoma tissue sample. We identified two subpopulations within these cells based on CNVs, DNA methylome, or transcriptome of individual cells. Our work offers a new avenue of dissecting the complex contribution of genomic and epigenomic heterogeneities to the transcriptomic heterogeneity within a population of cells.
DOI:10.1016/j.molcel.2020.09.025URLPMID:33068522 [本文引用: 1]
To address how genetic variation alters gene expression in complex cell mixtures, we developed direct nuclear tagmentation and RNA sequencing (DNTR-seq), which enables whole-genome and mRNA sequencing jointly in single cells. DNTR-seq readily identified minor subclones within leukemia patients. In a large-scale DNA damage screen, DNTR-seq was used to detect regions under purifying selection and identified genes where mRNA abundance was resistant to copy-number alteration, suggesting strong genetic compensation. mRNA sequencing (mRNA-seq) quality equals RNA-only methods, and the low positional bias of genomic libraries allowed detection of sub-megabase aberrations at ultra-low coverage. Each cell library is individually addressable and can be re-sequenced at increased depth, allowing multi-tiered study designs. Additionally, the direct tagmentation protocol enables coverage-independent estimation of ploidy, which can be used to identify cell singlets. Thus, DNTR-seq directly links each cell's state to its corresponding genome at scale, enabling routine analysis of heterogeneous tumors and other complex tissues.

{kind=link}
{kind=link}