 ,北京理工大学生命学院分子医学和生物诊疗工信部重点实验室,北京 100081
,北京理工大学生命学院分子医学和生物诊疗工信部重点实验室,北京 100081The roles of Rab protein family in neurological diseases
Anping Wu, Hong Qing, Zhenzhen Quan,Key Laboratory of Molecular Medicine and Biotherapy,School of Life Science, Beijing Institute of Technology, Beijing 100081, China通讯作者: 全贞贞,博士,副研究员,研究方向:阿尔茨海默病的分子机制。E-mail:qzzbit2015@bit.edu.cn
编委: 史岸冰
收稿日期:2020-11-17修回日期:2021-01-6网络出版日期:2021-01-20
| 基金资助: |
Received:2020-11-17Revised:2021-01-6Online:2021-01-20
| Fund supported: |
作者简介 About authors
吴安平,在读硕士研究生,专业方向:生物工程。E-mail:

摘要
细胞内膜囊泡运输是一个复杂的通路网络,Rab GTPases是膜囊泡运输的主要调节剂,通常被认为是细胞内吞和分泌系统中各种细胞器和囊泡的特异性标记和识别物。与Rab蛋白相关的轴突运输、内体运输发生障碍是造成神经退行性疾病的重要原因之一。本文主要介绍了Rab蛋白在多种神经退行性疾病病理机制中的作用机理与调控机制,同时讨论了线粒体和胶质细胞功能异常与Rab蛋白之间的关联。深入探究Rab蛋白的作用机制对人类神经性疾病的早期诊断和治疗具有潜在的指导意义。
关键词:
Abstract
The intracellular membrane trafficking is a complicated pathway network. Rab GTPases are key regulators of membrane trafficking that are generally considered as specific markers and indicators of various organelles and membrane trafficking in endocytic and secretory pathways. Dysfunction in axonal and endosomal transport related to Rab proteins is one of the most important causes of neurodegenerative diseases. In this review, we mainly introduce how the Rab proteins change in different neurodegenerative diseases and their regulatory roles in the pathological mechanisms of related diseases. We also discuss the relationships between mitochondrial and glial cell dysfunctions and Rab proteins. Further exploration of the regulatory roles of Rab proteins will shed lights on revealing the pathogenic mechanisms of neurological diseases and providing potential targets for the early diagnosis and treatment of neurological diseases.
Keywords:
PDF (867KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
吴安平, 庆宏, 全贞贞. Rab蛋白家族在神经类疾病中的作用. 遗传[J], 2021, 43(1): 16-29 doi:10.16288/j.yczz.20-318
Anping Wu.
中国是世界上痴呆症患者最多的国家,给公共和卫生保健系统带来了沉重负担。研究表明,2016年我国老年人口患痴呆症的概率是5.6%[1],目前我国的痴呆症患者大约有3100万,给患者及其家人带来沉重负担,将会引发越来越严重的公共卫生问题和社会问题[2]。
痴呆等认知障碍的形成是由于神经元结构和功能逐渐丧失,以及神经元死亡和胶质细胞失衡所导致的,主要包括阿尔兹海默病(Alzheimer’s disease, AD)、帕金森病(Parkinson’s disease, PD)、亨廷顿病(Huntington’s disease, HD)、肌萎缩性侧索硬化症(amyotrophic lateral sclerosis, ALS)、不同类型脊髓小脑共济失调病等神经类疾病。研究表明Rab蛋白能够调节神经元细胞的内吞和轴突运输,参与多种神经类疾病的病理进程。本文就Rab蛋白如何调控各类神经类疾病进行了归纳和分析,进一步阐述了Rab蛋白在的作用机制,为今后探究Rab蛋白在神经类疾病中的作用提供了思路和参考。
1 Rab蛋白结构
Rab蛋白是存在于细胞质膜和细胞器膜中的一类调节型小分子GTP结合蛋白,是Ras超家族中最大的亚家族。Rab蛋白在进化上高度保守,几乎存在于所有的真核生物中。Rab蛋白平均分子量大小为25 kDa,约由200个氨基酸组成。Rab蛋白含G结构域、N端与C端,其中,G结构域高度保守,N端与C端高度可变[3]。G结构域包含6个β折叠、5个α螺旋及5个多肽环,多肽环的氨基酸序列高度保守,是Mg2+和鸟嘌呤的结合位点,同时催化GTP水解。Rab蛋白的羧基末端包含-XXXCC-、-XXCCX-、-XCCXX-、-CCXXX-或-XXCXC-基本序列,其中两个半胱氨酸是异戊二烯化的底物。这种修饰对于膜结合至关重要。羧基末端高变区是必需的,但不足以将Rab蛋白正确靶向到细胞中的特定位置[3]。大多数Rab蛋白通过翻译后在羧基端附近的两个半胱氨酸上将两个香叶基(20碳聚异戊二烯基)基团与膜或膜蛋白复合物紧密结合,少数Rab蛋白只有一个香叶基[4]。N端的作用可能是参与指导C端半胱氨酸进行异戊二烯化修饰。G结构域的“开关”区域(称为开关1和2)通过与GTP结合产生稳定的构象,是Rab蛋白特异性识别其细胞效应子关键区域。不同的Rab蛋白在进行开“ON”和关“OFF”状态切换时,可能会发生比较灵活的构象变化[5]。对Rab蛋白突变体及嵌合体的研究表明,分子开关I和分子开关II、高度可变的N端和C端是Rab蛋白重要功能的决定因素。Rab蛋白的主要功能是调节细胞信号的传导、细胞的生长和分化[6]。Rab GTPases作为分子开关,在GTP结合的活性形式和GDP结合的非活性形式之间转换,控制细胞内囊泡运输的各个方面。Rab-GTP结合型和Rab-GDP结合型之间的循环受蛋白质调节剂严格控制:GDP解离抑制剂(GDP dissociation inhibitors, GDI)调控Rab蛋白与膜的连接及Rab蛋白从膜上解离,GDI作为循环因子起作用,只与异戊二烯化修饰的无活性的Rab蛋白结合,将Rab-GDP保持在胞液中。伴随着Rab蛋白与供体囊泡的膜或膜蛋白复合物结合,鸟嘌呤交换因子(guanine nucleotide exchange factor, GEF)催化Rab- GDP形式转化成Rab-GTP形式,Rab蛋白被激活,Rab-GTP与下游效应子相互作用,使效应子功能发生变化;GTPase活化蛋白(GTPase accelerating protein, GAP)催化GTP水解,Rab蛋白失活,不再与效应子相互作用;GDI从膜上抽出Rab-GDP,与其形成复合物,又循环到胞液中。通过激活/失活循环,Rab蛋白作为分子开关将上游信号传递给下游效应子(图1)。Rab蛋白在其活性形式时与许多不同的蛋白质相互作用,这些蛋白质被称为Rab蛋白的效应子。Rab蛋白的效应子在囊泡的形成、囊泡沿细胞骨架的转运及囊泡与靶膜的拴系锚定等阶段中发挥作用[7]。
图1
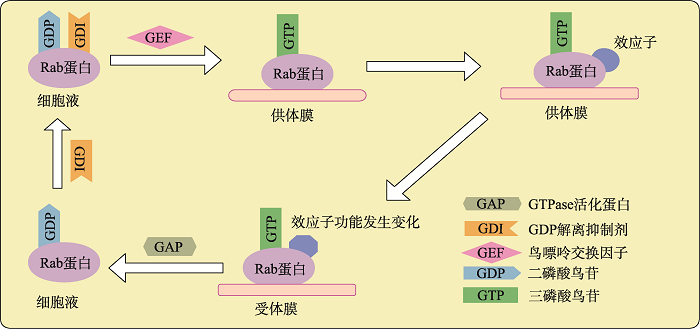 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1Rab蛋白的功能循环示意图
Rab蛋白与GDI、GDP结合保存在胞液中,由GEF激活转移到供体膜上转化成Rab-GTP形式,使下游效应子发挥作用,激活态的Rab-GTP蛋白被GAP催化水解,随后GDI与Rab-GDP又形成复合物,循环到胞液中,形成循环。
Fig. 1Schematic diagram of the functional cycle of Rab proteins
2 Rab蛋白在细胞中的调控途径
Rab蛋白最主要的功能是调控细胞内吞,内吞运输系统主要由内吞、循环、降解等子系统组成。其中,内吞循环运输负责将膜上大分子送回质膜回收再利用,在细胞极性形成和维持、细胞分裂及迁移、神经突触可塑性、免疫应答、生长因子受体调控等过程中不可或缺[8]。降解途径导向晚期内含体和溶酶体,货物在其中发生降解[9]。沿内吞途径的每个运输步骤均由不同的Rab蛋白介导:Rab5介导货物从质膜到早期内含体的运输并充当早期内含体的标记物[10];而Rab7则调节货物从早期到晚期内含体的运输并充当晚期内含体的标记物[11];Rab4,Rab11和Rab35介导回收途径;Rab4和Rab35调控货物从早期内含体、循环内含体直接返回质膜的快速回收[12,13];Rab11控制循环内含体的回收[14](图2)。研究表明,晚期内含体和溶酶体功能异常,在细胞衰老和神经退行性疾病(如帕金森病和阿尔茨海默病)中起着关键作用。图2
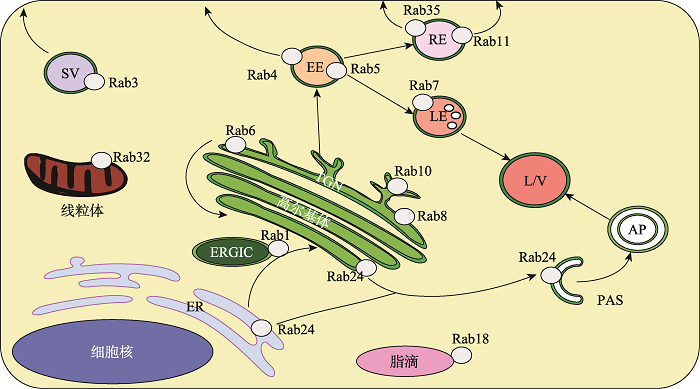 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2Rab蛋白在细胞中的分布
ER:内质网;TGN: 反高尔基网络;EE:早期内含体;LE:晚期内含体;RE:循环内含体;AP:自噬体;ERGIC:内质网一高尔基体中介组分;L/V:溶酶体/液泡;PAS:前自噬体;SV:分泌囊泡/颗粒。
Fig. 2Distribution of Rab proteins in cells
3 Rab蛋白在神经元中的相关功能
神经元的特殊形态和功能高度依赖于膜运输的严格调节。内吞功能障碍破坏了双向轴突运输和突触小泡与靶膜的对接,从而导致神经运输和神经营养信号发生异常。轴突运输的改变,特别是神经营养蛋白受体在轴突运输的改变与几种人类神经退行性疾病都有关联[15,16]。Rab3A是突触活性区的结构成分,与其他核心活性区蛋白RIM1和MUNC13一起形成三元复合物,调节突触小泡与膜的结合过程,以释放神经递质[17]。Rab3蛋白还参与淀粉样前体蛋白(amyloid protein precursor, APP)的快速轴突运输[18]。Rab7控制神经营养蛋白受体的逆行轴突运输[19],Rab7损伤可诱导神经营养因子TrkA在内含体中的积累,进而阻滞神经营养蛋白逆行转运[20]。Rab26位于突触小泡表面,自噬蛋白Atg16L1、LC3和Rab33B被募集到这些突触小泡中,从而连接了突触小泡与自噬机制[21]。与其他细胞类型相比,神经元可能对蛋白质水平的轻微升高或降低更敏感,特定蛋白质水平的微小变化也可能导致神经元功能障碍和死亡。4 Rab蛋白在神经退行性疾病中的作用
4.1 阿尔茨海默病
阿尔茨海默病是神经变性和痴呆症的最常见形式。超过90%~95%的AD病例是散发性(sporadic AD,SAD)且与已知的疾病突变无关。大约5%~10%的病例被归类为家族性AD (familial AD, FAD),与早老素1、早老素2和APP的突变有关。这些突变导致β-淀粉样蛋白(β-amyloid, Aβ)的产生发生异常。在Aβ形成途径中,APP首先被β-分泌酶切割,其中主要发挥作用的是β-分泌酶1 (β-site APP cleaving enzyme 1, BACE1),使APP 596~597位氨基酸之间发生裂解,释放一个可溶性的约100 kDa的N端片段sAPPβ,在膜上留下一个12 kDa的C端片段C99,C99片段被γ-分泌酶切割,从而产生Aβ。AD患者中Aβ产生过量,作为细胞外沉积物积聚在脑中,是形成老年斑的主要成分,其与由tau蛋白异常磷酸化导致的神经原纤维缠结是AD最主要的两大病理特征[22]。Rab11b位于早期和晚期内体,高尔基复合体和内质网中。通常与再循环内含体有关,调控着反面高尔基体管网状结构与内吞、在循环内含体或质膜之间的物质运输。通过共聚焦分析和免疫荧光染色显示,在Rab11B阳性循环内含体中观察到BACE1沿树突和轴突定位并运输。沉默Rab11导致内在的BACE1在体细胞中积累,伴随着轴突中BACE1的水平降低[23]。这些结果表明,Rab11是神经元中BACE1的分选调节剂,对于BACE1的轴突分选至关重要。另外,siRNA敲低Rab11的表达显著降低了sAPPβ和Aβ的水平,这提示Rab11功能障碍可能是部分散发性阿尔茨海默病病例的发病机制之一[24]。Rab21在蛋白从早期内含体向晚期内含体转运过程中发挥作用,研究发现过表达Rab21可以通过促进γ-分泌酶的内吞及向晚期内含体的迁移来影响其活性,进而促进Aβ的产生[25]。沉默Rab44、Rab6A、Rab10会降低Aβ水平,而不影响sAPPβ。这意味着这些Rab GTPases会影响γ-分泌酶对APP的水解或Aβ的运输[25,26]。同济大学生命科学与技术学院的裴刚院士课题组通过采用双分子荧光互补技术结合荧光共振能量转移技术发现ADAM10/BACE1二元复合物主要位于早期内含体,进一步观察到γ-分泌酶与α-/β-分泌酶二元复合物相互作用,表明α-、β-和γ-分泌酶可能形成三元复合物[27],这提示影响γ-分泌酶的Rab21蛋白可能同时调控α-分泌酶和β-分泌酶。Rab8A/Rab8是极化运输的重要调节剂,并参与反高尔基网络至基底外侧质膜的运输,突起形成和纤毛发生[28,29]。与对照相比,来自AD脑组织质膜部分的Rab8水平显著增加[30]。另有研究发现,沉默 Rab1A也可增加tau蛋白分泌。Rab1A与高尔基体膜相关,沉默 Rab1A会诱导高尔基体碎裂,这表明Rab介导的高尔基体动力学可能参与调节tau蛋白分泌[31]。Rab3蛋白参与调控APP在轴突的快速运输。沉默Rab3A和Rab3B能够降低总体APP蛋白水平,这表明Rab3在APP的运输和维持中发挥了作用[26]。
Ras and Rab Interactor 3 (RIN3)是Rab5 GTPase家族的鸟嘌呤核苷酸交换因子(GEF),美国加州大学圣地亚哥分校医学院的吴承标课题组发现,在AD发病早期,RIN3的mRNA水平在皮质和海马中均显著增加,这一现象先于β-淀粉样蛋白沉积产生[32]。RIN3在APP/PS1小鼠的基底前脑胆碱能神经元中选择性上调。由于RIN3是Rab5的GEF,因此增加的RIN3表达可能有助于APP/PS1小鼠基底前脑胆碱能神经元中Rab5早期内含体的增大。通过质谱分析发现,RIN3向Rab5早期内含体募集了另外两个AD危险因素BIN1和CD2AP,进而损害APP的运输和加工,导致具有神经元毒性的APP-CTF的产生增加。RIN3/BIN1复合物的增加还可以促进Tau蛋白过度磷酸化。Rab5在内含体运输、突触和突触功能中起重要作用[32]。它还参与了兴奋性突触传递的长期增强和抑制过程,这对于学习和记忆功能非常重要[33,34]。Rab5的激活可能会削弱突触功能和细胞之间的通信,导致信号传递到内含体的轴突运输减少和神经元萎缩。
在AD模型鼠中观察到明显增大的溶酶体聚集在一起,显示出溶酶体功能障碍。溶酶体呈现较低的pH值时,Aβ42可能被错误折叠成稳定的聚集体,该聚集体能够在细胞内繁殖并随后成核,并在胞外形成淀粉样斑块[35]。最近发现,参与内含体,自噬体和溶酶体运输的Rab7A可以调节tau蛋白分泌。抑制Rab7A的表达后,tau蛋白在Thr181和Ser422位点的磷酸化完全被消除,Ser 202和Ser404处的磷酸化也显著降低,tau蛋白磷酸化的减少可能有助于其分泌减少。与此一致的是Rab7A的显性负突变和组成型活性形式的过表达分别减少和增加了tau蛋白分泌[36]。AD患者的人脑和AD小鼠模型脑中可以检测到Rab7A水平升高,且 Rab7A和tau蛋白之间存在共定位现象,这进一步表明Rab7A可能促进tau蛋白在AD中的积累[36]。
4.2 帕金森病
帕金森病也是一种常见的神经退行性疾病,其临床表现为运动缺陷,包括静息性震颤和肌肉僵硬[37]。其病理特征主要是在黑质致密部中积累了由α-突触核蛋白(α-synuclein, α-syn)组成的路易小体和多巴胺能神经元的选择性变性。约95%的PD病例为散发性,其余为家族性。α-突触核蛋白和LRRK2的基因突变是引起家族性和散发性帕金森病的主要原因。人类遗传学的最新进展指出,膜运输缺陷是PD致病的关键途径[38]。α-Syn是神经元中高度丰富的蛋白质,溶酶体过程的破坏会影响其代谢[39]。在PD动物模型中已经证明,过量的α-syn会损害Rab蛋白依赖的细胞内运输,研究发现Rab1的过表达可能逆转细胞内毒性,从而阻止神经元的丢失[40]。α-syn的内吞与再循环可能由Rab11A和Rab13介导,同时Rab11A,Rab13和Rab8B可能也参与α-syn在包涵体中的清除[41]。Dinter等[42]发现在HEK293细胞中,Rab7的过表达能够增加α-syn的清除率,其效应子FYCO1需要活性Rab7才能发挥作用,继而刺激α-突触核蛋白的降解。在飞行模型中,Rab7和FYCO1可以挽救突变体α-syn诱导的运动功能障碍,因此Rab7可以作为治疗PD的潜在靶点[42]。
LRRK2位于高尔基体、高尔基相关囊泡、内质网、线粒体和溶酶体中,是囊泡运输的一般调节器。LRRK2可能通过调节与内含体分选相关的Rab GTPases (即Rab29、Rab8A、Rab10)在开关II区的保守苏氨酸残基处的磷酸化,进而调节货物的囊泡运输。反过来,LRRK2激酶的活性主要受高尔基复合体内Rab29和内含体中与逆转录子相关的VPS35的调控[43]。磷酸蛋白质组学的结果表明,包括Rab3A、Rab29、Rab8A、Rab10、Rab12和Rab43在内的几种Rab蛋白均是LRRK2的底物[44,45,46]。通过LRRK2激酶的体外活性测定还发现,Rab GTPase蛋白的一个子集,包括Rab1A、Rab3C、Rab35可以与LRRK2相互作用并被其磷酸化[47]。
越来越多的证据表明,沉默内源性LRRK2表达导致自噬缺陷,从而使α-syn的异常积累,进而导致PD的发生[39]。Rab29在溶酶体应激条件下,将LRRK2募集到较大的溶酶体中,易位至异常的溶酶体后,LRRK2募集其底物Rab8和Rab10,并通过介导其功能性下游调节子,EH结构域结合蛋白1 (EHBP1)和EHBP1样蛋白1 (EHBP1L1)来促进溶酶体分泌[48]。鉴于EHBP1和EHBP1L1参与内含体和制管膜曲率的形成[49,50],可以推测LRRK2-Rab途径调节溶酶体的形态。
研究证明Rab35是LRRK2的下游效应子。Rab GTPases上的磷酸化位点突变会引起原代皮层神经元中的神经毒性和多巴胺能神经元的变性,这在Rab35磷酸突变体中尤为严重[47]。韩国首尔国立大学医学院李胜在课题组在α-syn转基因小鼠中发现,LRRK2激酶的抑制降低了α-syn的病理特征并增强了α-syn向溶酶体的运输[51]。考虑到Rab35可以通过调节多囊泡体向质膜的运输和对接来调控外泌体的分泌[52],猜测LRRK2-RAB35途径的激活使α-syn避开了溶酶体降解途径,而是导致其进入分泌途径,从而促进了该蛋白的积累[53]。
4.3 肌萎缩性侧索硬化症
肌萎缩性侧索硬化症是一种进行性神经系统变性疾病,主要影响大脑和脊髓中的运动神经元[54]。与AD和PD相似,所有ALS病例中有90%是散发型的;有10%来自家族,是由编码多种蛋白质的基因突变引起的,这些基因包括ALS2 (Rab5的鸟嘌呤核苷酸交换因子),C9orf72 (染色体9上的开放读码框72),SOD1 (超氧化物歧化酶1),TDP-43 (交易反应性DNA结合蛋白43),FUS (肉瘤融合蛋白)和VAPB (VAMP (小突触泡蛋白)相关的蛋白B)等[55,56,57]。
ALS在儿童期发作是由于ALS2基因功能丧失的突变所致,ALS2通过激活Rab5在早期内含体中的成熟发挥关键作用。ALS2和Rab5相互作用可以调节神经营养蛋白的信号传导[58]。
