 ,西北大学生命科学学院,西部资源生物与现代生物技术教育部重点实验室,西安 710069
,西北大学生命科学学院,西部资源生物与现代生物技术教育部重点实验室,西安 710069Brachydactyly and the molecular mechanisms of digit formation
Zhaojie Lyu, Zhihao Wang, Shuxian Lu, Peirong Liu, Jing Tian,Key Laboratory of Resource Biology and Biotechnology in Western China(Ministry of Education), College of Life Sciences, Northwest University, Xi’an 710069, China通讯作者: 田静,博士,教授,研究方向:人类遗传与发育生物学。E-mail:tianjing@nwu.edu.cn
编委: 谢小冬
收稿日期:2019-06-27修回日期:2019-08-9网络出版日期:2019-12-20
| 基金资助: |
Editorial board:
Received:2019-06-27Revised:2019-08-9Online:2019-12-20
| Fund supported: |
作者简介 About authors
吕赵劼,硕士研究生,专业方向:人类遗传与发育生物学。E-mail:ZhaojieLyu@outlook.com。

摘要
短指(趾)症(brachydactyly, BD)是一类指(趾)骨或掌(跖)骨的异常缩短或缺失而造成的手/足畸形病变。从临床表型上短指(趾)症可以分为单纯型短指(趾)症以及包含短指(趾)症状的综合征,其中单纯型短指(趾)症又分为5种类型:BDA、BDB、BDC、BDD和BDE,而每一类型又分为不同的亚型。作为一类重要的分子疾病家族,随着对每种短指(趾)症的深入研究,大多数单纯型短指(趾)症和部分综合征的致病基因及其分子机制逐渐被发现。虽然短指(趾)症在表型上高度多样化,但在分子水平上这些致病基因主要影响Hedgehog、NOTCH、WNT和BMP等信号传导通路。这些信号传导通路组成了一个复杂的信号调控网络,在指(趾)骨及关节的不同发育阶段发挥着不同的作用,其中BMP信号传导通路扮演着至为关键的角色。本文在目前对短指(趾)症的分类基础上,详细综述了短指(趾)症相关致病基因及所影响的信号通路等方面的最新进展,旨在探讨指(趾)骨形成的分子机制,以期为短指(趾)症的临床诊断以及人类骨骼发育的分子调控机制研究提供参考。
关键词:
Abstract
Brachydactyly (BD) is a type of hand/foot malformation caused by the abnormal shortening or missing phalanges and/or metacarpals/metatarsals. BD most often occurs as an isolated trait, but can also occur as part of complex malformation syndromes. According to the patterns of affected digits, isolated BD can be divided into five groups: BDA, BDB, BDC, BDD, and BDE with individual subtypes. As an important molecular disease family, the pathogenic genes and molecular mechanisms of most isolated BD forms and some complicated syndromes are elucidated. Although BDs are highly diversified in phenotypes, at the molecular levels these pathogenic genes mainly affect several important signaling pathways: Hedgehog, NOTCH, WNT and BMP. These pathways form a complex signaling network and play different roles in different stages of the digit and joint development, in which BMP signaling pathway occupies a central position. Based on the current classification of BDs, this review summarizes the latest progress in the pathogenesis of BDs and the signaling pathways involved. The purpose of this review is to explore the molecular mechanisms of digit formation, which will provide references for the clinical diagnosis of BD, and the understanding of molecular mechanism of human bone development.
Keywords:
PDF (457KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
吕赵劼, 王志浩, 卢淑娴, 刘沛蓉, 田静. 短指(趾)症及指(趾)骨发育的分子调控机制. 遗传[J], 2019, 41(12): 1073-1083 doi:10.16288/j.yczz.19-100
Zhaojie Lyu.
