 ,1, 李玉斌,1,2
,1, 李玉斌,1,2Progress on Mutator superfamily
Chunsheng Cong,1, Yubin Li,1,2通讯作者: 李玉斌,研究员,博士生导师,研究方向:生物化学及分子生物学。E-mail:liyubin@caas.cn
第一联系人:
编委: 严建兵
收稿日期:2019-09-27修回日期:2019-12-17网络出版日期:2020-01-02
| 基金资助: |
Received:2019-09-27Revised:2019-12-17Online:2020-01-02
| Fund supported: |

摘要
转座子是一类可以在基因组中不同遗传位点间移动的DNA序列,在其转移过程中有时会伴随自身拷贝数的增加。作为基因组的重要组成部分,转座子可以通过多种方式影响宿主基因及基因组的结构与功能,进而在宿主的演化过程中扮演重要角色。目前依据转座过程中间体类型的不同可以将其分为I类转座子和II类转座子。Mutator超家族转座子是20世纪70年代在玉米(Zea may L.)中发现的一类特殊的转座子,其属于II类转座子,广泛存在于真核生物基因组中,包含遗传特征明晰可分的众多转座子家族。此外,该超家族转座子转座频率高,倾向于插入基因富含区及低拷贝序列区,可快速产生大量新的突变体,目前已被广泛应用于正向及反向遗传学研究。本文结合近年来相关研究结果,围绕Mutator超家族转座子的分类组成、结构特征、转座机制、插入偏好、靶位点重复序列以及玉米自主性MULEs元件展开综述,并对转座子研究面临的问题及未来研究方向进行了探讨,旨在与研究领域内的同行探讨相关研究的可能突破点、未来发展方向及可能产生的重大影响。
关键词:
Abstract
Transposable elements (TEs) are fragments of DNA sequence, which can mobile from one locus to another within a genome, often replication in the process. Occupying the main component of the genome, TEs can affect the structure and function of gene and/or genome in a variety of ways, and play an important role in the evolution of the host. Based on the transposition intermediate, eukaryotic TEs can be divided into two classes. The Mutator superfamily is found in maize (Zea may L.) in the 1970s. As the member of class II elements, Mutator superfamily transposons are found in all eukaryote genomes and contain many families with clearly distinguishable genetic characteristics. In addition, these TEs transpose at high rates and preferentially insert in gene-rich and low-repetitive genomic regions leading to the rapid generation of massive novel mutations, therefore, they are in great use of both forward and reverse genetics researches. In this review, we summarize the classification, structure characteristic, transposition mechanism, insertion preference and TSD sequence and other autonomous MULEs in maize. Moreover, we discuss the problems faced in TEs’ research and research directions in the future, with a view to discuss possible breakthroughs, future development directions and significant impacts with colleagues in the related research field..
Keywords:
PDF (870KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
从春生, 李玉斌. Mutator超家族转座子研究进展. 遗传[J], 2020, 42(2): 131-144 doi:10.16288/j.yczz.19-301
Chunsheng Cong.
20世纪40年代美国遗传学家Barbara McClintock在玉米中发现了一些在染色体上可以移动遗传位置的元件,并于50年代初提出可移动的遗传基因(即“跳跃基因”)学说,但直到30年后这一超越时代的学说才被科学界同行逐渐理解和接受,并将这些可以在基因组中移动的DNA序列统称为转座元件或转座子[1]。现有研究表明,转座子几乎存在于所有生物基因组中,是基因组的主要组成部分。由于其重复特性,转座子曾一度被认为是垃圾DNA,但越来越多的证据表明转座子是塑造基因组的重要因素[2,3,4,5]。在许多植物基因组中,半数以上的序列属于转座子[6,7,8,9,10,11,12],特别在玉米基因组中转座子序列的占比更高达85%[11],并且这些转座子序列通过不同方式影响了玉米的驯化[13]、传播[14,15]及优异农艺性状的形成[16,17]等。此外,转座子在自身序列、蛋白功能域和结构等方面具有十分丰富的变异[18]。按照转座过程中间体的类型,真核生物转座子可以划分为两大类:Ⅰ类转座子(RNA转座子或反转录转座子)和Ⅱ类转座子(DNA转座子)[19]。Ⅰ类转座子的转座反应通过DNA-RNA-DNA形式介导完成,通常以“copy- and-paste”方式进行转座,按照其长末端重复序列(long terminal repeats, LTR)的有无又可以分为LTR反转录转座子和non-LTR反转录转座子2个亚类;Ⅱ类转座子的转座反应通过DNA-DNA形式介导完成,绝大部分以“cut-and-paste”方式进行转座,少数转座子通过滚环模型或自我合成途径完成转座反应[20,21]。根据序列组成和基因结构特征,Ⅰ类和Ⅱ类转座子又都可分成不同的超家族[22],各超家族间既存在共性,同时又各有特性,尽管采用的分析方法不尽相同[23,24,25]。Mutator超家族转座子属于Ⅱ类转座子,广泛存在于真核生物基因组中,包含着遗传特征明晰可分的众多转座子家族,并且在转座子遗传特性方面的研究深入,在功能组学研究中的应用也十分广泛。本文结合近年来的研究结果,围绕Mutator超家族转座子的分类组成、结构特征、转座机制、插入偏好、靶位点重复序列以及玉米自主性MULEs元件进行了概述,同时对转座子研究面临的问题及未来研究方向进行了探讨,以便相关科研人员更充分、全面了解Mutator超家族转座子的研究进展。
1 Mutator超家族转座子分类组成
1978年,美国爱荷华州立大学(Iowa State University)的Donald Robertson博士报道了一份高突变频率玉米材料,其幼苗中的突变频率接近自发突变的30倍左右,而这一突变特性的遗传不符合经典的孟德尔遗传定律[26]并表现出明显的表观沉默[27]。这一遗传品系由于存在大量一类新型转座元件—Mutator或Mu[28,29,30],从而可以发生高频突变,也因此被称之为Mutator系[31]。目前这些转座元件同属Mutator家族或Mu家族,其中可以编码转座酶并使其自身发生转座的元件称为自主性Mu转座子—MuDR (Mutator-Donald Robertson),而在活性MuDR存在时才能进行转座的元件统称为非自主性Mu转座子[32,33,34]。MuDR与大量的非自主性Mu转座子组合可形成高效的突变系统,Mu系统是目前被广泛应用的致变能力极强的转座子插入突变体创制系统[35,36,37]。另外,minimal Mutator系是通过筛选获得的只含有单一MuDR和一个位于颜色基因中的非自主性Mu的遗传品系,成为研究玉米Mu转座子系统及调控的理想材料[38,39,40]。例如,从minimal Mutator系中发现了Mu killer (Muk) [41]。作为调控MuDR的显性遗传性位点,Muk可以沉默一个或多个活性MuDR,但Muk并不是维持MuDR沉默状态所必需的,在后代分离个体中,即使缺失Muk位点,MuDR仍无活性并且可以维持多代[42]。Muk的发现极大地促进了MuDR转座子表观沉默的研究,同时使用于突变体创制的Mu系统变得更为可控。
近年来,伴随着测序技术的发展以及被测序物种数量的不断增加,在植物[43,44,45,46,47]、真菌[48,49]、原生动物[50,51]以及多细胞动物[52,53]中均发现了与玉米MuDR序列相类似的转座子,统称为MULEs元件(Mutator-like transposable elements)。目前大部分鉴定出来的MULEs元件都属于非自主性转座子,它们自身不能编码功能完善的转座酶,只有极少数MULEs元件可以进行自主转座,例如尖孢镰刀菌(Fusarium oxysporum)中的Hop[49],拟南芥(Arabidopsis thaliana)中的AtMu1[54],玉米中的Jittery[55]和TED[56],水稻(Oryza sativa)中的Os3378[57]以及埃及伊蚊(Aedes aegypti)中的Muta1[58]等。另外,大量非自主性MULEs元件内部有时携带着来源于宿主的一个或多个不同基因的片段,这类元件被特别命名为Pack-MULEs。目前在拟南芥、水稻、玉米、百脉根(Lotus japonicus)、西红柿(Solanum lycopersicum)及荷花(Nelumbo nucifera Gaertn)的基因组中都发现了Pack-MULEs的存在[59,60,61,62,63,64,65],其中水稻中Pack- MULEs的数量巨大,有关研究也更为深入。水稻中有些Pack-MULEs元件所携带的多个宿主基因片段可形成崭新的开放阅读框并转录出嵌合转录本。氨基酸序列功能分析及蛋白组学研究表明,捕获的基因片段甚至可能具有特定的功能。结合以上研究结果及Pack-MULEs在植物中的普遍性,Jiang等[63]推想Pack-MULEs获取基因片段的方式很可能是高等植株基因进化的一种重要机制。虽然Pack-MULEs捕获宿主基因组片段的分子机制目前仍不清楚,但研究发现Pack-MULEs主要倾向于获得和保留GC含量高的序列,这种选择性捕获使Pack-MULEs更有可能捕获具有功能性的序列,进而为新基因的进化及现有基因的修饰提供新的遗传资源[66,67,68]。与此同时,相对于其他超家族转座元件,水稻中Pack- MULEs表现出独特的表观遗传学特性,其插入和表达不仅可以改变水稻染色体的表达模式,还可以抵消重组对染色体碱基组成的影响,进而对染色体结构进化产生影响[69]。
2 Mutator超家族转座子及其转座酶基本特征
与其他大多数DNA超家族转座子相比,Mutator超家族转座子两端具有较长的末端反向重复序列(terminal inverted repeats, TIR)。TIR序列中包含有转座酶结合位点[70],而携带单一TIR的转座子无法正常进行转座[60,71]。此外,TIR序列中还含有复杂的启动子序列,既可以启动转座酶或TIR间序列的转录,也可以调控转座酶在不同组织中的表达[63,72]。玉米Mu家族转座子的TIR比较保守,大多长约215 bp[73],根据两端TIR间序列的差异,又划分为不同亚家族(Mu1~Mu13)[74,75,76]。其中大部分为非自主性转座子,这些非自主性Mu转座子是MuDR内部片段缺失产生的衍生物或者是其他序列点突变导致转座酶功能丧失的MuDR同源序列(MuDR homologs, hMuDR)。hMuDR虽然不能催化转座反应,但可能在MuDR表观沉默中发挥增强作用[31]。相对于玉米Mu家族转座子,各种MULEs元件TIR序列变异丰富。有些MULEs元件TIR内含有串联重复序列,这些串联重复序列可能导致TIR自身形成特殊的二级结构,进而影响转座子的转座行为[77]。在植物和真菌中大部分MULEs元件具有较长的TIR (100~ 600 bp),但在拟南芥[65]、荷花[61]、玉米[78]和酵母(Yarrowia lipolytica)[48]基因组中鉴定到少数non-TIR MULEs元件(TIR<50 bp),这些元件虽分布较为广泛,但其与MULEs元件在进化中的关系仍不清楚。另外,在玉米、西红柿、水稻和拟南芥基因组中还检测到一些多TIR MULEs元件,这些TIR大多以串联形式分布,多TIR MULEs元件可能更有利于转座子转座和捕获宿主基因组序列[60]。MuDR作为Mu家族中的自主性转座子,同时也是Mutator超家族转座子研究的典型代表。MuDR编码两个转录方向相向的基因:mudrA和mudrB,各自转录起始于两端的TIR内部序列,两个转录本间没有重叠部分,在相距200 bp处终止转录[32] (图1A)。mudrA编码蛋白MURA (94 kDa),MURA与原核生物IS256转座子的转座酶序列相似[79],含有保守的蛋白结构域[80],被认为是转座酶,催化转座子转座。mudrB编码蛋白MURB (23 kDa),MURB并非体细胞组织转座剪切所必需,可能与生殖类细胞内转座子的重新插入相关[81,82],MURB调控方式及其在转座过程中的功能目前还没有更为详尽的报道。如前所述,在玉米及其他植物、动物、微生物中也已经鉴定到了少数几个自主性MULEs元件,但这些新发现的自主性MULEs元件均只含有mudrA同源基因,因此mudrB基因可能仅存在于玉米的MuDR中。Muk是MuDR转座插入并重排形成的2.2 kb反向重复序列,由两段反向加倍的TIRA及其下游mudrA相邻部分序列组成,不涉及任何mudrB基因序列(图1B)。Muk插入位点两翼残存的两个转录方向相向的基因(acm1和amy4)中仅有acm1启动子起始Muk转录,由此产生的发卡状转录本生成小RNA (主要是22 nt siRNAs),然后通过RNA介导的DNA甲基化方式沉默MuDR活性[41,42]。
图1
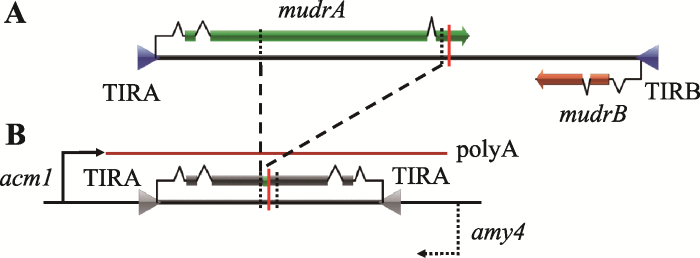 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1MuDR和Muk基因结构组成
A:转座子MuDR及其基因mudrA和mudrB的结构;B:Muk及其两翼残存基因的结构。
Fig. 1The gene structures of MuDR and Muk
转座酶是自然界中最丰富、最普遍存在的基因编码产物[83]。所有真核生物“cut-and-paste”类型超家族转座子其转座酶均具有DDE/D三氨基酸特征结构域[84],MuDR及其他自主性MULEs元件转座酶同样具有这样的特征(图2)。Liu等[58]根据保守的DDE/D结构域并通过生物信息学方法在埃及伊蚊基因组中发现了自主性MULEs元件—Muta1,通过定点突变首次证实了MULEs元件转座酶中DDE/D结构域3个特征氨基酸的重要性:其中任何单一氨基酸的改变都足以使转座酶的活性完全丧失。此外,与另一类DNA转座子超家族—hAT超家族的转座酶类似,大部分Mutator超家族成员的转座酶在DDE结构域的第2个D和E之间还含有一个保守的CXXH基序和一个色氨酸[85] (图2)。CXXH基序可能参与转座酶对TIR的识别,当CXXH基序中的组氨酸突变后,转座酶催化活性消失殆尽[58,86]。而色氨酸不仅与转座酶活性相关,还与转座酶的精确切割或修复相关。当转座酶中色氨酸突变为丙氨酸时,转座酶催化活性彻底消失;当其突变为其他芳香族氨基酸时,转座酶表现出一定活性,但转座子剪切频率变低,精确剪切比例也显著下降[58,85]。
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2不同自主性转座酶DDE结构域蛋白序列比对分析
黑色阴影表示氨基酸完全一致,粉色阴影表示同源性≥75%,绿色阴影表示同源性≥50%,保守的氨基酸及结构标注在序列底部。
Fig. 2The sequence alignment of DDE domain from different transposases
除了DDE/D三氨基酸这一特征结构域以外,在Mutator超家族转座子的转座酶中还可以鉴定到其他保守结构域(图3A)。例如,大部分转座酶的N端具有属于WRKY-GCM1超家族[87]的DNA结合结构域(DNA binding domain, DBD),可能通过结合转座子特定区段序列来调控转座酶活性及转座子的转座行为。水稻Os3378编码的转座酶DBD上游特定长度编码序列发生缺失突变后,转座子的剪切频率显著增高,进一步研究发现这部分片段中氨基酸组合的理化特性对Os3378转座酶活性至关重要[71]。 另外,大量MURA同源蛋白C端也具有相对保守的基序,如在拟南芥、玉米、水稻和甘蔗(Saccharum spp.)中先后鉴定到CX2CX4HX4 (或6) C基序[47,65],目前已知的自主性Mutator超家族转座子的C端 大多存在这些锌指基序(图3B),它们可能通过结 合核酸序列(DNA或RNA)参与调控转座子的转座行为。