在ALS患者的运动神经元中,C9orf72主要定位于Rab5阳性的早期内含体。在神经元细胞系和原代皮层神经元中,C9orf72与Rab1、Rab5、Rab7和Rab11共定位[57]。沉默C9orf72后,TrkB的内吞作用被抑制,这表明这些患者的内含体运输受到损害[59]。已有报道显示C9orf72与Rab蛋白的相互作用能够调节运动神经元中内含体的运输,该过程是溶酶体发生所必需。C9orf72的耗竭会损害自噬,并导致聚集体的异常聚集,而聚集体是ALS发病的主要特征。沉默C9orf72后,LC3II:LC3I比例显著增加,表明自噬体形成失调。
Rab1与突变型TDP-43,FUS和SOD1广泛共定位于神经元细胞中,并且Rab1在散发性ALS患者的脊髓运动神经元中参与形成包涵体。SOD1、TDP-43或FUS的突变会导致Rab1的定位出现错误,进而导致内质网-高尔基体网络中的蛋白质运输受损。而这些缺失可以通过Rab1的过表达来挽救[60]。Rab1的过表达对mSOD1、mTDP-43和mFUS诱导的内质网应激具有保护作用[55]。Rab1介导的内质网-高尔基体运输途径可能是ALS中的新型治疗靶点。
4.4 亨廷顿病
亨廷顿病是一种致命的神经退行性疾病,每100,000名居民中有5~10例患病,典型发病年龄为30~40岁。患者会遭受一系列复杂的精神、认知和运动障碍,直至死亡。亨廷顿病是由亨廷顿基因(Htt)中的聚谷氨酰胺(polyQ)重复扩增引起的常染色体显性遗传病。polyQ重复序列的长度与发病年龄成正比,但是遗传变异和环境因素改变了这种相关性[61]。它是由三核苷酸CAG在亨廷顿基因(Htt)基因5?末端附近的多态性区域发生异常扩增引起的。这种显性突变不仅耗尽了编码的亨廷顿蛋白,破坏了其生理功能,而且还产生了折叠错误的蛋白,即突变体Htt,并导致纹状体中的中棘神经元死亡[62]。Rab8 GTPase调节反面高尔基体网管状结构向质膜的运输,optineurin是其效应子,二者形成的复合物在高尔基复合体到基底外侧质膜的极化膜运输过程中以及神经元的树突中起重要作用[63]。Htt的突变与optineurin-Rab8复合物的相互作用减少,从而导致网格蛋白依赖性高尔基体到溶酶体区室的运输发生改变[64]。此外,Htt和HAP40(Htt相关蛋白40)形成的复合物是Rab5的效应子,可控制早期内含体的运动活性[65]。Rab5的过表达降低了Htt突变蛋白的聚集,而沉默Rab5会抑制内吞作用并阻断自噬,从而增加了polyQ的聚集[66]。Rab11是参与内含体再循环的Rab蛋白,内循环是细胞维持质膜成分的主要途径。突变体Htt抑制Rab11-GDP到Rab11-GTP的核苷酸交换,从而抑制Rab11的活性并导致细胞中转铁蛋白受体的再循环减慢。HD患者中Rab11活性和内体循环功能的异常可能与除转铁蛋白外的许多关键蛋白的转运有关,并对神经元的树突和轴突产生影响[67]。
在HD转基因小鼠中,观察到靠近Htt聚集体的神经元树突棘较少,提示树突棘损失可能是Htt聚集体清除率下降的早期结果。通过在海马神经元中过表达Rab11可消除表达Htt的原代鼠神经元中树突棘的丧失,这表明突触功能障碍与Rab11受损可能有早期HD的发病相关[68]。
HD与轴突运输的改变有关。在纹状体神经元中,Htt的表达破坏了轴突的快速运输[69]。在果蝇HD模型中,Rab11的过表达能够恢复早期的突触功能障碍,包括突触前囊泡大小的减少,数量振幅的减少和诱发的突触传递以及幼虫爬行的改变等[70]。
4.5 夏科特-玛丽-牙齿2B型
夏科特-玛丽-牙齿(Charcot-Marie-tooth, CMT)病是最常见的遗传性神经肌肉疾病,该病是由于几种不同基因的突变导致相似的表型。CMT主要分为1类(CMT1)和2类(CMT2)形式。CMT1是特征为神经传导速度降低的显性遗传性脱髓鞘神经病,而CMT2是特征为神经传导速度正常或略有降低的显性遗传性轴突神经病。除了这些主要类别外,CMT中还包括其他稀有形式[71]。在此着重介绍直接与Rab相关的CMT疾病,即CMT2B型,其表现包括严重的感觉丧失,远端肌肉无力以及由于反复感染引起的足部溃疡,感染甚至截肢的频繁发生。Rab7中高度保守的氨基酸残基的5个错义突变与CMT2B型表型相关。Rab7点突变引起Rab7蛋白水平升高或降低,进而影响Rab7控制的神经营养蛋白的转运和信号传导、神经突向外生长以及神经元迁移[20,72]。由于神经营养蛋白受体的内吞作用和逆行轴突运输对于神经营养蛋白信号的传导和控制神经元的分化、可塑性和存活至关重要,因此神经营养蛋白运输障碍可能会导致严重的神经变性[73,74]。
在一名CMT2B患者腓肠神经活检中,发现Rab7效应子-Rab相互作用溶酶体蛋白(Rab interacting lysosomal protein, RILP)被下调,导致受体降解和信号衰减。RILP不仅是晚期内体/溶酶体的正常分布所必需的,而且是晚期内体中腔内囊泡的形成所必需的[75,76]。但是,GTP水解不足可能会导致Rab7突变体隔离RILP,从而降低RILP作用于其他方面(如内含体)的能力。由于Rab7是普遍存在的RabGTPase,因此这种罕见疾病也显示了其神经元对膜运输的变化具有敏感性的特点。
5 Rab GTPases介导线粒体和星形胶质细胞的功能
Rab GTPas介导的膜运输与神经退行性疾病的关联是多种多样的。在神经退行性疾病中,在受损线粒体吞噬过程中Rab蛋白同样发挥重要的调控作用。线粒体的状态直接影响神经元的发育、功能和存活。神经元是长寿细胞,在整个生命周期中都存在,因此更容易受到线粒体功能障碍引起的累积损伤的影响。在AD患者神经元内的异常溶酶体中,发现了未消化的受损线粒体[77]。在PD的发病机理中LRRK2、α-Syn会损害线粒体和线粒体功能,因此造成的线粒体受损是必然的。ALS的致病基因涉及线粒体和线粒体调控基因(如OPTN、Ser/Thr蛋白激酶(Tbk1)、VCP和超氧化物歧化酶1 (SOD-1))[78]。功能障碍线粒体的积累已成为患者和动物模型中受累神经元的共同特征,可能出现在明显的认知缺陷之前。Rab7的活性受两个Rab7 GAPs (TBC1D15和TBC1D17)的调控,同时Rab7也被募集到线粒体外膜,在那里动员囊泡以建立自噬体。TBC1D15和TBC1D17的耗尽会导致Rab7在线粒体上积累,并导致自噬体样结构异常积累,从而延迟并阻碍受损线粒体的清除[79,80]。这些GAPs在空间上控制Rab7活性,Rab7活性本身控制了囊泡向自噬体膜的募集。处于其GTP结合活性状态的Rab32参与线粒体的分裂[81]。Rab35促进自噬受体NDP52的募集以及与泛素的结合,从而促进异种吞噬、线粒体吞噬和自噬体的成熟[82]。
许多研究表明AD、PD和HD等神经退行性疾病的发病机制中存在星形胶质细胞的功能异常,并伴有Rab蛋白水平的变化。神经炎症是所有神经退行性疾病的重要组成部分,其中小胶质细胞和星形胶质细胞通过释放多种促炎和抗炎细胞因子来发挥双向作用[83,84]。小胶质细胞可以通过介导神经炎症调节大脑免疫,并参与突触的连接和重塑;然而在AD早期小胶质细胞可以介导突触的异常丧失进而促进病理进程[85]。星形胶质细胞是大脑中存在较为丰富的细胞,可维持神经递质的稳态,引导突触的形成和成熟,调节活性氧和血脑屏障[86,87,88]。神经炎症和缺血可诱导出两种不同类型的反应性星形胶质细胞,称为A1和A2反应性星形胶质细胞。A1反应性星形胶质细胞的上调可能是有害的,而A2反应性星形胶质细胞的上调可能是有益的[89]。此外,衰老的星形胶质细胞具有神经炎性A1样反应性星形胶质细胞的反应性表型。除了释放有效的神经毒素外,A1星形胶质细胞还能促进新突触的形成,并导致中枢神经系统神经元的兴奋功能降低。除了星形胶质细胞反应性状态的改变外,星形胶质细胞中可能还会发生其他转录和功能性变化,这可以解释正常衰老过程中认知能力下降这一现象[90],而在PD模型中抑制A1星形胶质细胞的活性具有保护神经作用[91]。
研究发现,在AD中,星形胶质细胞是大脑在生理条件下表达ApoE的主要细胞,星形胶质细胞参与Aβ摄取和降解[92,93]。在AD早期阶段,BACE1活性增加伴随着BACE2活性的相应增加,BACE2主要定位于星形胶质细胞中[94]。在PD中,从神经元释放的α-Syn被星形胶质细胞吸收,进而影响其线粒体的完整性并导致神经毒性的产生[95,96,97]。研究发现星型胶质细胞在神经退行性疾病的病理性扩散中具有潜在的协同作用[98]。在HD中,随着疾病进展会增加相关星形细胞的活性,进而导致Htt的聚集[99,100]。在ALS中,星形胶质细胞显示出毒性表型,引起运动神经元变性。Rab31在表皮生长因子受体转运至晚期内含体的运输中发挥作用[101],Rab31沉默研究引起的EGFR信号转导增加可能会阻碍培养物中星形胶质细胞的完全发育,从而导致存活的星形胶质细胞百分比降低[102]。
在促炎条件下,智利大学Quest等[103]通过生物信息学发现,反应性星形胶质细胞中Rab的内吞途径发生了改变,其有利于蛋白水解的回收。特别是,Rab4、Rab5和Rab7的蛋白表达水平发生变化。另一方面,这种促炎环境增加了Rab5-GTP和Rac1- GTP的表达,并降低了Rab7-GTP的负荷。在促炎环境中,从早期(Rab5)到晚期内含体(Rab7)的货物运输也发生了变化。通过分析晚期和早期内体组分中的低密度脂蛋白LDL (注定要降解的货物)的分布,研究人员观察到LDL保留在TNFα刺激的细胞的外围,这表明由Rab7所介导的溶酶体降解能力下调[103]。在神经退行性疾病和脑损伤中,星形胶质细胞活化涉及Rab依赖性途径的分子机制尚待研究。
6 结语与展望
本文主要分析了一些Rab蛋白在神经类疾病中的运输和信号传导中的变化,Rab蛋白的异常表达、活性改变或定位错误可能与多种神经类疾病如(AD、PD和HD)的致病机制有关(表1)。参与内吞降解途径的Rab7A可以调节tau蛋白的分泌。Rab11和Rab21可以通过影响BACE1或PS1的定位进而影响Aβ的产生。Rab35可以促进α-syn的分泌。沉默Rab5加htt突变蛋白的聚集。在ALS和CMT2B中,Rab7参与调节轴突运输以及内吞途径,神经营养蛋白的信号传导。Rab蛋白主要通过调节内吞途径来影响蛋白聚集体的产生。另一方面,神经退行性疾病的致病机制,即线粒体和星形胶质细胞的功能异常也与Rab蛋白水平的改变密切相关。线粒体功能障碍发生在神经退行性疾病的早期阶段,是造成神经元死亡的重要原因。线粒体功能障碍的出现和加重可能是由病理过程发展中涉及的许多因素造成的。研究与受损线粒体自噬调节的相关Rab蛋白并分析Rab蛋白在其中的调节途径对神经退行性疾病的早期诊断将会有很大帮助。Table 1
表1
表1神经退行性疾病中的Rab蛋白及其功能
Table 1
| 名称 | 功能 | 影响的神经退行性疾病 |
|---|---|---|
| Rab 1A | 调节从内质网(ER)到高尔基体室以及细胞表面的囊泡蛋白运输 | AD、PD |
| Rab 3 | 参与胞吐作用,在神经递质释放和突触可塑性中起关键作用 | AD、PD |
| Rab 5 | 介导货物从质膜到早期内含体的运输并充当早期内含体的标记物,在内含体运输中、突触和突触功能中起重要作用 | AD、ALS、HD |
| Rab 7 | 调节货物从早期到晚期内含体的运输并充当晚期内含体的标记;控制神经营养蛋白受体的逆行轴突运输 | AD、PD 、ALS 、CMT2B |
| Rab 8 | 是极化运输的重要调节剂,并参与反高尔基网络至基底外侧质的膜运输,突起形成和纤毛发生 | AD、PD 、HD |
| Rab 10 | 调节小泡运输,参与反高尔基网络、内质网到基底膜的组织 | AD、PD |
| Rab 11 | 与再循环内含体有关,控制着反高尔基网络与内吞在循环室或质膜之间的物质运输 | AD、PD 、ALS 、HD |
| Rab 13 | 与生物合成和内含体再循环途径有关 | PD |
| Rab 21 | 在内吞和自噬中发挥作用 | AD |
| Rab 29 | 与溶酶体相关细胞器生物发生相关 | PD |
| Rab 31 | 在表皮生长因子受体转运至晚期内含体的运输中发挥作用 | 与星形胶质细胞相关 |
| Rab 35 | 控制货物从早期内含体、循环内含体直接返回质膜的快速回收 | PD、线粒体相关 |
新窗口打开|下载CSV
星形胶质细胞在神经退行性疾病中的作用机制是目前的研究热点,Rab蛋白在其中的作用尚不清楚。星形胶质细胞对于维持脑稳态和保护神经元至关重要。星形胶质细胞中的溶酶体降解能力不足,与包括PD在内的各种神经退行性疾病的发病机理有关。阻止或者减少A1反应性星形胶质细胞的形成可以减缓神经退行性疾病的病理进程。在成年啮齿动物的大脑中,Rab31仅在神经胶质纤维酸性蛋白阳性星形胶质细胞中表达[101]。Rab蛋白作为星型胶质细胞的生物标志物,具有作为神经退行性疾病诊断工具的巨大潜力。
尽管已经非常深入地研究了几种Rab蛋白,但是Rab蛋白家族仍有巨大的研究潜力,Rab蛋白在中枢神经系统不同组织中的特殊作用至关重要。神经退行性疾病是一些常见功能障碍导致的具有不同病理特征的疾病。Rab蛋白在不同的神经退行性疾病中显示出不同的调节作用,但是目前尚不清楚是Rab蛋白的变化引起功能障碍还是功能障碍导致Rab蛋白水平发生变化。发现和研究这些潜在的常见机制可以使我们更加了解神经元存活的基本要求,为深入探究这几种神经类疾病的病理学机制奠定基础,同时为开发有效的治疗策略开辟新的道路。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1016/S1474-4422(18)30403-4URLPMID:30497964 [本文引用: 1]

BACKGROUND: The number of individuals living with dementia is increasing, negatively affecting families, communities, and health-care systems around the world. A successful response to these challenges requires an accurate understanding of the dementia disease burden. We aimed to present the first detailed analysis of the global prevalence, mortality, and overall burden of dementia as captured by the Global Burden of Diseases, Injuries, and Risk Factors (GBD) Study 2016, and highlight the most important messages for clinicians and neurologists. METHODS: GBD 2016 obtained data on dementia from vital registration systems, published scientific literature and surveys, and data from health-service encounters on deaths, excess mortality, prevalence, and incidence from 195 countries and territories from 1990 to 2016, through systematic review and additional data-seeking efforts. To correct for differences in cause of death coding across time and locations, we modelled mortality due to dementia using prevalence data and estimates of excess mortality derived from countries that were most likely to code deaths to dementia relative to prevalence. Data were analysed by standardised methods to estimate deaths, prevalence, years of life lost (YLLs), years of life lived with disability (YLDs), and disability-adjusted life-years (DALYs; computed as the sum of YLLs and YLDs), and the fractions of these metrics that were attributable to four risk factors that met GBD criteria for assessment (high body-mass index [BMI], high fasting plasma glucose, smoking, and a diet high in sugar-sweetened beverages). FINDINGS: In 2016, the global number of individuals who lived with dementia was 43.8 million (95% uncertainty interval [UI] 37.8-51.0), increased from 20.2 million (17.4-23.5) in 1990. This increase of 117% (95% UI 114-121) contrasted with a minor increase in age-standardised prevalence of 1.7% (1.0-2.4), from 701 cases (95% UI 602-815) per 100 000 population in 1990 to 712 cases (614-828) per 100 000 population in 2016. More women than men had dementia in 2016 (27.0 million, 95% UI 23.3-31.4, vs 16.8 million, 14.4-19.6), and dementia was the fifth leading cause of death globally, accounting for 2.4 million (95% UI 2.1-2.8) deaths. Overall, 28.8 million (95% UI 24.5-34.0) DALYs were attributed to dementia; 6.4 million (95% UI 3.4-10.5) of these could be attributed to the modifiable GBD risk factors of high BMI, high fasting plasma glucose, smoking, and a high intake of sugar-sweetened beverages. INTERPRETATION: The global number of people living with dementia more than doubled from 1990 to 2016, mainly due to increases in population ageing and growth. Although differences in coding for causes of death and the heterogeneity in case-ascertainment methods constitute major challenges to the estimation of the burden of dementia, future analyses should improve on the methods for the correction of these biases. Until breakthroughs are made in prevention or curative treatment, dementia will constitute an increasing challenge to health-care systems worldwide. FUNDING: Bill & Melinda Gates Foundation.
