 ,2
,2Progress on the GntR family transcription regulators in bacteria
Guofang Liu1, Xinxin Wang2, Huizhao Su2, Guangtao Lu,2通讯作者: 陆光涛,博士,研究员,研究方向:植物保护。E-mail:lugt@gxu.edu.cn
编委: 梁海华
收稿日期:2020-09-18修回日期:2020-12-7网络出版日期:2021-01-20
| 基金资助: |
Received:2020-09-18Revised:2020-12-7Online:2021-01-20
| Fund supported: |
作者简介 About authors
刘国芳,博士,副教授,研究方向:植物保护。E-mail:

摘要
GntR家族转录调控因子是细菌中分布最为广泛的一类螺旋-转角-螺旋(helix-turn-helix,HTH)转录调控因子,此家族转录调控因子包含两个功能域,分别是N端的DNA结合结构域和C端的效应物结合结构域/寡聚化作用结构域。DNA结合结构域的氨基酸序列是非常保守的,但效应物结合结构域/寡聚化作用结构域的氨基酸序列却存在很大的差异性。目前许多GntR家族的转录调控因子已经被鉴定,这些转录因子调控细菌许多不同的细胞过程,如运动性、葡萄糖代谢、细菌的耐药性、病原细菌的致病力等。本文主要阐述了GntR家族转录调控因子的发现、二级结构、生物学功能、调控模式等方面的研究进展,旨在为研究者全面、深入地了解GntR家族转录调控因子的功能及作用机理提供帮助。
关键词:
Abstract
In bacteria, GntR family transcription regulators are the widespread family of transcription factors. Members of this family consist of two functional domains, a conserved N-terminal DNA-binding domain that contains a typical helix-turn-helix (HTH) motif and a C-terminal effector-binding or oligomerization domain. Usually, the amino acid sequences of N-terminal DNA-binding domains are highly conserved, but differ in the C-terminal effector-binding or oligomerization domains. In the past several decades, many GntR family transcription regulators have been characterized in a number of bacteria. These regulators control a variety of cellular processes such as cell motility, glucose metabolism, bacterial resistance, pathogenesis and virulence. In this review, we summarized the discovery, C-terminal domains, biological function and regulation mode of GntR family transcription regulators. This review will help researchers to obtain more knowledge about the functions and mechanisms of the GntR family transcriptional regulatory factors.
Keywords:
PDF (671KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
刘国芳, 王欣欣, 苏辉昭, 陆光涛. 细菌GntR家族转录调控因子的研究进展. 遗传[J], 2021, 43(1): 66-73 doi:10.16288/j.yczz.20-245
Guofang Liu.
细菌经常暴露在不断变化的环境中。为了生长和生存,细菌能够根据环境中可利用营养的数量和类型,开启不同的调控系统,使细胞代谢过程能够适应环境的变化。细菌的转录调控是细菌快速繁殖适应变幻莫测环境的主要机制之一[1],其中最常见的转录调控方式是通过转录调控因子与启动子的结合,来调控基因转录的起始,进而调控基因的转录水平。
在细菌中,已鉴定出许多转录调控因子,根据它们所结合DNA的模式以及自身保守的基序,人们将这些转录调控因子分为若干类群[2]。其中最为常见的是螺旋-转角-螺旋(helix-turn-helix, HTH)类群,此类群的转录调控因子含有一个保守的α螺旋-转角-α螺旋DNA结合结构域[3]。根据DNA结合结构域的序列,人们又将此类群的转录调控因子分为多个不同的家族[2],其中GntR家族转录调控因子是分布最为广泛的一类HTH转录调控因子。本文主要阐述了GntR家族转录调控因子的发现、二级结构、生物学功能、调控模式等方面的研究进展,将为转录调控因子调控机理的研究和动植物病原菌致病机理的研究提供新的知识和基础。
1 GntR家族转录调控因子的发现和基本结构
第一个发现的GntR家族转录调控因子是枯草芽孢杆菌(Bacillus subtilis)中的葡萄糖酸盐操纵子抑制子(gluconate operon repressor, GntR)[4]。GntR家族转录调控因子包含两个功能域(图1)分别是N端的DNA结合结构域(DNA-binding domain, DBD)和C端的效应物结合结构域/寡聚化作用结构域(effector- binding and oligomerization domains, EBD)。DNA结构域的氨基酸序列在GntR家族成员中是非常保守的,但效应物结合结构域/寡聚化作用结构域的氨基酸序列存在很大的差异性。根据C端氨基酸序列的不同,GntR家族转录调控因子分为7个亚家族(图2),分别是FadR (fatty acid degradation regulator)、HutC (histidine utilization repressor)、MocR (moc genes of GntR regulator)、YtrA (ytr operon transcriptional regulator)、AraR (L-arabinose operon transcriptional repressor)、DevA (development transcriptional regulator)、PlmA (plasmid maintenance)[5,6,7],其中FadR、HutC、MocR、YtrA亚家族是GntR家族中比较重要的亚家族。FadR亚家族是GntR家族转录转录调控因子中成员最多的一类,占约40%。其成员中的C端结构域包括6~7个α螺旋,由150~170个氨基酸组成。C端结构域可与效应物或者配体结合,如羧酸,结合后,蛋白的构象发生变化,进而影响与DNA的结合[8]。该亚家族转录调控因子是在大肠杆菌(Escherichia coli)中首次发现。E. coli中的FadR参与调控脂肪酸的合成与分解代谢,是一个酰基辅酶A结合蛋白,以二聚体的形式存在。其中N端是DNA结合结构域,C端是配体结合结构域,可以与酰基辅酶A结合[9]。
仅次于FadR亚家族的是HutC亚家族,约占GntR家族的30%。该亚家族成员的C端结构域由170个氨基酸组成,包括α螺旋和β折叠[10]。此亚家族的第一个转录调控因子是在恶臭假单胞菌(Pseudomonas putida)中发现。P. putida 的HutC是一个组氨酸利用操纵子调节因子[11]。结构研究发现,此亚家族的转录调控因子C端的效应物结合结构域/寡聚化作用结构域的折叠方式与E. coli中发现的单核分支酸裂解酶很像,均以二聚体的形式存在并且在HutC亚家族中是非常保守的[12]。