图3
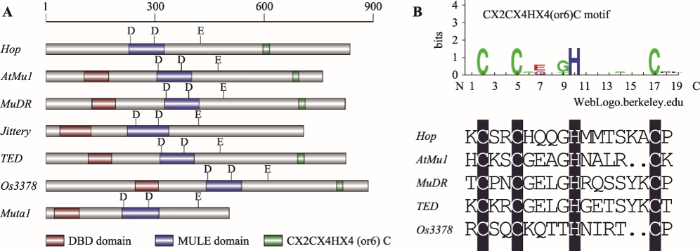 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3Mutator超家族转座酶的结构特征
A:MuDR及其他自主性MULEs元件转座酶的保守结构域;B:Mutator超家族转座酶C端的保守基序。
Fig. 3The characteristic of transposases of Mutator superfamily
3 Mutator超家族转座子转座机制
利用Mu家族转座子特有的遗传组成和转座特性,已经构建了多个玉米突变体资源库(如TUSC、MTM、RescueMu、UniformMu和ChinaMu等),为正向遗传学和反向遗传学的研究提供了丰富的突变体遗传材料[88,89]。然而,关于Mutator超家族转座子转座机制的认识仍缺乏直接的证据。基于与其他超家族DNA转座子的一些共性及大量转座事件分析,推测Mutator超家族转座子的剪切及再次插入可能与某些已知的转座机制存在相似之处。3.1 转座酶催化作用下的DNA双链断裂
真核生物DNA转座子的剪切过程一般以转座子两端某一条DNA单链的解离为起始,该过程为亲核裂解反应,通常H2O作为亲核试剂,在转座酶的作用下攻击转座子与侧翼序列连接处的磷酸二酯键而形成断裂口。某些超家族转座子会在转座子末端暴露出自由的3′-OH,而其他超家族转座子则在侧翼宿主序列末端暴露出自由的3′-OH,随后,不同类型超家族转座酶催化不同位置的3′-OH与不同类型DNA底物组合而使第二链断开,由此形成的DNA双链断裂(DNA double strand break, DSB)使转座子最终得以从供体位点释放出来[90]。最近研究发现,埃及伊蚊Muta1第二链断开方式与hAT、transib超家族转座子相类似[77]。在Muta1转座酶作用下,以H2O作为亲核试剂使转座子末端与侧翼DNA连接处的磷酸二酯键断开后在侧翼DNA的3′末端暴露出羟基,3′-OH进攻另一链而在侧翼DNA末端形成发卡结构,最后释放出转座子(图4)。剪切位点形成的DSB既可以通过非同源末端连接(non-homologous end joining, NHEJ)方式修复,留下不同类型转座印迹(footprint),还可能通过同源重组(homologous recombination, HR)方式,以一条姐妹染色单体或同系物作为模板进行修复。但是,侧翼DNA形成的发卡结构必须在修复前打开,相关的体外实验表明,这一过程并不是由转座酶催化完成,而可能是由宿主自身可以切割类似发卡结构的酶来完成[86]。目前对于Mutator超家族转座酶催化作用下的DNA双链断裂过程报道较少,该过程是否是Mutator超家族转座子的共同遗传特性亟待其他自主性Mutator超家族转座子相关研究加以验证。图4
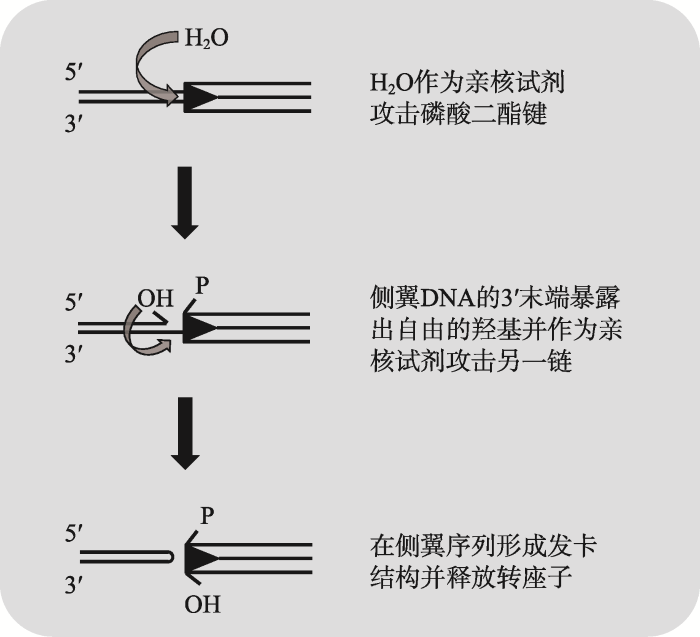 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4Muta1转座酶介导的DNA双链断裂过程
Fig. 4The progress of Muta1 transposase-mediated DSB
3.2 转座子剪切后的DSB修复
转座子剪切后导致DNA发生双链断裂,目前认为植物不同组织细胞中DSB修复方式不尽相同[91,92] (图5)。若剪切发生在生殖类细胞(包括配子体及配子体减数分裂前的有丝分裂细胞)的S期或G2期,此时转座子已经随着染色体发生了复制,细胞能够以姐妹染色单体为模板进行精确修复。由于剪切位点被完全修复,转座反应看似以“copy-and-paste”方式进行,但事实上是转座子剪切后又被重新修复的结果,而并非转座子的简单加倍。在DSB的修复过程中,由于模板内存在一些长短不一、散落分布的微同源序列,修复复制链发生位置滑移,便形成了大小有别、序列组成不同的多种缺陷型转座子。而在体细胞发育过程晚期,转座子剪切可能发生在细胞S期之前或者剪切形成的DSB主要通过易错易突变的NHEJ方式进行修复,结果导致转座子原插入位点产生多种类型的footprint序列[93,94]。图5
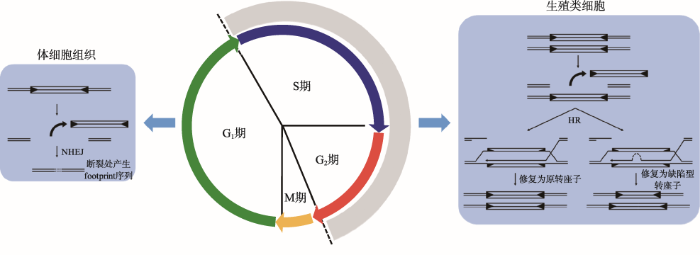 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5转座子剪切后的DNA双链断裂修复
Fig. 5DSB repairing after transposon excision
目前已有多方面证据支持上述假说。在MuDR和TED生殖类细胞转座研究中均可以检测到缺陷型转座子,并且这些缺陷型转座子在缺失序列的两翼存在微同源序列[56,91]。此外,研究还发现在一些缺陷型转座子序列内部含有填充序列(filler DNA),并且这些filler DNA均来自缺失位点附近序列[56,95]。这正是由于修复模板内存在多组微同源序列,修复复制时发生了多次复制链滑移造成的。由此可见,在生殖类细胞中转座子剪切后是通过依赖模板的方式进行修复的。另外,研究发现在玉米RAD51突变体中生殖类细胞内Mu转座子转座行为异常。RAD51在细胞减数分裂的DSB修复过程发挥重要作用,玉米中存在两个rad51同源基因,在含有活性MuDR的RAD51双突变材料中,生殖细胞转座反应中MuDR内部及侧翼序列缺失的频率比野生型材料高出数10倍,这也表明玉米生殖类细胞内Mu剪切 后需要RAD51介导的HR进行修复[96,97]。与生殖类细胞剪切修复相比,在体细胞组织转座过程中,转座子剪切后往往会形成多种类型footprint序列,大量Mu及TED体细胞组织转座事件研究已经证实了这一点[56,93,94]。但体细胞组织内转座子剪切部位绝大多数不存在微同源序列,因此,体细胞组织内转座子剪切后更可能是通过非模板修复方式进行修复的。
中间圆形表示细胞周期;左图表示转座子剪切发生在体细胞的S期前,转座子剪切后形成的DSB通过NHEJ方式修复,产生不同类型footprint序列;右图表示转座子剪切发生在生殖类细胞的S期或G2期,转座子剪切后形成的DSB通过HR方式以姐妹染色单体为模板进行修复,在断裂处修复为原转座子或由于微同源序列导致修复链滑移,修复为缺陷型转座子。
3.3 染色体外环形结构
原核IS256转座元件通过闭合环形结构介导转座过程[98],而早期研究发现,在携带活性MuDR的玉米材料中,Mu转座子也能够以染色体外共价闭合的环形结构形式存在,并且这些环形Mu的出现依赖于活性MuDR的存在,因此,其可能是MURA作用下的转座中间体或是Mu剪切后的产物[99]。但由于环形Mu序列信息的缺乏,难以明确其在转座反应中的作用及生物学意义。Li等[56]对玉米TED的研究发现,在含有活性TED的玉米体细胞中可以检测到共价闭合的环形TED或环形缺陷型TED结构;序列分析证实,这些环形结构的确是转座子两端共价连接的产物;同时酶切实验表明,所有检测的环形结构并不是两侧TIR末端完美的“头顶头(Head- to-Head)”共价连接,而可能是其他更为复杂的序列组成;除了预期大小的扩增产物,该研究还检测到一些其他扩增产物,克隆测序发现多数产物序列在连接点处缺失转座子单侧或两侧末端序列,缺失长度不等(<100 bp至>2 kb),有些涉及编码转座酶的区段。这些染色体外环形结构可能与转座子某些特性相关,例如转座子剪切后在非连锁位点的再次插入。目前,类似的转座子共价闭合环形结构在其他转座子研究中也有所报道[54,100,101],但不同转座子的染色体外环形结构是否参与转座反应,它们如何发挥作用及其生物学意义仍不明确。4 插入偏好
Mutator超家族转座子在基因组内的转座并非随机插入而具有一定的偏好性。目前通过多个Mu突变系已经获得了数万份玉米Mu插入突变体,这些Mu插入遍布整个玉米基因组[89,102,103],与Ac/Ds插入突变体不同,Mu新插入位点与原初插入位点并不连锁。值得注意的是,虽然玉米基因组大部分为反转录转座子序列,但绝大部分Mu插入在基因组低拷贝区域的基因内部或基因附近[103,104,105]。进一步研究发现,Mu更倾向于插入基因的5′末端,且插入区段序列GC含量较高,这与玉米及其他单子叶植物基因5′末端GC含量略高相对应[106]。另外,Mu插入位点与开放染色质表观遗传标记(如DNA甲基化和组蛋白修饰)紧密相关[107]。近期的研究也表明,真核生物转座子插入位点的选择受染色质结构影响[108]。在玉米W22基因组中,Ds插入位点与两翼序列染色质开放性无明显差异,而Mu插入位点染色质开放性显著增加。此外,Mu和Ds都倾向于插入CG和CHG甲基化程度极低的区域,但Ds插入位点通常与CG和CHG高度甲基化区域相距较远,而Mu插入位点与这些高度甲基化区段距离较近[109],这些特征有助于理解Mu更多插入在基因UTR区而Ds更倾向于插入基因编码区。5 靶位点重复序列
不同超家族转座子插入基因组后会在其两侧形成一定长度的正向重复序列,这些序列来自插入位点,被称为靶位点重复序列(target site duplication, TSD)。通常同一家族的转座子重新插入后形成相同长度甚至固定组成的TSD,因此,TSD序列长度、固定的序列组成也是进行转座子分类的依据之一[20,21]。Mutator超家族转座子转座主要形成长度为9 bp的TSD,并且这些TSD无明显的序列组成规律。近期研究表明,TSD与DNA转座子的转座行为之间关系密切。例如,在异源酵母系统中研究水稻Os3378的转座遗传特征时发现,当改变一侧TSD中紧邻Os3378的前3个碱基后,转座子的剪切频率显著下降,对于较长的非自主性转座子这种影响更为明显。并且不一致的TSD还会影响非自主性转座子Os3378NA剪切位点的精确修复,而对转座子重新插入频率并无显著影响[71]。另外,在异源酵母系统中,TSD同样影响埃及伊蚊Muta1的转座行为。当非自主性转座子Muta1AR携带有8 bp或9 bp TSD时,剪切频率较无TSD情况下显著提高,但与Os3378不同的是,当携带TSD时,相应转座子重新插入的频率也有所提高。此外,TSD的缺失同样影响相应Muta1AR转座子剪切位点的精确修复。当携带有8 bp或 9 bp TSD时,90%的回复突变均为精确剪切;当无TSD时,精确剪切频率仅占所有回复突变的10%。而对于TSD序列组成的研究表明,不同的TSD序列组成对于转座子的剪切和重新插入均无显著影响[58]。由此可见,TSD序列的有无、一致性及其长度对MULEs元件转座行为的影响更大,而TSD的序列组成对于转座行为的影响较小。在植物中Mutator超家族转座子的转座反应受到严格调控,而异源酵母系统中开展的研究可能不足以完全涵盖和揭示TSD在植物转座过程中的作用和调控机制。6 玉米自主性MULEs元件
除了MuDR以外,目前玉米中还克隆了另外两个自主性MULEs元件:Jittery [55]和TED[56]。这两个转座子与MuDR间存在一些共性,例如都具有mudrA同源序列,含有较长的TIR,插入位点形成9 bp TSD。而系统进化分析表明,这两个转座子各自作为独立的自主性转座子已经存在了数百万年[73]。与MuDR相比,Jittery和TED共享某些特性:(1)玉米中Jittery和TED的拷贝数很低;(2)它们在生殖类细胞内发生回复突变的频率均高于MuDR;(3)两者都不含有mudrB同源序列[55,56]。Jittery更为特殊,在生殖类细胞和体细胞组织内剪切后的修复都不留有任何footprint序列。此外,Jittery的自主性略显欠缺,目前只检测到玉米bronze位点的Jittery可以发生剪切,并未检测到其重新整合到基因组中,虽然不排除可能与缺少mudrB同源基因相关[55],但更可能是Jittery两端TIR序列微小差异影响了转座剪切后的再次插入。Jittery 3′端TIR比5′端TIR在末端少了4个核苷酸(GCTC),生物信息学分析发现,在其他已测序的玉米材料中,Jittery-like序列两侧TIR中均含有这4个核苷酸。因此,Jittery很可能原本两侧TIR序列一致,在转座到bronze位点过程中3′端TIR发生了序列丢失进而影响到Jittery的转座反应,导致其剪切后不能重新插入到基因组中。另外,其他已鉴定的自主性MULEs元件同样不含有mudrB同源基因,但转座后均可以重新插入到基因组其他位点。因此,即使mudrB基因确实与生殖类细胞内MuDR转座剪切后的重新插入相关,目前鉴定到的这些MULEs元件可能在转座剪切后的重新插入方面进化出了不同的机制,不再需要MURB功能蛋白。玉米中除了以上3个Mutator超家族转座子外,遗传学实验还鉴定到另外几个自主性MULEs元件,但这些转座子完整的基因组序列目前仍未被克隆,如玉米中Mrh家族转座子[110]。已有研究表明,缺陷型Mrh (rMrh)的TIR与Jittery TIR的序列在前50 bp高度同源,并且Jittery可以使rMrh发生移动。但关于自主性Mrh转座特性及其与Jittery间相互关系目前仍不清楚,尚需进行深入研究。7 结语与展望
转座子在真核生物基因和基因组的结构及进化过程中扮演着重要角色,众多农作物在其驯化过程中优异农艺形状和优良品质的形成以及对生物胁迫和非生物胁迫的不断适应的遗传基础都与转座子引发的变异密不可分。Mutator超家族转座子作为Ⅱ类转座子研究的重要方面,是转座子遗传学及功能基因组学的主要研究对象,而转座子研究中仍有许多科学问题亟待解决,转座子的开发应用更有待加强。因此,继续深入转座子基础遗传学研究并不断开发利用转座子资源必将发挥重要的学术及应用价值。随着高通量测序、生物信息学分析及机器深度学习等新技术的发展,转座子深入研究的成果势必更好地服务和推动生命科学的发展。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1073/pnas.1219372109URLPMID:23236127 [本文引用: 1]
DOI:10.1186/s13059-018-1577-zURLPMID:30454069 [本文引用: 1]

Transposable elements (TEs) are major components of eukaryotic genomes. However, the extent of their impact on genome evolution, function, and disease remain a matter of intense interrogation. The rise of genomics and large-scale functional assays has shed new light on the multi-faceted activities of TEs and implies that they should no longer be marginalized. Here, we introduce the fundamental properties of TEs and their complex interactions with their cellular environment, which are crucial to understanding their impact and manifold consequences for organismal biology. While we draw examples primarily from mammalian systems, the core concepts outlined here are relevant to a broad range of organisms.
DOI:10.1146/annurev-genet-110711-155616URL [本文引用: 1]
Transposons are DNA sequences capable of moving in genomes. Early evidence showed their accumulation in many species and suggested their continued activity in at least isolated organisms. In the past decade, with the development of various genomic technologies, it has become abundantly clear that ongoing activity is the rule rather than the exception. Active transposons of various classes are observed throughout plants and animals, including humans. They continue to create new insertions, have an enormous variety of structural and functional impact on genes and genomes, and play important roles in genome evolution. Transposon activities have been identified and measured by employing various strategies. Here, we summarize evidence of current transposon activity in various plant and animal genomes.