DOI:10.1016/S1474-4422(19)30290-XURLPMID:31494009 [本文引用: 1]
China has the largest population of patients with dementia in the world, imposing a heavy burden on the public and health care systems. More than 100 epidemiological studies on dementia have been done in China, but the estimates of the prevalence and incidence remain inconsistent because of the use of different sampling methods. Despite improved access to health services, inadequate diagnosis and management for dementia is still common, particularly in rural areas. The Chinese Government issued a new policy to increase care facilities for citizens older than 65 years, but most patients with dementia still receive care at home. Western medicines for dementia symptoms are widely used in China, but many patients choose Chinese medicines even though they have little evidence supporting efficacy. The number of clinical trials of Chinese and western medicines has substantially increased as a result of progress in research on new antidementia drugs but international multicentre studies are few in number. Efforts are needed to establish a national system of dementia care enhance training in dementia for health professionals, and develop global collaborations to prevent and cure this disease.
DOI:10.1038/362560a0URLPMID:8464498 [本文引用: 2]
The genes SEC4 and YPT1 encode Ras-related GTP-binding proteins in the yeast Saccharomyces cerevisiae. Ypt1 is necessary for vesicular transport from the endoplasmic reticulum to the Golgi, whereas Sec4 is required for fusion of post-Golgi secretory vesicles to the plasma membrane. Recently, three structural domains have been proposed to specify the stage in cellular transport at which members of the Sec4/Ypt1/Rab family act: the effector domain, the C-terminal hypervariable region, and a region corresponding to loop 7 in the structure of p21ras (ref. 8). Here we use Sec4/Ypt1 chimaeras to show that these three regions cooperate to specify Ypt1 function and that the C-terminal hypervariable region is needed for Ypt1 localization to the Golgi. Unexpectedly, we found that a single chimaera can function as either Ypt1 or Sec4 without missorting carboxypeptidase Y or invertase.
DOI:10.1016/S0014-5793(00)01143-1URL [本文引用: 1]
DOI:10.1126/science.2406906URLPMID:2406906 [本文引用: 1]
Ras proteins participate as a molecular switch in the early steps of the signal transduction pathway that is associated with cell growth and differentiation. When the protein is in its GTP complexed form it is active in signal transduction, whereas it is inactive in its GDP complexed form. A comparison of eight three-dimensional structures of ras proteins in four different crystal lattices, five with a nonhydrolyzable GTP analog and three with GDP, reveals that the
DOI:10.3390/cells8080909URL [本文引用: 1]
DOI:10.1016/j.ceb.2004.06.014URL [本文引用: 1]
Abstract
The remarkable degree of specificity with which Rab GTPases recognise distinct subsets of intracellular membranes forms the basis of their ability to act as key cellular regulators, determining the recruitment of downstream effectors to the right membrane at the right time. The molecular mechanisms controlling Rab localisation, however, have proved tricky issues to address. It is becoming increasingly apparent that multiple factors contribute to the specificity of Rab localisation and the close coordination of membrane targeting with Rab activation. With important new insights into the mode of action of the general Rab regulators REP and RabGDI, as well as the demonstration that novel factors such as Yip3/Pra1 act as GDI displacement factors and that signals within Rab proteins contribute to targeting specificity, a better understanding of the concepts governing Rab recruitment and function is now beginning to emerge. The diversity of cellular processes regulated by Rab family members is made possible, not only by the wide range of effectors they recruit, but also by the different mechanisms regulating their own targeting and activation.[本文引用: 1]
[本文引用: 1]
DOI:10.1016/0092-8674(91)90459-cURLPMID:1850321 [本文引用: 1]
Endocytosed proteins destined for degradation in lysosomes are targeted mainly to early endosomes following uptake. Late endosomes are the major site for entry of newly synthesized lysosomal hydrolases via the cation-independent mannose 6-phosphate receptor into the degradative pathway. No consensus exists as to the mechanism of transport from early to late endosomes. We used asialoorosomucoid and transferrin to label selected parts of the degradative and receptor-recycling pathways, respectively, in the human hepatoma cell line HepG2. Intracellular mixing of sequentially endocytosed 125I- and HRP-labeled ligands was monitored by using 3,3'-diaminobenzidine-mediated density perturbation. The entire endocytic pathway of asialoorosomucoid, except for the lysosomes, remained fully accessible to subsequently endocytosed transferrin conjugated to HRP with unchanged kinetics. These results together with immunoelectron microscopic data support a model in which early endosomes gradually mature into late endosomes.
DOI:10.1016/0092-8674(92)90306-wURLPMID:1516130 [本文引用: 1]
We have investigated the in vivo functional role of rab5, a small GTPase associated with the plasma membrane and early endosomes. Wild-type rab5 or rab5-ile133, a mutant protein defective in GTP binding, was overexpressed in baby hamster kidney cells. In cells expressing the rab5ile 133 protein, the rate of endocytosis was decreased by 50% compared with normal, while the rate of recycling was not significantly affected. The morphology of early endosomes was also drastically changed by the mutant protein, which induced accumulation of small tubules and vesicles at the periphery of the cell. Surprisingly, overexpression of wild-type rab5 accelerated the uptake of endocytic markers and led to the appearance of atypically large early endosomes. We conclude that rab5 is a rate-limiting component of the machinery regulating the kinetics of membrane traffic in the early endocytic pathway.
[本文引用: 1]
DOI:10.1016/j.cub.2006.07.020URL [本文引用: 1]
Summary
Cytokinesis is the final step of cell division and leads to the physical separation of the daughter cells. After the ingression of a cleavage membrane furrow that pinches the mother cell, future daughter cells spend much of the cytokinesis phase connected by an intercellular bridge. Rab proteins are major regulators of intracellular transport in eukaryotes [1], and here, we report an essential role for human Rab35 in both the stability of the bridge and its final abscission. We find that Rab35, whose function in membrane traffic was unknown, is localized to the plasma membrane and endocytic compartments and controls a fast endocytic recycling pathway. Consistent with a key requirement for Rab35-regulated recycling during cell division, inhibition of Rab35 function leads to the accumulation of endocytic markers on numerous cytoplasmic vacuoles in cells that failed cytokinesis. Moreover, Rab35 is involved in the intercellular bridge localization of two molecules essential for the postfurrowing steps of cytokinesis: the phosphatidylinositol 4,5-bis phosphate (PIP2) lipid [2], [3] and [4] and the septin SEPT2 [5]. We propose that the Rab35-regulated pathway plays an essential role during the terminal steps of cytokinesis by controlling septin and PIP2 subcellular distribution during cell division.DOI:10.1073/pnas.93.18.9559URL [本文引用: 1]
DOI:10.1083/jcb.135.4.913URLPMID:8922376 [本文引用: 1]
Small GTPases of the rab family are crucial elements of the machinery that controls membrane traffic. In the present study, we examined the distribution and function of rab11. Rab11 was shown by confocal immunofluorescence microscopy and EM to colocalize with internalized transferrin in the pericentriolar recycling compartment of CHO and BHK cells. Expression of rab11 mutants that are preferentially in the GTP- or GDP-bound state caused opposite effects on the distribution of transferrin-containing elements; rab11-GTP expression caused accumulation of labeled elements in the perinuclear area of the cell, whereas rab11-GDP caused a dispersion of the transferrin labeling. Functional studies showed that the early steps of uptake and recycling for transferrin were not affected by overexpression of rab11 proteins. However, recycling from the later recycling endosome was inhibited in cells overexpressing the rab11-GDP mutant. Rab5, which regulates early endocytic trafficking, acted before rab11 in the transferrin-recycling pathway as expression of rab5-GTP prevented transport to the rab11-positive recycling endosome. These results suggest a novel role for rab11 in controlling traffic through the recycling endosome.
DOI:10.1016/s0896-6273(03)00594-4URLPMID:14527431 [本文引用: 1]
We tested whether proteins implicated in Huntington's and other polyglutamine (polyQ) expansion diseases can cause axonal transport defects. Reduction of Drosophila huntingtin and expression of proteins containing pathogenic polyQ repeats disrupt axonal transport. Pathogenic polyQ proteins accumulate in axonal and nuclear inclusions, titrate soluble motor proteins, and cause neuronal apoptosis and organismal death. Expression of a cytoplasmic polyQ repeat protein causes adult retinal degeneration, axonal blockages in larval neurons, and larval lethality, but not neuronal apoptosis or nuclear inclusions. A nuclear polyQ repeat protein induces neuronal apoptosis and larval lethality but no axonal blockages. We suggest that pathogenic polyQ proteins cause neuronal dysfunction and organismal death by two non-mutually exclusive mechanisms. One mechanism requires nuclear accumulation and induces apoptosis; the other interferes with axonal transport. Thus, disruption of axonal transport by pathogenic polyQ proteins could contribute to early neuropathology in Huntington's and other polyQ expansion diseases.
DOI:10.1126/science.1105681URLPMID:15731448 [本文引用: 1]
We identified axonal defects in mouse models of Alzheimer's disease that preceded known disease-related pathology by more than a year; we observed similar axonal defects in the early stages of Alzheimer's disease in humans. Axonal defects consisted of swellings that accumulated abnormal amounts of microtubule-associated and molecular motor proteins, organelles, and vesicles. Impairing axonal transport by reducing the dosage of a kinesin molecular motor protein enhanced the frequency of axonal defects and increased amyloid-beta peptide levels and amyloid deposition. Reductions in microtubule-dependent transport may stimulate proteolytic processing of beta-amyloid precursor protein, resulting in the development of senile plaques and Alzheimer's disease.
DOI:10.1038/sj.emboj.7600753URLPMID:16052212 [本文引用: 1]
alpha-RIMs and Munc13s are active zone proteins that control priming of synaptic vesicles to a readily releasable state, and interact with each other via their N-terminal sequences. The alpha-RIM N-terminal sequence also binds to Rab3s (small synaptic vesicle GTPases), an interaction that regulates presynaptic plasticity. We now demonstrate that alpha-RIMs contain adjacent but separate Munc13- and Rab3-binding sites, allowing formation of a tripartite Rab3/RIM/Munc13 complex. Munc13 binding is mediated by the alpha-RIM zinc-finger domain. Elucidation of the three-dimensional structure of this domain by NMR spectroscopy facilitated the design of a mutation that abolishes alpha-RIM/Munc13 binding. Selective disruption of this interaction in the calyx of Held synapse decreased the size of the readily releasable vesicle pool. Our data suggest that the ternary Rab3/RIM/Munc13 interaction approximates synaptic vesicles to the priming machinery, providing a substrate for presynaptic plasticity. The modular architecture of alpha-RIMs, with nested binding sites for Rab3 and other targets, may be a general feature of Rab effectors that share homology with the alpha-RIM N-terminal sequence.
DOI:10.1523/JNEUROSCI.1546-09.2009URLPMID:19923287 [本文引用: 1]
The amyloid precursor protein (APP) is anterogradely transported by conventional kinesin in a distinct transport vesicle, but both the biochemical composition of such a vesicle and the specific kinesin-1 motor responsible for transport are poorly defined. APP may be sequentially cleaved by beta- and gamma-secretases leading to accumulation of beta-amyloid (Abeta) peptides in brains of Alzheimer's disease patients, whereas cleavage of APP by alpha-secretases prevents Abeta generation. Here, we demonstrate by time-lapse analysis and immunoisolations that APP is a cargo of a vesicle containing the kinesin heavy chain isoform kinesin-1C, the small GTPase Rab3A, and a specific subset of presynaptic protein components. Moreover, we report that assembly of kinesin-1C and APP in this vesicle type requires Rab3A GTPase activity. Finally, we show cleavage of APP in transport vesicles by alpha-secretase activity, likely mediated by ADAM10. Together, these data indicate that maturation of APP transport vesicles, including recruitment of conventional kinesin, requires Rab3 GTPase activity.
DOI:10.1523/JNEUROSCI.4322-12.2013URLPMID:23616551 [本文引用: 1]
Retrograde trophic signaling of nerve growth factor (NGF) supports neuronal survival and differentiation. Dysregulated trophic signaling could lead to various neurological disorders. Charcot-Marie-Tooth type 2B (CMT2B) is one of the most common inherited peripheral neuropathies characterized by severe terminal axonal loss. Genetic analysis of human CMT2B patients has revealed four missense point mutations in Rab7, a small GTPase that regulates late endosomal/lysosomal pathways, but the exact pathological mechanism remains poorly understood. Here, we show that these Rab7 mutants dysregulated axonal transport and diminished the retrograde signaling of NGF and its TrkA receptor. We found that all CMT2B Rab7 mutants were transported significantly faster than Rab7(wt) in the anterograde direction, accompanied with an increased percentile of anterograde Rab7-vesicles within axons of rat E15.5 dorsal root ganglion (DRG) neurons. In PC12M cells, the CMT2B Rab7 mutants drastically reduced the level of surface TrkA and NGF binding, presumably by premature degradation of TrkA. On the other hand, siRNA knock-down of endogenous Rab7 led to the appearance of large TrkA puncta in enlarged Rab5-early endosomes within the cytoplasm, suggesting delayed TrkA degradation. We also show that CMT2B Rab7 mutants markedly impaired NGF-induced Erk1/2 activation and differentiation in PC12M cells. Further analysis revealed that CMT2B Rab7 mutants caused axonal degeneration in rat E15.5 DRG neurons. We propose that Rab7 mutants induce premature degradation of retrograde NGF-TrkA trophic signaling, which may potentially contribute to the CMT2B disease.
DOI:10.1016/j.neuron.2006.08.018URLPMID:17046692 [本文引用: 2]
Vesicular pathways coupling the neuromuscular junction with the motor neuron soma are essential for neuronal function and survival. To characterize the organelles responsible for this long-distance crosstalk, we developed a purification strategy based on a fragment of tetanus neurotoxin (TeNT H(C)) conjugated to paramagnetic beads. This approach enabled us to identify, among other factors, the small GTPase Rab7 as a functional marker of a specific pool of axonal retrograde carriers, which transport neurotrophins and their receptors. Furthermore, Rab5 is essential for an early step in TeNT H(C) sorting but is absent from axonally transported vesicles. Our data demonstrate that TeNT H(C) uses a retrograde transport pathway shared with p75(NTR), TrkB, and BDNF, which is strictly dependent on the activities of both Rab5 and Rab7. Therefore, Rab7 plays an essential role in axonal retrograde transport by controlling a vesicular compartment implicated in neurotrophin traffic.
DOI:10.7554/eLife.05597URLPMID:25643395 [本文引用: 1]
Small GTPases of the Rab family not only regulate target recognition in membrane traffic but also control other cellular functions such as cytoskeletal transport and autophagy. Here we show that Rab26 is specifically associated with clusters of synaptic vesicles in neurites. Overexpression of active but not of GDP-preferring Rab26 enhances vesicle clustering, which is particularly conspicuous for the EGFP-tagged variant, resulting in a massive accumulation of synaptic vesicles in neuronal somata without altering the distribution of other organelles. Both endogenous and induced clusters co-localize with autophagy-related proteins such as Atg16L1, LC3B and Rab33B but not with other organelles. Furthermore, Atg16L1 appears to be a direct effector of Rab26 and binds Rab26 in its GTP-bound form, albeit only with low affinity. We propose that Rab26 selectively directs synaptic and secretory vesicles into preautophagosomal structures, suggesting the presence of a novel pathway for degradation of synaptic vesicles.
DOI:10.1007/BF00296367URLPMID:1831952 [本文引用: 1]
We have previously shown that abnormal Tau species are produced during the neurofibrillary degeneration of the Alzheimer type. These abnormal Tau proteins consist of a characteristic triplet named Tau 55, Tau 64 and Tau 69 which are constantly found in Alzheimer's disease (AD) and Downs syndrome brain regions with tangles. To determine if abnormal Tau species are also produced in other neurodegenerative conditions where intraneuronal filamentous Tau aggregates are observed, we have undertaken an immuno-blot study of brain homogenates from patients with progressive supranuclear palsy (PSP), a neurological disorder characterized by the presence of tangles in subcortical and cortical brain areas. We show here that abnormal Tau species are produced in PSP but that they are different from those in AD. Indeed, although Tau 64 and 69 were present in PSP brain homogenates, possibly as the result of an abnormal phosphorylation as in AD, they were detected in smaller amounts (three times lower) than in AD. In addition Tau 55 was undetected by the immunological tools, such as the absorbed anti-PHF antisera, which specifically label the abnormal Tau proteins. Also the two-dimensional analysis revealed different isoelectric properties. Our results suggest that the production of abnormal Tau species is a very important event during all types of neurofibrillary degeneration. The differences in the pathological Tau-variant profile that were observed between PSP and AD possibly reflect different etiopathogenetic pathways and might explain the formation of different types of filamentous Tau aggregates.