MocR亚家族中的转录调控因子的典型特征是C端有一个比较大的效应物结合结构域/寡聚化作用结构域,平均由350个氨基酸组成。折叠方式属于I型吡哆醛5?-磷酸盐(pyridoxal 5?-phosphate, PLP)依赖性酶,此折叠类型的原型酶为天冬氨酸转移酶(aspartate aminotransferase, AAT)[13]。此家族转录调控因子的N端DNA结合结构域和C端AAT结构域是通过多肽来连接的。多肽链的长度在MocR亚家族中不同成员之间是不相同的。
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1GntR家族蛋白结构示意图
Fig. 1Schematic diagram of GntR protein
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2GntR家族转录调控因子亚家族成员C端二级结构的示意图
Fig. 2Schematic diagram of C-terminal domains of the GntR sub-families transcription regulators
与GntR家族中其他6个亚家族相比,YtrA亚家族中的成员的肽链组成较短,一般由120~130个氨基酸组成。其中大约50个氨基酸是位于C端结构域,形成2个α螺旋[14]。YtrA最早是在枯草芽孢杆菌(Bacillus subtilis)中发现的[15]。B. subtilis中的 ytrA基因与其他5个基因构成一个操纵子ytrABCDEF。YtrA由130个氨基酸组成,负向调控ytrABCDEF操作子中基因的表达。绝大多数编码YtrA亚家族转录调控因子的基因与ATP-binding cassette (ABC)转运系统相关基因组成了操纵子,可以调控ABC (ATP-binding cassette)转运系统相关基因的表达[10]。由于YtrA亚家族成员氨基酸组成较少,肽链的长度较短,给其结构的研究造成了很大的困扰。
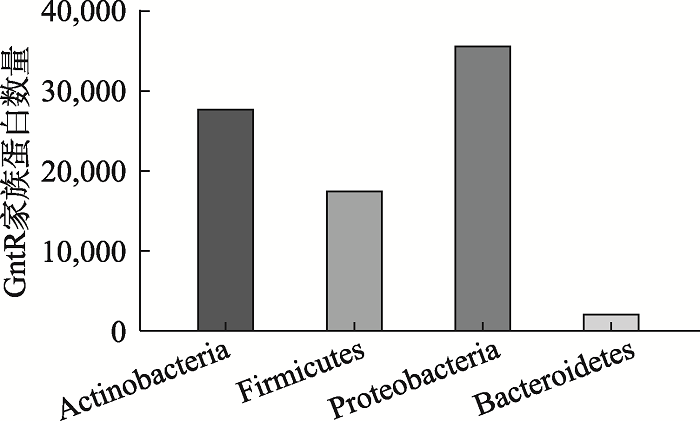
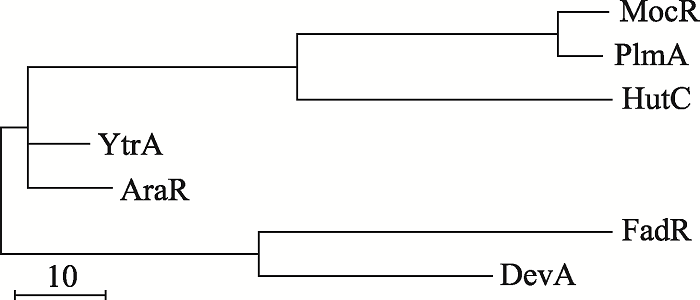
随着研究的深入,GntR家族蛋白越来越多的被鉴定出来。在PfAM数据库中,GntR家族序列由2008年的8561条增长到89733条。GntR家族在细菌中广泛分布,主要分布在变形菌门Proteobacteria,厚壁菌门Firmicutes 和放线菌门Actinobacteria等5958种细菌中(图3)。根据各亚家族C端的差异构建系统发育树,结果发现,MocR和PlmA在进化上虽然比较接近,但GntR家族各亚家族蛋白在整体进化上距离较远(图4),说明各亚家族在进化上变化较大。
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3GntR家族蛋白在细菌类群中的分布
放线菌门:Actinobacteria;厚壁菌门:Firmicutes;变形菌门:Proteobacteria;拟杆菌门:Bacteroidetes。
Fig. 3Distribution of GntR proteins throughout the bacterial group
图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4GntR家族蛋白系统发育树
Fig. 4GntR family protein phylogenetic tree
2 GntR家族转录调控因子的生物学功能
许多GntR家族转录调控因子已经被鉴定。这些转录因子调控细菌许多不同的细胞过程[5],包括运动性[16]、发育[17]、葡萄糖代谢[18]、细菌的耐药性[19]、细胞的毒性[20]、病原细菌的致病力[21,22]等。2.1 动物病原细菌中GntR家族转录调控因子的生物学功能
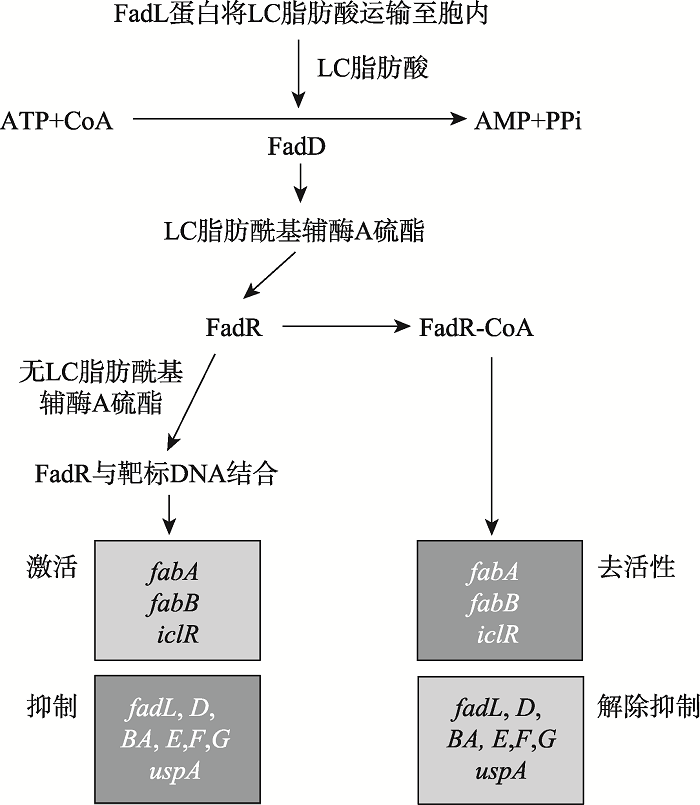
目前,与动物病原菌GntR家族转录调控因子相关的研究报道有很多。本文将以已报道的几个动物病原菌中的GntR转录调控因子为例,阐述动物病原菌中GntR家族转录调控因子的生物学功能。病原物E. coli中的FadR转录调控因子是一个全局调控因子,可调控脂肪酸合成、水解、转运等相关基因的表达。其N端的DNA结合结构域可与靶基因特定的DNA序列结合;C端可与配体结合,其生理配体是LC脂肪酰基辅酶A硫酯。当FadR与配体结合后,FadR就会从靶基因的结合位点上解离下来,进而调控靶基因的表达[23](图5)。FadR转录调控因子能够抑制脂肪酸转运相关基因(fadL和fadD)、编码分解代谢相关酶基因(fadE、fadF、fadG、fadBA、fadH)的表达[9]。
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5大肠杆菌中FadR调控脂肪酸代谢的模式图
Fig. 5Schematic overview of FadR control fatty acid metabolism in E. coli
铜绿假单胞菌(Pseudomonas aeruginosa)是一种条件致病菌,是一种医院内感染的主要致病菌,感染此菌后会引发一些疾病,如肺炎、细菌病、尿道和外科感染[24]。在此菌中发现,GntR家族转录调控因子不仅抑制自身基因的表达,而且参与该菌的许多代谢过程,包括抑制葡萄糖代谢过程中的关键酶葡萄糖酸盐透性酶GntP的表达[24];参与调控2-羟基喹啉和氨基苯甲酸盐的代谢[25]。
溶血弧菌(Vibrio parahaemolyticus)是一种食源性病原体,有时可在受污染的海鲜中发现,可引致人类急性肠胃炎和出血性败血症。此菌中存在两个鞭毛系统参与细菌的运动,在许多细菌中,运动性与病原细菌的致病、生物膜的形成和环境的适应相关[16]。在此菌中鉴定到的一个GntR家族转录调控因子参与调控细胞的运动性。变形链球菌(Streptococcus mutans)为革兰氏染色阳性球菌,是龋病的主要致病菌。在此菌中所鉴定到的GntR家族转录调控因子StsR,是通过调节糖转运相关基因的表达,来影响该菌早期生物膜的形成和胞外多糖(extracellular polysaccharide, EPS)的合成[26]。已知生物膜的形成和EPS往往与病原细菌的致病密切相关。所以,StsR极有可能参与调控变形链球菌的致病过程。肺炎链球菌(Streptococcus pneumoniae)是一种重要的人类病原体,通常存在于呼吸道上部,可引致肺炎、中耳炎、脑膜炎、败血症等疾病。在此菌中鉴定到的GntR家族转录调控因子CpsR可调控荚膜多糖的合成和致病过程[27]。金黄色葡萄球菌(Staphylococcus aureus)是一种重要的人类病原体,可引致多种感染,如脓肿、败血症、中毒性休克综合症和心内膜炎。在此菌中鉴定到的GntR家族转录调控因子NorG是一个全局转录调控因子,调控参与多种不同细胞过程的基因[28]。布鲁氏菌(Brucella spp.)能够引起布鲁氏菌病,此病是一种人畜共患的传染病,对公众健康及安全构成了极大危害。研究发现,布鲁氏菌侵染巨噬细胞期间,GntR转录调控因子影响IV型分泌系统(type IV secretion system, T4SS)和群体感应系统相关基因的表达[20]。
综上所述,动物病原菌中目前鉴定的GntR家族转录调控因子许多是全局转录调控因子,可调控许多细胞过程,包括运动性、生物膜的形成、胞外多糖的合成、群体感应等。这些全局性转录调控因子通过调控与动物病原菌致病相关的细胞过程,进而影响病原细菌的致病过程。
2.2 植物病原细菌中GntR家族转录调控因子的生物学功能
相较于动物病原菌,植物病原菌中关于GntR家族转录调控因子的研究相对较少。十字花科黑腐病菌(Xanthomonas campestris pv. Campestris, Xcc)能够引起十字花科植物发生黑腐病。在此菌中鉴定的GntR家族转录调控因子HpaR1是一个全局转录调控因子,调控许多细胞过程,包括致病过程、III型分泌系统(type III secretion system, T3SS)相关基因的表达、致病因子胞外多糖、胞外纤维素酶相关基因的表达等[29,30,31]。柑橘溃疡病菌(Xanthomonas citri ssp. citri)是一种对柑橘危害很大的致病菌,引发柑橘溃疡病,导致世界范围的柑橘产量减少。此菌中鉴定的GntR家族转录调控因子YtrA调控细菌致病过程、T3SS和III型效应物(type III effectors, T3Es)相关基因的表达、细菌在寄主和培养基中的生长等多个细胞过程[32]。除以上动植物病原菌之外,在一些非致病菌中也鉴定到了GntR家族转录调控因子。灰色链霉菌(Streptomyces griseus)中鉴定的GntR家族转录调控因子DasR参与调控N-乙酰氨基葡萄糖的代谢[33];天蓝色链霉菌(Streptomyces coelicolor)中鉴定的GntR家族转录调控因子DevA与该菌的发育相关[17];慢生型大豆根瘤菌(Bradyrhizobium japonicum)中鉴定的GntR家族转录调控因子MocR与该菌的运动性、生物膜的形成等相关[34]。