DOI:10.16288/j.yczz.15-140URLPMID:26399528 [本文引用: 1]
High throughput sequencing technology has dramatically improved the efficiency of DNA sequencing, and decreased the costs to a great extent. Meanwhile, this technology usually has advantages of better specificity, higher sensitivity and accuracy. Therefore, it has been applied to the research on genetic variations, transcriptomics and epigenomics. Recently, this technology has been widely employed in the studies of transposable elements and has achieved fruitful results. In this review, we summarize the application of high throughput sequencing technology in the fields of transposable elements, including the estimation of transposon content, preference of target sites and distribution, insertion polymorphism and population frequency, identification of rare copies, transposon horizontal transfers as well as transposon tagging. We also briefly introduce the major common sequencing strategies and algorithms, their advantages and disadvantages, and the corresponding solutions. Finally, we envision the developing trends of high throughput sequencing technology, especially the third generation sequencing technology, and its application in transposon studies in the future, hopefully providing a comprehensive understanding and reference for related scientific researchers.
DOI:10.16288/j.yczz.15-140URLPMID:26399528 [本文引用: 1]
High throughput sequencing technology has dramatically improved the efficiency of DNA sequencing, and decreased the costs to a great extent. Meanwhile, this technology usually has advantages of better specificity, higher sensitivity and accuracy. Therefore, it has been applied to the research on genetic variations, transcriptomics and epigenomics. Recently, this technology has been widely employed in the studies of transposable elements and has achieved fruitful results. In this review, we summarize the application of high throughput sequencing technology in the fields of transposable elements, including the estimation of transposon content, preference of target sites and distribution, insertion polymorphism and population frequency, identification of rare copies, transposon horizontal transfers as well as transposon tagging. We also briefly introduce the major common sequencing strategies and algorithms, their advantages and disadvantages, and the corresponding solutions. Finally, we envision the developing trends of high throughput sequencing technology, especially the third generation sequencing technology, and its application in transposon studies in the future, hopefully providing a comprehensive understanding and reference for related scientific researchers.
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/s41477-018-0349-9URLPMID:30692677 [本文引用: 1]
Snapdragon (Antirrhinum majus L.), a member of the Plantaginaceae family, is an important model for plant genetics and molecular studies on plant growth and development, transposon biology and self-incompatibility. Here we report a near-complete genome assembly of A. majus cultivar JI7 (A. majus cv.JI7) comprising 510?Megabases (Mb) of genomic sequence and containing 37,714 annotated protein-coding genes. Scaffolds covering 97.12% of the assembled genome were anchored on eight chromosomes. Comparative and evolutionary analyses revealed that a whole-genome duplication event occurred in the Plantaginaceae around 46-49 million years ago (Ma). We also uncovered the genetic architectures associated with complex traits such as flower asymmetry and self-incompatibility, identifying a unique duplication of TCP family genes dated to around 46-49 Ma and reconstructing a near-complete ψS-locus of roughly 2?Mb. The genome sequence obtained in this study not only provides a representative genome sequenced from the Plantaginaceae but also brings the popular plant model system of Antirrhinum into the genomic age.
DOI:10.1038/nature22043URLPMID:28447635 [本文引用: 1]
Cereal grasses of the Triticeae tribe have been the major food source in temperate regions since the dawn of agriculture. Their large genomes are characterized by a high content of repetitive elements and large pericentromeric regions that are virtually devoid of meiotic recombination. Here we present a high-quality reference genome assembly for barley (Hordeum vulgare L.). We use chromosome conformation capture mapping to derive the linear order of sequences across the pericentromeric space and to investigate the spatial organization of chromatin in the nucleus at megabase resolution. The composition of genes and repetitive elements differs between distal and proximal regions. Gene family analyses reveal lineage-specific duplications of genes involved in the transport of nutrients to developing seeds and the mobilization of carbohydrates in grains. We demonstrate the importance of the barley reference sequence for breeding by inspecting the genomic partitioning of sequence variation in modern elite germplasm, highlighting regions vulnerable to genetic erosion.
DOI:10.1101/gr.217117.116URLPMID:28420692 [本文引用: 1]
Advances in genome sequencing and assembly technologies are generating many high-quality genome sequences, but assemblies of large, repeat-rich polyploid genomes, such as that of bread wheat, remain fragmented and incomplete. We have generated a new wheat whole-genome shotgun sequence assembly using a combination of optimized data types and an assembly algorithm designed to deal with large and complex genomes. The new assembly represents &gt;78% of the genome with a scaffold N50 of 88.8 kb that has a high fidelity to the input data. Our new annotation combines strand-specific Illumina RNA-seq and Pacific Biosciences (PacBio) full-length cDNAs to identify 104,091 high-confidence protein-coding genes and 10,156 noncoding RNA genes. We confirmed three known and identified one novel genome rearrangements. Our approach enables the rapid and scalable assembly of wheat genomes, the identification of structural variants, and the definition of complete gene models, all powerful resources for trait analysis and breeding of this key global crop.
DOI:10.1038/nbt.3208URLPMID:25893780 [本文引用: 1]
Gossypium hirsutum has proven difficult to sequence owing to its complex allotetraploid (AtDt) genome. Here we produce a draft genome using 181-fold paired-end sequences assisted by fivefold BAC-to-BAC sequences and a high-resolution genetic map. In our assembly 88.5% of the 2,173-Mb scaffolds, which cover 89.6%~96.7% of the AtDt genome, are anchored and oriented to 26 pseudochromosomes. Comparison of this G. hirsutum AtDt genome with the already sequenced diploid Gossypium arboreum (AA) and Gossypium raimondii (DD) genomes revealed conserved gene order. Repeated sequences account for 67.2% of the AtDt genome, and transposable elements (TEs) originating from Dt seem more active than from At. Reduction in the AtDt genome size occurred after allopolyploidization. The A or At genome may have undergone positive selection for fiber traits. Concerted evolution of different regulatory mechanisms for Cellulose synthase (CesA) and 1-Aminocyclopropane-1-carboxylic acid oxidase1 and 3 (ACO1,3) may be important for enhanced fiber production in G. hirsutum.
DOI:10.1038/nature08670URLPMID:20075913 [本文引用: 1]
Soybean (Glycine max) is one of the most important crop plants for seed protein and oil content, and for its capacity to fix atmospheric nitrogen through symbioses with soil-borne microorganisms. We sequenced the 1.1-gigabase genome by a whole-genome shotgun approach and integrated it with physical and high-density genetic maps to create a chromosome-scale draft sequence assembly. We predict 46,430 protein-coding genes, 70% more than Arabidopsis and similar to the poplar genome which, like soybean, is an ancient polyploid (palaeopolyploid). About 78% of the predicted genes occur in chromosome ends, which comprise less than one-half of the genome but account for nearly all of the genetic recombination. Genome duplications occurred at approximately 59 and 13 million years ago, resulting in a highly duplicated genome with nearly 75% of the genes present in multiple copies. The two duplication events were followed by gene diversification and loss, and numerous chromosome rearrangements. An accurate soybean genome sequence will facilitate the identification of the genetic basis of many soybean traits, and accelerate the creation of improved soybean varieties.
DOI:10.1126/science.1178534URLPMID:19965430 [本文引用: 2]
We report an improved draft nucleotide sequence of the 2.3-gigabase genome of maize, an important crop plant and model for biological research. Over 32,000 genes were predicted, of which 99.8% were placed on reference chromosomes. Nearly 85% of the genome is composed of hundreds of families of transposable elements, dispersed nonuniformly across the genome. These were responsible for the capture and amplification of numerous gene fragments and affect the composition, sizes, and positions of centromeres. We also report on the correlation of methylation-poor regions with Mu transposon insertions and recombination, and copy number variants with insertions and/or deletions, as well as how uneven gene losses between duplicated regions were involved in returning an ancient allotetraploid to a genetically diploid state. These analyses inform and set the stage for further investigations to improve our understanding of the domestication and agricultural improvements of maize.
DOI:10.1038/nature07723URLPMID:19189423 [本文引用: 1]
Sorghum, an African grass related to sugar cane and maize, is grown for food, feed, fibre and fuel. We present an initial analysis of the approximately 730-megabase Sorghum bicolor (L.) Moench genome, placing approximately 98% of genes in their chromosomal context using whole-genome shotgun sequence validated by genetic, physical and syntenic information. Genetic recombination is largely confined to about one-third of the sorghum genome with gene order and density similar to those of rice. Retrotransposon accumulation in recombinationally recalcitrant heterochromatin explains the approximately 75% larger genome size of sorghum compared with rice. Although gene and repetitive DNA distributions have been preserved since palaeopolyploidization approximately 70 million years ago, most duplicated gene sets lost one member before the sorghum-rice divergence. Concerted evolution makes one duplicated chromosomal segment appear to be only a few million years old. About 24% of genes are grass-specific and 7% are sorghum-specific. Recent gene and microRNA duplications may contribute to sorghum's drought tolerance.
DOI:10.1038/ng.942URLPMID:21946354 [本文引用: 1]
Genetic diversity created by transposable elements is an important source of functional variation upon which selection acts during evolution. Transposable elements are associated with adaptation to temperate climates in Drosophila, a SINE element is associated with the domestication of small dog breeds from the gray wolf and there is evidence that transposable elements were targets of selection during human evolution. Although the list of examples of transposable elements associated with host gene function continues to grow, proof that transposable elements are causative and not just correlated with functional variation is limited. Here we show that a transposable element (Hopscotch) inserted in a regulatory region of the maize domestication gene, teosinte branched1 (tb1), acts as an enhancer of gene expression and partially explains the increased apical dominance in maize compared to its progenitor, teosinte. Molecular dating indicates that the Hopscotch insertion predates maize domestication by at least 10,000 years, indicating that selection acted on standing variation rather than new mutation.
DOI:10.1073/pnas.1310949110URLPMID:24089449 [本文引用: 1]
The postdomestication adaptation of maize to longer days required reduced photoperiod sensitivity to optimize flowering time. We performed a genome-wide association study and confirmed that ZmCCT, encoding a CCT domain-containing protein, is associated with the photoperiod response. In early-flowering maize we detected a CACTA-like transposable element (TE) within the ZmCCT promoter that dramatically reduced flowering time. TE insertion likely occurred after domestication and was selected as maize adapted to temperate zones. This process resulted in a strong selective sweep within the TE-related block of linkage disequilibrium. Functional validations indicated that the TE represses ZmCCT expression to reduce photoperiod sensitivity, thus accelerating maize spread to long-day environments.
DOI:10.1534/g3.114.010686URLPMID:24607887 [本文引用: 1]
One of the major quantitative trait loci for flowering time in maize, the Vegetative to generative transition 1 (Vgt1) locus, corresponds to an upstream (70 kb) noncoding regulatory element of ZmRap2.7, a repressor of flowering. At Vgt1, a miniature transposon (MITE) insertion into a conserved noncoding sequence was previously found to be highly associated with early flowering in independent studies. Because cytosine methylation is known to be associated with transposons and to influence gene expression, we aimed to investigate how DNA methylation patterns in wild-type and mutant Vgt1 correlate with ZmRap2.7 expression. The methylation state at Vgt1 was assayed in leaf samples of maize inbred and F1 hybrid samples, and at the syntenic region in sorghum. The Vgt1-linked conserved noncoding sequence was very scarcely methylated both in maize and sorghum. However, in the early maize Vgt1 allele, the region immediately flanking the highly methylated MITE insertion was significantly more methylated and showed features of methylation spreading. Allele-specific expression assays revealed that the presence of the MITE and its heavy methylation appear to be linked to altered ZmRap2.7 transcription. Although not providing proof of causative connection, our results associate transposon-linked differential methylation with allelic state and gene expression at a major flowering time quantitative trait locus in maize.
DOI:10.1111/nph.14688URLPMID:28722229 [本文引用: 1]
A major resistance quantitative trait locus, qRfg1, significantly enhances maize resistance to Gibberella stalk rot, a devastating disease caused by Fusarium graminearum. However, the underlying molecular mechanism remains unknown. We adopted a map-based cloning approach to identify the resistance gene at qRfg1 and examined the dynamic epigenetic changes during qRfg1-mediated maize resistance to the disease. A CCT domain-containing gene, ZmCCT, is the causal gene at the qRfg1 locus and a polymorphic CACTA-like transposable element (TE1) c. 2.4?kb upstream of ZmCCT is the genetic determinant of allelic variation. The non-TE1 ZmCCT allele is in a poised state, with predictive bivalent chromatin enriched for both repressive (H3K27me3/H3K9me3) and active (H3K4me3) histone marks. Upon pathogen challenge, this non-TE1 ZmCCT allele was promptly induced by a rapid yet transient reduction in H3K27me3/H3K9me3 and a progressive decrease in H3K4me3, leading to disease resistance. However, TE1 insertion in ZmCCT caused selective depletion of H3K4me3 and enrichment of methylated GC to suppress the pathogen-induced ZmCCT expression, resulting in disease susceptibility. Moreover, ZmCCT-mediated resistance to Gibberella stalk rot is not affected by photoperiod sensitivity. This chromatin-based regulatory mechanism enables ZmCCT to be more precise and timely in defense against F.?graminearum infection.
URLPMID:31690654 [本文引用: 1]
Stalk lodging, which is generally determined by stalk strength, results in considerable yield loss and has become a primary threat to maize (Zea mays) yield under high-density planting. However, the molecular genetic basis of maize stalk strength remains unclear, and improvement methods remain inefficient. Here, we combined map-based cloning and association mapping and identified the gene stiff1 underlying a major quantitative trait locus for stalk strength in maize. A 27.2-kb transposable element insertion was present in the promoter of the stiff1 gene, which encodes an F-box domain protein. This transposable element insertion repressed the transcription of stiff1, leading to the increased cellulose and lignin contents in the cell wall and consequently greater stalk strength. Furthermore, a precisely edited allele of stiff1 generated through the CRISPR/Cas9 system resulted in plants with a stronger stalk than the unedited control. Nucleotide diversity analysis revealed that the promoter of stiff1 was under strong selection in the maize stiff-stalk group. Our cloning of stiff1 reveals a case in which a transposable element played an important role in maize improvement. The identification of stiff1 and our edited stiff1 allele pave the way for efficient improvement of maize stalk strength.
[本文引用: 1]
DOI:10.1016/0168-9525(89)90039-5URLPMID:2543105 [本文引用: 1]
The changes in DNA sequence that have taken place during the evolution of eukaryotic genomes cannot be accounted for simply by base substitutions; some more complex mutations must have taken place as well. Transposable elements can affect gene structure and expression in several ways that suggest that they may have contributed to these evolutionary events.
DOI:10.1038/nrg2165-c1URLPMID:18421312 [本文引用: 2]
DOI:10.1038/nrg2165URLPMID:17984973 [本文引用: 2]
Our knowledge of the structure and composition of genomes is rapidly progressing in pace with their sequencing. The emerging data show that a significant portion of eukaryotic genomes is composed of transposable elements (TEs). Given the abundance and diversity of TEs and the speed at which large quantities of sequence data are emerging, identification and annotation of TEs presents a significant challenge. Here we propose the first unified hierarchical classification system, designed on the basis of the transposition mechanism, sequence similarities and structural relationships, that can be easily applied by non-experts. The system and nomenclature is kept up to date at the WikiPoson web site.
DOI:10.1186/s13100-015-0041-9URLPMID:26045719 [本文引用: 1]
Repbase Update (RU) is a database of representative repeat sequences in eukaryotic genomes. Since its first development as a database of human repetitive sequences in 1992, RU has been serving as a well-curated reference database fundamental for almost all eukaryotic genome sequence analyses. Here, we introduce recent updates of RU, focusing on technical issues concerning the submission and updating of Repbase entries and will give short examples of using RU data. RU sincerely invites a broader submission of repeat sequences from the research community.
DOI:10.3724/SP.J.1005.2012.01009URL [本文引用: 1]
Repetitive sequences (repeats) represent a significant fraction of the eukaryotic genomes and can be divided into tandem repeats, segmental duplications, and interspersed repeats on the basis of their sequence characteristics and how they are formed. Most interspersed repeats are derived from transposable elements (TEs). Eukaryotic TEs have been subdivided into two major classes according to the intermediate they use to move. The transposition and amplification of TEs have a great impact on the evolution of genes and the stability of genomes. However, identification and classification of TEs are complex and difficult due to the fact that their structure and classification are complex and diverse compared with those of other types of repeats. Here, we briefly introduced the function and classification of TEs, and summarized three different steps for identification, classification and annotation of TEs in eukaryotic genomes: (1) assembly of a repeat library, (2) repeat correction and classification, and (3) genome annotation. The existing computational approaches for each step were summarized and the advantages and disadvantages of the approaches were also highlighted in this review. To accurately identify, classify, and annotate the TEs in eukaryotic genomes requires combined methods. This review provides useful information for biologists who are not familiar with these approaches to find their way through the forest of programs.