DOI:10.1186/1750-1326-9-1URLPMID:24386896 [本文引用: 1]
BACKGROUND: BACE1 is one of the two enzymes that cleave amyloid precursor protein to generate Alzheimer's disease (AD) beta amyloid peptides. It is widely believed that BACE1 initiates APP processing in endosomes, and in the brain this cleavage is known to occur during axonal transport of APP. In addition, BACE1 accumulates in dystrophic neurites surrounding brain senile plaques in individuals with AD, suggesting that abnormal accumulation of BACE1 at presynaptic terminals contributes to pathogenesis in AD. However, only limited information is available on BACE1 axonal transport and targeting. RESULTS: By visualizing BACE1-YFP dynamics using live imaging, we demonstrate that BACE1 undergoes bi-directional transport in dynamic tubulo-vesicular carriers along axons in cultured hippocampal neurons and in acute hippocampal slices of transgenic mice. In addition, a subset of BACE1 is present in larger stationary structures, which are active presynaptic sites. In cultured neurons, BACE1-YFP is preferentially targeted to axons over time, consistent with predominant in vivo localization of BACE1 in presynaptic terminals. Confocal analysis and dual-color live imaging revealed a localization and dynamic transport of BACE1 along dendrites and axons in Rab11-positive recycling endosomes. Impairment of Rab11 function leads to a diminution of total and endocytosed BACE1 in axons, concomitant with an increase in the soma. Together, these results suggest that BACE1 is sorted to axons in endosomes in a Rab11-dependent manner. CONCLUSION: Our results reveal novel information on dynamic BACE1 transport in neurons, and demonstrate that Rab11-GTPase function is critical for axonal sorting of BACE1. Thus, we suggest that BACE1 transcytosis in endosomes contributes to presynaptic BACE1 localization.
DOI:10.1016/j.celrep.2013.12.005URLPMID:24373285 [本文引用: 1]
Alzheimer's disease (AD) is characterized by cerebral deposition of beta-amyloid (Abeta) peptides, which are generated from amyloid precursor protein (APP) by beta- and gamma-secretases. APP and the secretases are membrane associated, but whether membrane trafficking controls Abeta levels is unclear. Here, we performed an RNAi screen of all human Rab-GTPases, which regulate membrane trafficking, complemented with a Rab-GTPase-activating protein screen, and present a road map of the membrane-trafficking events regulating Abeta production. We identify Rab11 and Rab3 as key players. Although retromers and retromer-associated proteins control APP recycling, we show that Rab11 controlled beta-secretase endosomal recycling to the plasma membrane and thus affected Abeta production. Exome sequencing revealed a significant genetic association of Rab11A with late-onset AD, and network analysis identified Rab11A and Rab11B as components of the late-onset AD risk network, suggesting a causal link between Rab11 and AD. Our results reveal trafficking pathways that regulate Abeta levels and show how systems biology approaches can unravel the molecular complexity underlying AD.
[本文引用: 2]
DOI:10.1016/j.celrep.2013.12.005URLPMID:24373285 [本文引用: 2]
Alzheimer's disease (AD) is characterized by cerebral deposition of beta-amyloid (Abeta) peptides, which are generated from amyloid precursor protein (APP) by beta- and gamma-secretases. APP and the secretases are membrane associated, but whether membrane trafficking controls Abeta levels is unclear. Here, we performed an RNAi screen of all human Rab-GTPases, which regulate membrane trafficking, complemented with a Rab-GTPase-activating protein screen, and present a road map of the membrane-trafficking events regulating Abeta production. We identify Rab11 and Rab3 as key players. Although retromers and retromer-associated proteins control APP recycling, we show that Rab11 controlled beta-secretase endosomal recycling to the plasma membrane and thus affected Abeta production. Exome sequencing revealed a significant genetic association of Rab11A with late-onset AD, and network analysis identified Rab11A and Rab11B as components of the late-onset AD risk network, suggesting a causal link between Rab11 and AD. Our results reveal trafficking pathways that regulate Abeta levels and show how systems biology approaches can unravel the molecular complexity underlying AD.
DOI:10.3389/fnmol.2018.00431URLPMID:30538620 [本文引用: 1]
The competitive ectodomain shedding of amyloid-beta precursor protein (APP) by alpha-secretase and beta-secretase, and the subsequent regulated intramembrane proteolysis by gamma-secretase are the key processes in amyloid-beta peptides (Abeta) generation. Previous studies indicate that secretases form binary complex and the interactions between secretases take part in substrates processing. However, whether alpha-, beta- and gamma-secretase could form ternary complex remains to be explored. Here, we adopted bimolecular fluorescence complementation in combination with fluorescence resonance energy transfer (BiFC-FRET) to visualize the formation of triple secretase complex. We show that the interaction between alpha-secretase ADAM10 and beta-secretase BACE1 could be monitored by BiFC assay and the binding of APP to alpha-/beta-secretase binary complex was revealed by BiFC-FRET. Further, we observed that gamma-secretase interacts with alpha-/beta-secretase binary complex, providing evidence that alpha-, beta- and gamma-secretase might form a ternary complex. Thus our study extends the interplay among Alzheimer's disease (AD) related alpha-/beta-/gamma-secretase.
DOI:10.1083/jcb.123.1.35URLPMID:8408203 [本文引用: 1]
Small GTP-binding proteins of the rab family have been implicated as regulators of membrane traffic along the biosynthetic and endocytic pathways in eukaryotic cells. We have investigated the localization and function of rab8, closely related to the yeast YPT1/SEC4 gene products. Confocal immunofluorescence microscopy and immunoelectron microscopy on filter-grown MDCK cells demonstrated that, rab8 was localized to the Golgi region, vesicular structures, and to the basolateral plasma membrane. Two-dimensional gel electrophoresis showed that rab8p was highly enriched in immuno-isolated basolateral vesicles carrying vesicular stomatitis virus-glycoprotein (VSV-G) but was absent from vesicles transporting the hemagglutinin protein (HA) of influenza virus to the apical cell surface. Using a cytosol dependent in vitro transport assay in permeabilized MDCK cells we studied the functional role of rab8 in biosynthetic membrane traffic. Transport of VSV-G from the TGN to the basolateral plasma membrane was found to be significantly inhibited by a peptide derived from the hypervariable COOH-terminal region of rab8, while transport of the influenza HA from the TGN to the apical surface and ER to Golgi transport were unaffected. We conclude that rab8 plays a role in membrane traffic from the TGN to the basolateral plasma membrane in MDCK cells.
DOI:10.1016/j.cell.2007.03.053URLPMID:17574030 [本文引用: 1]
Primary cilium dysfunction underlies the pathogenesis of Bardet-Biedl syndrome (BBS), a genetic disorder whose symptoms include obesity, retinal degeneration, and nephropathy. However, despite the identification of 12 BBS genes, the molecular basis of BBS remains elusive. Here we identify a complex composed of seven highly conserved BBS proteins. This complex, the BBSome, localizes to nonmembranous centriolar satellites in the cytoplasm but also to the membrane of the cilium. Interestingly, the BBSome is required for ciliogenesis but is dispensable for centriolar satellite function. This ciliogenic function is mediated in part by the Rab8 GDP/GTP exchange factor, which localizes to the basal body and contacts the BBSome. Strikingly, Rab8(GTP) enters the primary cilium and promotes extension of the ciliary membrane. Conversely, preventing Rab8(GTP) production blocks ciliation in cells and yields characteristic BBS phenotypes in zebrafish. Our data reveal that BBS may be caused by defects in vesicular transport to the cilium.
DOI:10.3892/ijmm.3.6.597URLPMID:10341289 [本文引用: 1]
Alzheimer's disease (AD) is a neurodegenerative disease characterized by the progressive deterioration of cognitive function and memory in association with the wide-spread presence of senile plaques, neurofibrillary tangles and neuronal cell death. However, its pathophysiology remains unknown. GTP-binding proteins with molecular weights of approximately 20,000 are designated small G proteins. In the present study we quantitatively analyzed the small G proteins, Ras, Rap, Ral and Rab in brains removed at autopsy from controls and AD patients to examine whether small G proteins are equally or differentially affected in AD. Western blot analysis indicated that the protein level of Ras and RalB in both the cytosolic and membranous fractions and that of Rap2 in the cytosolic fraction was significantly decreased, while that of Rab8 in the membranous fraction was significantly increased in AD brains compared with controls. The protein level of other small G proteins was not different between control and AD brains. These results suggest a differential involvement of small G proteins in AD.
DOI:10.1371/journal.pone.0178288URLPMID:28552936 [本文引用: 1]
Tau protein can be released by neurons, an event linked to the propagation of Tau pathology in Alzheimer'disease (AD). Neuronal hyperexcitability was shown to significantly increase Tau release by neurons. We confirmed this in the present study. In a previous study, it was demonstrated that hyperexcitability induces Golgi apparatus dynamics resulting in its fragmentation. Our present results revealed that the increase of Tau secretion upon hyperexcitability could be significantly reduced by preventing Golgi dynamics through the inactivation of cdk5. We then verified whether a Golgi fragmentation not induced by hyperexcitability could also increase Tau secretion. The suppression of Rab1A, Rab GTPase associated with the Golgi membranes, known to induce a Golgi fragmentation increased Tau secretion by both neurons and HeLa cells. Although it remains to be demonstrated whether the Golgi is directly involved in Tau secretion, the present results demonstrate that its dynamics are correlated to a modulation of Tau secretion.
DOI:10.1186/s40035-020-00206-1URLPMID:32552912 [本文引用: 2]
BACKGROUND: In Alzheimer's Disease (AD), about one-third of the risk genes identified by GWAS encode proteins that function predominantly in the endocytic pathways. Among them, the Ras and Rab Interactor 3(RIN3) is a guanine nucleotide exchange factor (GEF) for the Rab5 small GTPase family and has been implicated to be a risk factor for both late onset AD (LOAD) and sporadic early onset AD (sEOAD). However, how RIN3 is linked to AD pathogenesis is currently undefined. METHODS: Quantitative PCR and immunoblotting were used to measure the RIN3 expression level in mouse brain tissues and cultured basal forebrain cholinergic neuron (BFCNs). Immunostaining was used to define subcellular localization of RIN3 and to visualize endosomal changes in cultured primary BFCNs and PC12 cells. Recombinant flag-tagged RIN3 protein was purified from HEK293T cells and was used to define RIN3-interactomes by mass spectrometry. RIN3-interacting partners were validated by co-immunoprecipitation, immunofluorescence and yeast two hybrid assays. Live imaging of primary neurons was used to examine axonal transport of amyloid precursor protein (APP) and beta-secretase 1 (BACE1). Immunoblotting was used to detect protein expression, processing of APP and phosphorylated forms of Tau. RESULTS: We have shown that RIN3 mRNA level was significantly increased in the hippocampus and cortex of APP/PS1 mouse brain. Basal forebrain cholinergic neurons (BFCNs) cultured from E18 APP/PS1 mouse embryos also showed increased RIN3 expression accompanied by early endosome enlargement. In addition, via its proline rich domain, RIN3 recruited BIN1(bridging integrator 1) and CD2AP (CD2 associated protein), two other AD risk factors, to early endosomes. Interestingly, overexpression of RIN3 or CD2AP promoted APP cleavage to increase its carboxyl terminal fragments (CTFs) in PC12 cells. Upregulation of RIN3 or the neuronal isoform of BIN1 increased phosphorylated Tau level. Therefore, upregulation of RIN3 expression promoted accumulation of APP CTFs and increased phosphorylated Tau. These effects by RIN3 was rescued by the expression of a dominant negative Rab5 (Rab5(S34N)) construct. Our study has thus pointed to that RIN3 acts through Rab5 to impact endosomal trafficking and signaling. CONCLUSION: RIN3 is significantly upregulated and correlated with endosomal dysfunction in APP/PS1 mouse. Through interacting with BIN1 and CD2AP, increased RIN3 expression alters axonal trafficking and procession of APP. Together with our previous studies, our current work has thus provided important insights into the role of RIN3 in regulating endosomal signaling and trafficking.
DOI:10.1016/j.neuron.2004.12.023URLPMID:15629704 [本文引用: 1]
The activity-dependent removal of AMPA receptors from synapses underlies long-term depression in hippocampal excitatory synapses. In this study, we have investigated the role of the small GTPase Rab5 during this process. We propose that Rab5 is a critical link between the signaling cascades triggered by LTD induction and the machinery that executes the activity-dependent removal of AMPA receptors. We have found that Rab5 activation drives the specific internalization of synaptic AMPA receptors in a clathrin-dependent manner and that this activity is required for LTD. Interestingly, Rab5 does not participate in the constitutive cycling of AMPA receptors. Rab5 is able to remove both GluR1 and GluR2 AMPA receptor subunits, leading to GluR1 dephosphorylation. Importantly, NMDA receptor-dependent LTD induction produces a rapid and transient increase of active (GTP bound) Rab5. We propose a model in which synaptic activity leads to Rab5 activation, which in turn drives the removal of AMPA receptors from synapses.
DOI:10.1073/pnas.0801174105URL [本文引用: 1]
DOI:10.1073/pnas.0911281106URLPMID:19910533 [本文引用: 1]
One of the neuropathological hallmarks of Alzheimer's disease (AD) is the amyloid plaque, primarily composed of aggregated amyloid-beta (Abeta) peptide. In vitro, Abeta(1-42), the major alloform of Abeta found in plaques, self-assembles into fibrils at micromolar concentrations and acidic pH. Such conditions do not exist in the extracellular fluid of the brain where the pH is neutral and Abeta concentrations are in the nanomolar range. Here, we show that extracellular soluble Abeta (sAbeta) at concentrations as low as 1 nM was taken up by murine cortical neurons and neuroblastoma (SHSY5Y) cells but not by human embryonic kidney (HEK293) cells. Following uptake, Abeta accumulated in Lysotracker-positive acidic vesicles (likely late endosomes or lysosomes) where effective concentrations (>2.5 microM) were greater than two orders of magnitude higher than that in the extracellular fluid (25 nM), as quantified by fluorescence intensity using laser scanning confocal microscopy. Furthermore, SHSY5Y cells incubated with 1 muM Abeta(1-42) for several days demonstrated a time-dependent increase in intracellular high molecular weight (HMW) (>200 kDa) aggregates, which were absent in cells grown in the presence of Abeta(1-40). Homogenates from these Abeta(1-42)-loaded cells were capable of seeding amyloid fibril growth. These results demonstrate that Abeta can be taken up by certain cells at low physiologically relevant concentrations of extracellular Abeta, and then concentrated into endosomes/lysosomes. At high concentrations, vesicular Abeta aggregates to form HMW species which are capable of seeding amyloid fibril growth. We speculate that extrusion of these aggregates may seed extracellular amyloid plaque formation during AD pathogenesis.
DOI:10.1111/jnc.13994URLPMID:28222213 [本文引用: 2]
The axonal microtubule-associated protein TAU, involved in Alzheimer's disease (AD), can be found in the extracellular space where it could be taken up by neurons, an event that is believed to contribute to the propagation of tau pathology in the brain. Since the small GTPase Rab7A is involved in the trafficking of endosomes, autophagosomes, and lysosomes, and RAB7A gene expression and protein levels are up-regulated in AD patients, we tested the hypothesis that Rab7A was involved in tau secretion. We previously reported that both primary cortical neurons and HeLa cells over-expressing human TAU can release tau. Using these two cellular systems, we demonstrated that Rab7A regulates tau secretion. Upon Rab7A deletion, tau secretion was decreased. Consistent with this, the over-expression of a dominant negative and a constitutively active form of Rab7A decreased and increased tau secretion, respectively. A partial co-localization of tau and Rab7-positive structures in both neurons and HeLa cells indicated that a late endosomal compartment could be involved in its secretion. Collectively, the present data indicate that Rab7A regulates tau secretion and therefore the up-regulation of RAB7A reported in AD, could contribute to the extracellular accumulation of pathological TAU species that could result in the propagation of tau pathology in the AD brain.
DOI:10.1136/jnnp-2017-317378URLPMID:29549192 [本文引用: 1]
OBJECTIVE: To ascertain demographic and clinical features of Parkinson disease (PD) associated with functional neurological features. METHODS: A standardised form was used to extract data from electronic records of 53 PD patients with associated functional neurological disorders (PD-FND) across eight movement disorders centres in the USA, Canada and Europe. These subjects were matched for age, gender and disease duration to PD patients without functional features (PD-only). Logistic regression analysis was used to compare both groups after adjusting for clustering effect. RESULTS: Functional symptoms preceded or co-occurred with PD onset in 34% of cases, nearly always in the most affected body side. Compared with PD-only subjects, PD-FND were predominantly female (68%), had longer delay to PD diagnosis, greater prevalence of dyskinesia (42% vs 18%; P=0.023), worse depression and anxiety (P=0.033 and 0.025, respectively), higher levodopa-equivalent daily dose (972+/-701 vs 741+/-559 mg; P=0.029) and lower motor severity (P=0.019). These patients also exhibited greater healthcare resource utilisation, higher use of [(123)I]FP-CIT SPECT and were more likely to have had a pre-existing psychiatric disorder (P=0.008) and family history of PD (P=0.036). CONCLUSIONS: A subtype of PD with functional neurological features is familial in one-fourth of cases and associated with more psychiatric than motor disability and greater use of diagnostic and healthcare resources than those without functional features. Functional manifestations may be prodromal to PD in one-third of patients.
URLPMID:31383252 [本文引用: 1]
Recent evidence from genetics, animal model systems and biochemical studies suggests that defects in membrane trafficking play an important part in the pathophysiology of Parkinson's disease (PD). Mutations in leucine-rich repeat kinase 2 (LRRK2) constitute the most frequent genetic cause of both familial and sporadic PD, and LRRK2 has been suggested as a druggable target for PD. Although the precise physiological function of LRRK2 remains largely unknown, mounting evidence suggests that LRRK2 controls membrane trafficking by interacting with key regulators of the endosomal-lysosomal pathway and synaptic recycling. In this review, we discuss the genetic, biochemical and functional links between LRRK2 and membrane trafficking. Understanding the mechanism by which LRRK2 influences such processes may contribute to the development of disease-modifying therapies for PD. [BMB Reports 2019; 52(9): 533-539].
DOI:10.1016/j.nbd.2017.12.005URLPMID:29246723 [本文引用: 2]
Missense mutations in the multi-domain kinase LRRK2 cause late onset familial Parkinson's disease. They most commonly with classic proteinopathy in the form of Lewy bodies and Lewy neurites comprised of insoluble alpha-synuclein, but in rare cases can also manifest tauopathy. The normal function of LRRK2 has remained elusive, as have the cellular consequences of its mutation. Data from LRRK2 null model organisms and LRRK2-inhibitor treated animals support a physiological role for LRRK2 in regulating lysosome function. Since idiopathic and LRRK2-linked PD are associated with the intraneuronal accumulation of protein aggregates, a series of critical questions emerge. First, how do pathogenic mutations that increase LRRK2 kinase activity affect lysosome biology in neurons? Second, are mutation-induced changes in lysosome function sufficient to alter the metabolism of alpha-synuclein? Lastly, are changes caused by pathogenic mutation sensitive to reversal with LRRK2 kinase inhibitors? Here, we report that mutation of LRRK2 induces modest but significant changes in lysosomal morphology and acidification, and decreased basal autophagic flux when compared to WT neurons. These changes were associated with an accumulation of detergent-insoluble alpha-synuclein and increased neuronal release of alpha-synuclein and were reversed by pharmacologic inhibition of LRRK2 kinase activity. These data demonstrate a critical and disease-relevant influence of native neuronal LRRK2 kinase activity on lysosome function and alpha-synuclein homeostasis. Furthermore, they also suggest that lysosome dysfunction, altered neuronal alpha-synuclein metabolism, and the insidious accumulation of aggregated protein over decades may contribute to pathogenesis in this late-onset form of familial PD.