3 调控模式
转录调控因子通过识别基因启动子区的特定序列并与其结合,进而调控基因的转录。根据调控机制的不同分为正转录调控(转录激活)和负转录调控(转录抑制)两类,GntR家族转录调控因子绝大多数是转录抑制子。基因转录抑制主要包括3种模式[35]:(1)转录因子结合在启动子区的-10区附近,使得RNA聚合酶(RNA polymerase, RNAP)无法与启动子结合,进而抑制基因的转录;(2)转录因子结合在启动子的特定位置,使启动子区的构象发生改变,形成一个环,封闭了启动子的-35区和-10区,使得RNAP无法与启动子结合,阻止了转录的起始;(3)某些基因的转录需要转录激活因子,而转录抑制因子通过与转录激活因子相互作用,使得激活因子无法起到激活转录的作用,进而阻止转录的起始。
在植物病原细菌Xcc中所鉴定的GntR家族转录调控因子HpaR1,其功能及调控模式不同于以往报道的GntR家族转录调控因子。HpaR1是一个全局转录调控因子,属于YtrA亚家族,正调控多种不同的细胞过程,包括胞外多糖、胞外酶活性、运动性以及抗逆性等多种细胞过程[29]。其调控Xcc的EPS的合成,是通过正调控一簇与EPS合成相关的基因gum操纵子来实现的。HpaR1结合在gumB基因启动子区的-35区和-10区之间。一般而言,转录调控因子结合在基因启动子的-35区和-10区内,会阻断RNAP沿DNA模板链的移动,从而抑制基因的转录,因而是起到一个转录抑制子的作用。但是HpaR1却是通过与RNAP蛋白间的相互作用,增强了RNAP与gumB基因启动子区的结合,从而增强了gumB基因的表达。因此,HpaR1对gumB基因的调控模式是一种新的调控模式[30]。
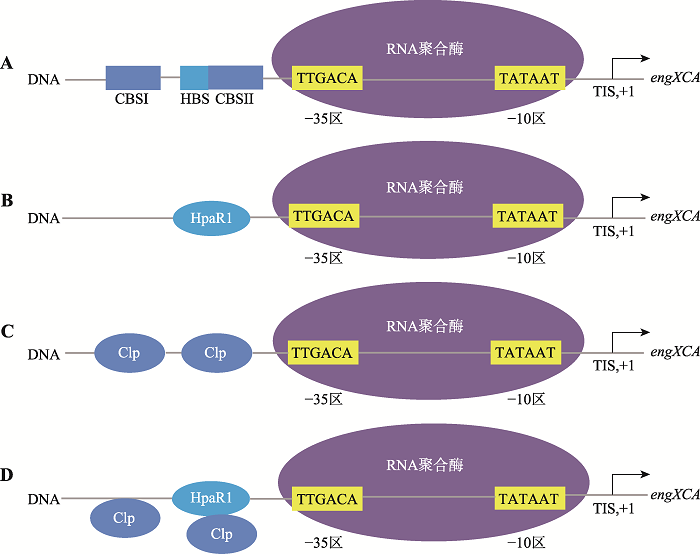
目前的研究报道主要集中在某个转录调控因子的调控模式的研究,但是细胞内的转录调控因子往往是形成一个调控网络发挥作用的。Xcc中的HpaR1和另一个全局性转录调控因子Clp,能够以一种特有的模式共同调控胞外纤维素酶主校基因XC_0639 (engXCA)表达(图6)。HpaR1和Clp均正调控engXCA的转录。HpaR1结合在engXCA启动子区的-35区上游包括-35区序列(HpaR1 binding site, HBS);Clp在engXCA启动子区中有两个结合位点,CBSI (Clp binding site I)位于engXCA启动子区的-35区上游,CBSII (Clp binding site II)位于engXCA启动子区的-35区上游包括-35区序列,其中CBSII与HBS是部分重叠。HpaR1对engXCA转录激活效率低于Clp,但对engXCA启动子区的结合能力比Clp更强。因此HpaR1能将Clp从CBSII结合位点驱逐出来,从而与Clp一起,精细地调控engXCA的转录水平[31]。
图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6HpaR1、Clp调控engXCA的模式图
A:HapR1、Clp均能与engXCA启动子区结合,Clp与engXCA启动子区有两个结合位点,其中CBSII与HBS是重叠的;B、C:HpaR1、Clp在体外均能激活基因engXCA的转录;D:HpaR1与Clp相比,HpaR1与engXCA启动子区有更高的亲和力,并且能将Clp从其结合位点CBSII驱逐下来[31]。
Fig. 6Model of regulation of gene engXCA by HpaR1 and Clp
4 结语与展望
随着功能基因组学的发展,转录调控因子的研究取得了极大的进展。越来越多的GntR家族转录调控因子被鉴定。但是目前关于GntR家族转录调控因子的研究主要还是集中在其生物学功能的研究,对于其结构、配体以及两个或者两个以上转录调控因子共同调控一个靶基因的研究相对比较少。因此,GntR家族转录调控因子与其它转录因子共同调控靶基因的作用机理是一个有趣的研究方向。我们对Xcc中两个全局转录调控因子HpaR1和Clp共同调控一个靶基因进行研究,已筛选出的一些候选靶基因与该菌的致病相关,包括编码致病因子胞外纤维素酶与胞外多糖等基因。这些研究将有利于病原菌致病网络的构建,给人们防治动植物病害提供理论基础。另外,目前只有很少的GntR家族蛋白的晶体结构被解析,如E. coli中的FadR、谷氨酸棒状杆菌(Corynebacterium glutamicum)中的LldR、B. subtilis中的YvoA等。GntR家族中的转录调控因子的晶体比较难获得,与靶标DNA共结合的晶体就更加难获得。结构得不到解析,对该家族转录调控因子功能的研究也造成了很大的影响。已知GntR家族转录调控因子C端结构域与配体结合后,使转录调控因子与靶基因结合或者解离,进而调控靶基因的转录水平。但目前只筛选到了很少的配体,主要是因为目前还没有快速大量筛选配体的方法,而细菌生活的环境又是复杂多变的,环境中的许多小分子物质都有可能是GntR家族转录调控因子的配体。只有快速筛选配体技术的发展,才能获得大量的配体。GntR家族转录调控因子往往可调控致病菌的致病过程。因此,通过鉴定其配体,可以此作为药物靶标,来防治动植物病害。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.2323/jgam.2016.05.004URLPMID:27829583 [本文引用: 1]

We modified GntR regulation in Bacillus subtilis to devise transient induction systems. GntR is the repressor antagonized by gluconate to induce transcription of the gntRKPZ operon for gluconate catabolism. On the other hand, the gnt operon is repressed by glucose via carbon catabolite repression involving CcpA/P-ser-HPr, which binds to two cre sites: one located in the gnt promoter region and the other within the gntR coding region. We initiated gntKPZ encoding of enzymes for gluconate catabolism expressed independently from the operon; this allowed constitutive degradation of gluconate. Both cre sites were mutated to abolish catabolite repression. The mutated gnt promoter was set up to drive the expression of the lacZ reporter under the control of GntR. Even in the presence of glucose, lacZ was induced upon the addition of gluconate and shut down again as gluconate was consumed. Thus, modified GntR regulation enables artificial transient induction. This will allow us to design a flexible metabolic engineering system with genes expressed only temporarily as desired.
