 ,1,2, 邱峰3, 张新安,1
,1,2, 邱峰3, 张新安,1Studies of congenital cataract-related TSR1 mutation and its expression in the lens
Yajie Yu,1,2, Feng Qiu3, Xin-an Zhang,1通讯作者: 张新安,博士,教授,研究方向:基础医学。E-mail:zhangxa2725@163.com
第一联系人:
编委: 徐湘民
收稿日期:2019-10-14修回日期:2020-01-1网络出版日期:2020-01-08
| 基金资助: |
Received:2019-10-14Revised:2020-01-1Online:2020-01-08
| Fund supported: |

摘要
先天性白内障(congenital cataract, CC)是一种罕见的晶状体发育异常疾病,主要表现为晶状体部分或完全浑浊。先天性白内障遗传异质性高,已鉴定的致病基因多达266个。本研究在一个中国先天性白内障家系中通过全基因组测序及Sanger测序验证,筛查到一个新的先天性白内障候选致病基因TSR1,与家系疾病表型共分离。通过minigene实验证实该变异影响TSR1基因mRNA剪接。Western blotting、免疫荧光和RT-PCR实验证实TSR1在人晶状体上皮细胞SRA01/04、年龄相关性白内障患者晶状体前囊膜组织、24周人胎眼晶状体和小鼠晶状体中表达。通过对iSyTE数据库的分析发现,Tsr1在小鼠的胚胎期和不同发育时期的晶状体中都有表达,且在晶状体特异性CBP:p300双敲除小鼠中Tsr1表达下调。提取在CBP:p300双敲除小鼠晶状体中与Tsr1具有相同表达模式的一组基因进行蛋白质-蛋白质相互作用网络(protein-protein interaction,PPI)分析,结果表明筛选出6个基因与Tsr1存在直接相互作用。GO功能分析表明Tsr1参与核糖体的组装,还可能在MAPK-Erk信号通路中发挥作用,为进一步明确Tsr1在晶状体中的功能提供了有价值的研究线索。
关键词:
Abstract
Congenital cataract (CC) is a rare disease with dysplasia of the lens, mainly characterized by partial or complete opacity of the lens. The molecular basis of the disease is complex, mutations in over 266 genes associated with congenital cataracts had been reported. In this study, a novel congenital cataract candidate gene TSR1 was identified by whole genome sequencing and Sanger sequencing in a Chinese congenital cataract family. The TSR1 c.202-1G>A substitution affected splicing of TSR1 mRNA was confirmed by a minigene assay. The expression of TSR1 in mouse lens, anterior lens capsule of age-related cataract patients and 24-week human fetal lens were determined by RT-PCR, Western blotting, and immunofluorescence assays. The expression of TSR1 in the embryonic and different developmental stages of the mouse lens was confirmed by analyzing the iSyTE database. The expression of TSR1 was down-regulated in the lens-specific CBP:p300 double knockout mouse, and a set of genes with the same expression pattern of Tsr1 in the CBP:p300 double knockout mouse lens were extracted for protein-protein interaction network analysis, and six proteins were screened for direct interaction with Tsr1. GO function analysis indicated that Tsr1 might play a role in the MAPK-Erk signaling pathway in addition to its involvement in ribosome assembly. This study provided valuable research clues to further clarify the function of Tsr1 in the lens.
Keywords:
PDF (1389KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
于雅洁, 邱峰, 张新安. TSR1突变导致先天性白内障及其在晶状体中的表达. 遗传[J], 2020, 42(2): 161-171 doi:10.16288/j.yczz.19-166
Yajie Yu.
先天性白内障(congenital cataract,CC)是指出生前即存在或出生后1年内才逐渐形成的白内障,发病率为0.1‰~0.6‰,是一种较为常见的儿童眼病,是小儿失明和弱视的主要原因[1,2]。先天性白内障通常表现为晶状体全部或部分浑浊,常伴随一种或多种其他的眼部并发症,如斜视、眼球震颤、先天性小眼球、眼内组织异常和缺失、视网膜和脉络膜病变等。先天性白内障发病受遗传和环境因素影响,遗传性先天性白内障多为单基因突变导致并且具有很强的遗传异质性。遗传方式包括常染色体显性遗传、常染色体隐性遗传、X连锁遗传,其中以常染色体显性遗传居多。目前已经报道的先天性白内障致病基因多达266个,其中32个基因可以导致单纯性先天性白内障,多数基因导致综合征性先天性白内障[3,4,5,6],除白内障表型外还包括一些眼部其他表型和身体其他组织和器官的异常和病变,如Warburg- Micro综合征、Zellweger综合征、Wolfram-like 综合征和Nance-Horan综合征等[7,8,9,10]。
本研究利用全基因组测序技术在一个中国先天性白内障家系中筛查致病基因突变,并利用minigene、RT-PCR、免疫荧光、Western blotting和生物信息学分析探讨TSR1致病可能机制。
1 材料与方法
1.1 材料
本研究的家系样本为沈阳市第四人民医院眼科收集,3代共8名患者,遗传方式为常染色体显性遗传,所有参与者均接受了裂隙灯检查。正常人群为炎黄中国人基因频率数据库(Chinese millionome database, CMDB)和北京诺禾致源科技股份有限公司(NovoDb)的中国正常人群样本库及中国医科大学附属第四医院体检中心的105例正常人,年龄相关性白内障患者晶状体前囊膜组织和24周人胎眼晶状体组织的石蜡切片由中国医科大学附属第四医院眼科中心提供,人晶状体上皮细胞SRA01/04为实验室自存,使用的小鼠为野生型C57BL/6,生物信息学数据分析来自于CBP:p300双敲除小鼠模型。本研究通过了沈阳市第四人民医院和中国医科大学的伦理审查,所有参与者均签署知情同意书。
1.2 基因组DNA提取及全外显子组测序
使用TIANamp Genomic DNA Kit(北京天根生化科技有限公司)提取先天性白内障家系成员基因组DNA,选择2名患者(III-3和III-5)和1名正常人(II-2)进行全外显子组测序。1.3 全外显子组测序结果分析
全外显子组测序原始数据由BWA软件mem命令比对到hg19参考基因组上,比对结果由samtools软件进行排序、标记重复和格式转换生成bam格式文件并构建索引,使用GATK4.0软件进行snp-calling生成vcf格式储存的变异信息文件,使用annovar软件对变异进行注释,首先将所有变异与ExAC、dbSNP、EVS、GnomAD、1000genomes、CMDB和NovoDb等数据库进行比对,排除正常人群中出现频率高于0.01%的变异,使用基因组可视化软件Integrative Genomics Viewer (IGV)对位于外显子、外显子非翻译区(untranslated region, UTR)和剪接位点的变异进行可视化,排除假阳性结果,剩余位点作为候选变异。1.4 PCR-Sanger测序及突变预测