DOI:10.3724/SP.J.1005.2012.01009URL [本文引用: 1]
Repetitive sequences (repeats) represent a significant fraction of the eukaryotic genomes and can be divided into tandem repeats, segmental duplications, and interspersed repeats on the basis of their sequence characteristics and how they are formed. Most interspersed repeats are derived from transposable elements (TEs). Eukaryotic TEs have been subdivided into two major classes according to the intermediate they use to move. The transposition and amplification of TEs have a great impact on the evolution of genes and the stability of genomes. However, identification and classification of TEs are complex and difficult due to the fact that their structure and classification are complex and diverse compared with those of other types of repeats. Here, we briefly introduced the function and classification of TEs, and summarized three different steps for identification, classification and annotation of TEs in eukaryotic genomes: (1) assembly of a repeat library, (2) repeat correction and classification, and (3) genome annotation. The existing computational approaches for each step were summarized and the advantages and disadvantages of the approaches were also highlighted in this review. To accurately identify, classify, and annotate the TEs in eukaryotic genomes requires combined methods. This review provides useful information for biologists who are not familiar with these approaches to find their way through the forest of programs.
DOI:10.3724/sp.j.1005.2012.01491URLPMID:23208147 [本文引用: 1]
LTR retrotransposons are an important class of eukaryotic transposable elements, which are ubiquitous and highly heterogeneous in plant and play a major role in genome evolution of eukaryote. They are now extensively employed in gene function and genetic diversity analyses. Identification of LTR retrotransposons is the precondition for its application. Therefore, it has important theoretical significance and practical application value in studying identification and analysis methods LTR retrotransposon sequences. Bioinformatic software of the sequence analysis, according to the work principle, can be classified roughly into two types: sequence alignment and sequence identification of conserved domains. Alignment software, such as BLAST and DNAstar, produce the corresponding sequence information through comparison of sequence similarity; however, this kind of software cannot be applied for full length sequences. According to the principle, LTR retro-transposon sequence identification software can be roughly sorted into four types: de novo repeat discovery method, com-parative genomic method, homology-based method, and structure-based method. For example, LTR_Finder based on de novo repeat discovery method can accurately predict and annotate LTR retrotransposons for full length sequences; Repeat-Masker, which is based on homology-based method, can discover LTR retrotransposons by comparing the similarity with known sequences in the database. In this article, different methods of identification and analysis of retrotransposon se-quences were compared and analyzed, and a set of flow of LTR retrotransposons sequence analysis was summarized in order to provide the reference for LTR retrotransposons sequence analysis.
DOI:10.3724/sp.j.1005.2012.01491URLPMID:23208147 [本文引用: 1]
LTR retrotransposons are an important class of eukaryotic transposable elements, which are ubiquitous and highly heterogeneous in plant and play a major role in genome evolution of eukaryote. They are now extensively employed in gene function and genetic diversity analyses. Identification of LTR retrotransposons is the precondition for its application. Therefore, it has important theoretical significance and practical application value in studying identification and analysis methods LTR retrotransposon sequences. Bioinformatic software of the sequence analysis, according to the work principle, can be classified roughly into two types: sequence alignment and sequence identification of conserved domains. Alignment software, such as BLAST and DNAstar, produce the corresponding sequence information through comparison of sequence similarity; however, this kind of software cannot be applied for full length sequences. According to the principle, LTR retro-transposon sequence identification software can be roughly sorted into four types: de novo repeat discovery method, com-parative genomic method, homology-based method, and structure-based method. For example, LTR_Finder based on de novo repeat discovery method can accurately predict and annotate LTR retrotransposons for full length sequences; Repeat-Masker, which is based on homology-based method, can discover LTR retrotransposons by comparing the similarity with known sequences in the database. In this article, different methods of identification and analysis of retrotransposon se-quences were compared and analyzed, and a set of flow of LTR retrotransposons sequence analysis was summarized in order to provide the reference for LTR retrotransposons sequence analysis.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:17246337 [本文引用: 1]
Mutator activity of the Mu mutator system of maize can be lost by either outcrossing or inbreeding Mu stocks. The nature of these two kinds of Mu-loss phenomena was analyzed by testing the results of crossing Mu-loss stocks by active Mu lines. Outcross- Mu-loss stocks are capable of supporting Mu activity if crossed by an active mutator line. Inbred-Mu-loss stocks, however, inactivate the active Mu system contributed by a Mu line. Also, inbred- Mu-loss lines do not regain Mu activity after at least three generations of outcrossing to non-Mu stocks. These results suggest that, once the Mu system is inactivated by inbreeding, it remains inactivated for at least three generations of outcrossing. Further, once the system responsible for inactivation is established, it will, in turn, inactivate an active Mu system contributed by crossing with Mu plants. The outcross-Mu-loss does not seem to involve such an inactivation system. These results are interpreted in the light of recent evidence that Mu inactivation results from the modification of Mu 1 transposable elements involved in the Mu phenotype.
DOI:10.1038/300539a0URLPMID:7144906 [本文引用: 1]
URLPMID:6099399 [本文引用: 1]
An approximately 1.4 kb fragment of DNA called Mu1, mutationally inserted into the Adh1 locus of maize in a Robertson's Mutator plant, has been cloned. The instability of the mutation induced by this element, the nature of the Robertson's Mutator system, and terminal inverted repeats indicate that the 1.4 kb insert is a transposable element. All Robertson's Mutator corn lines have Mu1-like elements, at copy numbers of 10-70 per diploid genome. The basic size of these multiple interspersed copies is generally the same. The elements are found on different genomic restriction fragments in closely related individuals, indicating a high degree of mobility. Aside from the one corn line identified by Robertson in the mid-1970s as Mutator, all maize lines tested, plus several near and remote corn relatives, have no detectable DNA which cross-hybridizes strongly with Mu1.
URLPMID:2444493 [本文引用: 1]
We have cloned and sequenced a 1.7-kb Mu element from a Mutator line of maize and compared its structure to Mu1, a 1.4-kb element. With the exception of a 385-bp block of DNA present in the 1.7-kb element, these transposable elements are structurally similar, sharing terminally inverted and internal direct repeated sequences. Derivation of 1.4-kb elements from the 1.7-kb class via deletion of internal sequence is suggested by the finding that a portion of the extra DNA in Mu1.7 is part of a truncated direct repeat sequence in the 1.4-kb element. An abundant poly(A)+ RNA homologous to a portion of this extra DNA is present in several tissues of both Mutator and non-Mutator lines. Analysis of transcripts from an unstable mutant bronze 1 (bz) allele containing a Mu1.7 element inserted in an exon of the gene detects three species of poly(A)+ RNA that hybridize to a Bz1 (Bronze) gene probe: the largest contains the entire Mu1.7 element in the Bz1 gene transcript; another appears to be a spliced, chimeric transcript; the smallest is normal size Bz1 mRNA. The latter is most likely encoded by the normal-size alleles detected by Southern analysis of tissue expressing purple pigment, suggesting that normal gene function is restored by excision of the Mu1.7 element.
[本文引用: 2]
DOI:10.1073/pnas.88.22.10198URLPMID:1719548 [本文引用: 2]
Mutator is a powerful system for generating new mutants in maize. Mutator activity is attributable to a family of transposable, multicopy Mu elements, but none of the known elements is an autonomous (regulatory) element. This paper reports the discovery of Mu9, a 4942-base-pair Mu element that was cloned after it transposed into the Bronze-2 locus. Like other Mu elements, Mu9 has approximately 215-base-pair terminal inverted repeats and creates a 9-base-pair host sequence duplication upon insertion. A small gene family of elements that cross-hybridize to Mu9 has been found in all maize lines, and one of the other known Mu elements, Mu5, probably arose as a deletion of Mu9. Mu9 has several of the properties expected for the proposed regulator of Mutator activity. (i) The presence of Mu9 parallels the presence of Mutator activity in individuals from a line that genetically segregates for the Mu regulator. (ii) Lines that transmit Mutator to greater than 90% of their progeny have multiple copies of Mu9. (iii) Most maize lines that lack Mutator activity and that are not descended from Mutator lines lack the Mu9 element. (iv) Transcripts that hybridize to Mu9 are abundant in active Mutator lines, but they are absent from lines that have epigenetically lost Mutator activity. These correlations suggest that Mu9 is a candidate for the autonomous Mutator element.
URLPMID:1657702 [本文引用: 1]
The Mutator system of maize consists of more than eight different classes of transposable elements each of which can be found in multiple copies. All Mu elements share the approximately 220-bp terminal inverted repeats, whereas each distinct element class is defined by its unique internal sequences. The regulation of instability of this system has been difficult to elucidate due to its multigenic inheritance. Here we present genetic experiments which demonstrate that there is a single locus, MuR1, which can regulate the transposition of Mu1 elements. We describe the cloning of members of a novel class of Mu elements, MuR, and demonstrate that a member of the class is the regulator of Mutator activity, MuR1. This conclusion is based on several criteria: MuR1 activity and a MuR-homologous restriction fragment cosegregate; when MuR1 undergoes a duplicative transposition, an additional MuR restriction fragment is observed, and MuR1 activity and the cosegregating MuR fragment are simultaneously lost within clonal somatic sectors. In addition, the MuR element hybridizes to transcripts in plants with Mutator activity. Our genetic experiments demonstrate that the MuR1 transposon is necessary to specify Mutator activity in our lines.
URLPMID:1661256 [本文引用: 1]
The identification of the autonomous or transposase-encoding element of the Mutator (Mu) transposable element system of maize is necessary to the characterization of the system. We reported previously that a transcript homologous to the internal region of the MuA element is associated with activity of the Mutator system. We describe here the cloning of another Mu element, designated MuA2, that cosegregates with Mutator activity as assayed by somatic instability of the a1-Mum2 allele. The MuA2 element has features typical of the transposable elements of the Mutator family, including the 210-bp terminal inverted repeats. Several lines of evidence suggest that MuA2 is an autonomous or transposase-encoding element of the Mu family: (1) MuA2 cosegregates with a genetically defined element that regulates somatic mutability of the a1-Mum2 allele; (2) MuA2 is hypomethylated while most other MuA2-hybridizing sequences in the genome are extensively methylated; (3) the increase of the copy number of MuA2 is concomitant with the increase of regulator elements; (4) MuA2-like elements are found in Mutator lines but not in non-Mutator inbreds. We propose that autonomous or transposase-encoding elements of the Mu family may be structurally conserved and MuA2-like.
DOI:10.1111/j.1365-313X.2005.02509.xURLPMID:16167895 [本文引用: 1]
We implement a novel strategy for harnessing the power of high-copy transposons for functional analysis of the maize genome, and report behavioral features of the Mutator system in a uniform inbred background. The unique UniformMu population and database facilitate high-throughput molecular analysis of Mu-tagged mutants and gene knockouts. Key features of the population include: (i) high mutation frequencies (7% independent seed mutations) and moderation of copy number (approximately 57 total Mu elements; 1-2 MuDR copies per plant) were maintained by continuous back-crossing into a phenotypically uniform inbred background; (ii) a bz1-mum9 marker enabled selection of stable lines (loss of MuDR), inhibiting further transpositions in lines selected for molecular analysis; (iii) build-up of mutation load was prevented by screening Mu-active parents to exclude plants carrying pre-existing seed mutations. To create a database of genomic sequences flanking Mu insertions, selected mutant lines were analyzed by sequencing of MuTAIL PCR clone libraries. These sequences were annotated and clustered to facilitate bioinformatic subtraction of ancestral elements and identification of insertions unique to mutant lines. New insertions targeted low-copy, gene-rich sequences, and in silico mapping revealed a random distribution of insertions over the genome. Our results indicate that Mu populations differ markedly in the occurrence of Mu insertion hotspots and the frequency of suppressible mutations. We suggest that controlled MuDR copy number in UniformMu lines is a key determinant of these differences. The public database (http://uniformmu.org; http://endosperm.info) includes pedigree and phenotypic data for over 2000 independent seed mutants selected from a population of 31 548 F2 lines and integrated with analyses of 34 255 MuTAIL sequences.
DOI:10.1073/pnas.1831119100URLPMID:12954979 [本文引用: 1]
We describe an efficient system for site-selected transposon mutagenesis in maize. A total of 43,776 F1 plants were generated by using Robertson's Mutator (Mu) pollen parents and self-pollinated to establish a library of transposon-mutagenized seed. The frequency of new seed mutants was between 10-4 and 10-5 per F1 plant. As a service to the maize community, maize-targeted mutagenesis selects insertions in genes of interest from this library by using the PCR. Pedigree, knockout, sequence, phenotype, and other information is stored in a powerful interactive database (maize-targeted mutagenesis database) that enables analysis of the entire population and the handling of knockout requests. By inhibiting Mu activity in most F1 plants, we sought to reduce somatic insertions that may cause false positives selected from pooled tissue. By monitoring the remaining Mu activity in the F2, however, we demonstrate that seed phenotypes depend on it, and false positives occur in lines that appear to lack it. We conclude that more than half of all mutations arising in this population are suppressed on losing Mu activity. These results have implications for epigenetic models of inbreeding and for functional genomics.
DOI:10.1385/1-59259-413-1:37URLPMID:14501057 [本文引用: 1]
RescueMu is a modified Mu1 transposon transformed into maize to permit mutagenesis and subsequent recovery of mutant alleles by plasmid rescue. RescueMu elements insert late in the germline as well as in terminally dividing somatic (e.g., leaf) cells. Germinal insertions may result in a mutant phenotype, and RescueMu permits recovery of 5-25 kb of transposon-flanking genomic DNA without having to construct and screen genomic DNA libraries. Late somatic insertions of RescueMu do not result in a visible phenotype, but they are instead used to construct plasmid libraries of gene-enriched maize genomic DNA to facilitate the identification and sequencing of the euchromatic portion of the maize genome. This is because maize leaves contain abundant independent RescueMu somatic insertions, and 70-90% of these insertions occur preferentially into genes and not repetitive DNA. This chapter describes detailed protocols on how to obtain, generate, and use RescueMu for maize genomics, including resources developed by the Maize Gene Discovery Project (MGDP) consortium available online at ZmDB.
DOI:10.1007/s00299-010-0922-9URL [本文引用: 1]
In maize, Mutator transposable elements are either active or silenced within the genome. In response to environmental stress, silenced Mutator elements could be reactivated, leading to changes in genome structure and gene function. However, there is no direct experimental evidence linking environmental stress and Mutator transposon reactivation. Using a maize line that contains a single inactive MuDR and a lone nonautonomous Mutator element, a Mu1 insertion in the recessive reporter allele a1-mum2 in an inactive Mutator background, we directly assessed Mutator reactivation following low-energy nitrogen ion implantation. We observed that N+ implantation decreased cytosine methylation in MuDR terminal inverted repeats and increased expression of mudrA and mudrB. Both changes were associated with increased transpositional activity of MuDR through reactivation of the inactive minimal Mutator transposable element system. This study provides direct evidence linking environmental stress agents and Mutator transposon mobilization in maize. In addition, the observed changes to DNA methylation suggest a new mechanism for mutations by low-energy ion implantation.
URLPMID:7789777 [本文引用: 1]
Most Mutator lines of maize harbor several different classes of Mu transposons, each of which may be present in high copy number. The regulatory element is also often found in high copy number, and it is this element's behavior that is presumed to cause the non-Mendelian inheritance of Mutator activity. Using a very simple Mutator line, we demonstrate tha MuDR-1, a regulator of the Mutator system, can functionally replace standard non-Mendelian Mutator activity and that MuDR-1 is associated with the loss of methylation of the termini of another Mu transposon. Further, we show that Mu transposons can transpose duplicatively, that reinsertion tends to be into unlinked sites, and that MuDR-1 frequently suffers deletions. Changes in chromosomal position and the mode of sexual transmission are shown to be associated with changes in the frequency of MuDR-1 duplication and with the activity of MuDR-1 as monitored by the excision frequency of a reporter transposon of the Mu family, Mu1. Our data are derived from a Minimal Mutator Line in which there are relatively few Mu transposons, including one MuDR-1 regulator and as few as one Mu1 reporter. The seemingly enigmatic results that have been obtained using more complicated Mu genotypes are reinterpreted using simple Mendelian principles. We have borrowed a gap-repair model from Drosophila biologists to explain both duplications and deletions of MuDR-1.
[本文引用: 1]
URLPMID:14573488 [本文引用: 2]
Mutations in a number of genes responsible for the maintenance of transposon silencing have been reported. However, the initiation of epigenetic silencing of transposable elements is poorly characterized. Here, we report the identification of a single dominant locus, Mu killer (Muk), that acts to silence MuDR, the autonomous regulatory transposon of the Mutator family of transposable elements in maize. Muk results in the methylation of MuDR TIRs and is competent to silence one or several active MuDR elements. Silencing by Muk is not dependent on the position of the MuDR element and occurs gradually during plant development. Transcript levels of the MuDR transposase, mudrA, decrease substantially when Muk is present. The other transcript encoded by MuDR, mudrB, also fails to accumulate in the poly(A) RNA fraction when MuDR and Muk are combined. Additionally, plants undergoing MuDR silencing produce small, mudrA-homologous approximately 26-nt RNAs, suggesting a role for RNA-directed DNA methylation in MuDR silencing. MuDR elements silenced by Muk remain silenced even in plants that do not inherit Muk, suggesting that Muk is required for the initiation of MuDR silencing but not for its maintenance.