DOI:10.1016/S0076-6879(07)00425-9URLPMID:18374176 [本文引用: 1]
Recent studies implicate a disruption in Rab-mediated protein trafficking as a possible contributing factor to neurodegeneration in Parkinson's disease (PD). Misfolding of the neuronal protein alpha-synuclein (asyn) is implicated in PD. Overexpression of asyn results in cell death in a wide variety of model systems, and in several organisms, including yeast, worms, flies, and rodent primary neurons, this toxicity is suppressed by the overproduction of Rab proteins. These and other findings suggest that asyn interferes with Rab function and provide new avenues for PD drug discovery. This chapter describes two assay formats that have been used successfully to identify small molecules that rescue asyn toxicity in yeast. The 96-well format monitors rescue by optical density and is suitable for screening thousands of compounds. A second format measures viable cells by reduction of the dye alamarBlue, a readout that is compatible with 96-, 384-, and 1536-well plates allowing the screening of large libraries (>100,000 compounds). A secondary assay to eliminate mechanistically undesirable hits is also described.
DOI:10.1371/journal.pgen.1005995URLPMID:27123591 [本文引用: 1]
Alpha-Synuclein (aSyn) misfolding and aggregation is common in several neurodegenerative diseases, including Parkinson's disease and dementia with Lewy bodies, which are known as synucleinopathies. Accumulating evidence suggests that secretion and cell-to-cell trafficking of pathological forms of aSyn may explain the typical patterns of disease progression. However, the molecular mechanisms controlling aSyn aggregation and spreading of pathology are still elusive. In order to obtain unbiased information about the molecular regulators of aSyn oligomerization, we performed a microscopy-based large-scale RNAi screen in living cells. Interestingly, we identified nine Rab GTPase and kinase genes that modulated aSyn aggregation, toxicity and levels. From those, Rab8b, Rab11a, Rab13 and Slp5 were able to promote the clearance of aSyn inclusions and rescue aSyn induced toxicity. Furthermore, we found that endocytic recycling and secretion of aSyn was enhanced upon Rab11a and Rab13 expression in cells accumulating aSyn inclusions. Overall, our study resulted in the identification of new molecular players involved in the aggregation, toxicity, and secretion of aSyn, opening novel avenues for our understanding of the molecular basis of synucleinopathies.
DOI:10.1111/jnc.13712URLPMID:27333324 [本文引用: 2]
Parkinson's disease can be caused by mutations in the alpha-synuclein gene and is characterized by aggregates of alpha-synuclein protein. Aggregates are degraded by the autophago-lysosomal pathway. Since Rab7 has been shown to regulate trafficking of late endosomes and autophagosomes, we hypothesized that over-expressing Rab7 might be beneficial in Parkinson's disease. To test this hypothesis, we expressed the pathogenic A53T mutant of alpha-synuclein in HEK293 cells and Drosophila melanogaster. In HEK293 cells, EGFP-Rab7-decorated vesicles contain alpha-synuclein. Rab7 over-expression reduced the percentage of cells with alpha-synuclein particles and the amount of alpha-synuclein protein. Time-lapse microscopy confirmed that particles frequently disappeared with Rab7 over-expression. Clearance of alpha-synuclein is explained by the increased occurrence of acidified alpha-synuclein vesicles with Rab7 over-expression, presumably representing autolysosomes. Rab7 over-expression reduced apoptosis and the percentage of dead cells in trypan blue staining. In the fly model, Rab7 rescued the locomotor deficit induced by neuronal expression of A53T-alpha-synuclein. These beneficial effects were not produced by Rab7 missense mutations causing Charcot Marie Tooth neuropathy, or by the related GTPases Rab5, Rab9, or Rab23. Using mass spectrometry, we identified Rab7 in neuromelanin granules purified from human substantia nigra, indicating that Rab7 might be involved in the biogenesis of these possibly protective, autophagosome-like organelles in dopaminergic neurons. Taken together, Rab7 increased the clearance of alpha-synuclein aggregates, reduced cell death, and rescued the phenotype in a fly model of Parkinson's disease. These findings indicate that Rab7 is rate-limiting for aggregate clearance, and that Rab7 activation may offer a therapeutic strategy for Parkinson's disease. Cells over-expressing aggregation-prone A53T alpha-synuclein develop cytoplasmic aggregates mimicking changes observed in Parkinson's disease. When following cells in time-lapse microscopy, some few cells are able to remove these aggregates (Opazo et al. 2008). We now show that the percentage of cells clearing all aggregates from their cytosol is greatly increased with Rab7 over-expression, indicating that availability of Rab7 is rate-limiting for autophagic clearance of aggregates. The functional significance of this effect in neurons was confirmed in a Drosophila melanogaster model of Parkinson's disease.
DOI:10.1016/j.ajhg.2011.06.001URL [本文引用: 1]
The identification of genetic causes for Mendelian disorders has been based on the collection of multi-incident families, linkage analysis, and sequencing of genes in candidate intervals. This study describes the application of next-generation sequencing technologies to a Swiss kindred presenting with autosomal-dominant, late-onset Parkinson disease (PD). The family has tremor-predominant dopa-responsive parkinsonism with a mean onset of 50.6 +/- 7.3 years. Exome analysis suggests that an aspartic-acid-to-asparagine mutation within vacuolar protein sorting 35 (VPS35 c.1858G>A; p.Asp620Asn) is the genetic determinant of disease. VPS35 is a central component of the retromer cargo-recognition complex, is critical for endosome-trans-golgi trafficking and membrane-protein recycling, and is evolutionarily highly conserved. VPS35 c.1858G>A was found in all affected members of the Swiss kindred and in three more families and one patient with sporadic PD, but it was not observed in 3,309 controls. Further sequencing of familial affected probands revealed only one other missense variant, VPS35 c.946C>T; (p.Pro316Ser), in a pedigree with one unaffected and two affected carriers, and thus the pathogenicity of this mutation remains uncertain. Retromer-mediated sorting and transport is best characterized for acid hydrolase receptors. However, the complex has many types of cargo and is involved in a diverse array of biologic pathways from developmental Wnt signaling to lysosome biogenesis. Our study implicates disruption of VPS35 and retromer-mediated trans-membrane protein sorting, rescue, and recycling in the neurodegenerative process leading to PD.
DOI:10.1093/hmg/ddx410URLPMID:29177506 [本文引用: 1]
Human genetic studies implicate LRRK2 and RAB7L1 in susceptibility to Parkinson disease (PD). These two genes function in the same pathway, as knockout of Rab7L1 results in phenotypes similar to LRRK2 knockout, and studies in cells and model organisms demonstrate LRRK2 and Rab7L1 interact in the endolysosomal system. Recently, a subset of Rab proteins have been identified as LRRK2 kinase substrates. Herein, we find that Rab8, Rab10, and Rab7L1 must be membrane and GTP-bound for LRRK2 phosphorylation. LRRK2 mutations that cause PD including R1441C, Y1699C, and G2019S all increase LRRK2 phosphorylation of Rab7L1 four-fold over wild-type LRRK2 in cells, resulting in the phosphorylation of nearly one-third the available Rab7L1 protein in cells. In contrast, the most common pathogenic LRRK2 mutation, G2019S, does not upregulate LRRK2-mediated phosphorylation of Rab8 or Rab10. LRRK2 interaction with membrane and GTP-bound Rab7L1, but not Rab8 or Rab10, results in the activation of LRRK2 autophosphorylation at the serine 1292 position, required for LRRK2 toxicity. Further, Rab7L1 controls the proportion of LRRK2 that is membrane-associated, and LRRK2 mutations enhance Rab7L1-mediated recruitment of LRRK2 to the trans-Golgi network. Interaction studies with the Rab8 and Rab10 GTPase-activating protein TBC1D4/AS160 demonstrate that LRRK2 phosphorylation may block membrane and GTP-bound Rab protein interaction with effectors. These results suggest reciprocal regulation between LRRK2 and Rab protein substrates, where Rab7L1-mediated upregulation of LRRK2 kinase activity results in the stabilization of membrane and GTP-bound Rab proteins that may be unable to interact with Rab effector proteins.
DOI:10.7554/eLife.12813URLPMID:26824392 [本文引用: 1]
Mutations in Park8, encoding for the multidomain Leucine-rich repeat kinase 2 (LRRK2) protein, comprise the predominant genetic cause of Parkinson's disease (PD). G2019S, the most common amino acid substitution activates the kinase two- to threefold. This has motivated the development of LRRK2 kinase inhibitors; however, poor consensus on physiological LRRK2 substrates has hampered clinical development of such therapeutics. We employ a combination of phosphoproteomics, genetics, and pharmacology to unambiguously identify a subset of Rab GTPases as key LRRK2 substrates. LRRK2 directly phosphorylates these both in vivo and in vitro on an evolutionary conserved residue in the switch II domain. Pathogenic LRRK2 variants mapping to different functional domains increase phosphorylation of Rabs and this strongly decreases their affinity to regulatory proteins including Rab GDP dissociation inhibitors (GDIs). Our findings uncover a key class of bona-fide LRRK2 substrates and a novel regulatory mechanism of Rabs that connects them to PD.
DOI:10.7554/eLife.31012URLPMID:29125462 [本文引用: 1]
We previously reported that Parkinson's disease (PD) kinase LRRK2 phosphorylates a subset of Rab GTPases on a conserved residue in their switch-II domains (Steger et al., 2016) (PMID: 26824392). Here, we systematically analyzed the Rab protein family and found 14 of them (Rab3A/B/C/D, Rab5A/B/C, Rab8A/B, Rab10, Rab12, Rab29, Rab35 and Rab43) to be specifically phosphorylated by LRRK2, with evidence for endogenous phosphorylation for ten of them (Rab3A/B/C/D, Rab8A/B, Rab10, Rab12, Rab35 and Rab43). Affinity enrichment mass spectrometry revealed that the primary ciliogenesis regulator, RILPL1 specifically interacts with the LRRK2-phosphorylated forms of Rab8A and Rab10, whereas RILPL2 binds to phosphorylated Rab8A, Rab10, and Rab12. Induction of primary cilia formation by serum starvation led to a two-fold reduction in ciliogenesis in fibroblasts derived from pathogenic LRRK2-R1441G knock-in mice. These results implicate LRRK2 in primary ciliogenesis and suggest that Rab-mediated protein transport and/or signaling defects at cilia may contribute to LRRK2-dependent pathologies.
DOI:10.1186/s13024-018-0240-1URLPMID:29439717 [本文引用: 2]
BACKGROUND: Mutations in leucine-rich repeat kinase 2 (LRRK2) are the most common cause of familial and sporadic Parkinson's disease (PD). Elevated kinase activity is associated with LRRK2 toxicity, but the substrates that mediate neurodegeneration remain poorly defined. Given the increasing evidence suggesting a role of LRRK2 in membrane and vesicle trafficking, here we systemically screened Rab GTPases, core regulators of vesicular dynamics, as potential substrates of LRRK2 and investigated the functional consequence of such phosphorylation in cells and in vivo. METHODS: In vitro LRRK2 kinase assay with forty-five purified human Rab GTPases was performed to identify Rab family proteins as substrates of LRRK2. We identified the phosphorylation site by tandem mass-spectrometry and confirmed it by assessing phosphorylation in the in vitro LRRK2 kinase assay and in cells. Effects of Rab phosphorylation on neurodegeneration were examined in primary cultures and in vivo by intracranial injection of adeno-associated viral vectors (AAV) expressing wild-type or phosphomutants of Rab35. RESULTS: Our screening revealed that LRRK2 phosphorylated several Rab GTPases at a conserved threonine residue in the switch II region, and by using the kinase-inactive LRRK2-D1994A and the pathogenic LRRK2-G2019S along with Rab proteins in which the LRRK2 site was mutated, we verified that a subset of Rab proteins, including Rab35, were authentic substrates of LRRK2 both in vitro and in cells. We also showed that phosphorylation of Rab regulated GDP/GTP-binding property in cells. Moreover, in primary cortical neurons, mutation of the LRRK2 site in several Rabs caused neurotoxicity, which was most severely induced by phosphomutants of Rab35. Furthermore, intracranial injection of the AAV-Rab35 -T72A or AAV-Rab35-T72D into the substantia nigra substantially induced degeneration of dopaminergic neurons in vivo. CONCLUSIONS: Here we show that a subset of Rab GTPases are authentic substrates of LRRK2 both in vitro and in cells. We also provide evidence that dysregulation of Rab phosphorylation in the LRRK2 site induces neurotoxicity in primary neurons and degeneration of dopaminergic neurons in vivo. Our study suggests that Rab GTPases might mediate LRRK2 toxicity in the progression of PD.
DOI:10.1073/pnas.1812196115URLPMID:30209220 [本文引用: 1]
Leucine-rich repeat kinase 2 (LRRK2) has been associated with a variety of human diseases, including Parkinson's disease and Crohn's disease, whereas LRRK2 deficiency leads to accumulation of abnormal lysosomes in aged animals. However, the cellular roles and mechanisms of LRRK2-mediated lysosomal regulation have remained elusive. Here, we reveal a mechanism of stress-induced lysosomal response by LRRK2 and its target Rab GTPases. Lysosomal overload stress induced the recruitment of endogenous LRRK2 onto lysosomal membranes and activated LRRK2. An upstream adaptor Rab7L1 (Rab29) promoted the lysosomal recruitment of LRRK2. Subsequent family-wide screening of Rab GTPases that may act downstream of LRRK2 translocation revealed that Rab8a and Rab10 were specifically accumulated on overloaded lysosomes dependent on their phosphorylation by LRRK2. Rab7L1-mediated lysosomal targeting of LRRK2 attenuated the stress-induced lysosomal enlargement and promoted lysosomal secretion, whereas Rab8 stabilized by LRRK2 on stressed lysosomes suppressed lysosomal enlargement and Rab10 promoted lysosomal secretion, respectively. These effects were mediated by the recruitment of Rab8/10 effectors EHBP1 and EHBP1L1. LRRK2 deficiency augmented the chloroquine-induced lysosomal vacuolation of renal tubules in vivo. These results implicate the stress-responsive machinery composed of Rab7L1, LRRK2, phosphorylated Rab8/10, and their downstream effectors in the maintenance of lysosomal homeostasis.
[本文引用: 1]
DOI:10.1371/journal.pgen.1006093URLPMID:27272733 [本文引用: 1]
EHBP-1 (Ehbp1) is a conserved regulator of endocytic recycling, acting as an effector of small GTPases including RAB-10 (Rab10). Here we present evidence that EHBP-1 associates with tubular endosomal phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2] enriched membranes through an N-terminal C2-like (NT-C2) domain, and define residues within the NT-C2 domain that mediate membrane interaction. Furthermore, our results indicate that the EHBP-1 central calponin homology (CH) domain binds to actin microfilaments in a reaction that is stimulated by RAB-10(GTP). Loss of any aspect of this RAB-10/EHBP-1 system in the C. elegans intestinal epithelium leads to retention of basolateral recycling cargo in endosomes that have lost their normal tubular endosomal network (TEN) organization. We propose a mechanism whereby RAB-10 promotes the ability of endosome-bound EHBP-1 to also bind to the actin cytoskeleton, thereby promoting endosomal tubulation.
DOI:10.1038/s41467-018-05958-zURLPMID:30150626 [本文引用: 1]
Propagation of alpha-synuclein aggregates has been suggested as a contributing factor in Parkinson's disease (PD) progression. However, the molecular mechanisms underlying alpha-synuclein aggregation are not fully understood. Here, we demonstrate in cell culture, nematode, and rodent models of PD that leucine-rich repeat kinase 2 (LRRK2), a PD-linked kinase, modulates alpha-synuclein propagation in a kinase activity-dependent manner. The PD-linked G2019S mutation in LRRK2, which increases kinase activity, enhances propagation efficiency. Furthermore, we show that the role of LRRK2 in alpha-synuclein propagation is mediated by RAB35 phosphorylation. Constitutive activation of RAB35 overrides the reduced alpha-synuclein propagation phenotype in lrk-1 mutant C. elegans. Finally, in a mouse model of synucleinopathy, administration of an LRRK2 kinase inhibitor reduced alpha-synuclein aggregation via enhanced interaction of alpha-synuclein with the lysosomal degradation pathway. These results suggest that LRRK2-mediated RAB35 phosphorylation is a potential therapeutic target for modifying disease progression.
[本文引用: 1]
DOI:10.1016/j.bbadis.2019.165632URLPMID:31812666 [本文引用: 1]
LRRK2 and SNCA, the gene for alpha-synuclein, are the two of the most important genetic factors of Parkinson's disease (PD). A-synuclein is aggregated and accumulated in neurons and glia in PD and considered the pathogenic culprit of the disease. A-synuclein aggregates spread from a few discrete regions of the brain to larger areas as the disease progresses through cell-to-cell propagation mechanism. LRRK2 is involved in the regulation of vesicle trafficking, in particular in the endolysosomal and autophagic pathways. Studies also suggest that LRRK2 might regulate the pathogenic actions of alpha-synuclein. However, the relationship between these two proteins in the pathogenesis of PD remains elusive. Here, we review the current literature on the pathophysiological function of LRRK2 with an emphasis on its role in the endolysosomal and autophagic pathways. We also propose a potential mechanism by which LRRK2 is involved in the regulation of aggregation and the propagation of alpha-synuclein.
DOI:10.4103/2152-7806.169561URLPMID:26629397 [本文引用: 1]
Amyotrophic lateral sclerosis (ALS) is a late-onset fatal neurodegenerative disease affecting motor neurons with an incidence of about 1/100,000. Most ALS cases are sporadic, but 5-10% of the cases are familial ALS. Both sporadic and familial ALS (FALS) are associated with degeneration of cortical and spinal motor neurons. The etiology of ALS remains unknown. However, mutations of superoxide dismutase 1 have been known as the most common cause of FALS. In this study, we provide a comprehensive review of ALS. We cover all aspects of the disease including epidemiology, comorbidities, environmental risk factor, molecular mechanism, genetic factors, symptoms, diagnostic, treatment, and even the available supplement and management of ALS. This will provide the reader with an advantage of receiving a broad range of information about the disease.