对全外显子组测序数据进行分析筛选,设计特异性引物扩增所有候选位点并进行Sanger测序验证。使用MEGA-X软件进行位点保守性分析,Human splicing finder 3.1在线软件预测突变对剪接的影响。1.5 minigene实验
设计引物扩增TSR1第1内含子3°端150 bp序列以及第2外显子、第2内含子、第3外显子、第3内含子、第4外显子和第4内含子5°端150 bp序列,上游引物TSR1-BamF:5°-CGCGGATCCGCTATTTGGCCGTTATTAACTCTT-3°,下游引物TSR1- MluR:5°-CGACGCGTAAGCCCAACACTGAACTACCC-3°,以先证者基因组DNA为模板,PCR反应条件为98℃预变性2 min,98℃变性20 s、60℃退火20 s、72℃延伸90 s,35个循环,72℃延伸5 min。经BamHⅠ和MluⅠ酶切后连入pCAS2质粒,构建野生型和突变型minigene质粒。pCAS2空载、TSR1野生型和TSR1突变型质粒转染至SRA01/04细 胞,24 h后收集细胞使用Trizol(北京全式金生物 技术有限公司)提取总RNA并反转录成cDNA(北京全式金生物技术有限公司),使用载体特异性引物(pCAS2-RTF:5°-CTGACCCTGCTGACCCTCCT-3°,pCAS2-RTR:5°-TTGCTGAGAAGGCGTGGTAGAG-3°)进行PCR扩增,PCR产物切胶回收纯化,进行Sanger测序。1.6 小鼠组织RNA提取及反转录
采用断颈法处死实验小鼠,依次分离出晶状体、大脑、肝脏、脾脏、肺、肾脏组织,PBS冲洗后剪取0.1 g组织使用Trizol提取组织总RNA。分别取1 μg总RNA进行反转录,cDNA于-20℃冰箱保存备用。1.7 PCR检测小鼠组织TSR1表达
利用PCR检测小鼠各组织TSR1表达情况,TSR1上游引物为:5°-CGGTGTATTTGAGTGAACGGG-3°,下游引物为:5°-CAGATCCCCTGGTCTTGCAT-3°,内参GAPDH上游引物为:5°-AGGTCGGTGTGAACGGATTTG-3°,下游引物为:5°-GGGGTCGTTGATGGCAACA-3°。反应条件为94℃预变性5 min,94℃变性30 s、60℃退火30 s、72℃延伸30 s,共30个循环,72℃延伸5 min。1.8 人晶状体上皮细胞SRA01/04蛋白提取
利用RAPI裂解液提取细胞总蛋白,细胞浆蛋白和细胞核蛋白提取按照ProteinExtTM Mammalian Nuclear and Cytoplasmic Protein Extraction Kit (DE201,北京全式金生物技术有限公司)说明书进行,于-80℃保存。1.9 Western blotting检测TSR1蛋白表达
SRA01/04细胞总蛋白、细胞浆蛋白和细胞核蛋白12% SDS-PAGE电泳并转移至PVDF膜,5%脱脂奶粉室温封闭1 h,TSR1一抗(1∶1000) (ab220639, abcam, UK)和β-actin一抗(1∶1000) (KGAA001-1,江苏凯基生物技术股份有限公司,南京市)4℃孵育过夜,辣根过氧化物酶标记的二抗(KGAA35和KGAA37,江苏凯基生物技术股份有限公司)室温孵育,TBST洗涤后滴加显色液在化学发光凝胶检测系统中显影检测。1.10 免疫荧光实验
年龄相关性白内障患者晶状体前囊膜组织手术取出后立刻包埋于组织包埋剂O.T.C中,冰冻切片厚度为5 μm,切片立刻置于预冷的丙酮中固定;24周人胎眼石蜡切片置于二甲苯Ⅰ和二甲苯Ⅱ中脱蜡处理,再依次置于体积分数100%、95%、90%、80%和70%的梯度酒精中浸泡。PBS冲洗后将切片浸泡于抗原修复液中进行抗原修复,封闭后滴加TSR1抗体(1∶200稀释)和ACTB抗体(1∶100稀释) 4℃孵育过夜。次日使用PBS冲洗后滴加两种二抗混合液避光孵育,DAPI染液(100 mg/mL)复染,封片剂封片,使用尼康A1R激光共聚焦显微镜观察拍照。1.11 生物信息学分析
使用iSyTE数据库检索Tsr1和相关基因在不同时期的小鼠晶状体组织中的表达变化情况,使用在线软件DAVID进行GO分析和KEGG分析,使用bioGRID在线软件绘制蛋白质-蛋白质相互作用网络PPI图。2 结果与分析
2.1 先天性白内障家系基因突变分析
本研究共收集了3代18人血样,其中患者8人,正常人10人(图1A)。全外显子组测序数据经筛选后,人群频率小于万分之一的杂合位点共14个(表1),候选位点在家系中进行Sanger测序验证,结果显示TSR1 c.202-1G>A杂合突变在家系中与患者白内障表型共分离(图1B)。ExAC、dbSNP、EVS、GnomAD、1000genomes、CMDB和NovoDb等正常人数据库中均未见该变异位点,HGMD和ClinVar等疾病数据库也未见该位点的报道,家系正常人和105例正常对照不携带该变异位点。TSR1 c.202-1G>A位点在物种间高度保守(图1C),Human splicing finder 3.1在线软件预测突变会影响外显子的正常剪接(图1D)。图1
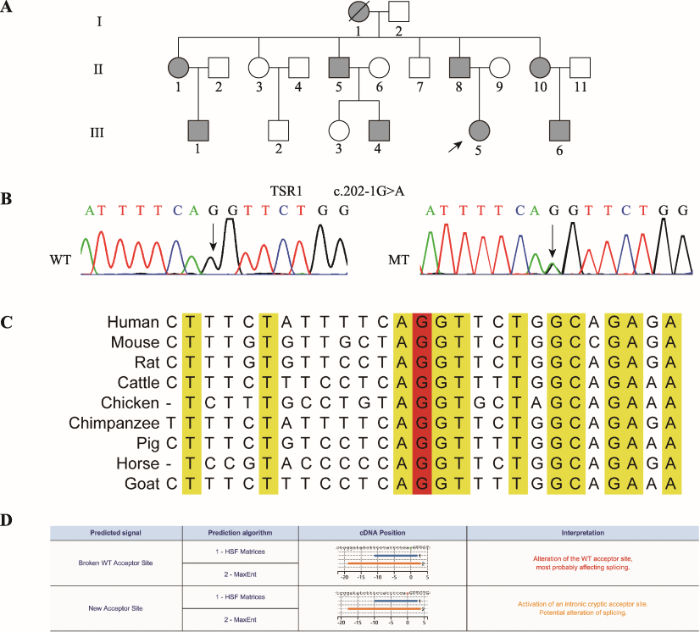 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1先天性白内障家系TSR1变异分析
A:先天性白内障家系系谱图。实心圆圈表示女性患者,实心方块表示男性患者,空心圆圈表示女性正常人,空心方块表示男性正常人,黑色箭头表示先证者,黑色斜线表示个体已死亡。B:TSR1 Sanger测序图。WT为野生型,MT为c.202-1G>A突变型,箭头所指为突变位点。C:位点保守性分析结果。红色G表示突变位点。D:Human splicing finder预测结果。突变破坏了正常受体位点,形成了一个新的剪接受体位点。
Fig. 1TSR1 mutation analysis of the family with CC
Table 1
表1
表1全外显子组测序可疑位点Sanger测序验证情况
Table 1
| 编号 | 染色体 | 基因名 | NM号 | 变异 | 功能 | 是否共分离 |
|---|---|---|---|---|---|---|
| 1 | Chr.1 | PDE4DIP | NM_001002811 | c.A622G:p.T208A | exonic | 否 |
| 2 | Chr.1 | ATP1B1 | NM_001677 | c.*464_*465insT | UTR3 | 否 |
| 3 | Chr.2 | C2orf68 | NM_001013649 | c.*243_*239delTTTTT | UTR3 | 否 |
| 4 | Chr.5 | CXCL14 | NM_004887 | c.*110_*92delAAAAAAAAAAAAAAAAAAA | UTR3 | 否 |
| 5 | Chr.8 | ARHGAP39 | NM_025251 | c.T1352G:p.M451R | exonic | 否 |
| 6 | Chr.10 | NCOA4 | NM_001145260 | c.T22G:p.F8V | exonic | 否 |
| 7 | Chr.12 | KDM5A | NM_001042603 | c.-12delC | UTR5 | 否 |
| 8 | Chr.12 | TPCN1 | NM_001301214 | c.G854C:p.R285T | exonic | 否 |
| 9 | Chr.15 | MEF2A | NM_001130928 | c.1027_1029del:p.Q352del | exonic | 否 |
| 10 | Chr.17 | TSR1 | NM_018128 | c.202-1G>A | splicing | 是 |
| 11 | Chr.17 | ABCA10 | NM_080282 | c.A1297G:p.K433E | exonic | 否 |
| 12 | Chr.18 | TPGS2 | NM_001330572 | c.G734A:p.S245N | exonic | 否 |
| 13 | Chr.21 | TMPRSS15 | NM_002772 | c.T1509A:p.N503K | exonic | 否 |