DOI:10.1038/ng1576URLPMID:15908951 [本文引用: 2]
It has been suggested that gene silencing evolved as a defense against genomic parasites such as transposons. This idea is based on analysis of mutations that reactivate transposons that are stably silenced: they affect maintenance rather than initiation of silencing. Here we describe the cloning and characterization of a naturally occurring locus able to heritably silence the otherwise highly active MuDR transposon in maize. This locus, Mu killer (Muk), results from the inverted duplication of a partially deleted autonomous MuDR element located at the breakpoint of a genomic deletion. Muk produces a hybrid hairpin transcript that is processed into small RNAs, which are amplified when the target MuDR transcript is present. Muk provides the first example of a naturally occurring transposon derivative capable of initiating the heritable silencing of an active transposon family. Further, transposon-generated inverted duplications may be important for the generation of double-stranded RNAs used in gene silencing.
DOI:10.1007/s10709-015-9842-5URLPMID:25981486 [本文引用: 1]
Transposable elements (TEs) are mobile DNA segments, abundant and dynamic in plant genomes. Because their mobility can be potentially deleterious to the host, a variety of mechanisms evolved limiting that negative impact, one of them being preference for a specific target insertion site. Here, we describe a family of Mutator-like DNA transposons in Medicago truncatula targeting TA microsatellites. We identified 218 copies of MuTAnTs and an element carrying a complete ORF encoding a mudrA-like transposase. Most insertion sites are flanked by a variable number of TA tandem repeats, indicating that MuTAnTs are specifically targeting TA microsatellites. Other TE families flanked by TA repeats (e.g. TAFT elements in maize) were described previously, however we identified the first putative autonomous element sharing that characteristics with a related group of short non-autonomous transposons.
DOI:10.1371/journal.pone.0090895URLPMID:24608103 [本文引用: 1]
Transposable elements (TEs) are the most abundant genomic components in eukaryotes and affect the genome by their replications and movements to generate genetic plasticity. Sweet potato performs asexual reproduction generally and the TEs may be an important genetic factor for genome reorganization. Complete identification of TEs is essential for the study of genome evolution. However, the TEs of sweet potato are still poorly understood because of its complex hexaploid genome and difficulty in genome sequencing. The recent availability of the sweet potato transcriptome databases provides an opportunity for discovering and characterizing the expressed TEs.
DOI:10.1007/s00438-008-0366-xURLPMID:18636276 [本文引用: 1]
Cassava (Manihot esculenta Crantz), though a major world crop with enormous potential, is very under studied. Little is known about its genome structure and organisation. Transposable elements have a key role in the evolution of genome structure, and can be used as important tools in applied genetics. This paper sets out to survey the diversity of members of three major classes of transposable element within the cassava genome and in relation to similar elements in other plants. Members of two classes of LTR-retrotransposons, Ty1/copia-like and Ty3/gypsy-like, and of Enhancer/Suppressor Mutator (En/Spm)-like transposons were isolated and characterised. Analyses revealed 59 families of Ty1/copia, 26 families of Ty3/gypsy retrotransposons, and 40 families of En/Spm in the cassava genome. In the comparative analyses, the predicted amino acid sequences for these transposon classes were compared with those of related elements from other plant species. These revealed that there were multiple lineages of Ty1/copia-like retrotransposons in the genome of cassava and suggested that vertical and horizontal transmission as the source of cassava Mecops may not be mutually exclusive. For the Ty3/gypsy elements network, two groups of cassava Megyps were evident including the Arabidopsis Athila lineage. However, cassava En/Spm-like elements (Meens) constituted a single group within a network of plant En/Spm-like elements. Hybridisation analysis supported the presence of transposons in the genome of cassava in medium (Ty3/gypsy and En/Spm) to high (Ty1/copia) copy numbers. Thus the cassava genome was shown to contain diverse members of three major classes of transposable element; however, the different classes exhibited contrasting evolutionary histories.
DOI:10.1007/s00438-006-0194-9URLPMID:17136348 [本文引用: 1]
The transposon Mutator was first identified in maize, and is one of the most active mobile elements in plants. The Arabidopsis thaliana genome contains at least 200 Mutator-like elements (MULEs), which contain the Mutator-like transposase gene, and often additional genes. We have detected a novel type of MULEs in melon (CUMULE), which, besides the transposase, contains two ubiquitin-like specific protease-like sequences (ULP1). This element is not present in the observed location in some melon cultivars. Multiple copies of this element exist in the Cucumis melo genome, and it has been detected in other Cucurbitaceae species. Analysis of the A. thaliana genome revealed more than 90 CUMULE-like elements, containing one or two Ulp1-like sequences, although no evidence of mobility exists for these elements. We detected various putative transposable elements containing ULP1-like sequences in rice. The discovery of these MULEs in melon and Arabidopsis, and the existence of similar elements in rice and maize, suggest that a proteolytic function may be important for this subset of the MULE transposable elements.
DOI:10.1007/s00438-004-1036-2URLPMID:15338280 [本文引用: 2]
The maize Mutator ( Mu) system has been described as the most active and mutagenic plant transposon so far discovered. Mu -like elements (MULEs) are widespread among plants, and many and diverse variants can coexist in a particular genome. The autonomous regulatory element MuDR contains two genes: mudrA encodes the transposase, while the function of the mudrB gene product remains unknown. Although mudrA -like sequences are ubiquitous in plants, mudrB seems to be restricted to the genus Zea. In the SUCEST (the Brazilian Sugarcane EST Sequencing Project) database, several mudrA -like cDNAs have been identified, suggesting the presence of a transcriptionally active Mu system in sugarcane. Phylogenetic studies have revealed the presence in plants of four classes of mudrA -like sequences, which arose prior to the monocot/eudicot split. At least three of the four classes are also found in the progenitors of the sugarcane hybrid (Saccharum spp.), Saccharum officinarum and S. spontaneum. The frequency of putatively functional transposase ORFs varies among the classes, as revealed at both cDNA and genomic levels. The predicted products of some sugarcane mudrA -like transcripts contain both a DNA-binding domain and a transposase catalytic-site motif, supporting the idea that an active Mu system exists in this hybrid genome.
DOI:10.1128/EC.4.3.615-624.2005URLPMID:15755923 [本文引用: 2]
A new type of DNA transposon, Mutyl, has been identified in the sequenced genome of the yeast Yarrowia lipolytica. This transposon is 7,413 bp long and carries two open reading frames (ORFs) which potentially encode proteins of 459 and 1,178 amino acids, respectively. Whereas the first ORF shows no significant homology to previously described proteins, the second ORF shows sequence similarities with various Mutator-like element (MULE)-encoded transposases, including the bacterial transposase signature sequence. Other MULE features shared by Mutyl include a zinc finger motif in the putative transposase, a 22-bp-long imperfect inverted repeat at each end, and a 9- to 10-bp duplication of its target site in the chromosome. Of the five copies of Mutyl present in the genome, one has a deletion of the first 8 bases, and the others are full length with a single base change in one element. The first potential gene of Mutyl, mutB, was shown to be expressed in exponentially growing cells. Its sequence contains a predicted intron with two 5' splice sites, a single branch point, and two 3' splice sites. Its mRNA is alternatively spliced, as judged by reverse transcription-PCR, and generates four mRNAs corresponding to protein-coding sequences of 128, 156, 161, and 190 amino acids. Of the three distinct lineages characterized in Y. lipolytica, strains from the German lineage and the French lineage do not carry Mutyl. A study of the distribution of Mutyl in strains of the French lineage evidenced a recent transposition event. Taken together, these results indicate that Mutyl is still active.
DOI:10.1093/molbev/msg155URLPMID:12777515 [本文引用: 2]
A new type of active DNA transposon has been identified in the genome of Fusarium oxysporum by its transposition into the niaD target gene. Two insertions within the final exon, in opposite orientations at the same nucleotide site, have been characterized. These elements, called Hop, are 3,299 bp long, with perfect terminal inverted repeats (TIRs) of 99 bp. The sequencing of genomic copies reveals a 9-bp target site duplication and no apparent sequence specificity at the insertion sites. The sequencing of a cDNA indicates that Hop does not contain an intron and encodes a putative transposase of 836 amino acids. The structural features (length, TIRs size, and 9-bp duplication), together with the presence of conserved domains in the transposase, strongly suggest that Hop is a Mutator-like element (MULE). Hop is thus the first active member of this family found beyond plants. The high rate of excision observed indicates that Hop is very active and thus represents a promising efficient tagging system for the isolation of fungal genes. The distribution of Hop elements within the Fusarium genus revealed that they are present in different species, suggesting that related elements could be present in other fungal genomes. In fact, Hop-related sequences have been identified in the survey of the entire genome sequence of three other ascomycetes, Magnaporthe grisea, Neurospora crassa, and Aspergillus fumigatus.
DOI:10.1186/1471-2164-10-330URLPMID:19622157 [本文引用: 1]
For three decades the Mutator system was thought to be exclusive of plants, until the first homolog representatives were characterized in fungi and in early-diverging amoebas earlier in this decade.
DOI:10.1093/molbev/msi169URLPMID:15901838 [本文引用: 1]
We report the first comprehensive analysis of transposable element content in the compact genomes (approximately 20 Mb) of four species of Entamoeba unicellular protozoans for which draft sequences are now available. Entamoeba histolytica and Entamoeba dispar, two human parasites, have many retrotransposons, but few DNA transposons. In contrast, the reptile parasite Entamoeba invadens and the free-living Entamoeba moshkovskii contain few long interspersed elements but harbor diverse and recently amplified populations of DNA transposons. Representatives of three DNA transposase superfamilies (hobo/Activator/Tam3, Mutator, and piggyBac) were identified for the first time in a protozoan species in addition to a variety of members of a fourth superfamily (Tc1/mariner), previously reported only from ciliates and Trichomonas vaginalis among protozoans. The diversity of DNA transposons and their differential amplification among closely related species with similar compact genomes are discussed in the context of the biology of Entamoeba protozoans.
DOI:10.1017/S0031182011000886URL [本文引用: 1]
Transposons of the Mutator superfamily have been widely described in plants, but only recently have metazoan organisms been shown to harbour them. In this work we describe novel Mutator superfamily transposons from the genomes of the human parasites Schistosoma mansoni and S. japonicum, which we name Curupira-1 and Curupira-2. Curupira elements do not have Terminal Inverted Repeats (TIRs) at their extremities and generate Target Site Duplications (TSDs) of 9 base pairs. Curupira-2 transposons code for a conserved transposase and SWIM zinc finger domains, while Curupira-1 elements comprise these same domains plus a WRKY zinc finger. Alignment of transcript sequences from both elements back to the genomes indicates that they are subject to splicing to produce mature transcripts. Phylogenetic analyses indicate that these transposons represent a new lineage of metazoan Mutator-like elements with characteristics that are distinct from the recently described Phantom elements. Description of these novel schistosome transposons provides new insights in the evolution of transposable elements in schistosomes.
DOI:10.1534/genetics.110.116673URLPMID:20457878 [本文引用: 1]
Transposons of the Mutator (Mu) superfamily have been shown to play a critical role in the evolution of plant genomes. However, the identification of Mutator transposons in other eukaryotes has been quite limited. Here we describe a previously uncharacterized group of DNA transposons designated Phantom identified in the genomes of a wide range of eukaryotic taxa, including many animals, and provide evidence for its inclusion within the Mutator superfamily. Interestingly three Phantom proteins were also identified in two insect viruses and phylogenetic analysis suggests horizontal movement from insect to virus, providing a new line of evidence for the role of viruses in the horizontal transfer of DNA transposons in animals. Many of the Phantom transposases are predicted to harbor a FLYWCH domain in the amino terminus, which displays a WRKY-GCM1 fold characteristic of the DNA binding domain (DBD) of Mutator transposases and of several transcription factors. While some Phantom elements have terminal inverted repeats similar in length and structure to Mutator elements, some display subterminal inverted repeats (sub-TIRs) and others have more complex termini reminiscent of so-called Foldback (FB) transposons. The structural plasticity of Phantom and the distant relationship of its encoded protein to known transposases may have impeded the discovery of this group of transposons and it suggests that structure in itself is not a reliable character for transposon classification.
DOI:10.1101/gad.193701URLPMID:11238379 [本文引用: 2]
Robertson's Mutator transposable elements in maize undergo cycles of activity and then inactivity that correlate with changes in cytosine methylation. Mutator-like elements are present in the Arabidopsis genome but are heavily methylated and inactive. These elements become demethylated and active in the chromatin-remodeling mutant ddm1 (Decrease in DNA Methylation), which leads to loss of heterochromatic DNA methylation. Thus, DNA transposons in plants appear to be regulated by chromatin remodeling. In inbred ddm1 strains, transposed elements may account, in part, for mutant phenotypes unlinked to ddm1. Gene silencing and paramutation are also regulated by DDM1, providing support for the proposition that epigenetic silencing is related to transposon regulation.
DOI:10.1105/tpc.019802URLPMID:15075398 [本文引用: 4]
The unstable mutation bz-m039 arose in a maize (Zea mays) stock that originated from a plant infected with barley stripe mosaic virus. The instability of the mutation is caused by a 3.9-kb mobile element that has been named Jittery (Jit). Jit has terminal inverted repeats (TIRs) of 181 bp, causes a 9-bp direct duplication of the target site, and appears to excise autonomously. It is predicted to encode a single 709-amino acid protein, JITA, which is distantly related to the MURA transposase protein of the Mutator system but is more closely related to the MURA protein of Mutator-like elements (MULEs) from Arabidopsis thaliana and rice (Oryza sativa). Like MULEs, Jit resembles Mutator in the length of the element's TIRs, the size of the target site duplication, and in the makeup of its transposase but differs from the autonomous element Mutator-Don Robertson in that it encodes a single protein. Jit also differs from Mutator elements in the high frequency with which it excises to produce germinal revertants and in its copy number in the maize genome: Jit-like TIRs are present at low copy number in all maize lines and teosinte accessions examined, and JITA sequences occur in only a few maize inbreds. However, Jit cannot be considered a bona fide transposon in its present host line because it does not leave footprints upon excision and does not reinsert in the genome. These unusual mobile element properties are discussed in light of the structure and gene organization of Jit and related elements.
DOI:10.1105/tpc.113.116517URL [本文引用: 7]
Mutator (Mu) elements, one of the most diverse superfamilies of DNA transposons, are found in all eukaryotic kingdoms, but are particularly numerous in plants. Most of the present knowledge on the transposition behavior of this superfamily comes from studies of the maize (Zea mays) Mu elements, whose transposition is mediated by the autonomous Mutator-Don Robertson (MuDR) element. Here, we describe the maize element TED (for Transposon Ellen Dempsey), an autonomous cousin that differs significantly from MuDR. Element excision and reinsertion appear to require both proteins encoded by MuDR, but only the single protein encoded by TED. Germinal excisions, rare with MuDR, are common with TED, but arise in one of the mitotic divisions of the gametophyte, rather than at meiosis. Instead, transposition-deficient elements arise at meiosis, suggesting that the double-strand breaks produced by element excision are repaired differently in mitosis and meiosis. Unlike MuDR, TED is a very low-copy transposon whose number and activity do not undergo dramatic changes upon inbreeding or outcrossing. Like MuDR, TED transposes mostly to unlinked sites and can form circular transposition products. Sequences closer to TED than to MuDR were detected only in the grasses, suggesting a rather recent evolutionary split from a common ancestor.
DOI:10.1007/s00438-012-0676-xURL [本文引用: 1]
Transposable elements (TEs) represent an important fraction of plant genomes and play a significant role in gene and genome evolution. Among all TE super-families discovered in plants, Mutator from maize (Zea mays) is the most active and mutagenic element. Mutator-like elements (MULEs) were identified in a wide range of plants. However, only few active MULEs have been reported, and the transposition mechanism of the elements is still poorly understood. In this study, an active MULE named Os3378 was discovered in rice (Oryza sativa) by a combination of computational and experimental approaches. The four newly identified Os3378 elements share more than 98% sequence identity between each other, and all of them encode transposases without any deletion derivatives, indicating their capability of autonomous transposition. Os3378 is present in the rice species with AA genome type but is absent in other non-AA genome species. A new insertion of Os3378 was identified in a rice somaclonal mutant Z418, and the element remained active in the descendants of the mutant for more than ten generations. Both germinal and somatic excision events of Os3378 were observed, and no footprint was detected after excision. Furthermore, the occurrence of somatic excision of Os3378 appeared to be associated with plant developmental stages and tissue types. Taken together, Os3378 is a unique active element in rice, which provides a valuable resource for further studying of transposition mechanism and evolution of MULEs.