DOI:10.1007/s00401-015-1468-2URLPMID:26298469 [本文引用: 2]
Several diverse proteins are linked genetically/pathologically to neurodegeneration in amyotrophic lateral sclerosis (ALS) including SOD1, TDP-43 and FUS. Using a variety of cellular and biochemical techniques, we demonstrate that ALS-associated mutant TDP-43, FUS and SOD1 inhibit protein transport between the endoplasmic reticulum (ER) and Golgi apparatus in neuronal cells. ER-Golgi transport was also inhibited in embryonic cortical and motor neurons obtained from a widely used animal model (SOD1(G93A) mice), validating this mechanism as an early event in disease. Each protein inhibited transport by distinct mechanisms, but each process was dependent on Rab1. Mutant TDP-43 and mutant FUS both inhibited the incorporation of secretory protein cargo into COPII vesicles as they bud from the ER, and inhibited transport from ER to the ER-Golgi intermediate (ERGIC) compartment. TDP-43 was detected on the cytoplasmic face of the ER membrane, whereas FUS was present within the ER, suggesting that transport is inhibited from the cytoplasm by mutant TDP-43, and from the ER by mutant FUS. In contrast, mutant SOD1 destabilised microtubules and inhibited transport from the ERGIC compartment to Golgi, but not from ER to ERGIC. Rab1 performs multiple roles in ER-Golgi transport, and over-expression of Rab1 restored ER-Golgi transport, and prevented ER stress, mSOD1 inclusion formation and induction of apoptosis, in cells expressing mutant TDP-43, FUS or SOD1. Rab1 also co-localised extensively with mutant TDP-43, FUS and SOD1 in neuronal cells, and Rab1 formed inclusions in motor neurons of spinal cords from sporadic ALS patients, which were positive for ubiquitinated TDP-43, implying that Rab1 is misfolded and dysfunctional in sporadic disease. These results demonstrate that ALS-mutant forms of TDP-43, FUS, and SOD1 all perturb protein transport in the early secretory pathway, between ER and Golgi compartments. These data also imply that restoring Rab1-mediated ER-Golgi transport is a novel therapeutic target in ALS.
DOI:10.1016/j.cell.2008.04.039URLPMID:18555774 [本文引用: 1]
VAP proteins (human VAPB/ALS8, Drosophila VAP33, and C. elegans VPR-1) are homologous proteins with an amino-terminal major sperm protein (MSP) domain and a transmembrane domain. The MSP domain is named for its similarity to the C. elegans MSP protein, a sperm-derived hormone that binds to the Eph receptor and induces oocyte maturation. A point mutation (P56S) in the MSP domain of human VAPB is associated with Amyotrophic lateral sclerosis (ALS), but the mechanisms underlying the pathogenesis are poorly understood. Here we show that the MSP domains of VAP proteins are cleaved and secreted ligands for Eph receptors. The P58S mutation in VAP33 leads to a failure to secrete the MSP domain as well as ubiquitination, accumulation of inclusions in the endoplasmic reticulum, and an unfolded protein response. We propose that VAP MSP domains are secreted and act as diffusible hormones for Eph receptors. This work provides insight into mechanisms that may impact the pathogenesis of ALS.
DOI:10.1093/hmg/ddu068URL [本文引用: 2]
Intronic expansion of a hexanucleotide GGGGCC repeat in the chromosome 9 open reading frame 72 (C9ORF72) gene is the major cause of familial amyotrophic lateral sclerosis (ALS) and frontotemporal dementia. However, the cellular function of the C9ORF72 protein remains unknown. Here, we demonstrate that C9ORF72 regulates endosomal trafficking. C9ORF72 colocalized with Rab proteins implicated in autophagy and endocytic transport: Rab1, Rab5, Rab7 and Rab11 in neuronal cell lines, primary cortical neurons and human spinal cord motor neurons, consistent with previous predictions that C9ORF72 bears Rab guanine exchange factor activity. Consistent with this notion, C9ORF72 was present in the extracellular space and as cytoplasmic vesicles. Depletion of C9ORF72 using siRNA inhibited transport of Shiga toxin from the plasma membrane to Golgi apparatus, internalization of TrkB receptor and altered the ratio of autophagosome marker light chain 3 (LC3) II:LC3I, indicating that C9ORF72 regulates endocytosis and autophagy. C9ORF72 also colocalized with ubiquilin-2 and LC3-positive vesicles, and co-migrated with lysosome-stained vesicles in neuronal cell lines, providing further evidence that C9ORF72 regulates autophagy. Investigation of proteins interacting with C9ORF72 using mass spectrometry identified other proteins implicated in ALS; ubiquilin-2 and heterogeneous nuclear ribonucleoproteins, hnRNPA2/B1 and hnRNPA1, and actin. Treatment of cells overexpressing C9ORF72 with proteasome inhibitors induced the formation of stress granules positive for hnRNPA1 and hnRNPA2/B1. Immunohistochemistry of C9ORF72 ALS patient motor neurons revealed increased colocalization between C9ORF72 and Rab7 and Rab11 compared with controls, suggesting possible dysregulation of trafficking in patients bearing the C9ORF72 repeat expansion. Hence, this study identifies a role for C9ORF72 in Rab-mediated cellular trafficking.
DOI:10.1074/jbc.M313504200URLPMID:15033976 [本文引用: 1]
ALS2 is the gene mutated in a recessive juvenile form of amyotrophic lateral sclerosis (ALS2). ALS2 encodes a large protein termed alsin, which contains a number of predicted cell signaling and protein trafficking sequence motifs. To gain insight into the overall function of alsin and to begin to evaluate its role in motor neuron maintenance, we examined the subcellular localization of alsin and the biochemical activities associated with its individual subdomains. We found that the Vps9p domain of alsin has Rab5 guanine nucleotide exchange activity. In addition, alsin interacted specifically with and acted as a guanine nucleotide exchange factor for Rac1. Immunofluorescence and fractionation experiments in both fibroblasts and neurons revealed that alsin is a cytosolic protein, with a significant portion associated with small, punctate membrane structures. Many of these membrane structures also contained Rab5 or Rac1. Upon overexpression of full-length alsin, the overexpressed material was largely cytosolic, indicating that the association with membrane structures could be saturated. We also found that alsin was present in membrane ruffles and lamellipodia. These data suggest that alsin is involved in membrane transport events, potentially linking endocytic processes and actin cytoskeleton remodeling.
DOI:10.1093/hmg/ddu068URL [本文引用: 1]
Intronic expansion of a hexanucleotide GGGGCC repeat in the chromosome 9 open reading frame 72 (C9ORF72) gene is the major cause of familial amyotrophic lateral sclerosis (ALS) and frontotemporal dementia. However, the cellular function of the C9ORF72 protein remains unknown. Here, we demonstrate that C9ORF72 regulates endosomal trafficking. C9ORF72 colocalized with Rab proteins implicated in autophagy and endocytic transport: Rab1, Rab5, Rab7 and Rab11 in neuronal cell lines, primary cortical neurons and human spinal cord motor neurons, consistent with previous predictions that C9ORF72 bears Rab guanine exchange factor activity. Consistent with this notion, C9ORF72 was present in the extracellular space and as cytoplasmic vesicles. Depletion of C9ORF72 using siRNA inhibited transport of Shiga toxin from the plasma membrane to Golgi apparatus, internalization of TrkB receptor and altered the ratio of autophagosome marker light chain 3 (LC3) II:LC3I, indicating that C9ORF72 regulates endocytosis and autophagy. C9ORF72 also colocalized with ubiquilin-2 and LC3-positive vesicles, and co-migrated with lysosome-stained vesicles in neuronal cell lines, providing further evidence that C9ORF72 regulates autophagy. Investigation of proteins interacting with C9ORF72 using mass spectrometry identified other proteins implicated in ALS; ubiquilin-2 and heterogeneous nuclear ribonucleoproteins, hnRNPA2/B1 and hnRNPA1, and actin. Treatment of cells overexpressing C9ORF72 with proteasome inhibitors induced the formation of stress granules positive for hnRNPA1 and hnRNPA2/B1. Immunohistochemistry of C9ORF72 ALS patient motor neurons revealed increased colocalization between C9ORF72 and Rab7 and Rab11 compared with controls, suggesting possible dysregulation of trafficking in patients bearing the C9ORF72 repeat expansion. Hence, this study identifies a role for C9ORF72 in Rab-mediated cellular trafficking.
DOI:10.1091/mbc.E16-07-0519URLPMID:27535427 [本文引用: 1]
TAR DNA-binding protein 43 (TDP-43) is genetically and functionally linked to amyotrophic lateral sclerosis (ALS) and regulates transcription, splicing, and transport of thousands of RNA targets that function in diverse cellular pathways. In ALS, pathologically altered TDP-43 is believed to lead to disease by toxic gain-of-function effects on RNA metabolism, as well as by sequestering endogenous TDP-43 and causing its loss of function. However, it is unclear which of the numerous cellular processes disrupted downstream of TDP-43 dysfunction lead to neurodegeneration. Here we found that both loss and gain of function of TDP-43 in Drosophila cause a reduction of synaptic growth-promoting bone morphogenic protein (BMP) signaling at the neuromuscular junction (NMJ). Further, we observed a shift of BMP receptors from early to recycling endosomes and increased mobility of BMP receptor-containing compartments at the NMJ. Inhibition of the recycling endosome GTPase Rab11 partially rescued TDP-43-induced defects in BMP receptor dynamics and distribution and suppressed BMP signaling, synaptic growth, and larval crawling defects. Our results indicate that defects in receptor traffic lead to neuronal dysfunction downstream of TDP-43 misregulation and that rerouting receptor traffic may be a viable strategy for rescuing neurological impairment.
DOI:10.1002/ajmg.a.20190URLPMID:12784292 [本文引用: 1]
Huntington disease (HD) is a neurodegenerative disorder caused by the abnormal expansion of CAG repeats in the HD gene on chromosome 4p16.3. Past studies have shown that the size of expanded CAG repeat is inversely associated with age at onset (AO) of HD. It is not known whether the normal Huntington allele size influences the relation between the expanded repeat and AO of HD. Data collected from two independent cohorts were used to test the hypothesis that the unexpanded CAG repeat interacts with the expanded CAG repeat to influence AO of HD. In the New England Huntington Disease Center Without Walls (NEHD) cohort of 221 HD affected persons and in the HD-MAPS cohort of 533 HD affected persons, we found evidence supporting an interaction between the expanded and unexpanded CAG repeat sizes which influences AO of HD (P = 0.08 and 0.07, respectively). The association was statistically significant when both cohorts were combined (P = 0.012). The estimated heritability of the AO residual was 0.56 after adjustment for normal and expanded repeats and their interaction. An analysis of tertiles of repeats sizes revealed that the effect of the normal allele is seen among persons with large HD repeat sizes (47-83). These findings suggest that an increase in the size of the normal repeat may mitigate the expression of the disease among HD affected persons with large expanded CAG repeats.
DOI:10.1016/j.bbadis.2016.06.007URLPMID:27288730 [本文引用: 1]
Huntington's disease (HD) is an autosomal neurodegenerative disorder affecting approximately 5-10 persons per 100,000 worldwide. The pathophysiology of HD is not fully understood but the age of onset is known to be highly dependent on the number of CAG triplet repeats in the huntingtin gene. Using (1)H NMR spectroscopy this study biochemically profiled 39 brain metabolites in post-mortem striatum (n=14) and frontal lobe (n=14) from HD sufferers and controls (n=28). Striatum metabolites were more perturbed with 15 significantly affected in HD cases, compared with only 4 in frontal lobe (p<0.05; q<0.3). The metabolite which changed most overall was urea which decreased 3.25-fold in striatum (p<0.01). Four metabolites were consistently affected in both brain regions. These included the neurotransmitter precursors tyrosine and l-phenylalanine which were significantly depleted by 1.55-1.58-fold and 1.48-1.54-fold in striatum and frontal lobe, respectively (p=0.02-0.03). They also included l-leucine which was reduced 1.54-1.69-fold (p=0.04-0.09) and myo-inositol which was increased 1.26-1.37-fold (p<0.01). Logistic regression analyses performed with MetaboAnalyst demonstrated that data obtained from striatum produced models which were profoundly more sensitive and specific than those produced from frontal lobe. The brain metabolite changes uncovered in this first (1)H NMR investigation of human HD offer new insights into the disease pathophysiology. Further investigations of striatal metabolite disturbances are clearly warranted.
DOI:10.1083/jcb.200501162URLPMID:15837803 [本文引用: 1]
Myosin VI plays a role in the maintenance of Golgi morphology and in exocytosis. In a yeast 2-hybrid screen we identified optineurin as a binding partner for myosin VI at the Golgi complex and confirmed this interaction in a range of protein interaction studies. Both proteins colocalize at the Golgi complex and in vesicles at the plasma membrane. When optineurin is depleted from cells using RNA interference, myosin VI is lost from the Golgi complex, the Golgi is fragmented and exocytosis of vesicular stomatitis virus G-protein to the plasma membrane is dramatically reduced. Two further binding partners for optineurin have been identified: huntingtin and Rab8. We show that myosin VI and Rab8 colocalize around the Golgi complex and in vesicles at the plasma membrane and overexpression of constitutively active Rab8-Q67L recruits myosin VI onto Rab8-positive structures. These results show that optineurin links myosin VI to the Golgi complex and plays a central role in Golgi ribbon formation and exocytosis.
DOI:10.1091/mbc.e08-07-0726URLPMID:19144827 [本文引用: 1]
Huntingtin regulates post-Golgi trafficking of secreted proteins. Here, we studied the mechanism by which mutant huntingtin impairs this process. Colocalization studies and Western blot analysis of isolated Golgi membranes showed a reduction of huntingtin in the Golgi apparatus of cells expressing mutant huntingtin. These findings correlated with a decrease in the levels of optineurin and Rab8 in the Golgi apparatus that can be reverted by overexpression of full-length wild-type huntingtin. In addition, immunoprecipitation studies showed reduced interaction between mutant huntingtin and optineurin/Rab8. Cells expressing mutant huntingtin produced both an accumulation of clathrin adaptor complex 1 at the Golgi and an increase of clathrin-coated vesicles in the vicinity of Golgi cisternae as revealed by electron microscopy. Furthermore, inverse fluorescence recovery after photobleaching analysis for lysosomal-associated membrane protein-1 and mannose-6-phosphate receptor showed that the optineurin/Rab8-dependent post-Golgi trafficking to lysosomes was impaired in cells expressing mutant huntingtin or reducing huntingtin levels by small interfering RNA. Accordingly, these cells showed a lower content of cathepsin D in lysosomes, which led to an overall reduction of lysosomal activity. Together, our results indicate that mutant huntingtin perturbs post-Golgi trafficking to lysosomal compartments by delocalizing the optineurin/Rab8 complex, which, in turn, affects the lysosomal function.
DOI:10.1083/jcb.200509091URLPMID:16476778 [本文引用: 1]
The molecular mechanisms underlying the targeting of Huntingtin (Htt) to endosomes and its multifaceted role in endocytosis are poorly understood. In this study, we have identified Htt-associated protein 40 (HAP40) as a novel effector of the small guanosine triphosphatase Rab5, a key regulator of endocytosis. HAP40 mediates the recruitment of Htt by Rab5 onto early endosomes. HAP40 overexpression caused a drastic reduction of early endosomal motility through their displacement from microtubules and preferential association with actin filaments. Remarkably, endogenous HAP40 was up-regulated in fibroblasts and brain tissue from human patients affected by Huntington's disease (HD) as well as in STHdhQ(111) striatal cells established from a HD mouse model. These cells consistently displayed altered endosome motility and endocytic activity, which was restored by the ablation of HAP40. In revealing an unexpected link between Rab5, HAP40, and Htt, we uncovered a new mechanism regulating cytoskeleton-dependent endosome dynamics and its dysfunction under pathological conditions.
DOI:10.1242/jcs.025726URLPMID:18430781 [本文引用: 1]
Huntington disease (HD) is caused by a polyglutamine-expansion mutation in huntingtin (HTT) that makes the protein toxic and aggregate-prone. The subcellular localisation of huntingtin and many of its interactors suggest a role in endocytosis, and recently it has been shown that huntingtin interacts indirectly with the early endosomal protein Rab5 through HAP40. Here we show that Rab5 inhibition enhanced polyglutamine toxicity, whereas Rab5 overexpression attenuated toxicity in our cell and fly models of HD. We tried to identify a mechanism for the Rab5 effects in our HD model systems, and our data suggest that Rab5 acts at an early stage of autophagosome formation in a macromolecular complex that contains beclin 1 (BECN1) and Vps34. Interestingly chemical or genetic inhibition of endocytosis also impeded macroautophagy, and enhanced aggregation and toxicity of mutant huntingtin. However, in contrast to Rab5, inhibition of endocytosis by various means suppressed autophagosome-lysosome fusion (the final step in the macroautophagy pathway) similar to bafilomycin A1. Thus, Rab5, which has previously been thought to be exclusively involved in endocytosis, has a new role in macroautophagy. We have previously shown that macroautophagy is an important clearance route for several aggregate-prone proteins including mutant huntingtin. Thus, better understanding of Rab5-regulated autophagy might lead to rational therapeutic targets for HD and other protein-conformation diseases.
DOI:10.1128/MCB.00420-09URLPMID:19752198 [本文引用: 1]
Huntingtin (Htt) localizes to endosomes, but its role in the endocytic pathway is not established. Recently, we found that Htt is important for the activation of Rab11, a GTPase involved in endosomal recycling. Here we studied fibroblasts of healthy individuals and patients with Huntington's disease (HD), which is a movement disorder caused by polyglutamine expansion in Htt. The formation of endocytic vesicles containing transferrin at plasma membranes was the same in control and HD patient fibroblasts. However, HD fibroblasts were delayed in recycling biotin-transferrin back to the plasma membrane. Membranes of HD fibroblasts supported less nucleotide exchange on Rab11 than did control membranes. Rab11-positive vesicular and tubular structures in HD fibroblasts were abnormally large, suggesting that they were impaired in forming vesicles. We used total internal reflection fluorescence imaging of living fibroblasts to monitor fluorescence-labeled transferrin-carrying transport intermediates that emerged from recycling endosomes. HD fibroblasts had fewer small vesicles and more large vesicles and long tubules than did control fibroblasts. Dominant active Rab11 expressed in HD fibroblasts normalized the recycling of biotin-transferrin. We propose a novel mechanism for cellular dysfunction by the HD mutation arising from the inhibition of Rab11 activity and a deficit in vesicle formation at recycling endosomes.