DOI:10.1146/annurev.bi.61.070192.005201URLPMID:1497306 [本文引用: 2]
DOI:10.1016/0378-1097(91)90101-fURLPMID:2060763 [本文引用: 1]
A new family of bacterial regulatory proteins has been identified by sequence similarity. The family contains the repressor of the Bacillus subtilis gluconate operon (GntR), the regulators for histidine utilization in Pseudomonas putida (HutCPp) and Klebsiella aerogenes (HutCKa), the repressor (FadR) of fatty acid degradation in Escherichia coli, a regulator involved in the conjugal transfer of the broad host range plasmid pIJ101 (KorA), and three proteins of unidentified function in E. coli (GenA, P30 and PhnF). The proteins share amino acid sequence similarities in a 69-residue N-terminal region. A helix-turn-helix motif is predicted in the most highly-conserved segment of each protein suggesting that they are members of a new family of helix-turn-helix DNA-binding proteins.
URLPMID:3669339 [本文引用: 1]
DOI:10.1016/S0065-2164(09)69001-8URLPMID:19729089 [本文引用: 2]
One of the most abundant and widely distributed groups of Helix-turn-helix (HTH) transcription factors is the metabolite-responsive GntR family of regulators (>8500 members in the Pfam database; Jan 2009). These proteins contain a DNA-binding HTH domain at the N terminus of the protein and an effector-binding and/or oligomerisation domain at the C terminus, where upon on binding an effector molecule, a conformational change occurs in the protein which influences the DNA-binding properties of the regulator resulting in repression or activation of transcription. This review summarises what we know about the distribution, structure, function and classification of these regulators and suggests that they may have a future role in biotechnology.
DOI:10.1371/journal.pone.0132618URLPMID:26151451 [本文引用: 1]
The GNTR family of transcription factors (TFs) is a large group of proteins present in diverse bacteria and regulating various biological processes. Here we use the comparative genomics approach to reconstruct regulons and identify binding motifs of regulators from three subfamilies of the GNTR family, FADR, HUTC, and YTRA. Using these data, we attempt to predict DNA-protein contacts by analyzing correlations between binding motifs in DNA and amino acid sequences of TFs. We identify pairs of positions with high correlation between amino acids and nucleotides for FADR, HUTC, and YTRA subfamilies and show that the most predicted DNA-protein interactions are quite similar in all subfamilies and conform well to the experimentally identified contacts formed by FadR from E. coli and AraR from B. subtilis. The most frequent predicted contacts in the analyzed subfamilies are Arg-G, Asn-A, Asp-C. We also analyze the divergon structure and preferred site positions relative to regulated genes in the FADR and HUTC subfamilies. A single site in a divergon usually regulates both operons and is approximately in the middle of the intergenic area. Double sites are either involved in the co-operative regulation of both operons and then are in the center of the intergenic area, or each site in the pair independently regulates its own operon and tends to be near it. We also identify additional candidate TF-binding boxes near palindromic binding sites of TFs from the FADR, HUTC, and YTRA subfamilies, which may play role in the binding of additional TF-subunits.
DOI:10.1186/1471-2164-8-289URLPMID:17714599 [本文引用: 1]
BACKGROUND: Mycobacterium smegmatis is fast growing non-pathogenic mycobacteria. This organism has been widely used as a model organism to study the biology of other virulent and extremely slow growing species like Mycobacterium tuberculosis. Based on the homology of the N-terminal DNA binding domain, the recently sequenced genome of M. smegmatis has been shown to possess several putative GntR regulators. A striking characteristic feature of this family of regulators is that they possess a conserved N-terminal DNA binding domain and a diverse C-terminal domain involved in the effector binding and/or oligomerization. Since the physiological role of these regulators is critically dependent upon effector binding and operator sites, we have analysed and classified these regulators into their specific subfamilies and identified their potential binding sites. RESULTS: The sequence analysis of M. smegmatis putative GntRs has revealed that FadR, HutC, MocR and the YtrA-like regulators are encoded by 45, 8, 8 and 1 genes respectively. Further out of 45 FadR-like regulators, 19 were classified into the FadR group and 26 into the VanR group. All these proteins showed similar secondary structural elements specific to their respective subfamilies except MSMEG_3959, which showed additional secondary structural elements. Using the reciprocal BLAST searches, we further identified the orthologs of these regulators in Bacillus subtilis and other mycobacteria. Since the expression of many regulators is auto-regulatory, we have identified potential operator sites for a number of these GntR regulators by analyzing the upstream sequences. CONCLUSION: This study helps in extending the annotation of M. smegmatis GntR proteins. It identifies the GntR regulators of M. smegmatis that could serve as a model for studying orthologous regulators from virulent as well as other saprophytic mycobacteria. This study also sheds some light on the nucleotide preferences in the target-motifs of GntRs thus providing important leads for initiating the experimental characterization of these proteins, construction of the gene regulatory network for these regulators and an understanding of the influence of these proteins on the physiology of the mycobacteria.
DOI:10.1107/S0907444909004727URLPMID:19307717 [本文引用: 1]
The GntR superfamily of dimeric transcription factors, with more than 6200 members encoded in bacterial genomes, are characterized by N-terminal winged-helix DNA-binding domains and diverse C-terminal regulatory domains which provide a basis for the classification of the constituent families. The largest of these families, FadR, contains nearly 3000 proteins with all-alpha-helical regulatory domains classified into two related Pfam families: FadR_C and FCD. Only two crystal structures of FadR-family members, those of Escherichia coli FadR protein and LldR from Corynebacterium glutamicum, have been described to date in the literature. Here, the crystal structure of TM0439, a GntR regulator with an FCD domain found in the Thermotoga maritima genome, is described. The FCD domain is similar to that of the LldR regulator and contains a buried metal-binding site. Using atomic absorption spectroscopy and Trp fluorescence, it is shown that the recombinant protein contains bound Ni(2+) ions but that it is able to bind Zn(2+) with K(d) < 70 nM. It is concluded that Zn(2+) is the likely physiological metal and that it may perform either structural or regulatory roles or both. Finally, the TM0439 structure is compared with two other FadR-family structures recently deposited by structural genomics consortia. The results call for a revision in the classification of the FadR family of transcription factors.
DOI:10.1093/emboj/19.19.5167URLPMID:11013219 [本文引用: 2]
FadR is a dimeric acyl coenzyme A (acyl CoA)-binding protein and transcription factor that regulates the expression of genes encoding fatty acid biosynthetic and degrading enzymes in Escherichia coli. Here, the 2.0 A crystal structure of full-length FadR is described, determined using multi-wavelength anomalous dispersion. The structure reveals a dimer and a two-domain fold, with DNA-binding and acyl-CoA-binding sites located in an N-terminal and C-terminal domain, respectively. The N-terminal domain contains a winged helix-turn-helix prokaryotic DNA-binding fold. Comparison with known structures and analysis of mutagenesis data delineated the site of interaction with DNA. The C-terminal domain has a novel fold, consisting of a seven-helical bundle with a crossover topology. Careful analysis of the structure, together with mutational and biophysical data, revealed a putative hydrophobic acyl-CoA-binding site, buried in the core of the seven-helical bundle. This structure aids in understanding FadR function at a molecular level, provides the first structural scaffold for the large GntR family of transcription factors, which are keys in the control of metabolism in bacterial pathogens, and could thus be a possible target for novel chemotherapeutic agents.
DOI:10.1074/jbc.M110968200URLPMID:11756427 [本文引用: 2]
Haydon and Guest (Haydon, D. J, and Guest, J. R. (1991) FEMS Microbiol. Lett. 63, 291-295) first described the helix-turn-helix GntR family of bacterial regulators. They presented them as transcription factors sharing a similar N-terminal DNA-binding (d-b) domain, but they observed near-maximal divergence in the C-terminal effector-binding and oligomerization (E-b/O) domain. To elucidate this C-terminal heterogeneity, structural, phylogenetic, and functional analyses were performed on a family that now comprises about 270 members. Our comparative study first focused on the C-terminal E-b/O domains and next on DNA-binding domains and palindromic operator sequences, has classified the GntR members into four subfamilies that we called FadR, HutC, MocR, and YtrA. Among these subfamilies a degree of similarity of about 55% was observed throughout the entire sequence. Structure/function associations were highlighted although they were not absolutely stringent. The consensus sequences deduced for the DNA-binding domain were slightly different for each subfamily, suggesting that fusion between the D-b and E-b/O domains have occurred separately, with each subfamily having its own D-b domain ancestor. Moreover, the compilation of the known or predicted palindromic cis-acting elements has highlighted different operator sequences according to our subfamily subdivision. The observed C-terminal E-b/O domain heterogeneity was therefore reflected on the DNA-binding domain and on the cis-acting elements, suggesting the existence of a tight link between the three regions involved in the regulating process.