DOI:10.1186/s13100-016-0084-6URLPMID:28096902 [本文引用: 5]
Mutator-like transposable elements (MULEs) are widespread with members in fungi, plants, and animals. Most of the research on the MULE superfamily has focused on plant MULEs where they were discovered and where some are extremely active and have significant impact on genome structure. The maize MuDR element has been widely used as a tool for both forward and reverse genetic studies because of its high transposition rate and preference for targeting genic regions. However, despite being widespread, only a few active MULEs have been identified, and only one, the rice Os3378, has demonstrated activity in a non-host organism.
DOI:10.1186/gb-2013-14-5-r41URLPMID:23663246 [本文引用: 1]
Sacred lotus is a basal eudicot with agricultural, medicinal, cultural and religious importance. It was domesticated in Asia about 7,000 years ago, and cultivated for its rhizomes and seeds as a food crop. It is particularly noted for its 1,300-year seed longevity and exceptional water repellency, known as the lotus effect. The latter property is due to the nanoscopic closely packed protuberances of its self-cleaning leaf surface, which have been adapted for the manufacture of a self-cleaning industrial paint, Lotusan.
DOI:10.1155/2012/695827URLPMID:22474413 [本文引用: 3]
Mutator-like transposable elements (MULEs) are widespread in plants and the majority have long terminal inverted repeats (TIRs), which distinguish them from other DNA transposons. It is known that the long TIRs of Mutator elements harbor transposase binding sites and promoters for transcription, indicating that the TIR sequence is critical for transposition and for expression of sequences between the TIRs. Here, we report the presence of MULEs with multiple TIRs mostly located in tandem. These elements are detected in the genomes of maize, tomato, rice, and Arabidopsis. Some of these elements are present in multiple copies, suggesting their mobility. For those elements that have amplified, sequence conservation was observed for both of the tandem TIRs. For one MULE family carrying a gene fragment, the elements with tandem TIRs are more prevalent than their counterparts with a single TIR. The successful amplification of this particular MULE demonstrates that MULEs with tandem TIRs are functional in both transposition and duplication of gene sequences.
DOI:10.1534/genetics.106.062752URLPMID:17028332 [本文引用: 2]
The largest component of plant and animal genomes characterized to date is transposable elements (TEs). The availability of a significant amount of Lotus japonicus genome sequence has permitted for the first time a comprehensive study of the TE landscape in a legume species. Here we report the results of a combined computer-assisted and experimental analysis of the TEs in the 32.4 Mb of finished TAC clones. While computer-assisted analysis facilitated a determination of TE abundance and diversity, the availability of complete TAC sequences permitted identification of full-length TEs, which facilitated the design of tools for genomewide experimental analysis. In addition to containing all TE types found in previously characterized plant genomes, the TE component of L. japonicus contained several surprises. First, it is the second species (after Oryza sativa) found to be rich in Pack-MULEs, with &gt;1000 elements that have captured and amplified gene fragments. In addition, we have identified what appears to be a legume-specific MULE family that was previously identified only in fungal species. Finally, the L. japonicus genome contains many hundreds, perhaps thousands of Sireviruses: Ty1/copia-like elements with an extra ORF. Significantly, several of the L. japonicus Sireviruses have recently amplified and may still be actively transposing.
DOI:10.1093/molbev/msk015URLPMID:16581939 [本文引用: 1]
Transposons comprise a major component of eukaryotic genomes, yet it remains controversial whether they are merely genetic parasites or instead significant contributors to organismal function and evolution. In plants, thousands of DNA transposons were recently shown to contain duplicated cellular gene fragments, a process termed transduplication. Although transduplication is a potentially rich source of novel coding sequences, virtually all appear to be pseudogenes in rice. Here we report the results of a genome-wide survey of transduplication in Mutator-like elements (MULEs) in Arabidopsis thaliana, which shows that the phenomenon is generally similar to rice transduplication, with one important exception: KAONASHI (KI). A family of more than 97 potentially functional genes and apparent pseudogenes, evidently derived at least 15 MYA from a cellular small ubiquitin-like modifier-specific protease gene, KI is predominantly located in potentially autonomous non-terminal inverted repeat MULEs and has evolved under purifying selection to maintain a conserved peptidase domain. Similar to the associated transposase gene but unlike cellular genes, KI is targeted by small RNAs and silenced in most tissues but has elevated expression in pollen. In an Arabidopsis double mutant deficient in histone and DNA methylation with elevated KI expression compared to wild type, at least one KI-MULE is mobile. The existence of KI demonstrates that transduplicated genes can retain protein-coding capacity and evolve novel functions. However, in this case, our evidence suggests that the function of KI may be selfish rather than cellular.
DOI:10.1038/nature02953URLPMID:15457261 [本文引用: 3]
Mutator-like transposable elements (MULEs) are found in many eukaryotic genomes and are especially prevalent in higher plants. In maize, rice and Arabidopsis a few MULEs were shown to carry fragments of cellular genes. These chimaeric elements are called Pack-MULEs in this study. The abundance of MULEs in rice and the availability of most of the genome sequence permitted a systematic analysis of the prevalence and nature of Pack-MULEs in an entire genome. Here we report that there are over 3,000 Pack-MULEs in rice containing fragments derived from more than 1,000 cellular genes. Pack-MULEs frequently contain fragments from multiple chromosomal loci that are fused to form new open reading frames, some of which are expressed as chimaeric transcripts. About 5% of the Pack-MULEs are represented in collections of complementary DNA. Functional analysis of amino acid sequences and proteomic data indicate that some captured gene fragments might be functional. Comparison of the cellular genes and Pack-MULE counterparts indicates that fragments of genomic DNA have been captured, rearranged and amplified over millions of years. Given the abundance of Pack-MULEs in rice and the widespread occurrence of MULEs in all characterized plant genomes, gene fragment acquisition by Pack-MULEs might represent an important new mechanism for the evolution of genes in higher plants.
DOI:10.1016/s1360-1385(02)02347-6URLPMID:12417150 [本文引用: 1]
Mutator (Mu) element insertion has become the main way of mutating and cloning maize genes, but we are only beginning to understand how this transposon system is regulated. Mu elements are under tight developmental control and are subject to a form of epigenetic regulation that shares some features with the regulation of paramutable maize genes. Mu-like elements (MULEs) are widespread among angiosperms, and multiple diverged functional variants appear to have coexisted in genomes for long periods. In addition to its utility, the means by which this widespread and highly mutagenic system is held in check should help us to address fundamental issues concerning the stability of genomes.
URLPMID:11102392 [本文引用: 3]
While genome-wide surveys of abundance and diversity of mobile elements have been conducted for some class I transposable element families, little is known about the nature of class II transposable elements on this scale. In this report, we present the results from analysis of the sequence and structural diversity of Mutator-like elements (MULEs) in the genome of Arabidopsis thaliana (Columbia). Sequence similarity searches and subsequent characterization suggest that MULEs exhibit extreme structure, sequence, and size heterogeneity. Multiple alignments at the nucleotide and amino acid levels reveal conserved, potentially transposition-related sequence motifs. While many MULEs share common structural features to Mu elements in maize, some groups lack characteristic long terminal inverted repeats. High sequence similarity and phylogenetic analyses based on nucleotide sequence alignments indicate that many of these elements with diverse structural features may remain transpositionally competent and that multiple MULE lineages may have been evolving independently over long time scales. Finally, there is evidence that MULEs are capable of the acquisition of host DNA segments, which may have implications for adaptive evolution, both at the element and host levels.
DOI:10.1186/s13059-016-0954-8URLPMID:27154274 [本文引用: 1]
Mutator-like transposable elements, a class of DNA transposons, exist pervasively in both prokaryotic and eukaryotic genomes, with more than 10,000 copies identified in the rice genome. These elements can capture ectopic genomic sequences that lead to the formation of new gene structures. Here, based on whole-genome comparative analyses, we comprehensively investigated processes and mechanisms of the evolution of putative genes derived from Mutator-like transposable elements in ten Oryza species and the outgroup Leersia perieri, bridging ~20 million years of evolutionary history.
DOI:10.1104/pp.113.223271URL [本文引用: 1]
The process of gene duplication followed by sequence and functional divergence is important for the generation of new genes. Pack-MULEs, nonautonomous Mutator-like elements (MULEs) that carry genic sequence(s), are potentially involved in generating new open reading frames and regulating parental gene expression. These elements are identified in many plant genomes and are most abundant in rice (Oryza sativa). Despite the abundance of Pack-MULEs, the mechanism by which parental genes are captured by Pack-MULEs remains largely unknown. In this study, we identified all MULEs in rice and examined factors likely important for sequence acquisition. Terminal inverted repeat MULEs are the predominant MULE type and account for the majority of the Pack-MULEs. In addition to genic sequences, rice MULEs capture guanine-cytosine (GC)-rich intergenic sequences, albeit at a much lower frequency. MULEs carrying nontransposon sequences have longer terminal inverted repeats and higher GC content in terminal and subterminal regions. An overrepresentation of genes with known functions and genes with orthologs among parental genes of Pack-MULEs is observed in rice, maize (Zea mays), and Arabidopsis (Arabidopsis thaliana), suggesting preferential acquisition for bona fide genes by these elements. Pack-MULEs selectively acquire/retain parental sequences through a combined effect of GC content and breadth of expression, with GC content playing a stronger role. Increased GC content and number of tissues with detectable expression result in higher chances of a gene being acquired by Pack-MULEs. Such selective acquisition/retention provides these elements greater chances of carrying functional sequences that may provide new genetic resources for the evolution of new genes or the modification of existing genes.
DOI:10.1073/pnas.1010814108URLPMID:21220310 [本文引用: 1]
In monocots, many genes demonstrate a significant negative GC gradient, meaning that the GC content declines along the orientation of transcription. Such a gradient is not observed in the genes of the dicot plant Arabidopsis. In addition, a lack of homology is often observed when comparing the 5' end of the coding region of orthologous genes in rice and Arabidopsis. The reasons for these differences have been enigmatic. The presence of GC-rich sequences at the 5' end of genes may influence the conformation of chromatin, the expression level of genes, as well as the recombination rate. Here we show that Pack-Mutator-like transposable elements (Pack-MULEs) that carry gene fragments specifically acquire GC-rich fragments and preferentially insert into the 5' end of genes. The resulting Pack-MULEs form independent, GC-rich transcripts with a negative GC gradient. Alternatively, the Pack-MULEs evolve into additional exons at the 5' end of existing genes, thus altering the GC content in those regions. We demonstrate that Pack-MULEs modify the 5' end of genes and are at least partially responsible for the negative GC gradient of genes in grasses. Such a unique and global impact on gene composition and gene structure has not been observed for any other transposable elements.
DOI:10.1093/nar/gky025URLPMID:29365184 [本文引用: 1]
Acquisition and rearrangement of host genes by transposable elements (TEs) is an important mechanism to increase gene diversity as exemplified by the ~3000 Pack-Mutator-like TEs in the rice genome which have acquired gene sequences (Pack-MULEs), yet remain enigmatic. To identify signatures of functioning Pack-MULEs and Pack-MULE evolution, we generated transcriptome, translatome, and epigenome datasets and compared Pack-MULEs to genes and other TE families. Approximately 40% of Pack-MULEs were transcribed with 9% having translation evidence, clearly distinguishing them from other TEs. Pack-MULEs exhibited a unique expression profile associated with specificity in reproductive tissues that may be associated with seed traits. Expressed Pack-MULEs resemble regular protein-coding genes as exhibited by a low level of DNA methylation, association with active histone marks and DNase I hypersensitive sites, and an absence of repressive histone marks, suggesting that a substantial fraction of Pack-MULEs are potentially functional in vivo. Interestingly, the expression capacity of Pack-MULEs is independent of the local genomic environment, and the insertion and expression of Pack-MULEs may have altered the local chromosomal expression pattern as well as counteracted the impact of recombination on chromosomal base composition, which has profound consequences on the evolution of chromosome structure.
DOI:10.1128/mcb.17.9.5165URLPMID:9271394 [本文引用: 1]
The autonomous MuDR element of the Mutator (Mu) transposable element family of maize encodes at least two proteins, MURA and MURB. Based on amino acid sequence similarity, previous studies have reported that MURA is likely to be a transposase. The functional characterization of MURA has been hindered by the instability of its cDNA, mudrA, in Escherichia coli. In this study, we report the first successful stabilization and expression of MURA in Saccharomyces cerevisiae. Gel mobility shift assays demonstrate that MURA is a DNA-binding protein that specifically binds to sequences within the highly conserved Mu element terminal inverted repeats (TIRs). DNase I and 1,10-phenanthroline-copper footprinting of MURA-Mu1 TIR complexes indicate that MURA binds to a conserved approximately 32-bp region in the TIR of Mu1. In addition, MURA can bind to the same region in the TIRs of all tested actively transposing Mu elements but binds poorly to the diverged Mu TIRs of inactive elements. Previous studies have reported a correlation between Mu transposon inactivation and methylation of the Mu element TIRs. Gel mobility shift assays demonstrate that MURA can interact differentially with unmethylated, hemimethylated, and homomethylated TIR substrates. The significance of MURA's interaction with the TIRs of Mu elements is discussed in the context of what is known about the regulation and mechanisms of Mutator activities in maize.
DOI:10.1105/tpc.114.128488URLPMID:25587002 [本文引用: 3]
Mutator-like transposable elements (MULEs) are widespread in plants and are well known for their high transposition activity as well as their ability to duplicate and amplify host gene fragments. Despite their abundance and importance, few active MULEs have been identified. In this study, we demonstrated that a rice (Oryza sativa) MULE, Os3378, is capable of excising and reinserting in yeast (Saccharomyces cerevisiae), suggesting that yeast harbors all the host factors for the transposition of MULEs. The transposition activity induced by the wild-type transposase is low but can be altered by modification of the transposase sequence, including deletion, fusion, and substitution. Particularly, fusion of a fluorescent protein to the transposase enhanced the transposition activity, representing another approach to manipulate transposases. Moreover, we identified a critical region in the transposase where the net charge of the amino acids seems to be important for activity. Finally, transposition efficiency is also influenced by the element and its flanking sequences (i.e., small elements are more competent than their large counterparts). Perfect target site duplication is favorable, but not required, for precise excision. In addition to the potential application in functional genomics, this study provides the foundation for further studies of the transposition mechanism of MULEs.
DOI:10.1046/j.1365-313x.2001.00939.xURLPMID:11169184 [本文引用: 1]
The Mu transposons of maize are under stringent developmental control. Elements excise at high frequencies in terminally dividing somatic cells, but not in meristems. Mu elements in germinal cells amplify, without excision, and insert throughout the genome. All activities require MuDR, which encodes two genes, mudrA and mudrB, whose near-identical promoters are located in the transposon terminal inverted repeats (TIR). We have fused the 216 bp TIR of the mudrB gene to GUS and luciferase reporters. We demonstrate that TIRB programs reporter expression in diverse, meristematic somatic cells, paradoxically in those cells in which Mu excisions are repressed. In germinal cells, immature tassel and mature pollen, reporter expression increases up to 20-fold compared to leaf. By RNA blot hybridization, we demonstrate that endogenous mudrB and mudrA transcripts increase significantly in mature pollen; sequence comparisons demonstrate that the MuDR TIRs contain plant cell-cycle enhancer motifs and functionally defined pollen enhancers. Therefore, the MuDR TIR promoters are developmentally regulated in both somatic and germinal tissues. Because database sequence analysis suggests that the MuDR TIR enhancers should be functional in both monocots and dicots, we suggest that the native MuDR promoter be used in attempts to transfer the unique behavior of Mu transposition to heterologous hosts.
[本文引用: 2]
[本文引用: 1]
URLPMID:11861572 [本文引用: 1]
The widespread use of the maize Mutator (Mu) system to generate mutants exploits the preference of Mu transposons to insert into genic regions. However, little is known about the specificity of Mu insertions within genes. Analysis of 79 independently isolated Mu-induced alleles at the gl8 locus established that at least 75 contain Mu insertions. Analysis of the terminal inverted repeats (TIRs) of the inserted transposons defined three new Mu transposons: Mu10, Mu 11, and Mu12. A large percentage (&gt;80%) of the insertions are located in the 5' untranslated region (UTR) of the gl8 gene. Ten positions within the 5' UTR experienced multiple independent Mu insertions. Analyses of the nucleotide composition of the 9-bp TSD and the sequences directly flanking the TSD reveals that the nucleotide composition of Mu insertion sites differs dramatically from that of random DNA. In particular, the frequencies at which C's and G's are observed at positions -2 and +2 (relative to the TSD) are substantially higher than expected. Insertion sites of 315 RescueMu insertions displayed the same nonrandom nucleotide composition observed for the gl8-Mu alleles. Hence, this study provides strong evidence for the involvement of sequences flanking the TSD in Mu insertion-site selection.