DOI:10.1038/cdd.2010.127URL [本文引用: 1]
Huntington's disease (HD) is a fatal neurodegenerative disorder caused by expansion of a polyglutamine tract in the huntingtin protein (htt) that mediates formation of intracellular protein aggregates. In the brains of HD patients and HD transgenic mice, accumulation of protein aggregates has been causally linked to lesions in axo-dendritic and synaptic compartments. Here we show that dendritic spines - sites of synaptogenesis - are lost in the proximity of htt aggregates because of functional defects in local endosomal recycling mediated by the Rab11 protein. Impaired exit from recycling endosomes (RE) and association of endocytosed protein with intracellular structures containing htt aggregates was demonstrated in cultured hippocampal neurons cells expressing a mutant htt fragment. Dendrites in hippocampal neurons became dystrophic around enlarged amphisome-like structures positive for Rab11, LC3 and mutant htt aggregates. Furthermore, Rab11 overexpression rescues neurodegeneration and dramatically extends lifespan in a Drosophila model of HD. Our findings are consistent with the model that mutant htt aggregation increases local autophagic activity, thereby sequestering Rab11 and diverting spine-forming cargo from RE into enlarged amphisomes. This mechanism may contribute to the toxicity caused by protein misfolding found in a number of neurodegenerative diseases. Cell Death and Differentiation (2011) 18, 191-200; doi: 10.1038/cdd.2010.127; published online 19 November 2010
DOI:10.1523/JNEUROSCI.4144-08.2008URLPMID:19074039 [本文引用: 1]
Huntington's disease (HD) is an autosomal dominant neurodegenerative disease linked to a polyQ (polyglutamine) expansion in the huntingtin protein. Although general brain atrophy is found in HD patients, the striatum is the most severely affected region. Loss or mutant forms of huntingtin were reported to disrupt fast axonal transport in Drosophila, squid, and mice. However, previous work did not resolve whether mutant huntingtin affects global axonal transport or only a subset of cargoes, nor did it resolve whether striatal neurons are preferentially sensitive to huntingtin-mediated defects. We used amyloid precursor protein (APP)-yellow fluorescent protein and brain-derived neurotrophic factor (BDNF)-mCherry fusion proteins as markers for fast axonal transport when huntingtin is altered. We found that movement of APP and BDNF is impaired in striatal and hippocampal, but not cortical, neurons from presymptomatic homozygous mutant mice carrying 150Q huntingtin knock-in mutations. In addition, loss of huntingtin disrupts APP axonal transport, whereas overexpression of wild-type, but not mutant, huntingtin enhances APP transport in all three types of neurons tested. These data suggest that a loss of wild-type huntingtin function in fast axonal transport plays important roles in the development of cell-type-specific defects in HD.
DOI:10.1038/cdd.2010.127URL [本文引用: 1]
Huntington's disease (HD) is a fatal neurodegenerative disorder caused by expansion of a polyglutamine tract in the huntingtin protein (htt) that mediates formation of intracellular protein aggregates. In the brains of HD patients and HD transgenic mice, accumulation of protein aggregates has been causally linked to lesions in axo-dendritic and synaptic compartments. Here we show that dendritic spines - sites of synaptogenesis - are lost in the proximity of htt aggregates because of functional defects in local endosomal recycling mediated by the Rab11 protein. Impaired exit from recycling endosomes (RE) and association of endocytosed protein with intracellular structures containing htt aggregates was demonstrated in cultured hippocampal neurons cells expressing a mutant htt fragment. Dendrites in hippocampal neurons became dystrophic around enlarged amphisome-like structures positive for Rab11, LC3 and mutant htt aggregates. Furthermore, Rab11 overexpression rescues neurodegeneration and dramatically extends lifespan in a Drosophila model of HD. Our findings are consistent with the model that mutant htt aggregation increases local autophagic activity, thereby sequestering Rab11 and diverting spine-forming cargo from RE into enlarged amphisomes. This mechanism may contribute to the toxicity caused by protein misfolding found in a number of neurodegenerative diseases. Cell Death and Differentiation (2011) 18, 191-200; doi: 10.1038/cdd.2010.127; published online 19 November 2010
DOI:10.1016/j.bbrc.2008.05.060URLPMID:18501189 [本文引用: 1]
Four missense mutations, that target highly conserved amino acid residues in the small GTPase Rab7, have been associated with the Charcot-Marie-Tooth (CMT) type 2B phenotype. CMT2B peripheral axonal neuropathies are characterized by severe sensory loss, often complicated by infections, arthropathy, and amputations. Here, we have investigated the biochemical and functional properties of the Rab7 K157N mutated protein. Interestingly, Rab7 K157N showed altered nucleotide exchange rate and GTP hydrolysis compared to the wild type protein. Consistently, the majority of the expressed protein in HeLa cells was bound to GTP. In addition, Rab7 K157N was able to restore EGF degradation, previously inhibited by Rab7 silencing. Altogether these data indicate that Rab7 K157N, similarly to the other three mutated proteins causative of CMT2B, is predominantly in the GTP-bound form and behaves as an active mutant. Therefore, activated forms of Rab7 protein cause the CMT2B disease.
DOI:10.1523/JNEUROSCI.2029-05.2005URLPMID:16306406 [本文引用: 1]
Nerve growth factor (NGF) and its TrkA receptor exert important bioactivities on neuronal cells such as promoting survival and neurite outgrowth. Activated TrkA receptors are not only localized on the cell surface but also in signaling endosomes, and internalized TrkA receptors are important for the mediation of neurite outgrowth. The regulation of the endosomal trafficking of TrkA is so far unknown. Because the endosome-associated GTPase Rab7 coimmunoprecipitated with TrkA, we examined whether the endosomal trafficking of TrkA might be under the control of Rab7. Inhibiting Rab7 by expression of a green fluorescent protein-tagged, dominant-negative Rab7 variant resulted in endosomal accumulation of TrkA and pronounced enhancement of TrkA signaling in response to limited stimulations with NGF, such as increased activation of Erk1/2 (extracellular signal-regulated kinase 1/2), neurite outgrowth, and expression of GAP-43 (growth-associated protein 43). Our studies show that the endosomal GTPase Rab7 controls the endosomal trafficking and neurite outgrowth signaling of TrkA. Because mutations of Rab7 are found in patients suffering from hereditary polyneuropathies, dysfunction of Rab7 might contribute to neurodegenerative conditions by affecting the trafficking of neurotrophins. Moreover, strategies aimed at controlling Rab7 activity might be useful for the treatment of neurodegenerative diseases.
DOI:10.1002/dneu.20513URLPMID:17514710 [本文引用: 1]
The internalization and retrograde axonal transport of neurotrophin receptors is important for their retrograde signal transduction supporting neuronal differentiation, plasticity, and survival. To influence transcription, neurotrophin signals initiated at synapses have to be conveyed retrogradely to the cell body. Signaling endosomes containing neurotrophin receptor signaling complexes mediate retrograde neurotrophin signaling from synapses to the nucleus. Interestingly, many neurodegenerative diseases, including Alzheimer's disease, Niemann Pick disease Type C, and Charcot-Marie-Tooth neuropathies, show alterations of vesicular transport, suggesting that traffic jams within neuronal processes may cause neurodegeneration. Although most of these diseases are complex and may be modulated by diverse pathways contributing to neuronal death, altered neurotrophin transport is emerging as a strong candidate influence on neurodegeneration. In this article, we review the mechanisms of internalization and endocytic trafficking of neurotrophin receptors, and discuss the potential roles of perturbations in neurotrophin trafficking in a number of neurodegenerative diseases.
DOI:10.1007/s12035-007-8000-1URL [本文引用: 1]
Growth factors such as the neurotrophins promote neuronal survival and shape neuronal morphology. Neurotrophin receptors are located on the surface of axons and dendrites and must convey their signal retrogradely to the nucleus to influence transcription of target genes. The distance between the site of receptor activation and the nucleus is tremendous. How is the retrograde transmission of survival signals being achieved? Recent work showed that signaling endosomes containing neurotrophin receptors and associated downstream kinases undergo retrograde vesicular transport along microtubules, propelled by the molecular motor dynein. The next objective in the “neurotrophin receptor trafficking meets signal transduction field” will be to elucidate the traffic control mechanisms governing the directed movement of signaling endosomes. Much is already known on the trafficking of the receptor for epidermal growth factor, EGFR. We will summarize the known traffic control mechanisms for EGFR and hypothesize whether EGFR-relevant traffic control mechanisms might also be relevant for neurotrophin receptor traffic control. Moreover, we speculate about potential implications of neurotrophin receptor traffic jams for neurodegenerative diseases.
DOI:10.1242/jcs.051474URL [本文引用: 1]
DOI:10.1242/jcs.017301URL [本文引用: 1]
DOI:10.1093/hmg/ddv056URLPMID:25678552 [本文引用: 1]
Accumulation of dysfunctional mitochondria is one of the hallmarks in Alzheimer's disease (AD). Mitophagy, a selective autophagy for eliminating damaged mitochondria, constitutes a key cellular pathway in mitochondrial quality control. Recent studies established that acute depolarization of mitochondrial membrane potential (Deltapsim) using Deltapsim dissipation reagents in vitro induces Parkin-mediated mitophagy in many non-neuronal cell types or neuronal cell lines. However, neuronal pathways inducing mitophagy, particularly under pathophysiological relevant context in AD mouse models and patient brains, are largely unknown. Here, we reveal, for the first time, that Parkin-mediated mitophagy is robustly induced in mutant hAPP neurons and AD patient brains. In the absence of Deltapsim dissipation reagents, hAPP neurons exhibit increased recruitment of cytosolic Parkin to depolarized mitochondria. Under AD-linked pathophysiological conditions, Parkin translocation predominantly occurs in the somatodendritic regions; such distribution is associated with reduced anterograde and increased retrograde transport of axonal mitochondria. Enhanced mitophagy was further confirmed in AD patient brains, accompanied with depletion of cytosolic Parkin over disease progression. Thus, aberrant accumulation of dysfunctional mitochondria in AD-affected neurons is likely attributable to inadequate mitophagy capacity in eliminating increased numbers of damaged mitochondria. Altogether, our study provides the first line of evidence that AD-linked chronic mitochondrial stress under in vitro and in vivo pathophysiological conditions effectively triggers Parkin-dependent mitophagy, thus establishing a foundation for further investigations into cellular pathways in regulating mitophagy to ameliorate mitochondrial pathology in AD.
DOI:10.1073/pnas.1523810113URLPMID:27247382 [本文引用: 1]
Mitochondria play an essential role in maintaining cellular homeostasis. The removal of damaged or depolarized mitochondria occurs via mitophagy, in which damaged mitochondria are targeted for degradation via ubiquitination induced by PTEN-induced putative kinase 1 (PINK1) and Parkin. Mitophagy receptors, including optineurin (OPTN), nuclear dot 52 kDa protein (NDP52), and Tax1-binding protein 1 (TAX1BP1), are recruited to mitochondria via ubiquitin binding and mediate autophagic engulfment through their association with microtubule-associated protein light chain 3 (LC3). Here, we use live-cell imaging to demonstrate that OPTN, NDP52, and TAX1BP1 are recruited to mitochondria with similar kinetics following either mitochondrial depolarization or localized generation of reactive oxygen species, leading to sequestration by the autophagosome within approximately 45 min after insult. Despite this corecruitment, we find that depletion of OPTN, but not NDP52, significantly slows the efficiency of sequestration. OPTN is phosphorylated by the kinase TANK-binding kinase 1 (TBK1) at serine 177; we find that TBK1 is corecruited with OPTN to depolarized mitochondria. Inhibition or depletion of TBK1, or expression of amyotrophic lateral sclerosis (ALS)-associated OPTN or TBK1 mutant blocks efficient autophagosome formation. Together, these results indicate that although there is some functional redundancy among mitophagy receptors, efficient sequestration of damaged mitochondria in response to mitochondrial stress requires both TBK1 and OPTN. Notably, ALS-linked mutations in OPTN and TBK1 can interfere with mitophagy, suggesting that inefficient turnover of damaged mitochondria may represent a key pathophysiological mechanism contributing to neurodegenerative disease.
DOI:10.7554/eLife.01612URLPMID:24569479 [本文引用: 1]
Damaged mitochondria can be selectively eliminated by mitophagy. Although two gene products mutated in Parkinson's disease, PINK1, and Parkin have been found to play a central role in triggering mitophagy in mammals, how the pre-autophagosomal isolation membrane selectively and accurately engulfs damaged mitochondria remains unclear. In this study, we demonstrate that TBC1D15, a mitochondrial Rab GTPase-activating protein (Rab-GAP), governs autophagosome biogenesis and morphology downstream of Parkin activation. To constrain autophagosome morphogenesis to that of the cargo, TBC1D15 inhibits Rab7 activity and associates with both the mitochondria through binding Fis1 and the isolation membrane through the interactions with LC3/GABARAP family members. Another TBC family member TBC1D17, also participates in mitophagy and forms homodimers and heterodimers with TBC1D15. These results demonstrate that TBC1D15 and TBC1D17 mediate proper autophagic encapsulation of mitochondria by regulating Rab7 activity at the interface between mitochondria and isolation membranes. DOI: http://dx.doi.org/10.7554/eLife.01612.001.
DOI:10.7554/eLife.31326URLPMID:29360040 [本文引用: 1]
Damaged mitochondria are selectively eliminated by mitophagy. Parkin and PINK1, gene products mutated in familial Parkinson's disease, play essential roles in mitophagy through ubiquitination of mitochondria. Cargo ubiquitination by E3 ubiquitin ligase Parkin is important to trigger selective autophagy. Although autophagy receptors recruit LC3-labeled autophagic membranes onto damaged mitochondria, how other essential autophagy units such as ATG9A-integrated vesicles are recruited remains unclear. Here, using mammalian cultured cells, we demonstrate that RABGEF1, the upstream factor of the endosomal Rab GTPase cascade, is recruited to damaged mitochondria via ubiquitin binding downstream of Parkin. RABGEF1 directs the downstream Rab proteins, RAB5 and RAB7A, to damaged mitochondria, whose associations are further regulated by mitochondrial Rab-GAPs. Furthermore, depletion of RAB7A inhibited ATG9A vesicle assembly and subsequent encapsulation of the mitochondria by autophagic membranes. These results strongly suggest that endosomal Rab cycles on damaged mitochondria are a crucial regulator of mitophagy through assembling ATG9A vesicles.
DOI:10.1083/jcb.200204081URLPMID:12186851 [本文引用: 1]
A-kinase anchoring proteins (AKAPs) tether the cAMP-dependent protein kinase (PKA) and other signaling enzymes to distinct subcellular organelles. Using the yeast two-hybrid approach, we demonstrate that Rab32, a member of the Ras superfamily of small molecular weight G-proteins, interacts directly with the type II regulatory subunit of PKA. Cellular and biochemical studies confirm that Rab32 functions as an AKAP inside cells. Anchoring determinants for PKA have been mapped to sites within the conserved alpha5 helix that is common to all Rab family members. Subcellular fractionation and immunofluorescent approaches indicate that Rab32 and a proportion of the cellular PKA pool are associated with mitochondria. Transient transfection of a GTP binding-deficient mutant of Rab32 promotes aberrant accumulation of mitochondria at the microtubule organizing center. Further analysis of this mutant indicates that disruption of the microtubule cytoskeleton results in aberrantly elongated mitochondria. This implicates Rab32 as a participant in synchronization of mitochondrial fission. Thus, Rab32 is a dual function protein that participates in both mitochondrial anchoring of PKA and mitochondrial dynamics.
DOI:10.15252/embj.201796463URLPMID:28848034 [本文引用: 1]
Autophagy targets intracellular molecules, damaged organelles, and invading pathogens for degradation in lysosomes. Recent studies have identified autophagy receptors that facilitate this process by binding to ubiquitinated targets, including NDP52. Here, we demonstrate that the small guanosine triphosphatase Rab35 directs NDP52 to the corresponding targets of multiple forms of autophagy. The active GTP-bound form of Rab35 accumulates on bacteria-containing endosomes, and Rab35 directly binds and recruits NDP52 to internalized bacteria. Additionally, Rab35 promotes interaction of NDP52 with ubiquitin. This process is inhibited by TBC1D10A, a GAP that inactivates Rab35, but stimulated by autophagic activation via TBK1 kinase, which associates with NDP52. Rab35, TBC1D10A, and TBK1 regulate NDP52 recruitment to damaged mitochondria and to autophagosomes to promote mitophagy and maturation of autophagosomes, respectively. We propose that Rab35-GTP is a critical regulator of autophagy through recruiting autophagy receptor NDP52.
DOI:10.1002/glia.22499URL [本文引用: 1]
Central nervous system (CNS) trauma involves extensive cellular damage that is due, in part, to an innate inflammatory response induced by extracellular ATP. The innate immune response is regulated by pattern recognition receptors (PRRs), which include NOD-like receptors (NLRs). The PRRs and signaling cascades that regulate innate glial responses to CNS injury remain largely undefined. In this report, we show that human astrocytes express the NLR protein 2 (NLRP2) inflammasome that is activated by the danger associated molecular pattern (DAMP) ATP. The NLRP2 inflammasome is a multiprotein complex that consists of NLRP2, the adaptor protein apoptosis-speck-like protein containing a caspase recruitment domain (ASC) and caspase-1. NLRP2 also interacts with the P2X7 receptor and the pannexin 1 channel. Stimulation of human astrocytes with ATP resulted in activation of the NLRP2 inflammasome leading to the processing of inflammatory caspase-1 and interleukin-1 (IL-1). ATP-induced activation of the NLRP2 inflammasome was inhibited by the pannexin 1 inhibitor probenecid and by the P2X7 receptor antagonist Brilliant Blue G (BBG). siRNA knockdown of NLRP2 significantly decreased NLRP2 levels and caspase-1 processing in human astrocytes in response to ATP. Our findings suggest that the astrocytic NLRP2 inflammasome is an important component of the CNS inflammatory response and that the NLRP2 inflammasome may be a therapeutic target to inhibit inflammation induced by CNS injury.
DOI:10.1523/JNEUROSCI.2557-04.2004URLPMID:15525768 [本文引用: 1]
Microglia are the principle immune effector and phagocytic cells in the CNS. These cells are associated with fibrillar beta-amyloid (fAbeta)-containing plaques found in the brains of Alzheimer's disease (AD) patients. The plaque-associated microglia undergo a phenotypic conversion into an activated phenotype and are responsible for the development of a focal inflammatory response that exacerbates and accelerates the disease process. Paradoxically, despite the presence of abundant activated microglia in the brain of AD patients, these cells fail to mount a phagocytic response to Abeta deposits but can efficiently phagocytose Abeta fibrils and plaques in vitro. We report that exposure of microglia to fAbeta in vitro induces phagocytosis through mechanisms distinct from those used by the classical phagocytic receptors, the Ig receptors (FcRgammaI and FcgammaRIII) or complement receptors. Microglia interact with fAbeta through a recently characterized Abeta cell surface receptor complex comprising the B-class scavenger receptor CD36, alpha6beta1 integrin, and CD47 (integrin-associated protein). Antagonists specific for each component of the receptor complex blocks fAbeta-stimulated phagocytosis. These data demonstrated that engagement of this ensemble of receptors is required for induction of phagocytosis. The phagocytic response stimulated by this receptor complex is driven principally by a beta(1) integrin-linked process that is morphologically and mechanistically distinct from the classical type I and type II phagocytic mechanisms. These data provide evidence for phagocytic uptake of fAbeta through a receptor-mediated, nonclassical phagocytic mechanism.