DOI:10.1128/jb.172.9.5470-5476.1990URLPMID:2203753 [本文引用: 1]
The hutC gene of Pseudomonas putida encodes a repressor which, in combination with the inducer urocanate, regulates expression of the five structural genes necessary for conversion of histidine to glutamate, ammonia, and formate. The nucleotide sequence of the hutC region was determined and found to contain two open reading frames which overlapped by one nucleotide. The first open reading frame (ORF1) appeared to encode a 27,648-dalton protein of 248 amino acids whose sequence strongly resembled that of the hut repressor of Klebsiella aerogenes (A. Schwacha and R. A. Bender, J. Bacteriol. 172:5477-5481, 1990) and contained a helix-turn-helix motif that could be involved in operator binding. The gene was preceded by a sequence which was nearly identical to that of the operator site located upstream of hutU which controls transcription of the hutUHIG genes. The operator near hutC would presumably allow the hut repressor to regulate its own synthesis as well as the expression of the divergent hutF gene. A second open reading frame (ORF2) would encode a 21,155-dalton protein, but because this region could be deleted with only a slight effect on repressor activity, it is not likely to be involved in repressor function or structure.
DOI:10.1371/journal.pone.0157691URLPMID:27337024 [本文引用: 1]
Small molecule effectors regulate gene transcription in bacteria by altering the DNA-binding affinities of specific repressor proteins. Although the GntR proteins represent a large family of bacterial repressors, only little is known about the allosteric mechanism that enables their function. DasR from Streptomyces coelicolor belongs to the GntR/HutC subfamily and specifically recognises operators termed DasR-responsive elements (dre-sites). Its DNA-binding properties are modulated by phosphorylated sugars. Here, we present several crystal structures of DasR, namely of dimeric full-length DasR in the absence of any effector and of only the effector-binding domain (EBD) of DasR without effector or in complex with glucosamine-6-phosphate (GlcN-6-P) and N-acetylglucosamine-6-phosphate (GlcNAc-6-P). Together with molecular dynamics (MD) simulations and a comparison with other GntR/HutC family members these data allowed for a structural characterisation of the different functional states of DasR. Allostery in DasR and possibly in many other GntR/HutC family members is best described by a conformational selection model. In ligand-free DasR, an increased flexibility in the EBDs enables the attached DNA-binding domains (DBD) to sample a variety of different orientations and among these also a DNA-binding competent conformation. Effector binding to the EBDs of DasR significantly reorganises the atomic structure of the latter. However, rather than locking the orientation of the DBDs, the effector-induced formation of beta-strand beta* in the DBD-EBD-linker segment merely appears to take the DBDs 'on a shorter leash' thereby impeding the 'downwards' positioning of the DBDs that is necessary for a concerted binding of two DBDs of DasR to operator DNA.
DOI:10.1016/j.biopen.2016.07.002URLPMID:29450126 [本文引用: 1]
Peptide inter-domain linkers are peptide segments covalently linking two adjacent domains within a protein. Linkers play a variety of structural and functional roles in naturally occurring proteins. In this work we analyze the sequence properties of the predicted linker regions of the bacterial transcriptional regulators belonging to the recently discovered MocR subfamily of the GntR regulators. Analyses were carried out on the MocR sequences taken from the phyla Actinobacteria, Firmicutes, Alpha-, Beta- and Gammaproteobacteria. The results suggest that MocR linkers display phylum-specific characteristics and unique features different from those already described for other classes of inter-domain linkers. They show an average length significantly higher: 31.8 +/- 14.3 residues reaching a maximum of about 150 residues. Compositional propensities displayed general and phylum-specific trends. Pro is dominating in all linkers. Dyad propensity analysis indicate Pro-Pro as the most frequent amino acid pair in all linkers. Physicochemical properties of the linker regions were assessed using amino acid indices relative to different features: in general, MocR linkers are flexible, hydrophilic and display propensity for beta-turn or coil conformations. Linker sequences are hypervariable: only similarities between MocR linkers from organisms related at the level of species or genus could be found with sequence searches. The results shed light on the properties of the linker regions of the new MocR subfamily of bacterial regulators and may provide knowledge-based rules for designing artificial linkers with desired properties.
DOI:10.1110/ps.072976907URLPMID:17766384 [本文引用: 1]
Among the transcription factors, the helix-turn-helix (HTH) GntR family comprised of FadR, HutC, MocR, YtrA, AraR, and PlmA subfamilies regulates the most varied biological processes. Generally, proteins belonging to this family contain an N-terminal DNA-binding domain and a C-terminal effector-binding/oligomerization domain. The members of the YtrA subfamily are much shorter than other members of this family, with chain lengths of 120-130 residues with about 50 residues located in the C-terminal domain. Because of this length, the mode of dimerization and the ability to bind effectors by the C-terminal domain are puzzling. Here, we first report the structure of the transcription factor CGL2947 from Corynebacterium glutamicum, which belongs to the YtrA family. The monomer is composed of a DNA-binding domain containing a winged HTH motif in the N terminus and two helices (alpha4 and alpha5) with a fishhook-shaped arrangement in the C terminus. Helices alpha4 and alpha5 of two monomers intertwine together to form a novel homodimer assembly. The effector-accommodating pocket with 2-methyl-2,4-pentanediol (MPD) docked was located, and it was suggested to represent a novel mode of effector binding. The structures in two crystal forms (MPD-free and -bound in the proposed effector-binding pocket) were solved. The structural variations have implications regarding how the effector-induced conformational change modulates DNA affinity for YtrA family members.
DOI:10.1128/jb.182.19.5454-5461.2000URLPMID:10986249 [本文引用: 1]
The ytrABCDEF operon of Bacillus subtilis was deduced to encode a putative ATP-binding cassette (ABC) transport system. YtrB and YtrE could be the ABC subunits, and YtrC and YtrD are highly hydrophobic and could form a channel through the cell membrane, while YtrF could be a periplasmic lipoprotein for substrate binding. Expression of the operon was examined in cells grown in a minimal medium. The results indicate that the expression was induced only early in the stationary phase. The six ytr genes form a single operon, transcribed from a putative sigma(A)-dependent promoter present upstream of ytrA. YtrA, which possesses a helix-turn-helix motif of the GntR family, acts probably as a repressor and regulates its own transcription. Inactivation of the operon led to a decrease in maximum cell yield and less-efficient sporulation, suggesting its involvement in the growth in stationary phase and sporulation. It is known that B. subtilis produces acetoin as an external carbon storage compound and then reuses it later during stationary phase and sporulation. When either the entire ytr operon or its last gene, ytrF, was inactivated, the production of acetoin was not affected, but the reuse of acetoin became less efficient. We suggest that the Ytr transport system plays a role in acetoin utilization during stationary phase and sporulation.
DOI:10.3390/pathogens8040235URL [本文引用: 2]
DOI:10.1128/JB.00307-06URLPMID:16816174 [本文引用: 2]
The gram-positive filamentous bacterium Streptomyces coelicolor has a complex developmental cycle with three distinct phases: growth of the substrate mycelium, development of reproductive structures called aerial hyphae, and differentiation of these aerial filaments into long chains of exospores. During a transposon mutagenesis screen, we identified a novel gene (devA) required for proper development. The devA mutant produced only rare aerial hyphae, and those that were produced developed aberrant spore chains that were much shorter than wild-type chains and had misplaced septa. devA encodes a member of the GntR superfamily, a class of transcriptional regulators that typically respond to metabolite effector molecules. devA forms an operon with the downstream gene devB, which encodes a putative hydrolase that is also required for aerial mycelium formation on R5 medium. S1 nuclease protection analysis showed that transcription from the single devA promoter was temporally associated with vegetative growth, and enhanced green fluorescent protein transcriptional fusions showed that transcription was spatially confined to the substrate hyphae in the wild type. In contrast, devAB transcript levels were dramatically upregulated in a devA mutant and the devA promoter was also active in aerial hyphae and spores in this background, suggesting that DevA might negatively regulate its own production. This suggestion was confirmed by gel mobility shift assays that showed that DevA binds its own promoter region in vitro.