| 14 | Chr.22 | SEPT3 | NM_019106 | c.*129_*130insTGTG | UTR3 | 否 |
新窗口打开|下载CSV
2.2 Minigene实验检测突变对外显子剪接的影响
野生型和突变型minigene质粒转染SRA01/04细胞,24 h后提取总RNA进行RT-PCR,琼脂糖凝胶电泳结果表明pCAS2空载和野生型minigene可以正常剪接,野生型和突变型条带大小无明显差异(图2A)。Sanger测序表明突变型TSR1 c.202-1G>A激活了外显子3的隐蔽剪接位点,导致突变型mRNA缺少一个碱基G,预测TSR1蛋白第68位氨基酸后发生移码,并于其后46位形成终止密码子,p.(Phe68Valfs*46) (图2B),与human splicing finder 3.1在线软件预测结果一致。图2
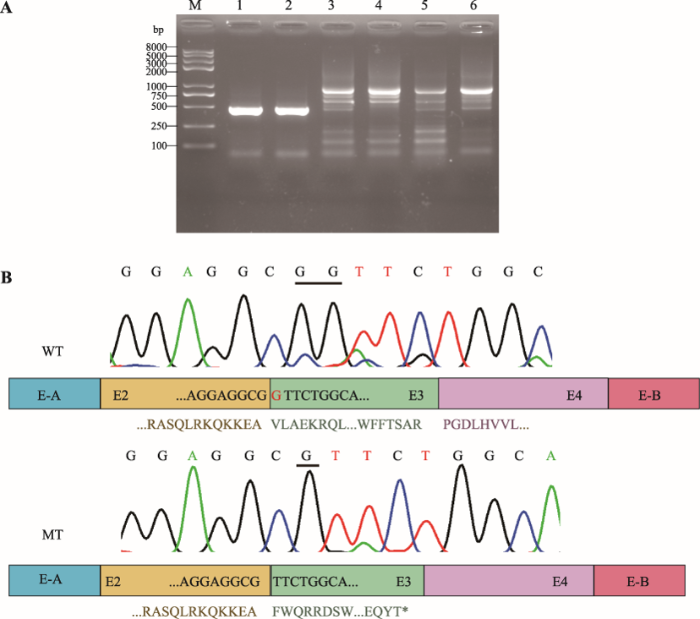 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2Minigene分析
A:RT-PCR产物电泳结果。M:DNA Marker trans2k plusII;1:pCAS2空白载体;2:pCAS2空白载体;3:野生型pCAS2-TSR1质粒;4:野生型pCAS2-TSR1质粒;5:突变型pCAS2-TSR1c.202-1G>A质粒;6:突变型pCAS2-TSR1c.202-1G>A质粒。B:野生型和突变型RT-PCR产物Sanger测序结果。WT:野生型;MT:突变型。野生型TSR1可以正常转录,突变型产生缺失一个碱基“G”的转录本,导致TSR1蛋白移码。*形成终止密码子。
Fig. 2Minigene assays
2.3 RT-PCR检测小鼠各组织Tsr1表达
以小鼠晶状体、大脑、肝脏、脾脏、肺和肾脏组织cDNA为模板进行RT-PCR,结果显示Tsr1基因在所选取的6种小鼠组织中均有表达(图3),表达量并无明显差异。图3
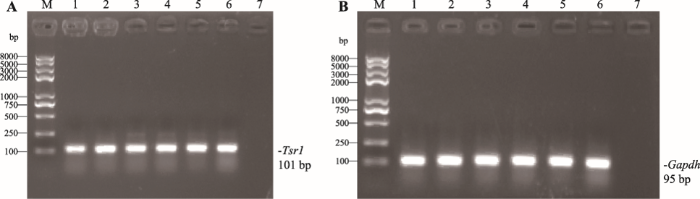 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3Tsr1在小鼠不同组织中的表达
A:Tsr1在小鼠各组织中的表达。B: GAPDH在小鼠各组织中的表达。M:DNA Marker trans2k plusⅡ;1:晶状体;2:脑;3:肝;4:脾;5:肺;6:肾;7:空白对照。
Fig. 3Expression of Tsr1 in mouse different tissues
2.4 Western blotting检测人晶状体上皮细胞SRA01/04中TSR1表达
Western blotting结果显示TSR1在人晶状体上皮细胞SRA01/04细胞核和细胞质中均有表达,且表达量都很高(图4)。图4
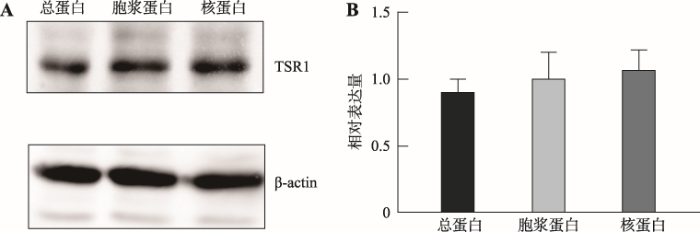 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4TSR1在SRA01/04细胞中的表达
A:Western blotting检测TSR1在SRA01/04中的表达。;B:定量分析TSR1在SRA01/04中的表达。TSR1在细胞质和细胞核中均有表达,没有显著差异。
Fig. 4Expression of TSR1 in SRA 01/04 cells
2.5 免疫荧光实验检测人晶状体中TSR1表达和定位
免疫荧光结果显示TSR1在年龄相关性白内障患者晶状体前囊膜组织细胞有表达,在细胞核和细胞质中均有表达(图5A)。在24周人胎眼晶状体纤维细胞、晶状体上皮细胞以及晶状体前囊组织中均有表达(图5B)。图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5免疫荧光法检测人晶状体TSR1的表达及定位
A:年龄相关性白内障患者晶状体前囊免疫荧光结果。绿色荧光:TSR1;红色荧光:β-actin;蓝色:DAPI。B:24周人胎眼晶状体组织免疫荧光结果。绿色荧光:TSR1;蓝色:DAPI。
Fig. 5Expression and intracellular localization of TSR1 detected by immunofluorescence
2.6 生物信息学分析结果
Tsr1在胚胎期和出生后的小鼠晶状体组织中持续表达(图6A),晶状体特异性CBP:p300双敲除的小鼠晶状体中表达下调(图6B)。使用iSyTE的Co-expression功能获取双敲除小鼠中与Tsr1表达模式相同的一组基因进行GO功能、KEGG信号通路分析及PPI分析,GO分析结果显示,与Tsr1表达模式相同的基因主要参与了代谢、生物调节、应激图6
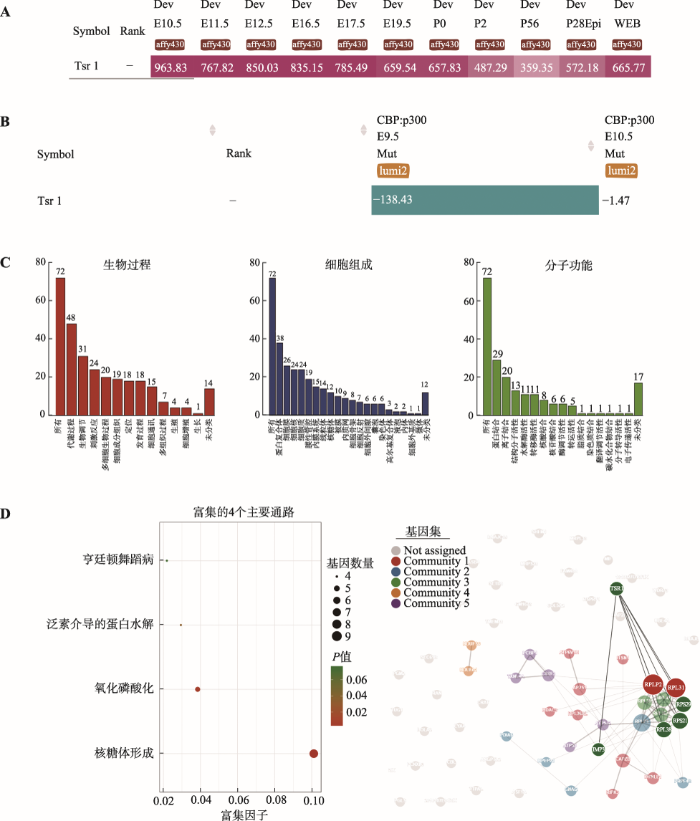 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6TSR1表达模式、GO功能、KEGG信号通路及PPI等生物信息学分析
A:iSyTE数据库中Tsr1表达模式。检索iSyTE数据库,发现Tsr1在小鼠晶状体发育各个阶段均持续表达。B:CBP: p300双敲除小鼠晶状体Tsr1表达。CBP: p300双敲除小鼠晶状体Tsr1表达下降。C:GO功能分析。提取与Tsr1表达模式相同的基因进行GO分析。D:KEGG分析。KEGG分析结果表明基因主要被富集到核糖体形成、氧化磷酸化、泛素介导的蛋白水解和亨廷顿舞蹈病4个通路。E:PPI分析。提取与Tsr1表达模式相同的基因进行蛋白-蛋白相互作用网络分析,发现表达下降的基因其编码蛋白Rps21、Rps29、Rpl38、Rplp2、Rpl31和Imp3与Tsr1直接相互作用。
Fig. 6Bioinformatics analysis of TSR1 expression, GO function, KEGG signal pathways and PPI
等生物过程。在分子功能方面,主要参与核糖体构成、金属离子结合等相关功能(图6C)。KEGG信号通路分析显示与Tsr1表达模式相同的基因在核糖体形成、氧化磷酸化、泛素介导的蛋白水解和亨廷顿舞蹈病4个通路中富集(图6D)。PPI结果显示基因被聚类到5个不同的信号通路上,其中Rps21、Rps29、Rpl38、Rplp2、Rpl31和Imp3与Tsr1存在直接相互作用(图6E)。
3 讨论
先天性白内障遗传异质性高,致病基因众多,致病机制复杂,同一基因突变在不同家系中的表型可能不同,即使在一个家系内,患者的表型也可能不尽相同。这也导致临床诊断和遗传学诊断困难重重。随着高通量测序技术的发展和人类基因组数据库的完善,已经有越来越多的先天性白内障致病基因和突变位点被发现[11,12,13,14,15,16]。先天性白内障是最早被发现呈常染色体显性遗传的人类疾病。近年来,国内多个研究小组利用中国人群的白内障家系资源优势,在致病基因定位克隆和突变基因鉴定方面取得了突出成绩。2002年,中国科学院上海生命科学研究院孔祥银团队发现编码热休克转录因子家族成员的HSF4基因突变可引起常染色体显性板层白内障和Marner白内障[17]。2005年,中国医学科学院沈岩团队报道CRYGS基因突变导致皮质性白内障[18]。2009年,中国医学科学院张学团队发现EPHA2基因突变导致后极性白内障[19]。2014年,青岛眼科医院谢立信团队报道ABCA3基因突变导致白内障小角膜综合征[20]。2019年,中国人民解放军第三军医大学大坪医院叶剑团队报道PANK4基因突变导致后极性白内障[21]。
本研究通过全基因组测序技术,在一个中国常染色体显性遗传先天性白内障家系检测到一个可疑的候选基因变异TSR1 c.202-1G>A,p.(Phe68Valfs*46),该变异在家系中与白内障表型共分离,正常人数据库未见该位点的报道,疾病数据库中未见该基因与人类疾病的相关报道,105名正常对照个体未携带此突变,因此TSR1可能是一个新先天性白内障致病基因。