[本文引用: 1]
DOI:10.1093/nar/gkx357URLPMID:28482040 [本文引用: 2]
Mutator-like transposable elements (MULEs) are widespread across fungal, plant and animal species. Despite their abundance and importance as genetic tools in plants, the transposition mechanism of the MULE superfamily was previously unknown. Discovery of the Muta1 element from Aedes aegypti and its successful transposition in yeast facilitated the characterization of key steps in Muta1 transposition. Here we show that purified transposase binds specifically to the Muta1 ends and catalyzes excision through double strand breaks (DSB) and the joining of newly excised transposon ends with target DNA. In the process, the DSB forms hairpin intermediates on the flanking DNA side. Analysis of transposase proteins containing site-directed mutations revealed the importance of the conserved DDE motif and a W residue. The transposition pathway resembles that of the V(D)J recombination reaction and the mechanism of hAT and Transib transposases including the importance of the conserved W residue in both MULEs and hATs. In addition, yeast transposition and in vitro assays demonstrated that the terminal motif and subterminal repeats of the Muta1 terminal inverted repeat also influence Muta1 transposition. Collectively, our data provides new insights to understand the evolutionary relationships between MULE, hAT and Transib elements and the V(D)J recombinase.
DOI:10.1073/pnas.0603080103URLPMID:17101975 [本文引用: 1]
Maize is probably the most diverse of all crop species. Unexpectedly large differences among haplotypes were first revealed in a comparison of the bz genomic regions of two different inbred lines, McC and B73. Retrotransposon clusters, which comprise most of the repetitive DNA in maize, varied markedly in makeup, and location relative to the genes in the region and genic sequences, later shown to be carried by two helitron transposons, also differed between the inbreds. Thus, the allelic bz regions of these Corn Belt inbreds shared only a minority of the total sequence. To investigate further the variation caused by retrotransposons, helitrons, and other insertions, we have analyzed the organization of the bz genomic region in five additional cultivars selected because of their geographic and genetic diversity: the inbreds A188, CML258, and I137TN, and the land races Coroico and NalTel. This vertical comparison has revealed the existence of several new helitrons, new retrotransposons, members of every superfamily of DNA transposons, numerous miniature elements, and novel insertions flanked at either end by TA repeats, which we call TAFTs (TA-flanked transposons). The extent of variation in the region is remarkable. In pairwise comparisons of eight bz haplotypes, the percentage of shared sequences ranges from 25% to 84%. Chimeric haplotypes were identified that combine retrotransposon clusters found in different haplotypes. We propose that recombination in the common gene space greatly amplifies the variability produced by the retrotransposition explosion in the maize ancestry, creating the heterogeneity in genome organization found in modern maize.
DOI:10.1093/nar/22.13.2634URLPMID:8041625 [本文引用: 1]
The Mutator transposable element system of maize is the most active transposable element system characterized in higher plants. While Mutator has been used to generate and tag thousands of new maize mutants, the mechanism and regulation of its transposition are poorly understood. The Mutator autonomous element, MuDR, encodes two proteins: MURA and MURB. We have detected an amino acid sequence motif shared by MURA and the putative transposases of a group of bacterial insertion sequences. Based on this similarity we believe that MURA is the transposase of the Mutator system. In addition we have detected two rice cDNAs in genbank with extensive similarity to MURA. This sequence similarity suggests that a Mutator-like element is present in rice. We believe that Mutator, a group of bacterial insertion sequences, and an uncharacterized rice transposon represent members of a family of transposable elements.
DOI:10.1007/s00239-008-9178-1URL [本文引用: 1]
The eukaryotic Mutator family of transposable elements is widespread in plants. Active or potentially active copies are also found in fungi and protozoans, and sequences related to this family have been detected in metazoans as well. Members of this family are called Mutator-like elements (MULE s). They encode transposases, which contain a region conserved with transposases of the IS256 prokaryotic family, known to harbor a DDE catalytic domain. Different DDE or D34E motifs have been proposed in some groups of eukaryotic MULEs based on primary sequence conservation. On a large number of protein sequences related to, and representative of, all MULE families, we analyzed global conservation, the close environment of different acidic residues and the secondary structure. This allowed us to identify a potential DDE motif that is likely to be homologous to the one in IS256-like transposases. The characteristics of this motif are depicted in each known family of MULEs. Different hypotheses about the evolution of this triad are discussed.
DOI:10.1105/tpc.12.1.5URLPMID:10634904 [本文引用: 1]
The MuDR element responsible for Mutator activities in maize encodes two genes, mudrA and mudrB. Each encodes multiple transcripts hypothesized to regulate, directly or indirectly, the unique late timing and switch in transposition mechanism during maize development. mudrA, which encodes the MURA transposase, is unstable in bacterial plasmids, a technical problem solved by using phage M13 as a vector to prepare DNA for biolistic transformation. In transgenic maize, a single 2.7-kb mudrA cDNA predicted to encode an 823-amino acid protein is sufficient to catalyze late somatic excisions, despite removal of the native promoter, alternative transcription start sites, known introns, polymorphic 5' and 3' untranslated sequences, and the mudrB gene. These results suggest that post-translational regulation confers Mu excision timing. The transgene is active in lines containing silencing MuDR elements. This suggests that endogenous MuDR transposons do not measurably immunize the host against expression of a homologous transgene.
URLPMID:9872971 [本文引用: 1]
The regulatory transposon of the Mutator system of transposable elements in maize is MuDR. MuDR elements produce two transcripts, from genes mudrA and mudrB, encoding proteins MURA and MURB, respectively. Like many other transposons, MuDR elements often undergo deletions, usually of internal sequences. Analysis of a deletion that is restricted to the region encoding MURB demonstrates that this gene is not required to cause excisions of a reporter element, although it may be required for transposition or suppression of suppressible alleles. Conversely, a derivative that lacks the region encoding MURA but that produces MURB is nonfunctional for all aspects of Mutator activity. Northern analysis of these derivatives reveals that each of the two transcripts can be independently transcribed, and analysis using an antibody specific for MURB reveals that mudrB transcript can also be successfully translated and its product appropriately localized in the absence of mudrA. A third deletion derivative provides evidence for a source of previously reported antisense transcript.
DOI:10.1093/nar/gkq140URLPMID:20215432 [本文引用: 1]
Genes, like organisms, struggle for existence, and the most successful genes persist and widely disseminate in nature. The unbiased determination of the most successful genes requires access to sequence data from a wide range of phylogenetic taxa and ecosystems, which has finally become achievable thanks to the deluge of genomic and metagenomic sequences. Here, we analyzed 10 million protein-encoding genes and gene tags in sequenced bacterial, archaeal, eukaryotic and viral genomes and metagenomes, and our analysis demonstrates that genes encoding transposases are the most prevalent genes in nature. The finding that these genes, classically considered as selfish genes, outnumber essential or housekeeping genes suggests that they offer selective advantage to the genomes and ecosystems they inhabit, a hypothesis in agreement with an emerging body of literature. Their mobile nature not only promotes dissemination of transposable elements within and between genomes but also leads to mutations and rearrangements that can accelerate biological diversification and--consequently--evolution. By securing their own replication and dissemination, transposases guarantee to thrive so long as nucleic acid-based life forms exist.
DOI:10.1073/pnas.1104208108URLPMID:21518873 [本文引用: 1]
Cut-and-paste DNA transposable elements are major components of eukaryotic genomes and are grouped into superfamilies (e.g., hAT, P) based on sequence similarity of the element-encoded transposase. The transposases from several superfamilies possess a protein domain containing an acidic amino acid triad (DDE or DDD) that catalyzes the &quot;cut and paste&quot; transposition reaction. However, it was unclear whether this domain was shared by the transposases from all superfamilies. Through multiple-alignment of transposase sequences from a diverse collection of previously identified and recently annotated elements from a wide range of organisms, we identified the putative DDE/D triad for all superfamilies. Furthermore, we identified additional highly conserved amino acid residues or motifs within the DDE/D domain that together form a &quot;signature string&quot; that is specific to each superfamily. These conserved residues or motifs were exploited as phylogenetic characters to infer evolutionary relationships among all superfamilies. The phylogenetic analysis revealed three major groups that were not previously discerned and led us to revise the classification of several currently recognized superfamilies. Taking the data together, this study suggests that all eukaryotic cut-and-paste transposable element superfamilies have a common evolutionary origin and establishes a phylogenetic framework for all future cut-and-paste transposase comparisons.
DOI:10.1016/j.cell.2014.05.037URL [本文引用: 2]
Hermes is a member of the hAT transposon superfamily that has active representatives, including McClintock's archetypal Ac mobile genetic element, in many eukaryotic species. The crystal structure of the Hermes transposase-DNA complex reveals that Hermes forms an octameric ring organized as a tetramer of dimers. Although isolated dimers are active in vitro for all the chemical steps of transposition, only octamers are active in vivo. The octamer can provide not only multiple specific DNA-binding domains to recognize repeated subterminal sequences within the transposon ends, which are important for activity, but also multiple nonspecific DNA binding surfaces for target capture. The unusual assembly explains the basis of bipartite DNA recognition at hAT transposon ends, provides a rationale for transposon end asymmetry, and suggests how the avidity provided by multiple sites of interaction could allow a transposase to locate its transposon ends amidst a sea of chromosomal DNA.
DOI:10.1038/nature03157URLPMID:15616554 [本文引用: 2]
Transposons are DNA sequences that encode functions that promote their movement to new locations in the genome. If unregulated, such movement could potentially insert additional DNA into genes, thereby disrupting gene expression and compromising an organism's viability. Transposable elements are classified by their transposition mechanisms and by the transposases that mediate their movement. The mechanism of movement of the eukaryotic hAT superfamily elements was previously unknown, but the divergent sequence of hAT transposases from other elements suggested that these elements might use a distinct mechanism. Here we have analysed transposition of the insect hAT element Hermes in vitro. Like other transposons, Hermes excises from DNA via double-strand breaks between the donor-site DNA and the transposon ends, and the newly exposed transposon ends join to the target DNA. Interestingly, the ends of the donor double-strand breaks form hairpin intermediates, as observed during V(D)J recombination, the process which underlies the combinatorial formation of antigen receptor genes. Significant similarities exist in the catalytic amino acids of Hermes transposase, the V(D)J recombinase RAG, and retroviral integrase superfamily transposases, thereby linking the movement of transposable elements and V(D)J recombination.
DOI:10.1093/nar/gkl888URLPMID:17130173 [本文引用: 1]
WRKY and GCM1 are metal chelating DNA-binding domains (DBD) which share a four stranded fold. Using sensitive sequence searches, we show that this WRKY-GCM1 fold is also shared by the FLYWCH Zn-finger domain and the DBDs of two classes of Mutator-like element (MULE) transposases. We present evidence that they share a stabilizing core, which suggests a possible origin from a BED finger-like intermediate that was in turn ultimately derived from a C2H2 Zn-finger domain. Through a systematic study of the phyletic pattern, we show that this WRKY-GCM1 superfamily is a widespread eukaryote-specific group of transcription factors (TFs). We identified several new members across diverse eukaryotic lineages, including potential TFs in animals, fungi and Entamoeba. By integrating sequence, structure, gene expression and transcriptional network data, we present evidence that at least two major global regulators belonging to this superfamily in Saccharomyces cerevisiae (Rcs1p and Aft2p) have evolved from transposons, and attained the status of transcription regulatory hubs in recent course of ascomycete yeast evolution. In plants, we show that the lineage-specific expansion of WRKY-GCM1 domain proteins acquired functional diversity mainly through expression divergence rather than by protein sequence divergence. We also use the WRKY-GCM1 superfamily as an example to illustrate the importance of transposons in the emergence of new TFs in different lineages.
[本文引用: 1]
DOI:10.1104/pp.19.00894URLPMID:31636104 [本文引用: 2]
Sequence-indexed insertional libraries are important resources for functional gene study in model plants. However, the maize (Zea mays) UniformMu library covers only 36% of the annotated maize genes. Here, we generated a new sequence-indexed maize Mutator insertional library named ChinaMu through high-throughput sequencing of enriched Mu-tagged sequences. A total of 2,581 Mu F2 lines were analyzed, and 311,924 nonredundant Mu insertion sites were obtained. Based on experimental validation, ChinaMu contains about 97,000 germinal Mu insertions, about twice as many as UniformMu. About two-thirds (66,565) of the insertions are high-quality germinal insertions (positive rate > 90%), 89.6% of which are located in genic regions. Furthermore, 45.7% (20,244) of the 44,300 annotated maize genes are effectively tagged and about two-thirds (13,425) of these genes harbor multiple insertions. We tested the utility of ChinaMu using pentatricopeptide repeat (PPR) genes. For published PPR genes with defective kernel phenotypes, 17 out of 20 were tagged, 11 of which had the previously reported mutant phenotype. For 16 unstudied PPR genes with both Mu insertions and defective kernel phenotypes, 6 contained insertions that cosegregated with the mutant phenotype. Our sequence-indexed Mu insertional library provides an important resource for functional genomics study in maize.
DOI:10.3109/10409230903505596URLPMID:20067338 [本文引用: 1]
DNA rearrangements are important in genome function and evolution. Genetic material can be rearranged inadvertently during processes such as DNA repair, or can be moved in a controlled manner by enzymes specifically dedicated to the task. DNA transposases comprise one class of such enzymes. These move DNA segments known as transposons to new locations, without the need for sequence homology between transposon and target site. Several biochemically distinct pathways have evolved for DNA transposition, and genetic and biochemical studies have provided valuable insights into many of these. However, structural information on transposases - particularly with DNA substrates - has proven elusive in most cases. On the other hand, large-scale genome sequencing projects have led to an explosion in the number of annotated prokaryotic and eukaryotic mobile elements. Here, we briefly review biochemical and mechanistic aspects of DNA transposition, and propose that integrating sequence information with structural information using bioinformatics tools such as secondary structure prediction and protein threading can lead not only to an additional level of understanding but possibly also to testable hypotheses regarding transposition mechanisms. Detailed understanding of transposition pathways is a prerequisite for the long-term goal of exploiting DNA transposons as genetic tools and as a basis for genetic medical applications.
URLPMID:8852857 [本文引用: 2]
Previous research has demonstrated that the autonomous Cy transposon can activate the excision of Mu transposons. To determine the relationship between Cy and the more recently described autonomous Mu transposon, MuDR, a Cy transposon inserted at the mutable a1 allele, a1-m5216, was isolated and cloned. DNA sequence analyses established that this Cy insertion is identical to MuDR (Mu9, GenBank accession No.: m76978.gb-pl). Therefore, Cy will henceforth be termed MuDR:Cy. Defective derivatives of MuDR:Cy were isolated that had lost their capacity to activate their own excision or the excision of a Mu7 transposon. Most of these derivatives are nonautonomous transposons because they can excise, but only in the presence of unlinked MuDR:Cy transposons. Physical mapping and DNA sequence analyses have established that six of these defective derivatives carry internal deletions. It has been proposed previously that such deletions arise via interrupted gap repair. The DNA sequences of the break points associated with all four sequenced deletions are consistent with this model. The finding that three of the excision-defective derivatives carry deletions that disrupt the coding region of the mudrA (but not the mudrB) transcript supports the view that mudrA plays a role in the excision of Mu transposons.
DOI:10.1105/tpc.7.12.1989URLPMID:8718617 [本文引用: 1]
The Mutator (Mu) system of transposable elements is highly mutagenic and can maintain high levels of activity through multiple generations due to frequent transpositions of both its autonomous and nonautonomous components. This family also shows pronounced developmental regulation. Most notable is the very low frequency of germinal reversions, despite the high levels of somatic transpositions and excisions, and the high frequency of germinally transmitted duplication events. Here, we report the production of antibodies raised against MURB, one of two proteins encoded by MuDR, the autonomous regulator of the Mu family. Immunolocalizations performed using anti-MURB antibodies reveal that this protein is present in specific tissues during male inflorescence development. Throughout much of development, MURB is detected at the highest levels in cell lineages that may find themselves in the germ line, but no MURB is detected in microspore mother cells. These cells are the direct precursors to pollen. Based on these observations as well as previous data, we discuss the relationship between the expression of MURB and developmental regulation of Mu activity.
DOI:10.1007/bf00259680URLPMID:1648169 [本文引用: 2]
Germinal and somatic excision products of Mu1 from the insertion allele bz::mu1 were selectively amplified from maize cob tissue. The sequence of these &quot;footprints&quot; often included deletions at the target site, suggesting that substantial exonucleolytic degradation occurs upon excision of the element. In addition to deletions of target site sequences, single base insertions were also found. The isolation of an excision product including a 4 bp inverted duplication of the target site provides evidence that the double-stranded chromosomal break generated by Mu excision may be terminated by a covalently closed hairpin structure. The majority of excision products, however, do not include inverted duplications of target site sequences, suggesting that such structures are the result of occasional repair activities, rather than an essential step in the mechanism of Mu excision. The sequence of the Mu insertion sites of the bz::mu1 and bz::mu2 alleles is also presented.