DOI:10.1111/1462-2920.13871URLPMID:28752954 [本文引用: 1]
In contrast to Escherichia coli, glucose metabolism in pseudomonads occurs exclusively through the Entner-Doudoroff (ED) pathway. This pathway, as well as the three routes to generate the initial ED pathway substrate, 6-phosphogluconate, is regulated by the PtxS, HexR and GtrS/GltR systems. With GntR (PA2320) we report here the identification of an additional regulator in Pseudomonas aeruginosa PAO1. GntR repressed its own expression as well as that of the GntP gluconate permease. In contrast to PtxS and GtrS/GltR, GntR did not modulate expression of the toxA gene encoding the exotoxin A virulence factor. GntR was found to bind to promoters PgntR and PgntP and the consensus sequence of its operator was defined as 5'-AC-N-AAG-N-TAGCGCT-3'. Both operator sites overlapped with the RNA polymerase binding site and we show that GntR employs an effector mediated de-repression mechanism. The release of promoter bound GntR is induced by gluconate and 6-phosphogluconate that bind with similar apparent affinities to the GntR/DNA complex. GntR and PtxS are paralogous and may have evolved from a common ancestor. The concerted action of four regulatory systems in the regulation of glucose metabolism in Pseudomonas can be considered as a model to understand complex regulatory circuits in bacteria.
DOI:10.1128/JB.01819-06URLPMID:17277059 [本文引用: 1]
MgrA is a known regulator of the expression of several multidrug transporters in Staphylococcus aureus. We identified another regulator of multiple efflux pumps, NorG, by its ability, like that of MgrA, to bind specifically to the promoter of the gene encoding the NorA efflux pump. NorG is a member of the family of the GntR-like transcriptional regulators, and it binds specifically to the putative promoters of the genes encoding multidrug efflux pumps NorA, NorB, NorC, and AbcA. Overexpression of norG produces a threefold increase in norB transcripts associated with a fourfold increase in the level of resistance to quinolones. In contrast, disruption of norG produces no change in the level of transcripts of norA, norB, and norC but causes an increase of at least threefold in the transcript level of abcA, associated with a fourfold increase in resistance to methicillin, cefotaxime, penicillin G, and nafcillin. Overexpression of cloned abcA caused an 8- to 128-fold increase in the level of resistance to all four beta-lactam antibiotics. Furthermore, MgrA and NorG have opposite effects on norB and abcA expression. MgrA acts as an indirect repressor for norB and a direct activator for abcA, whereas NorG acts as a direct activator for norB and a direct repressor for abcA.
DOI:10.1007/s11274-017-2230-9URLPMID:28243986 [本文引用: 2]
The pathogenic mechanisms of Brucella are still poorly understood. GntR is a transcriptional regulator and plays an important role in the intracellular survival of Brucella. To investigate whether GntR is involved in the cytotoxicity of Brucella abortus (B. abortus), we created a 2308DeltagntR mutant of B. abortus 2308 (S2308). Lactate dehydrogenase (LDH) cytotoxicity assays using a murine macrophage cell line (RAW 264.7) show that high-dose infection with the parental strain produces a high level of cytotoxicity to macrophages, but the 2308DeltagntR mutant exhibits a very low level of cytotoxicity, indicating that mutation of GntR impairs the cytotoxicity of B. abortus to macrophages. After the macrophages are infected with 2308DeltagntR, the levels of tumor necrosis factor-alpha (TNF-alpha), interleukin-6 (IL-6) and interleukin-8 (IL-8) increase and are slightly higher than that for the S2308 infected group, indicating that the 2308DeltagntR mutant could induce the secretion of inflammatory cytokines. The virulence factor detection experiments indicate that genes involved in the type IV secretion system (T4SS) and quorum sensing system (QSS) are down-regulated in 2308DeltagntR. The lower levels of survival of 2308DeltagntR under various stress conditions and the increased sensitivity of 2308DeltagntR to polymyxin B suggest that GntR is a virulence factor and that deletion of gntR reduces of B. abortus to stress conditions. Taken together, our results demonstrate that GntR is involved in the cytotoxicity, virulence and intracellular survival of B. abortus during its infection.
DOI:10.1111/mpp.12397URLPMID:26972728 [本文引用: 1]
Xanthomonas contains a large group of plant-associated species, many of which cause severe diseases on important crops worldwide. Six gluconate-operon repressor (GntR) family transcriptional regulators are predicted in Xanthomonas, one of which, belonging to the YtrA subfamily, plays a prominent role in bacterial virulence. However, the direct targets and comprehensive regulatory profile of YtrA remain unknown. Here, we performed microarray and high-resolution chromatin immunoprecipitation-exonuclease (ChIP-exo) experiments to identify YtrA direct targets and its DNA binding motif in X. citri ssp. citri (Xac), the causal agent of citrus canker. Integrative microarray and ChIP-exo data analysis revealed that YtrA directly regulates three operons by binding to a palindromic motif GGTG-N16 -CACC at the promoter region. A similar palindromic motif and YtrA homologues were also identified in many other bacteria, including Stenotrophomonas, Pseudoxanthomonas and Frateuria, indicating a widespread phenomenon. Deletion of ytrA in Xac abolishes bacterial virulence and induction of the hypersensitive response (HR). We found that YtrA regulates the expression of hrp/hrc genes encoding the bacterial type III secretion system (T3SS) and controls multiple biological processes, including motility and adhesion, oxidative stress, extracellular enzyme production and iron uptake. YtrA represses the expression of its direct targets in artificial medium or in planta. Importantly, over-expression of yro3, one of the YtrA directly regulated operons which contains trmL and XAC0231, induced weaker canker symptoms and down-regulation of hrp/hrc gene expression, suggesting a negative regulation in Xac virulence and T3SS. Our study has significantly advanced the mechanistic understanding of YtrA regulation and its contribution to bacterial virulence.
DOI:10.1093/femsle/fny052URLPMID:29514248 [本文引用: 1]
Magnetotactic bacteria (MTB) can biosynthesise magnetosomes, which have great potential for commercial applications. A new MTB strain, Magnetospirillum sp. ME-1, was isolated and cultivated from freshwater sediments of East Lake (Wuhan, China) using the limiting dilution method. ME-1 had a chain of 17 +/- 4 magnetosomes in the form of cubooctahedral crystals with a shape factor of 0.89. ME-1 was closest to Magnetospirillum sp. XM-1 according to 16S rRNA gene sequence similarity. Compared with XM-1, ME-1 possessed an additional copy of mamPA and a larger mamO in magnetosome-specific genes. ME-1 had an intact citric acid cycle, and complete pathway models of ammonium assimilation and dissimilatory nitrate reduction. Potential carbon and nitrogen sources in these pathways were confirmed to be used in ME-1. Adipate was determined to be used in the fermentation medium as a new kind of dicarboxylic acid. The optimised fermentation medium was determined by orthogonal tests. The large-scale production of magnetosomes was achieved and the magnetosome yield (wet weight) reached 120 mg L-1 by fed-batch cultivation of ME-1 at 49 h in a 10-L fermenter with the optimised fermentation medium. This study may provide insights into the isolation and cultivation of other new MTB strains and the production of magnetosomes.
DOI:10.3389/fcimb.2017.00513URLPMID:29312893 [本文引用: 1]
The structure of Vibrio cholerae FadR (VcFadR) complexed with the ligand oleoyl-CoA suggests an additional ligand-binding site. However, the fatty acid metabolism and its regulation is poorly addressed in Vibrio alginolyticus, a species closely-related to V. cholerae. Here, we show crystal structures of V. alginolyticus FadR (ValFadR) alone and its complex with the palmitoyl-CoA, a long-chain fatty acyl ligand different from the oleoyl-CoA occupied by VcFadR. Structural comparison indicates that both VcFadR and ValFadR consistently have an additional ligand-binding site (called site 2), which leads to more dramatic conformational-change of DNA-binding domain than that of the E. coli FadR (EcFadR). Isothermal titration calorimetry (ITC) analyses defines that the ligand-binding pattern of ValFadR (2:1) is distinct from that of EcFadR (1:1). Together with surface plasmon resonance (SPR), electrophoresis mobility shift assay (EMSA) demonstrates that ValFadR binds fabA, an important gene of unsaturated fatty acid (UFA) synthesis. The removal of fadR from V. cholerae attenuates fabA transcription and results in the unbalance of UFA/SFA incorporated into membrane phospholipids. Genetic complementation of the mutant version of fadR (Delta42, 136-177) lacking site 2 cannot restore the defective phenotypes of DeltafadR while the wild-type fadR gene and addition of exogenous oleate can restore them. Mice experiments reveals that VcFadR and its site 2 have roles in bacterial colonizing. Together, the results might represent an additional example that illustrates the Vibrio FadR-mediated lipid regulation and its role in pathogenesis.