TSR1(NM_018128.4)位于17p13.3,含15个外显子,编码804个氨基酸。TSR1编码前体rRNA加工蛋白,在40S核糖体成熟过程中发挥重要作用[22]。TSR1基因c.202-1G位点高度保守,minigene实验证实该变异影响了TSR1基因mRNA剪接导致蛋白移码。RT-PCR、免疫荧光和Western blotting实验证实了TSR1在小鼠和人的晶状体组织和细胞系中均有表达。通过检索iSyTE数据库,本研究发现Tsr1在胚胎期和出生后的小鼠晶状体组织中持续表达,且Tsr1在CBP:p300双敲除的白内障小鼠晶状体中表达下调[23],推测其可能在晶状体的发育过程中发挥重要作用并参与了敲除小鼠白内障表型的形成。通过敲除小鼠晶状体中与Tsr1表达模式相同基因的蛋白质-蛋白质相互作用(PPI)分析发现,表达下调的基因中参与核糖体形成的Rps21、Rps29、Rps38、Rplp2、Rpl31和Imp3均与Tsr1有直接相互作用,其中Rps21、Rps29、Rps38、Rplp2和Rpl31均为核糖体蛋白,参与核糖体的组装和成熟[24,25,26,27]。目前已有报道核糖体蛋白L21、L15、L13a和L7a表达下调可以导致年龄相关性白内障,可能原因是核糖体异常导致的晶状体蛋白合成障碍[28]。由于没有匹配的相同年龄性别的正常人晶状体前囊膜组织做对照,本研究只检测了TSR1在年龄相关性白内障患者前囊膜组织有表达,没有进行定量分析其表达变化情况。在CBP:p300双敲除小鼠晶状体中,晶状体蛋白α、β和γ家族基因和多个晶状体发育的关键转录因子表达均有不同程度的下调[23],推测这些基因的表达下调可能是由核糖体的组装异常引起的。TSR1作为核糖体组装的重要成分之一,可能与其他表达下调的核糖体蛋白一同参与双敲除小鼠晶状体表型形成。在表达下调的基因中,与Tsr1存在直接相互作用的Imp3蛋白参与MAPK-Erk信号通路,Imp3通过失活衔接蛋白Ksr1进而抑制MAPK-Erk通路[29]。在晶状体中,MAPK信号通路的增强可以诱导晶状体上皮细胞的凋亡,进而抑制白内障的发生[30]。当Imp3基因下调时,Ksr1对MAPK通路的抑制作用增强,晶状体上皮细胞凋亡减少,细胞过量增殖和迁移,最终可能导致白内障的发生。Tsr1与Imp3的相互作用机制尚不明确,Tsr1是否能与Imp3一同调节MAPK通路,进而影响晶状体细胞的凋亡和增殖还需要进一步研究确定。
综上所述,本研究在一个中国先天性白内障家系中通过全基因组测序及Sanger测序验证,检测到TSR1变异c.202-1G>A,p.(Phe68Valfs*46),与家系疾病表型共分离。通过minigene实验证实该变异影响了TSR1基因mRNA剪接。Western blotting、免疫荧光和RT-PCR实验证实了TSR1在人晶状体上皮细胞SRA01/04、年龄相关性白内障患者晶状体前囊膜组织、24周人胎眼晶状体和小鼠晶状体中表达。利用iSyTE、GO分析、KEGG分析和PPI数据库分析,为进一步明确TSR1在晶状体中的功能提供了有价值的研究线索。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1016/s0886-3350(97)80040-5URLPMID:9278811 [本文引用: 1]

Cataract is the most important cause of treatable childhood blindness. There are an estimated 200,000 children blind from cataract worldwide; 20,000 to 40,000 children with developmental bilateral cataract are born each year. Rubella is still an important cause of preventable disease in many countries. In the developing world, there is a need to improve early case detection and referral services and to establish centers with expertise in the assessment, surgical treatment, and long-term management of the child with cataract.
URLPMID:11285667 [本文引用: 1]
The major causes of blindness in children vary widely from region to region, being largely determined by socioeconomic development, and the availability of primary health care and eye care services. In high-income countries, lesions of the optic nerve and higher visual pathways predominate as the cause of blindness, while corneal scarring from measles, vitamin A deficiency, the use of harmful traditional eye remedies, and ophthalmia neonatorum are the major causes in low-income countries. Retinopathy of prematurity is an important cause in middle-income countries. Other significant causes in all countries are cataract, congenital abnormalities, and hereditary retinal dystrophies. It is estimated that, in almost half of the children who are blind today, the underlying cause could have been prevented, or the eye condition treated to preserve vision or restore sight. The control of blindness in children is a priority within the World Health Organization's VISION 2020 programme. Strategies need to be region specific, based on activities to prevent blindness in the community--through measles immunization, health education, and control of vitamin A deficiency--and the provision of tertiary-level eye care facilities for conditions that require specialist management.
DOI:10.1038/eye.1995.137URLPMID:8543070 [本文引用: 1]
It is estimated that at least 200,000 children in India have severe visual impairment or blindness and approximately 15,000 are in schools for the blind. Although this represents a small percentage of the estimated 5 million blind in India, it is significant in terms of 'blind-years'. Strategies to combat childhood blindness require accurate data on the causes to allocate resources to appropriate preventive and curative services. Since socio-economic factors vary in different areas of this industrializing country data should be representative of the country as a whole. This is the first multi-state study to be undertaken in India using the Record for Children with Blindness and Low Vision from the World Health Organization/PBL Programme. A total of 1411 