DOI:10.1093/nar/19.3.579URLPMID:1849263 [本文引用: 2]
The Mu transposons of the Robertsons's Mutator transposable element system in maize are unusual in many respects, when compared to the other known plant transposon systems. The excision of these elements occurs late in somatic tissues and very rarely in the germ line. Unlike the other plant transposons, there is no experimental evidence directly linking Mu element excision and integration. We have analyzed the excision products generated by a Mu1 transposon inserted into the bronze 1 locus of maize. We find that the excision products or 'footprints' left by the Mu1 element resemble those of the other plant transposable elements, rather than those of the animal transposable element systems. We also find some novel types of footprints resembling recombinational events. We suggest that the Mu1 element can promote intrachromosomal crossovers and conversions near its site of insertion, and that this may be another mechanism by which transposons can accelerate the evolution of genomes.
DOI:10.1105/tpc.010002URLPMID:11449053 [本文引用: 1]
RescueMu, a Mu1 element containing a bacterial plasmid, is mobilized by MuDR in transgenic maize. Somatic excision from a cell-autonomous marker gene yields &gt;90% single cell sectors; empty donor sites often have deletions and insertions, including up to 210 bp of RescueMu/Mu1 terminal DNA. Late somatic insertions are contemporaneous with excisions, suggesting that &quot;cut-and-paste&quot; transposition occurs in the soma. During reproduction, RescueMu transposes infrequently from the initial transgene array, but once transposed, RescueMu is suitable for high throughput gene mutation and cloning. As with MuDR/Mu elements, heritable RescueMu insertions are not associated with excisions. Both somatic and germinal RescueMu insertions occur preferentially into genes and gene-like sequences, but they exhibit weak target site preferences. New insights into Mu behaviors are discussed with reference to two models proposed to explain the alternative outcomes of somatic and germinal events: a switch from somatic cut-and-paste to germinal replicative transposition or to host-mediated gap repair from sister chromatids.
DOI:10.1534/genetics.106.062604URLPMID:17507687 [本文引用: 1]
In Saccharomyces cerevisiae, Rad51p plays a central role in homologous recombination and the repair of double-strand breaks (DSBs). Double mutants of the two Zea mays L. (maize) rad51 homologs are viable and develop well under normal conditions, but are male sterile and have substantially reduced seed set. Light microscopic analyses of male meiosis in these plants reveal reduced homologous pairing, synapsis of nonhomologous chromosomes, reduced bivalents at diakinesis, numerous chromosome breaks at anaphase I, and that &gt;33% of quartets carry cells that either lack an organized nucleolus or have two nucleoli. This indicates that RAD51 is required for efficient chromosome pairing and its absence results in nonhomologous pairing and synapsis. These phenotypes differ from those of an Arabidopsis rad51 mutant that exhibits completely disrupted chromosome pairing and synapsis during meiosis. Unexpectedly, surviving female gametes produced by maize rad51 double mutants are euploid and exhibit near-normal rates of meiotic crossovers. The finding that maize rad51 double mutant embryos are extremely susceptible to radiation-induced DSBs demonstrates a conserved role for RAD51 in the repair of mitotic DSBs in plants, vertebrates, and yeast.
DOI:10.1105/tpc.11.5.809URLPMID:10330467 [本文引用: 1]
An open question in meiosis is whether the Rad51 recombination protein functions solely in meiotic recombination or whether it is also involved in the chromosome homology search. To address this question, we have performed three-dimensional high-resolution immunofluorescence microscopy to visualize native Rad51 structures in maize male meiocytes. Maize has two closely related RAD51 genes that are expressed at low levels in differentiated tissues and at higher levels in mitotic and meiotic tissues. Cells and nuclei were specially fixed and embedded in polyacrylamide to maintain both native chromosome structure and the three dimensionality of the specimens. Analysis of Rad51 in maize meiocytes revealed that when chromosomes condense during leptotene, Rad51 is diffuse within the nucleus. Rad51 foci form on the chromosomes at the beginning of zygotene and rise to approximately 500 per nucleus by mid-zygotene when chromosomes are pairing and synapsing. During chromosome pairing, we consistently found two contiguous Rad51 foci on paired chromosomes. These paired foci may identify the sites where DNA sequence homology is being compared. During pachytene, the number of Rad51 foci drops to seven to 22 per nucleus. This higher number corresponds approximately to the number of chiasmata in maize meiosis. These observations are consistent with a role for Rad51 in the homology search phase of chromosome pairing in addition to its known role in meiotic recombination.
DOI:10.1128/jb.184.17.4709-4714.2002URLPMID:12169594 [本文引用: 1]
IS256 is a highly active insertion sequence (IS) element of multiresistant staphylococci and enterococci. Here we show that, in a Staphylococcus epidermidis clinical isolate, as well as in recombinant Staphylococcus aureus and Escherichia coli carrying a single IS256 insertion on a plasmid, IS256 excises as an extrachromosomal circular DNA molecule. First, circles were identified that contained a complete copy of IS256. In this case, the sequence connecting the left and right ends of IS256 was derived from flanking DNA sequences of the parental genetic locus. Second, circle junctions were detected in which one end of IS256 was truncated. Nucleotide sequencing of circle junctions revealed that (i) either end of IS256 can attack the opposite terminus and (ii) the circle junctions vary significantly in size. Upon deletion of the IS256 open reading frame at the 3' end and site-directed mutageneses of the putative DDE motif, circular IS256 molecules were no longer detectable, which implicates the IS256-encoded transposase protein with the circularization of the element.
DOI:10.1073/pnas.84.14.4924URLPMID:3037528 [本文引用: 1]
Maize lines known as Robertson's Mutator (Mu) lines generate unstable recessive mutations at high frequencies. These lines carry actively transposing copies of the transposons (Tn) Mu1 and Mu1.7. TnMu1 and TnMu1.7 are approximately 1400 and 1700 base pairs long, respectively, and they have 210-base-pair terminal inverted repeats. We report here extrachromosomal forms of TnMu1 and TnMu1.7. The extrachromosomal Mu1 and Mu1.7 molecules are resistant to alkaline denaturation and to proteinase treatment and have circular restriction maps; therefore, they are probably covalently closed circular DNA. Further, we show that their occurrence is correlated with Mu activity, so they are probably generated during Mu transposition as transposition intermediates or as products of Mu excision. When the total extrachromosomal supercoiled DNA from immature male flowers of a Mu line was examined by electron microscopy, the Mu transposons appeared to constitute a significant fraction of the extrachromosomal DNA circles in Mu lines.
URLPMID:10790408 [本文引用: 1]
The mechanism of transposition of the maize Ac/Ds elements is not well understood. The true transposition intermediates are not known and it has not been possible to distinguish between excision models involving 8-bp staggered cuts or 1-bp staggered cuts followed by hairpin formation. In this work, we have analyzed extrachromosomal excision products to gain insight into the excision mechanism. Plasmid rescue was used to demonstrate that Ds excision is associated with the formation of circular molecules. In addition, we present evidence for the formation of linear extrachromosomal species during Ds excision. Sequences found at the termini of circular and linear elements showed a broad range of nucleotide additions or deletions, suggesting that these species are not true intermediates. Additional nucleotides adjacent to the termini in extrachromosomal elements were compared to the sequence of the original donor site. This analysis showed that: (1) the first nucleotide adjacent to the transposon end was significantly more similar to the first nucleotide flanking the element in the donor site than to a random sequence and (2) the second and farther nucleotides did not resemble the donor site. The implications of these findings for excision models are discussed.
URLPMID:9093867 [本文引用: 1]
The maize Ac/Ds transposable elements are thought to transpose via a cut-and-paste mechanism, but the intermediates formed during transposition are still unknown. In this work we present evidence that circular Ac molecules are formed in plants containing actively transposing elements. In these circles, transposon ends are joined head-to-head. The sequence at the ends' junction is variable, containing small deletions or insertions. Circles containing deleted Ac ends are probably unable to successfully reintegrate. To test the ability of circles with intact transposon ends to integrate into the genome, an artificial Ds circle was constructed by cloning the joined ends of Ac into a plasmid carrying a plant selectable marker. When such a circular Ds was introduced into tobacco protoplasts in the presence of Ac-transposase, no efficient transposase-mediated integration was observed. Although a circular transposition intermediate cannot be ruled out, the findings of circles with deleted transposon ends and the absence of transposase-mediated integration of the circular Ds suggest that some of the joined-ends-carrying elements are not transposition intermediates, but rather abortive excision products. The formation of Ac circles might account for the previously described phenomenon of Ac-loss. The origin of Ac circles and the implications for models of Ac transposition are discussed.
DOI:10.1186/1471-2164-8-116URLPMID:17490480 [本文引用: 1]
Gene knockouts are a critical resource for functional genomics. In Arabidopsis, comprehensive knockout collections were generated by amplifying and sequencing genomic DNA flanking insertion mutants. These Flanking Sequence Tags (FSTs) map each mutant to a specific locus within the genome. In maize, FSTs have been generated using DNA transposons. Transposable elements can generate unstable insertions that are difficult to analyze for simple knockout phenotypes. Transposons can also generate somatic insertions that fail to segregate in subsequent generations.
DOI:10.1186/gb-2004-5-10-r82URLPMID:15461800 [本文引用: 2]
Derived from the maize Mu1 transposon, RescueMu provides strategies for maize gene discovery and mutant phenotypic analysis. 9.92 Mb of gene-enriched sequences next to RescueMu insertion sites were co-assembled with expressed sequence tags and analyzed. Multiple plasmid recoveries identified probable germinal insertions and screening of RescueMu plasmid libraries identified plants containing probable germinal insertions. Although frequently recovered parental insertions and insertion hotspots reduce the efficiency of gene discovery per plasmid, RescueMu targets a large variety of genes and produces knockout mutants.
URLPMID:7635296 [本文引用: 1]
The Mutator transposable element system of maize was originally identified through its induction of mutations at an exceptionally high frequency and at a wide variety of loci. The Mu1 subfamily of transposable elements within this system are responsible for the majority of Mutator-induced mutations. Mu 1-related elements were isolated from active Mutator plants and their flanking DNA was characterized. Sequence analyses revealed perfect nine base target duplications directly flanking the insert for 13 of the 14 elements studied. Hybridizational studies indicated that Mu1-like elements insert primarily into regions of the maize genome that are of low copy number. This preferential selection of low copy number DNA as targets for Mu element insertion was not directed by any specific secondary structure(s) that could be detected in this study, but the 9-bp target duplications exhibited a discernibly higher than random match with the consensus sequence 5'-G-T-T-G-G/C-A-G-G/A-G-3'.
DOI:10.1046/j.1365-313x.2000.00830.xURLPMID:10972882 [本文引用: 1]
We have used a universal adaptor amplification procedure to isolate random Mutator-tagged fragments from Mutator-active maize plants. Direct sequence characterization of 761 Mutator-tagged fragments indicated that a significant number were homologous to sequences within the public databases. The ability of Mutator-tagged fragments to detect homology was not related to the length of the sequence within the range 100-400 bp. However, fragments above this size did show an increased chance of detecting homology to either expressed sequence tags or genes. Characterization of the insertion sites of the Mutator elements suggested that while it does target transcribed regions, Mutator does not appear to have any site preference within the transcription unit. Hybridization of previously unidentified Mutator-tagged fragments to arrayed cDNA libraries confirmed that many of these also showed homology to transcribed regions of the genome. Examination of back-crossed progeny confirmed that all the insertions examined were germinal; however, in all but one case, selfing five individual Mutator-tagged lines failed to reveal an obvious phenotype. This study suggests that the random sequencing of Mutator-tagged fragments is capable of producing both a significant number of interesting transposon tagged genes and mutant plant lines, all of which could be extremely valuable in future gene discovery and functional genomics programmes.
DOI:10.1101/gr.189102URLPMID:12045139 [本文引用: 1]
In this study, we describe a property of Gramineae genes, and perhaps all monocot genes, that is not observed in eudicot genes. Along the direction of transcription, beginning at the junction of the 5'-UTR and the coding region, there are gradients in GC content, codon usage, and amino-acid usage. The magnitudes of these gradients are large enough to hinder the annotation of the rice genome and to confound the detection of protein homologies across the monocot-eudicot divide.
DOI:10.1371/journal.pgen.1000733URLPMID:19936291 [本文引用: 1]
The Mu transposon system of maize is highly active, with each of the approximately 50-100 copies transposing on average once each generation. The approximately one dozen distinct Mu transposons contain highly similar approximately 215 bp terminal inverted repeats (TIRs) and generate 9-bp target site duplications (TSDs) upon insertion. Using a novel genome walking strategy that uses these conserved TIRs as primer binding sites, Mu insertion sites were amplified from Mu stocks and sequenced via 454 technology. 94% of approximately 965,000 reads carried Mu TIRs, demonstrating the specificity of this strategy. Among these TIRs, 21 novel Mu TIRs were discovered, revealing additional complexity of the Mu transposon system. The distribution of &gt;40,000 non-redundant Mu insertion sites was strikingly non-uniform, such that rates increased in proportion to distance from the centromere. An identified putative Mu transposase binding consensus site does not explain this non-uniformity. An integrated genetic map containing more than 10,000 genetic markers was constructed and aligned to the sequence of the maize reference genome. Recombination rates (cM/Mb) are also strikingly non-uniform, with rates increasing in proportion to distance from the centromere. Mu insertion site frequencies are strongly correlated with recombination rates. Gene density does not fully explain the chromosomal distribution of Mu insertion and recombination sites, because pronounced preferences for the distal portion of chromosome are still observed even after accounting for gene density. The similarity of the distributions of Mu insertions and meiotic recombination sites suggests that common features, such as chromatin structure, are involved in site selection for both Mu insertion and meiotic recombination. The finding that Mu insertions and meiotic recombination sites both concentrate in genomic regions marked with epigenetic marks of open chromatin provides support for the hypothesis that open chromatin enhances rates of both Mu insertion and meiotic recombination.
DOI:10.1038/nrg.2017.7URLPMID:28286338 [本文引用: 1]
Transposable elements and retroviruses are found in most genomes, can be pathogenic and are widely used as gene-delivery and functional genomics tools. Exploring whether these genetic elements target specific genomic sites for integration and how this preference is achieved is crucial to our understanding of genome evolution, somatic genome plasticity in cancer and ageing, host-parasite interactions and genome engineering applications. High-throughput profiling of integration sites by next-generation sequencing, combined with large-scale genomic data mining and cellular or biochemical approaches, has revealed that the insertions are usually non-random. The DNA sequence, chromatin and nuclear context, and cellular proteins cooperate in guiding integration in eukaryotic genomes, leading to a remarkable diversity of insertion site distribution and evolutionary strategies.
DOI:10.1038/s41588-018-0158-0URLPMID:30061736 [本文引用: 1]
The maize W22 inbred has served as a platform for maize genetics since the mid twentieth century. To streamline maize genome analyses, we have sequenced and de novo assembled a W22 reference genome using short-read sequencing technologies. We show that significant structural heterogeneity exists in comparison to the B73 reference genome at multiple scales, from transposon composition and copy number variation to single-nucleotide polymorphisms. The generation of this reference genome enables accurate placement of thousands of Mutator (Mu) and Dissociation (Ds) transposable element insertions for reverse and forward genetics studies. Annotation of the genome has been achieved using RNA-seq analysis, differential nuclease sensitivity profiling and bisulfite sequencing to map open reading frames, open chromatin sites and DNA methylation profiles, respectively. Collectively, the resources developed here integrate W22 as a community reference genome for functional genomics and provide a foundation for the maize pan-genome.
DOI:10.1002/dvg.1020100610URLPMID:2557991 [本文引用: 1]
A new allele of the maize A1 gene, a gene required for anthocyanin pigment biosynthesis, was identified in a genetic stock exhibiting a high frequency of chromosome breakage at the second microspore mitosis. This allele, a-mrh, is unstable in both somatic and germinal tissue when an independent locus, Mrh, is present in the genome. a-mrh was molecularly cloned, and a 246 bp DNA insertion with characteristics of a transposable element was identified within the fourth exon of the gene. Southern blot analysis of germinal derivatives of a-mrh suggests that the DNA insert rMrh is excised from the locus when a wild-type phenotype is restored. Genetic crosses with components of other two-element mutable systems of maize failed to induce mutability. We therefore conclude that rMrh is a member of a new, two-element transposon system of maize. The genetic and molecular characteristics of the elements involved are discussed with respect to stress-activated transposition, response of an element to developmental signals, and a possible new role of plant transposons in gene evolution.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}