DOI:10.1186/s12866-017-1112-5URLPMID:28927382 [本文引用: 2]
BACKGROUND: Pseudomonas aeruginosa is a model organism for the study of quorum sensing, biofilm formation, and also leading cause of nosocomial infections in immune compromised patients. As such P. aeruginosa is one of the most well studied organisms in terms of its genetics. However, the construction of gene deletions and replacements in Pseudomonas aeruginosa is relatively time-consuming, requiring multiple steps including suicide vector construction, conjugation, inactivation with insertion of antibiotic resistance cassettes and allelic exchange. Even employing Gateway recombineering techniques with direct transformation requires a minimum two weeks. METHODS: We have developed a rapid streamlined method to create clean deletion mutants in P. aeruginosa through direct transformation, eliminating the need for the creation of Gateway-compatible suicide vectors. In this method, upstream and downstream sequences of the gene/locus to be deleted are amplified by polymerase chain reaction (PCR) and seamlessly fused with the linearized pEX18Tc sacB suicide plasmid by Gibson assembly. The resulting deletion plasmid is transformed into P. aeruginosa by an electroporation method optimized in this study. The plasmid is then integrated into the chromosome by homologous recombination, and deletion mutants are identified via sacB mediated sucrose counter-selection. RESULTS: The current method was employed to generate clean gene deletions of the heme assimilation system anti-sigma factor, hasS and the virulence regulator involving ECF system anti-sigma and sigma factors vreA and vreI, respectively. The process from plasmid construction to confirmation by DNA sequencing of the gene deletion was completed in one week. Furthermore, the utility of the method is highlighted in the construction of the vreA and vreI deletions, where the start codon of vreA and the stop codon of vreI overlap. Utilizing Gibson assembly deletion mutants were constructed with single base pair precision to generate the respective vreA and vreI deletions, while maintaining the start and stop codon of the respective genes. Overall, this method allows for rapid construction of gene deletions in P. aeruginosa with base pair precision. CONCLUSION: This method from the construction of the suicide vector to sequence confirmation of the unmarked gene deletion can be performed in one week, without the requirement for expensive proprietary reagents or instruments. The precision of Gibson assembly and the fact the accuracy in generating the desirable construct is 95%, makes this a viable and attractive alternative to previous methods.
DOI:10.1111/mmi.14584URLPMID:32748556 [本文引用: 1]
The GntR family regulators are widely distributed in bacteria and play critical roles in metabolic processes and bacterial pathogenicity. In this study, we describe a GntR family protein encoded by PA4132 that we named MpaR (MvfR-mediated PQS and anthranilate regulator) for its regulation of Pseudomonas quinolone signal (PQS) production and anthranilate metabolism in Pseudomonas aeruginosa. The deletion of mpaR increased biofilm formation and reduced pyocyanin production. RNA sequencing analysis revealed that the mRNA levels of antABC encoding enzymes for the synthesis of catechol from anthranilate, a precursor of the PQS, were most affected by mpaR deletion. Data showed that MpaR directly activates the expression of mvfR, a master regulator of pqs system, and subsequently promotes PQS production. Accordingly, deletion of mpaR activates the expression of antABC genes, and thus, increases catechol production. We also demonstrated that MpaR represses the rhl quorum-sensing (QS) system, which has been shown to control antABC activity. These results suggested that MpaR function is integrated into the QS regulatory network. Moreover, mutation of mpaR promotes bacterial survival in a mouse model of acute pneumonia infection. Collectively, this study identified a novel regulator of pqs system, which coordinately controls anthranilate metabolism and bacterial virulence in P. aeruginosa.
DOI:10.3389/fmicb.2018.03224URLPMID:30692967 [本文引用: 1]
GntR family transcription factors have been implicated in the regulation of carbohydrate transport and metabolism in many bacteria. However, the function of this transcription factor family is poorly studied in Streptococcus mutans, which is a commensal bacterium in the human oral cavity and a well-known cariogenic pathogen. One of the most important virulence traits of S. mutans is its ability to transport and metabolize carbohydrates. In this study, we identified a GntR transcription factor in S. mutans named StsR (Sugar Transporter Systems Regulator). The deletion of the stsR gene in S. mutans caused a decrease in both the formation of biofilm and the production of extracellular polysaccharides (EPS) at early stage. Global gene expression profiling revealed that the expression levels of 188 genes were changed in the stsR mutant, which could be clustered with the sugar PTS and ABC transporters. Furthermore, StsR protein was purified and its conserved DNA binding motif was determined using electrophoretic mobility shift assays (EMSA) and DNase I footprinting assays. Collectively, the results of this research indicate that StsR is an important transcription factor in S. mutans that regulates the expression of sugar transporter genes, production of EPS and formation of biofilm.
DOI:10.1038/srep29255URLPMID:27386955 [本文引用: 1]
Transcriptional regulation of capsule expression is critical for pneumococcal transition from carriage to infection, yet the underlying mechanism remains incompletely understood. Here, we describe the regulation of capsular polysaccharide, one of the most important pneumococcal virulence factor by a GntR family regulator, CpsR. Electrophoretic mobility-shift assays have shown the direct interaction between CpsR and the cps promoter (cpsp), and their interaction could be competitively interfered by glucose. DNase I footprinting assays localized the binding site to a region -146 to -114 base pairs relative to the transcriptional start site of the cps locus in S. pneumoniae D39. We found that CpsR negatively controlled the transcription of the cps locus and hence CPS production, which was confirmed by fine-tuning expression of CpsR in a DeltacpsR complemented strain. Increased expression of CpsR in complemented strain led to a decreased resistance to the whole-blood-mediated killing, suggesting a protective role for CpsR-cpsp interaction in the establishment of invasive infection. Finally, animal experiments showed that CpsR-cpsp interaction was necessary for both pneumococcal colonization and invasive infection. Taken together, our results provide a thorough insight into the regulation of capsule production mediated by CpsR and its important roles in pneumococcal pathogenesis.
DOI:10.1128/JB.05847-11URL [本文引用: 1]
The GntR-like protein NorG has been shown to affect Staphylococcus aureus genes involved in resistance to quinolones and beta-lactams, such as those encoding the NorB and AbcA transporters. To identify the target genes regulated by NorG, we carried out transcriptional-profiling assays using S. aureus RN6390 and its isogenic norG::cat mutant. Our data showed that NorG positively affected the transcription of global regulators mgrA, arlS, and sarZ. The three putative drug efflux pump genes most positively affected by NorG were the NorB efflux pump (5.1-fold), the MmpL-like protein SACOL2566 (5.2-fold), and the BcrA-like drug transporter SACOL2525 (5.7-fold) genes. The S. aureus predicted MmpL protein showed 53% homology with the MmpL lipid transporter of Mycobacterium tuberculosis, and the putative SACOL2525 protein showed 87% homology with the bacitracin drug transporter BcrA of Staphylococcus hominis. Two pump genes most negatively affected by NorG were the NorC (4-fold) and AbcA (6-fold) genes. Other categories of genes, such as those participating in amino acid, inorganic ion, or nucleotide transporters and metabolism, were also affected by NorG. Real-time reverse transcription (RT)-PCR assays for mgrA, arlS, sarZ, norB, norC, abcA, mmpL, and bcrA-like were carried out to verify microarray data and showed the same level of up-or downregulation by NorG. The norG mutant showed a 2-fold increase in resistance to norfloxacin and rhodamine, both substrates of the NorC transporter, which is consistent with the resistance phenotype conferred by overexpression of norC on a plasmid. These data indicate that NorG has broad regulatory function in S. aureus.