children in 22 schools from nine states in different geographical zones were examined by an ophthalmologist and optometrist. Of these, 1318 children were severely visually impaired or blind (SVI/BL). The major causes of SVI/BL in this study were: (1) corneal staphyloma, scar and phthisis bulbi (mainly attributable to vitamin A deficiency) in 26.4%; (2) microphthalmos, anophthalmos and coloboma in 20.7%; (3) retinal dystrophies and albinism in 19.3%; and (4) cataract, uncorrected aphakia and amblyopia in 12.3%. This mixed pattern of causes lies in an intermediate position between the patterns seen in developing countries and those seen in industrialised countries. The causes identified indicate the importance both of preventive public health strategies and of specialist paediatric ophthalmic and optical services in the management of childhood blindness in India.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/hgv.2015.34URLPMID:27081543 [本文引用: 1]
Warburg micro syndrome is an autosomal recessive disease where patients present with optic, neurologic and genital symptoms. Until now, four disease genes for Warburg micro syndrome, RAB3GAP1, RAB3GAP2, RAB18 and TBC1D20, have been identified. Here, we report two novel homozygous RAB3GAP1 mutations (c.22G>T, p.Glu8* and c.1353delA, p.Pro452Hisfs*5) in two consanguineous families by whole-exome sequencing.
DOI:10.1016/j.jns.2017.02.058URLPMID:28320181 [本文引用: 1]
Peroxisome biogenesis factor 10 (PEX10) is involved in the import of peroxisomal matrix proteins, and the mutation of this gene causes 3 subtypes of peroxisome biogenesis disorders, namely Zellweger syndrome (severe), neonatal adrenoleukodystrophy (moderate) and an ataxic form (mild). Here, we report 3 siblings of the ataxic form with cerebellar ataxia, mild mental retardation, and 3 additional characteristic features: mydriasis, hyperreflexia and involuntary head movement. All 3 siblings are compound heterozygous for a previously reported mutation, c.2T>C (p.M1T), and a novel mutation, c.920G>A, causing a missense change (p.C307Y) located in the RING finger domain of PEX10. The present cases suggest that these PEX10 mutations involve not only cerebellar but also more multiple nervous systems including pupillary autonomic, pyramidal and extrapyramidal systems.
DOI:10.2337/db16-1296URLPMID:28468959 [本文引用: 1]
Neonatal diabetes is frequently part of a complex syndrome with extrapancreatic features: 18 genes causing syndromic neonatal diabetes have been identified to date. There are still patients with neonatal diabetes who have novel genetic syndromes. We performed exome sequencing in a patient and his unrelated, unaffected parents to identify the genetic etiology of a syndrome characterized by neonatal diabetes, sensorineural deafness, and congenital cataracts. Further testing was performed in 311 patients with diabetes diagnosed before 1 year of age in whom all known genetic causes had been excluded. We identified 5 patients, including the initial case, with three heterozygous missense mutations in WFS1 (4/5 confirmed de novo). They had diabetes diagnosed before 12 months (2 before 6 months) (5/5), sensorineural deafness diagnosed soon after birth (5/5), congenital cataracts (4/5), and hypotonia (4/5). In vitro studies showed that these WFS1 mutations are functionally different from the known recessive Wolfram syndrome-causing mutations, as they tend to aggregate and induce robust endoplasmic reticulum stress. Our results establish specific dominant WFS1 mutations as a cause of a novel syndrome including neonatal/infancy-onset diabetes, congenital cataracts, and sensorineural deafness. This syndrome has a discrete pathophysiology and differs genetically and clinically from recessive Wolfram syndrome.