[本文引用: 1]
DOI:10.1074/jbc.M112.380824URLPMID:23055518 [本文引用: 1]
Assembly of synapses requires proper coordination between pre- and postsynaptic elements. Identification of cellular and molecular events in synapse formation and maintenance is a key step to understand human perception, learning, memory, and cognition. A key role for astrocytes in synapse formation and function has been proposed. Here, we show that transforming growth factor beta (TGF-beta) signaling is a novel synaptogenic pathway for cortical neurons induced by murine and human astrocytes. By combining gain and loss of function approaches, we show that TGF-beta1 induces the formation of functional synapses in mice. Further, TGF-beta1-induced synaptogenesis involves neuronal activity and secretion of the co-agonist of the NMDA receptor, D-serine. Manipulation of D-serine signaling, by either genetic or pharmacological inhibition, prevented the TGF-beta1 synaptogenic effect. Our data show a novel molecular mechanism that might impact synaptic function and emphasize the evolutionary aspect of the synaptogenic property of astrocytes, thus shedding light on new potential therapeutic targets for synaptic deficit diseases.
DOI:10.1016/s0891-5849(98)00050-1URLPMID:9667499 [本文引用: 1]
The survival of cultured neurons is promoted by the presence of antioxidants or astrocytes. This indicates that extracellular reactive oxygen species (ROS) impair neuronal survival and suggests that astrocytes exert their survival-enhancing effect through inactivation of these toxicants. However, to our knowledge, data supporting this hypothesis are lacking. Previously, we showed that loss of the antioxidant glutathione abolishes the neuronal survival-stimulating action of astrocytes in cocultures, consisting of rat striatal astrocytes and mesencephalic, dopaminergic neurons. Using uptake of [3H]dopamine as marker of neuronal survival, we presently investigated whether this effect of glutathione depletion is mediated by extracellular ROS. For this purpose, we incubated glutathione-depleted cocultures with superoxide dismutase, catalase or both. Whereas superoxide dismutase had no effect and catalase only partially protected, addition of the enzymes together completely prevented the impairment of neuronal survival caused by glutathione loss. No change in neuronal survival occurred upon exposure of control cocultures to superoxide dismutase and/or catalase. These data strongly implicate scavenging of extracellular ROS in astrocyte-stimulated neuronal survival and moreover suggest a crucial role for glutathione in this process.
DOI:10.1038/nature02827URLPMID:15356633 [本文引用: 1]
Cerebral blood flow (CBF) is coupled to neuronal activity and is imaged in vivo to map brain activation. CBF is also modified by afferent projection fibres that release vasoactive neurotransmitters in the perivascular region, principally on the astrocyte endfeet that outline cerebral blood vessels. However, the role of astrocytes in the regulation of cerebrovascular tone remains uncertain. Here we determine the impact of intracellular Ca(2+) concentrations ([Ca(2+)](i)) in astrocytes on the diameter of small arterioles by using two-photon Ca(2+) uncaging to increase [Ca(2+)](i). Vascular constrictions occurred when Ca(2+) waves evoked by uncaging propagated into the astrocyte endfeet and caused large increases in [Ca(2+)](i). The vasoactive neurotransmitter noradrenaline increased [Ca(2+)](i) in the astrocyte endfeet, the peak of which preceded the onset of arteriole constriction. Depressing increases in astrocyte [Ca(2+)](i) with BAPTA inhibited the vascular constrictions in noradrenaline. We find that constrictions induced in the cerebrovasculature by increased [Ca(2+)](i) in astrocyte endfeet are generated through the phospholipase A(2)-arachidonic acid pathway and 20-hydroxyeicosatetraenoic acid production. Vasoconstriction by astrocytes is a previously unknown mechanism for the regulation of CBF.
DOI:10.1016/j.immuni.2017.06.006URLPMID:28636962 [本文引用: 1]
Astrocytes constitute approximately 30% of the cells in the mammalian central nervous system (CNS). They are integral to brain and spinal-cord physiology and perform many functions important for normal neuronal development, synapse formation, and proper propagation of action potentials. We still know very little, however, about how these functions change in response to immune attack, chronic neurodegenerative disease, or acute trauma. In this review, we summarize recent studies that demonstrate that different initiating CNS injuries can elicit at least two types of
DOI:10.1073/pnas.1800165115URLPMID:29437957 [本文引用: 1]
The decline of cognitive function occurs with aging, but the mechanisms responsible are unknown. Astrocytes instruct the formation, maturation, and elimination of synapses, and impairment of these functions has been implicated in many diseases. These findings raise the question of whether astrocyte dysfunction could contribute to cognitive decline in aging. We used the Bac-Trap method to perform RNA sequencing of astrocytes from different brain regions across the lifespan of the mouse. We found that astrocytes have region-specific transcriptional identities that change with age in a region-dependent manner. We validated our findings using fluorescence in situ hybridization and quantitative PCR. Detailed analysis of the differentially expressed genes in aging revealed that aged astrocytes take on a reactive phenotype of neuroinflammatory A1-like reactive astrocytes. Hippocampal and striatal astrocytes up-regulated a greater number of reactive astrocyte genes compared with cortical astrocytes. Moreover, aged brains formed many more A1 reactive astrocytes in response to the neuroinflammation inducer lipopolysaccharide. We found that the aging-induced up-regulation of reactive astrocyte genes was significantly reduced in mice lacking the microglial-secreted cytokines (IL-1alpha, TNF, and C1q) known to induce A1 reactive astrocyte formation, indicating that microglia promote astrocyte activation in aging. Since A1 reactive astrocytes lose the ability to carry out their normal functions, produce complement components, and release a toxic factor which kills neurons and oligodendrocytes, the aging-induced up-regulation of reactive genes by astrocytes could contribute to the cognitive decline in vulnerable brain regions in normal aging and contribute to the greater vulnerability of the aged brain to injury.
DOI:10.1038/s41591-018-0051-5URLPMID:29892066 [本文引用: 1]
Activation of microglia by classical inflammatory mediators can convert astrocytes into a neurotoxic A1 phenotype in a variety of neurological diseases(1,2). Development of agents that could inhibit the formation of A1 reactive astrocytes could be used to treat these diseases for which there are no disease-modifying therapies. Glucagon-like peptide-1 receptor (GLP1R) agonists have been indicated as potential neuroprotective agents for neurologic disorders such as Alzheimer's disease and Parkinson's disease(3-13). The mechanisms by which GLP1R agonists are neuroprotective are not known. Here we show that a potent, brain-penetrant long-acting GLP1R agonist, NLY01, protects against the loss of dopaminergic neurons and behavioral deficits in the alpha-synuclein preformed fibril (alpha-syn PFF) mouse model of sporadic Parkinson's disease(14,15). NLY01 also prolongs the life and reduces the behavioral deficits and neuropathological abnormalities in the human A53T alpha-synuclein (hA53T) transgenic mouse model of alpha-synucleinopathy-induced neurodegeneration(16). We found that NLY01 is a potent GLP1R agonist with favorable properties that is neuroprotective through the direct prevention of microglial-mediated conversion of astrocytes to an A1 neurotoxic phenotype. In light of its favorable properties, NLY01 should be evaluated in the treatment of Parkinson's disease and related neurologic disorders characterized by microglial activation.
DOI:10.1038/nm1058URLPMID:15195085 [本文引用: 1]
We have previously shown that apolipoprotein E (Apoe) promotes the formation of amyloid in brain and that astrocyte-specific expression of APOE markedly affects the deposition of amyloid-beta peptides (Abeta) in a mouse model of Alzheimer disease. Given the capacity of astrocytes to degrade Abeta, we investigated the potential role of Apoe in this astrocyte-mediated degradation. In contrast to cultured adult wild-type mouse astrocytes, adult Apoe(-/-) astrocytes do not degrade Abeta present in Abeta plaque-bearing brain sections in vitro. Coincubation with antibodies to either Apoe or Abeta, or with RAP, an antagonist of the low-density lipoprotein receptor family, effectively blocks Abeta degradation by astrocytes. Phase-contrast and confocal microscopy show that Apoe(-/-) astrocytes do not respond to or internalize Abeta deposits to the same extent as do wild-type astrocytes. Thus, Apoe seems to be important in the degradation and clearance of deposited Abeta species by astrocytes, a process that may be impaired in Alzheimer disease.
DOI:10.1016/j.neuron.2018.05.008URLPMID:29861287 [本文引用: 1]
The apolipoprotein E4 (APOE4) variant is the single greatest genetic risk factor for sporadic Alzheimer's disease (sAD). However, the cell-type-specific functions of APOE4 in relation to AD pathology remain understudied. Here, we utilize CRISPR/Cas9 and induced pluripotent stem cells (iPSCs) to examine APOE4 effects on human brain cell types. Transcriptional profiling identified hundreds of differentially expressed genes in each cell type, with the most affected involving synaptic function (neurons), lipid metabolism (astrocytes), and immune response (microglia-like cells). APOE4 neurons exhibited increased synapse number and elevated Abeta42 secretion relative to isogenic APOE3 cells while APOE4 astrocytes displayed impaired Abeta uptake and cholesterol accumulation. Notably, APOE4 microglia-like cells exhibited altered morphologies, which correlated with reduced Abeta phagocytosis. Consistently, converting APOE4 to APOE3 in brain cell types from sAD iPSCs was sufficient to attenuate multiple AD-related pathologies. Our study establishes a reference for human cell-type-specific changes associated with the APOE4 variant. VIDEO ABSTRACT.
DOI:10.1016/j.ajpath.2011.09.034URLPMID:22074738 [本文引用: 1]
beta-Secretase, the rate-limiting enzymatic activity in the production of the amyloid-beta (Abeta) peptide, is a major target of Alzheimer's disease (AD) therapeutics. There are two forms of the enzyme: beta-site Abeta precursor protein cleaving enzyme (BACE) 1 and BACE2. Although BACE1 increases in late-stage AD, little is known about BACE2. We conducted a detailed examination of BACE2 in patients with preclinical to late-stage AD, including amnestic mild cognitive impairment, and age-matched controls, cases of frontotemporal dementia, and Down's syndrome. BACE2 protein and enzymatic activity increased as early as preclinical AD and were found in neurons and astrocytes. Although the levels of total BACE2 mRNA were unchanged, the mRNA for BACE2 splice form C (missing exon 7) increased in parallel with BACE2 protein and activity. BACE1 and BACE2 were strongly correlated with each other at all levels, suggesting that their regulatory mechanisms may be largely shared. BACE2 was also elevated in frontotemporal dementia but not in Down's syndrome, even in patients with substantial Abeta deposition. Thus, expression of both forms of beta-secretase are linked and may play a combined role in human neurologic disease. A better understanding of the normal functions of BACE1 and BACE2, and how these change in different disease states, is essential for the future development of AD therapeutics.
DOI:10.1016/j.mcn.2017.04.009URLPMID:28450268 [本文引用: 1]
The presence of Lewy bodies, mainly consisting of aggregated alpha-synuclein, is a pathological hallmark of Parkinson's disease (PD) and dementia with Lewy bodies (DLB). The alpha-synuclein inclusions are predominantly found in neurons, but also appear frequently in astrocytes. However, the pathological significance of alpha-synuclein inclusions in astrocytes and the capacity of glial cells to clear toxic alpha-synuclein species remain unknown. In the present study we investigated uptake, degradation and toxic effects of oligomeric alpha-synuclein in a co-culture system of primary neurons, astrocytes and oligodendrocytes. Alpha-synuclein oligomers were found to co-localize with the glial cells and the astrocytes were found to internalize particularly large amounts of the protein. Following ingestion, the astrocytes started to degrade the oligomers via the lysosomal pathway but, due to incomplete digestion, large intracellular deposits remained. Moreover, the astrocytes displayed mitochondrial abnormalities. Taken together, our data indicate that astrocytes play an important role in the clearance of toxic alpha-synuclein species from the extracellular space. However, when their degrading capacity is overburdened, alpha-synuclein deposits can persist and result in detrimental cellular processes.
DOI:10.1186/2047-9158-2-20URLPMID:24093918 [本文引用: 1]
The accumulation and aggregation of alpha-synuclein (alpha-syn) in several tissue including the brain is a major pathological hallmark in Parkinson's disease (PD). In this study, we show that alpha-syn can be taken up by primary human cortical neurons, astrocytes and skin-derived fibroblasts in vitro. Our findings that brain and peripheral cells exposed to alpha-syn can lead to impaired mitochondrial function, leading to cellular degeneration and cell death, provides additional evidence for the involvement of mitochondrial dysfunction as a mechanism of toxicity of alpha-syn in human cells.
DOI:10.1074/jbc.M109.081125URLPMID:20071342 [本文引用: 1]
Abnormal neuronal aggregation of alpha-synuclein is implicated in the development of many neurological disorders, including Parkinson disease and dementia with Lewy bodies. Glial cells also show extensive alpha-synuclein pathology and may contribute to disease progression. However, the mechanism that produces the glial alpha-synuclein pathology and the interaction between neurons and glia in the disease-inflicted microenvironment remain unknown. Here, we show that alpha-synuclein proteins released from neuronal cells are taken up by astrocytes through endocytosis and form inclusion bodies. The glial accumulation of alpha-synuclein through the transmission of the neuronal protein was also demonstrated in a transgenic mouse model expressing human alpha-synuclein. Furthermore, astrocytes that were exposed to neuronal alpha-synuclein underwent changes in the gene expression profile reflecting an inflammatory response. Induction of pro-inflammatory cytokines and chemokines correlated with the extent of glial accumulation of alpha-synuclein. Together, these results suggest that astroglial alpha-synuclein pathology is produced by direct transmission of neuronal alpha-synuclein aggregates, causing inflammatory responses. This transmission step is thus an important mediator of pathogenic glial responses and could qualify as a new therapeutic target.
DOI:10.1523/JNEUROSCI.3020-14.2014URLPMID:25471560 [本文引用: 1]
Traumatic brain injury (TBI) is an established risk factor for the early development of dementia, including Alzheimer's disease, and the post-traumatic brain frequently exhibits neurofibrillary tangles comprised of aggregates of the protein tau. We have recently defined a brain-wide network of paravascular channels, termed the
DOI:10.1006/exnr.1998.6778URLPMID:9527890 [本文引用: 1]
Huntington's disease (HD) is an autosomal dominant neurodegenerative disease associated with a CAG trinucleotide repeat expansion in a large gene on chromosome 4. The gene encodes the protein huntingtin with a polyglutamine tract encoded by the CAG repeat at the N-terminus. The number of CAG repeats in HD are significantly increased (36 to 120+) compared with the normal population (8-39). The pathological mechanism associated with the expanded CAG repeat in HD is not clear but there is evidence that polyglutamine is directly neurotoxic. We have immunolocalized huntingtin with an in-house, well-characterised, polyclonal antibody in HD, Alzheimer's disease (AD), and Picks disease (PiD) brains. Control brain tissue sections were from head injured and cerebral ischaemia cases. In HD, huntingtin was immunopositive in the surviving but damaged neurons and reactive astrocytes of the caudate and putamen. However, in AD and PiD the immunostaining was largely restricted to the characteristic intracellular inclusion bodies associated with the disease process in each case. In AD, huntingtin was localized only in the intracellular neurofibrillary tangles and dystrophic neurites within the neuritic amyloid plaques but not with the amyloid. In PiD, strongly positive huntingtin immunostaining was present within cytoplasmic Pick bodies. Our findings suggest huntingtin selectively accumulates in association with abnormal intracytoplasmic and cytoskeletal filaments of neurons and glia in neurodegenerative diseases such as HD, AD, and PiD. Cells in the CNS appear sensitive to damage by the aggregated, toxic levels of huntingtin and evidence of its interaction with neurofilaments could provide information about its potential role in the aetiology of HD.
DOI:10.1083/jcb.200508072URLPMID:16365166 [本文引用: 1]
Huntington disease (HD) is characterized by the preferential loss of striatal medium-sized spiny neurons (MSNs) in the brain. Because MSNs receive abundant glutamatergic input, their vulnerability to excitotoxicity may be largely influenced by the capacity of glial cells to remove extracellular glutamate. However, little is known about the role of glia in HD neuropathology. Here, we report that mutant huntingtin accumulates in glial nuclei in HD brains and decreases the expression of glutamate transporters. As a result, mutant huntingtin (htt) reduces glutamate uptake in cultured astrocytes and HD mouse brains. In a neuron-glia coculture system, wild-type glial cells protected neurons against mutant htt-mediated neurotoxicity, whereas glial cells expressing mutant htt increased neuronal vulnerability. Mutant htt in cultured astrocytes decreased their protection of neurons against glutamate excitotoxicity. These findings suggest that decreased glutamate uptake caused by glial mutant htt may critically contribute to neuronal excitotoxicity in HD.
DOI:10.1002/jcp.21911URLPMID:19725050 [本文引用: 2]
The expression profile and functions of the brain-enriched Rab22B/Rab31 small GTPase had remained uncharacterized. Using specific antibodies against Rab22B, we found the protein to be exceptionally enriched in nestin and RC2-positive radial glia of the embryonic mouse brain. In the adult brain, Rab22B is rather specifically expressed in glial fibrillary acidic protein (GFAP)-positive mature astrocytes, but is not clearly detectable in either 2',3'-cyclic nucleotide 3'-phosphodiesterase (CNPase)-positive mature oligodendrocytes or betaIII-tubulin (TuJ)-positive neurons. In probing for specific functions of Rab22B, we found that Rab22B silencing in A431 cells resulted in abnormal trafficking of the epidermal growth factor receptor (EGFR), Texas-red-labeled EGF, and the cation-independent mannose 6-phosphate receptor (M6PR). Affinity pull-down assays and co-immunoprecipitation analysis indicated that Rab22B could associate with EGFR in a GTP-dependent manner. Rab22B is thus a Rab protein specifically expressed in the astroglia lineage and may have a role in regulating EGFR trafficking in some cell types. Given that EGFR signaling modulates astrocyte development and oncogenesis of multiple cell types, Rab22B may thus have specific developmental or pathophysiological roles in cell types which it is enriched in.
DOI:10.1016/j.febslet.2014.06.060URL [本文引用: 1]
Rab31 is expressed in both GFAP- and nestin- positive fibres in regions of neurogenic potential in the adult mouse brain. To investigate the role of Rab31 in neural progenitor cells (NPCs), we cultured NPCs and found significant levels of Rab31 expression in these cells. Rab31 levels showed a sharp initial decrease and then reappeared gradually in a subpopulation of astrocytes when NPCs were induced to differentiate. Silencing of Rab31 hindered, while overexpression enhanced, the differentiation of NPCs to astrocytes. Our results suggest a previously unrecognised role for Rab31 in influencing the differentiation and fate of NPCs. (C) 2014 Federation of European Biochemical Societies. Published by Elsevier B. V.
[本文引用: 2]

{kind=link}
{kind=link}
{kind=link}
{kind=link}