DOI:10.1094/MPMI-08-10-0180URLPMID:21615202 [本文引用: 2]
The GntR family is one of the most abundant and widely distributed groups of helix-turn-helix transcriptional regulators in bacteria. Six open reading frames in the genome of the plant pathogen Xanthomonas campestris pv. campestris were predicted to encode GntR regulators. All six of the predicted GntR-encoding genes were individually mutagenized and mutants from five of them were successfully obtained. Plant disease response assays revealed that one, whose product belongs to the YtrA subfamily and has been named HpaR1, is involved in the hypersensitive response (HR) and virulence. Electrophoretic mobility shift assays and in vitro transcription assays revealed that HpaR1 could repress its own transcription level through binding to its promoter sequence, indicating an autoregulatory feedback inhibition mechanism for HpaR1 expression. Promoter-gusA reporter and reverse-transcription polymerase chain reaction analyses revealed that HpaR1 positively and negatively affects the expression of HR and pathogenicity (hrp) genes in host plant and standard media, respectively. Constitutive expression of the key hrp regulator, hrpG, in the hpaR1 mutant could bypass the requirement of HpaR1 for the induction of wild-type HR, suggesting that HpaR1 regulates the expression of hrp genes that encode the type III secretion system via hrpG.
DOI:10.1038/srep19862URLPMID:26818230 [本文引用: 2]
The GntR family transcription regulator HpaR1 identified from Xanthomonas campestris pv. campestris has been previously shown to positively regulate the genes responsible for hypersensitive reaction and pathogenicity and to autorepress its own expression. Here, we demonstrated that HpaR1 is a global regulator that positively regulates diverse biological processes, including xanthan polysaccharide production, extracellular enzyme activity, cell motility and tolerance to various stresses. To investigate the regulatory mechanisms of HpaR1, we began with xanthan polysaccharide production, which is governed by a cluster of gum genes. These are directed by the gumB promoter. Disruption of HpaR1 significantly reduced gumB transcription and an electrophoretic mobility shift assay demonstrated that HpaR1 interacts directly with gumB promoter. DNase I footprint analysis revealed that HpaR1 and RNA polymerase were bound to the sequences extending from -21 to +10 and -41 to +29 relative to the transcription initiation site of gumB, respectively. Furthermore, in vitro transcription assays showed that HpaR1 facilitated the binding of RNA polymerase to gumB promoter, leading to an enhancement of its transcription. These results suggest that HpaR1 regulates gumB transcription via a mechanism similar but different to what was found, until now, to only be used by some MerR family transcription activators.
DOI:10.1111/mpp.12739URLPMID:30091270 [本文引用: 3]
Transcriptional regulators are key players in pathways that allow bacteria to alter gene expression in response to environmental conditions. However, work to understand how such transcriptional regulatory networks interact in bacterial plant pathogens is limited. Here, in the phytopathogen Xanthomonas campestris, we demonstrate that the global transcriptional regulator HpaR1 influences many of the same genes as another global regulator Clp, including the engXCA gene that encodes extracellular endoglucanase. We demonstrate that HpaR1 facilitates the binding of RNA polymerase to the engXCA promoter. In addition, we show that HpaR1 binds directly to the engXCA promoter. Furthermore, our in vitro tests characterize two binding sites for Clp within the engXCA promoter. Interestingly, one of these sites overlaps with the HpaR1 binding site. Mobility shift assays reveal that HpaR1 has greater affinity for binding to the engXCA promoter. This observation is supported by promoter activity assays, which show that the engXCA expression level is lower when both HpaR1 and Clp are present together, rather than alone. The data also reveal that HpaR1 and Clp activate engXCA gene expression by binding directly to its promoter. This transcriptional activation is modulated as both regulators compete to bind to overlapping sites on the engXCA promoter. Bioinformatics analysis suggests that this mechanism may be used broadly in Xanthomonas campestris pv. campestris (Xcc) and is probably widespread in Xanthomonads and, potentially, other bacteria. Taken together, these data support a novel mechanism of competitive activation by two global regulators of virulence gene expression in Xcc which is probably widespread in Xanthomonads and, potentially, other bacteria.
DOI:10.1111/mpp.12397URLPMID:26972728 [本文引用: 1]
Xanthomonas contains a large group of plant-associated species, many of which cause severe diseases on important crops worldwide. Six gluconate-operon repressor (GntR) family transcriptional regulators are predicted in Xanthomonas, one of which, belonging to the YtrA subfamily, plays a prominent role in bacterial virulence. However, the direct targets and comprehensive regulatory profile of YtrA remain unknown. Here, we performed microarray and high-resolution chromatin immunoprecipitation-exonuclease (ChIP-exo) experiments to identify YtrA direct targets and its DNA binding motif in X. citri ssp. citri (Xac), the causal agent of citrus canker. Integrative microarray and ChIP-exo data analysis revealed that YtrA directly regulates three operons by binding to a palindromic motif GGTG-N16 -CACC at the promoter region. A similar palindromic motif and YtrA homologues were also identified in many other bacteria, including Stenotrophomonas, Pseudoxanthomonas and Frateuria, indicating a widespread phenomenon. Deletion of ytrA in Xac abolishes bacterial virulence and induction of the hypersensitive response (HR). We found that YtrA regulates the expression of hrp/hrc genes encoding the bacterial type III secretion system (T3SS) and controls multiple biological processes, including motility and adhesion, oxidative stress, extracellular enzyme production and iron uptake. YtrA represses the expression of its direct targets in artificial medium or in planta. Importantly, over-expression of yro3, one of the YtrA directly regulated operons which contains trmL and XAC0231, induced weaker canker symptoms and down-regulation of hrp/hrc gene expression, suggesting a negative regulation in Xac virulence and T3SS. Our study has significantly advanced the mechanistic understanding of YtrA regulation and its contribution to bacterial virulence.
DOI:10.4014/jmb.2011.11029URLPMID:33263336 [本文引用: 1]
Vaccination is the most effective way to prevent influenza virus infections. However, conventional vaccines based on hemagglutinin (HA) have to be annually updated because the HA of influenza viruses constantly mutates. In this study, we produced a 3M2e-3HA2-NP chimeric protein as a vaccine antigen candidate using an Escherichia coli expression system. The vaccination of chimeric protein (15 mug) conferred complete protection against A/Puerto Rico/8/1934 (H1N1; PR8) in mice. It strongly induced influenza virus-specific antibody responses, cytotoxic T lymphocyte activity, and antibody-dependent cellular cytotoxicity. To spare the dose and enhance the cross-reactivity of the chimeric, we used a complex of poly-gamma-glutamic acid and alum (PGA/alum) as an adjuvant. PGA/alum-adjuvanted low-dose chimeric protein (1 or 5 mug) exhibited higher cross-protective effects against influenza A viruses (PR8, CA04, and H3N2) compared with those of chimeric alone or alum-adjuvanted proteins in vaccinated mice. Moreover, the depletion of CD4(+) T, CD8(+) T, and NK cells reduced the survival rate and efficacy of the PGA/alum-adjuvanted chimeric protein. Collectively, the vaccination of PGA/alum-adjuvanted chimeric protein induced strong protection efficacy against homologous and heterologous influenza viruses in mice, which suggests that it may be a promising universal influenza vaccine candidate.
DOI:10.1007/s12275-015-5313-zURLPMID:26224454 [本文引用: 1]
Bradyrhizobium japonicum is a Gram-negative soil bacterium that can fix nitrogen into ammonia by developing a symbiotic relationship with the soybean plant. MocR proteins make up a subfamily of GntR superfamily, one of the most widely distributed and prolific groups of the helix-turn-helix transcription factors. In this study, we constructed a mutant strain for mocR (blr6977) to investigate its role in cellular processes and symbiosis in B. japonicum. Although growth rate and morphology of the mutant were indistinguishable from those of the wild type, the mutant showed significant differences in motility and attachment (i.e., biofilm formation) from the wild type. The mutant displayed a decrease in biofilm formation, but was more motile than the wild type. The inactivation of mocR did not affect the number of nodules on soybean roots, but caused delayed nodulation. Delayed nodulation intrigued us to study competitiveness of the mutant infecting soybeans. The mutant was less competitive than the wild type, indicating that delayed nodulation might be due to competitiveness. Gene expressions of other MocR subfamily members were also compared between the wild type and mutant strains. None of the mocR-like genes examined in this study were differentially expressed between both strains.
DOI:10.1038/nrmicro787URLPMID:15035009 [本文引用: 1]
Bacteria use their genetic material with great effectiveness to make the right products in the correct amounts at the appropriate time. Studying bacterial transcription initiation in Escherichia coli has served as a model for understanding transcriptional control throughout all kingdoms of life. Every step in the pathway between gene and function is exploited to exercise this control, but for reasons of economy, it is plain that the key step to regulate is the initiation of RNA-transcript formation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}