URLPMID:17417607 [本文引用: 1]
Nance-Horan Syndrome (NHS) is an infrequent and often overlooked X-linked disorder characterized by dense congenital cataracts, microphthalmia, and dental abnormalities. The syndrome is caused by mutations in the NHS gene, whose function is not known. The purpose of this study was to identify the frequency and distribution of NHS gene mutations and compare genotype with Nance-Horan phenotype in five North American NHS families.
DOI:10.1002/humu.22948URLPMID:26694549 [本文引用: 1]
Congenital cataracts are a significant cause of lifelong visual loss. They may be isolated or associated with microcornea, microphthalmia, anterior segment dysgenesis (ASD) and glaucoma, and there can be syndromic associations. Genetic diagnosis is challenging due to marked genetic heterogeneity. In this study, next-generation sequencing (NGS) of 32 cataract-associated genes was undertaken in 46 apparently nonsyndromic congenital cataract probands, around half sporadic and half familial cases. We identified pathogenic variants in 70% of cases, and over 68% of these were novel. In almost two-thirds (20/33) of these cases, this resulted in new information about the diagnosis and/or inheritance pattern. This included identification of: new syndromic diagnoses due to NHS or BCOR mutations; complex ocular phenotypes due to PAX6 mutations; de novo autosomal-dominant or X-linked mutations in sporadic cases; and mutations in two separate cataract genes in one family. Variants were found in the crystallin and gap junction genes, including the first report of severe microphthalmia and sclerocornea associated with a novel GJA8 mutation. Mutations were also found in rarely reported genes including MAF, VIM, MIP, and BFSP1. Targeted NGS in presumed nonsyndromic congenital cataract patients provided significant diagnostic information in both familial and sporadic cases.
DOI:10.1007/s00439-016-1747-6URLPMID:27878435 [本文引用: 1]
Pediatric cataract is highly heterogeneous clinically and etiologically. While mostly isolated, cataract can be part of many multisystem disorders, further complicating the diagnostic process. In this study, we applied genomic tools in the form of a multi-gene panel as well as whole-exome sequencing on unselected cohort of pediatric cataract (166 patients from 74 families). Mutations in previously reported cataract genes were identified in 58% for a total of 43 mutations, including 15 that are novel. GEMIN4 was independently mutated in families with a syndrome of cataract, global developmental delay with or without renal involvement. We also highlight a recognizable syndrome that resembles galactosemia (a fulminant infantile liver disease with cataract) caused by biallelic mutations in CYP51A1. A founder mutation in RIC1 (KIAA1432) was identified in patients with cataract, brain atrophy, microcephaly with or without cleft lip and palate. For non-syndromic pediatric cataract, we map a novel locus in a multiplex consanguineous family on 4p15.32 where exome sequencing revealed a homozygous truncating mutation in TAPT1. We report two further candidates that are biallelically inactivated each in a single cataract family: TAF1A (cataract with global developmental delay) and WDR87 (non-syndromic cataract). In addition to positional mapping data, we use iSyTE developmental lens expression and gene-network analysis to corroborate the proposed link between the novel candidate genes and cataract. Our study expands the phenotypic, allelic and locus heterogeneity of pediatric cataract. The high diagnostic yield of clinical genomics supports the adoption of this approach in this patient group.
DOI:10.1534/g3.117.300109URLPMID:28839118 [本文引用: 1]
Pediatric cataract is a leading cause of childhood blindness. This study aimed to determine the genetic cause of pediatric cataract in Australian families by screening known disease-associated genes using massively parallel sequencing technology. We sequenced 51 previously reported pediatric cataract genes in 33 affected individuals with a family history (cases with previously known or published mutations were excluded) using the Ion Torrent Personal Genome Machine. Variants were prioritized for validation if they were predicted to alter the protein sequence and were absent or rare with minor allele frequency <1% in public databases. Confirmed mutations were assessed for segregation with the phenotype in all available family members. All identified novel or previously reported cataract-causing mutations were screened in 326 unrelated Australian controls. We detected 11 novel mutations in GJA3, GJA8, CRYAA, CRYBB2, CRYGS, CRYGA, GCNT2, CRYGA, and MIP; and three previously reported cataract-causing mutations in GJA8, CRYAA, and CRYBB2 The most commonly mutated genes were those coding for gap junctions and crystallin proteins. Including previous reports of pediatric cataract-associated mutations in our Australian cohort, known genes account for >60% of familial pediatric cataract in Australia, indicating that still more causative genes remain to be identified.
DOI:10.1016/j.ophtha.2014.06.006URL [本文引用: 1]
Purpose: To assess the utility of integrating genomic data from next-generation sequencing and phenotypic data to enhance the diagnosis of bilateral congenital cataract (CC).
Design: Evaluation of diagnostic technology.
Participants: Thirty-six individuals diagnosed with nonsyndromic or syndromic bilateral congenital cataract were selected for investigation through a single ophthalmic genetics clinic.
Methods: Participants underwent a detailed ophthalmic examination, accompanied by dysmorphology assessment where appropriate. Lenticular, ocular, and systemic phenotypes were recorded. Mutations were detected using a custom-designed target enrichment that permitted parallel analysis of 115 genes associated with CC by high-throughput, next-generation DNA sequencing (NGS). Thirty-six patients and a known positive control were tested. Suspected pathogenic variants were confirmed by bidirectional Sanger sequencing in relevant probands and other affected family members.
Main Outcome Measures: Molecular genetic results and details of clinical phenotypes were identified.
Results: Next-generation DNA sequencing technologies are able to determine the precise genetic cause of CC in 75% of individuals, and 85% patients with nonsyndromic CC were found to have likely pathogenic mutations, all of which occurred in highly conserved domains known to be vital for normal protein function. The pick-up rate in patients with syndromic CC also was high, with 63% having potential disease-causing mutations.
Conclusions: This analysis demonstrates the clinical utility of this test, providing examples where it altered clinical management, directed care pathways, and enabled more accurate genetic counseling. This comprehensive screen will extend access to genetic testing and lead to improved diagnostic and management outcomes through a stratified medicine approach. Establishing more robust genotype-phenotype correlations will advance knowledge of cataract-forming mechanisms. (C) 2014 by the American Academy of Ophthalmology.
[本文引用: 1]
[本文引用: 1]
DOI:10.1360/yc-007-0137URLPMID:17369166 [本文引用: 1]
Cataract is a serious public health problem. It is usually inherited as an autosomal dominant trait, although autosomal recessive and X-linked inheritance are seen less commonly. With the development of molecular biology techniques, a large number of inherited cataract models are produced, which will reveal the pathogenesis of cataracts and provide a new view for the development and physiology of the lens. In addition, these models also facilitate our understanding of the manner of inheritance, the effect of the environment and nutrition on the lens and provide clues for the diagnosis and treatment of cataracts. Here we presented the relative genes of the animal models for inherited cataract, mutation forms and its progress.
DOI:10.1360/yc-007-0137URLPMID:17369166 [本文引用: 1]
Cataract is a serious public health problem. It is usually inherited as an autosomal dominant trait, although autosomal recessive and X-linked inheritance are seen less commonly. With the development of molecular biology techniques, a large number of inherited cataract models are produced, which will reveal the pathogenesis of cataracts and provide a new view for the development and physiology of the lens. In addition, these models also facilitate our understanding of the manner of inheritance, the effect of the environment and nutrition on the lens and provide clues for the diagnosis and treatment of cataracts. Here we presented the relative genes of the animal models for inherited cataract, mutation forms and its progress.
DOI:10.1038/ng921URLPMID:12089525 [本文引用: 1]
Congenital cataracts cause 10-30% of all blindness in children, with one-third of cases estimated to have a genetic cause. Lamellar cataract is the most common type of infantile cataract. We carried out whole-genome linkage analysis of Chinese individuals with lamellar cataract, and found that the disorder is associated with inheritance of a 5.11-cM locus on chromosome 16. This locus coincides with one previously described for Marner cataract. We screened individuals of three Chinese families for mutations in HSF4 (a gene at this locus that encodes heat-shock transcription factor 4) and discovered that in each family, a distinct missense mutation, predicted to affect the DNA-binding domain of the protein, segregates with the disorder. We also discovered an association between a missense mutation and Marner cataract in an extensive Danish family. We suggest that HSF4 is critical to lens development.
DOI:10.1136/jmg.2004.028274URLPMID:16141006 [本文引用: 1]
Congenital or childhood cataract is clinically and genetically a highly heterogeneous lens disorder in children. Autosomal dominant inheritance is most common.
DOI:10.1002/humu.20995URLPMID:19306328 [本文引用: 1]
Congenital cataracts (CCs) are clinically and genetically heterogeneous. Mutations in the same gene may lead to CCs differing in inheritance, morphology and severity. Loci for autosomal dominant posterior polar CC and total CC have both been mapped to the chromosomal 1p36 region harboring the EPHA2 receptor tyrosine kinase gene. Here, we report mutations of EPHA2 in three CC families from different ancestral groups. In a Chinese family with posterior polar CC, we identified a missense mutation, c.2819C&gt;T (p.T940I), replacing a critical amino acid that functions at the receptor oligomerization interface. In a British family with posterior polar CC and an Australian family with total CC, we found a frameshift mutation (c.2915_2916delTG) and a splicing mutation (c.2826-9G&gt;A), respectively. These two mutations are predicted to produce novel C-terminal polypeptides with 39 identical amino acids. Yeast two-hybrid analysis showed stronger interaction between the total CC-associated mutant EPHA2 and low molecular weight protein-tyrosine phosphatase, a negative regulator of EPHA2 signaling. Our results implicate the Eph-ephrin signaling system in development of human cataract and provide a novel insight into the molecular mechanism underlying the pathogenesis of human CCs.
DOI:10.1167/iovs.14-14098URLPMID:25406294 [本文引用: 1]
Cataract-microcornea syndrome (CCMC) is an autosomal dominant inherited disease characterized by the association of congenital cataract and microcornea without any other systemic anomaly or dysmorphism. Although mutations of several genes have been shown to cause dominant CCMC, in many patients the causative gene has not yet been identified. Our aim was to identify the disease-associated gene in Chinese patients with CCMC.
DOI:10.1002/humu.23696URLPMID:30585370 [本文引用: 1]
Though many mutations have been identified to be associated with the occurrence of congenital cataract, pathogenic loci in some affected families are still unknown. Clinical data and genomic DNA were collected from a four-generation Chinese family. Candidate mutations were independently verified for cosegregation in the whole pedigree. Linkage analysis showed that the disease-causing mutation was located between 1p36.21 and 1p36.33. Analysis of the whole-exome sequencing data combined with linkage analysis identified a novel pathogenic variant (g.2451906C>T) at intron 4 of Pantothenate kinase 4 (PANK4 protein, PANK4 gene) in 1p36.32|606162. This variant showed complete cosegregation with the phenotype in the pedigree. The mutation was not detected in 106 normal controls nor in 40 sporadic congenital cataract patients. The mutation was demonstrated to significantly reduce the expression of the PANK4 protein level in the blood of cataract patients than that in normal individuals by ELISA. Pank4-/- mice showed a cataract phenotype with increased numbers of apoptotic lens epithelial cells, fiber cell aggregation, and significant mRNA variation of crystallin family members. Thus, the association of a new entity of an autosomal dominant cataract with mutations in PANK4, which influences cell proliferation, apoptosis of lens epithelial cells, crystallin abnormalities, and fiber cell derangement, subsequently induces cataract.
DOI:10.15252/embj.201798499URLPMID:29459436 [本文引用: 1]
Final maturation of eukaryotic ribosomes occurs in the cytoplasm and requires the sequential removal of associated assembly factors and processing of the immature 20S pre-RNA Using cryo-electron microscopy (cryo-EM), we have determined the structure of a yeast cytoplasmic pre-40S particle in complex with Enp1, Ltv1, Rio2, Tsr1, and Pno1 assembly factors poised to initiate final maturation. The structure reveals that the pre-rRNA adopts a highly distorted conformation of its 3' major and 3' minor domains stabilized by the binding of the assembly factors. This observation is consistent with a mechanism that involves concerted release of the assembly factors orchestrated by the folding of the rRNA in the head of the pre-40S subunit during the final stages of maturation. Our results provide a structural framework for the coordination of the final maturation events that drive a pre-40S particle toward the mature form capable of engaging in translation.
DOI:10.1093/nar/gkt824URLPMID:24038357 [本文引用: 2]
Lens induction is a classical embryologic model to study cell fate determination. It has been proposed earlier that specific changes in core histone modifications accompany the process of cell fate specification and determination. The lysine acetyltransferases CBP and p300 function as principal enzymes that modify core histones to facilitate specific gene expression. Herein, we performed conditional inactivation of both CBP and p300 in the ectodermal cells that give rise to the lens placode. Inactivation of both CBP and p300 resulted in the dramatic discontinuation of all aspects of lens specification and organogenesis, resulting in aphakia. The CBP/p300(-/-) ectodermal cells are viable and not prone to apoptosis. These cells showed reduced expression of Six3 and Sox2, while expression of Pax6 was not upregulated, indicating discontinuation of lens induction. Consequently, expression of αB- and αA-crystallins was not initiated. Mutant ectoderm exhibited markedly reduced levels of histone H3 K18 and K27 acetylation, subtly increased H3 K27me3 and unaltered overall levels of H3 K9ac and H3 K4me3. Our data demonstrate that CBP and p300 are required to establish lens cell-type identity during lens induction, and suggest that posttranslational histone modifications are integral to normal cell fate determination in the mammalian lens.
DOI:10.1002/ajh.23807URLPMID:25042156 [本文引用: 1]
Diamond Blackfan anemia (DBA), a syndrome primarily characterized by anemia and physical abnormalities, is one among a group of related inherited bone marrow failure syndromes (IBMFS) which share overlapping clinical features. Heterozygous mutations or single-copy deletions have been identified in 12 ribosomal protein genes in approximately 60% of DBA cases, with the genetic etiology unexplained in most remaining patients. Unlike many IBMFS, for which functional screening assays complement clinical and genetic findings, suspected DBA in the absence of typical alterations of the known genes must frequently be diagnosed after exclusion of other IBMFS. We report here a novel deletion in a child that presented such a diagnostic challenge and prompted development of a novel functional assay that can assist in the diagnosis of a significant fraction of patients with DBA. The ribosomal proteins affected in DBA are required for pre-rRNA processing, a process which can be interrogated to monitor steps in the maturation of 40S and 60S ribosomal subunits. In contrast to prior methods used to assess pre-rRNA processing, the assay reported here, based on capillary electrophoresis measurement of the maturation of rRNA in pre-60S ribosomal subunits, would be readily amenable to use in diagnostic laboratories. In addition to utility as a diagnostic tool, we applied this technique to gene discovery in DBA, resulting in the identification of RPL31 as a novel DBA gene.
DOI:10.1042/BJ20080049URLPMID:18422483 [本文引用: 1]
The 'stalk' is a large ribosomal subunit domain that regulates translation. In the present study the role of the ribosomal stalk P proteins in modulating ribosomal activity has been investigated in human cells using RNA interference. A strong down-regulation of P2 mRNA and a drastic decrease in P2 protein in a stable human cell line was achieved using a doxycycline-inducible system. Interestingly, the amount of P1 protein was similarly decreased in these cells, in contrast with the expression of P1 mRNA. The loss of P1/P2 proteins produced a decrease in the growth rate of these cells, as well as an altered polysome pattern with reduced translation efficiency, but without affecting the free 40 S/60 S subunit ratio. A decrease in the ribosomal-subunit joining capacity was also observed. These data indicate that P1/P2 proteins modulate cytoplasmic translation by influencing the interaction between subunits, thereby regulating the rate of cell proliferation.
DOI:10.1182/blood-2013-11-540278URLPMID:24829207 [本文引用: 1]
Diamond-Blackfan anemia (DBA) is a cancer-prone inherited bone marrow failure syndrome. Approximately half of DBA patients have a germ-line mutation in a ribosomal protein gene. We used whole-exome sequencing to identify disease-causing genes in 2 large DBA families. After filtering, 1 nonsynonymous mutation (p.I31F) in the ribosomal protein S29 (RPS29[AUQ1]) gene was present in all 5 DBA-affected individuals and the obligate carrier, and absent from the unaffected noncarrier parent in 1 DBA family. A second DBA family was found to have a different nonsynonymous mutation (p.I50T) in RPS29. Both mutations are amino acid substitutions in exon 2 predicted to be deleterious and resulted in haploinsufficiency of RPS29 expression compared with wild-type RPS29 expression from an unaffected control. The DBA proband with the p.I31F RPS29 mutation had a pre-ribosomal RNA (rRNA) processing defect compared with the healthy control. We demonstrated that both RPS29 mutations failed to rescue the defective erythropoiesis in the rps29(-/-) mutant zebra fish DBA model. RPS29 is a component of the small 40S ribosomal subunit and essential for rRNA processing and ribosome biogenesis. We uncovered a novel DBA causative gene, RPS29, and showed that germ-line mutations in RPS29 can cause a defective erythropoiesis phenotype using a zebra fish model.
DOI:10.1371/journal.pone.0186047URLPMID:29016636 [本文引用: 1]
Few quantifiable tissue biomarkers for the diagnosis and prognosis of prostate cancer exist. Using an unbiased, quantitative approach, this study evaluates the potential of three proteins of the 40S ribosomal protein complex as putative biomarkers of malignancy in prostate cancer. Prostate tissue arrays, constructed from 82 patient samples (245 tissue cores, stage pT3a or pT3b), were stained for antibodies against three ribosomal proteins, RPS19, RPS21 and RPS24. Semi-automated Ox-DAB signal quantification using ImageJ software revealed a significant change in expression of RPS19, RPS21 and RPS24 in malignant vs non-malignant tissue (p&lt;0.0001). Receiver operating characteristics curves were calculated to evaluate the potential of each protein as a biomarker of malignancy in prostate cancer. Positive likelihood ratios for RPS19, RPS21 and RPS24 were calculated as 2.99, 4.21, and 2.56 respectively, indicating that the overexpression of the protein is correlated with the presence of disease. Triple-labelled, quantitative, immunofluorescence (with RPS19, RPS21 and RPS24) showed significant changes (p&lt;0.01) in the global intersection coefficient, a measure of how often two fluorophore signals intersect, for RPS19 and RPS24 only. No change was observed in the co-localization of any other permutations of the three proteins. Our results show that RPS19, RPS21 or RPS24 are upregulated in malignant tissue and may serve as putative biomarkers for prostate cancer.
URLPMID:11773032 [本文引用: 1]
To identify lens epithelial genes with altered expression levels in age-related human cataract.
DOI:10.1074/jbc.M112.386938URLPMID:23105109 [本文引用: 1]
The opposing regulators of ubiquitylation status, E3 ligases and deubiquitylases, are often found to be associated in complexes. Here we report on a novel interaction between the E3 ligase BRAP (also referred to as IMP), a negative regulator of the MAPK scaffold protein KSR, and two closely related deubiquitylases, USP15 and USP4. We map the interaction to the N-terminal DUSP-UBL domain of USP15 and the coiled coil region of BRAP. USP15 as well as USP4 oppose the autoubiquitylation of BRAP, whereas BRAP promotes the ubiquitylation of USP15. Importantly, USP15 but not USP4 depletion destabilizes BRAP by promoting its proteasomal degradation, and BRAP-protein levels can be rescued by reintroducing catalytically active but not inactive mutant USP15. Unexpectedly, USP15 depletion results in a decrease in amplitude of MAPK signaling in response to EGF and PDGF. We provide evidence for a model in which the dominant effect of prolonged USP15 depletion upon signal amplitude is due to a decrease in CRAF levels while allowing for the possibility that USP15 may also function to dampen MAPK signaling through direct stabilization of a negative regulator, the E3 ligase BRAP.
DOI:10.3892/mmr.2019.10087URLPMID:30942394 [本文引用: 1]
Posterior capsular opacification (PCO) remains a major complication of cataract surgery and is the most common reason for loss of vision. PCO is primarily associated with uncontrolled proliferation of residual human lens epithelial cells (HLEs). Sanguinarine is a type of benzophenanthridine alkaloid extracted from the herbaceous plant Sanguinaria?canadensis, which is widely used for its anti?microbial, anti?inflammatory, anti?oxidative and anti?proliferative properties. However, studies examining the effect of sanguinarine on HLEs and the underlying mechanism are scarce. The present study aimed to investigate the effects of sanguinarine on HLEs. An MTT assay was used to determine the effect of sanguinarine on cell viability. Flow cytometry was used to evaluate cell apoptosis, and the mitochondrial membrane potential and reactive oxygen species (ROS) levels. A caspase 3/7 activity assay was also used to evaluate cell apoptosis, while western blotting was performed to determine protein levels. The results demonstrated that sanguinarine exerted an anti?proliferative effect by inducing ROS, and caused cell apoptosis via mitochondrial and caspase?dependent pathways. Treatment with sanguinarine led to the loss of mitochondrial membrane potential. Sanguinarine also significantly increased the phosphorylation levels of c?Jun N?terminal kinase and p38, which indicated the involvement of the mitogen?activated protein kinase signaling pathway. These results suggested that sanguinarine may have a noteworthy pro?apoptotic effect on HLEs, and may be used as a potential drug for PCO or even cataract prevention.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}