人的手指(脚趾)是人类进化过程中最复杂的骨骼结构之一。手指的灵活操作使人类具有处理更精细物品的能力,脚趾的灵活则给人类带来了更多的运动姿势。拥有精细复杂结构的指(趾)骨发育和功能,如同人体其他组织结构一样,受一系列复杂的分子机制调控,并对不同的病理变化高度敏感。指(趾)骨疾病在日常生活有多种呈现形式,如短(趾)指、并(趾)指、多(趾)指、软骨发育异常等,其中短指(趾)症及其综合征家族对研究指(趾)骨的正常发育有着极其重要的意义。
短指(趾)症(brachydactyly, BD)是一类指(趾)骨或掌(跖)骨的异常缩短、缺失或融合而造成的畸形病变。近年研究表明关于短指(趾)症的致病基因已达10余种并涉及到Hedgehog信号通路(hedgehog signaling pathway)、NOTCH信号通路(NOTCH signaling pathway)、WNT信号通路(wingless-related MMTV integration site signaling pathway)、BMP通路(bone morphogenetic protein signaling pathway)。本文结合Bell分类[1]和人类孟德尔遗传目录[2]对短指(趾)症的分类以及所涉及的分子信号通路进行讨论,以期为短指(趾)症的临床诊断以及人类指(趾)骨发育的分子调控机制研究提供参考。
1 短指(趾)症的分类及分子遗传学基础
随着短指(趾)症的不断研究,短指(趾)症的分类也越来越详细[3]。指(趾)骨畸形作为短指(趾)症的典型表征可在人群中表现为单纯型,也可成为复杂综合征表征的一部分[3,4]。1.1 单纯型短指(趾)症
根据Bell分类并结合不同指(趾)骨的受累情况,单纯型短指(趾)症目前可分为A、B、C、D、E 5种分型[1,2]。BDA表现为中节指(趾)骨缩短,其中BDA1所有中节指(趾)骨均缩短或退化,偶尔与远节指(趾)骨融合,拇指和大脚趾的近节(趾)骨均有缩短;BDA2表现为第2指(趾)的中节指(趾)骨发育不全,表现出缩短或退化;BDA3的表现为第5指的中节指(趾)骨缩短,其余指(趾)骨均正常;BDA4表现为第2和第5指(趾)中节指(趾)骨短,且由于中节指(趾)骨形状异常导致远节指(趾)骨桡侧偏离,下肢受累的患者通常为第4趾中节趾骨缺失[1]。BDB具有两种分型:BDB1和BDB2。BDB1主要表现为远节和(或)中节指(趾)骨缩短或发育不全,并伴有指甲缺失;BDB2表现远节指(趾)骨发育不全,伴有远端交感神经管炎、腕/跗骨融合和局部皮肤性并指畸形[5,6]。BDC主要表现为第2、3、5指(趾)中节指(趾)骨缩短并伴有第2和/或3指(趾)指(趾)骨多节化,第1掌骨也出现缩短[7]。BDD型拇指(趾)远节指(趾)骨有不同程度的缩短,并且宽于正常表型[8]。BDE具体可分为BDE1和BDE2:BDE1限于第4掌(跖)骨的缩短;BDE2为多种掌(跖)骨缩短畸形,并且第1、3指(趾)远节指(趾)骨和第 2、5指(趾)中节指(趾)骨缩短[8,9]。目前,已发现的单纯型短指(趾)症大多为常染色体显性遗传模式。除BDA3和BDD相对常见之外,其余单纯型短指(趾)症均极为罕见。短指(趾)症的致病基因在肢端发育过程中具有极其重要的作用,目前的研究已对部分致病基因及其分子机制有一定了解(表1)。现已发现BDA1 (MIM:112 500)致病基因为IHH (India Hedgehog)[10]、GDF5 (Growth differentiation factor-5)[11]以及BMPR1B (Bone Morphogenetic Protein Receptor 1B)[12]。BDA2 (MIM:112 600) 致病基因有BMPR1B[13]、GDF5[14]、BMP2 (Bone Morphogenetic Protein 2)[15]等。BDB1 (MIM:113000)和BDB2 (MIM:611 377)的致病基因分别为ROR2 (Receptor tyrosine kinase-like orphan receptor 2)和NOG (Noggin)[5,6]。BDC (MIM:113 100)、BDD (MIM:113 200)的致病基因分别为GDF5[7]、HOXD13 (the homeobox D13)[8]。BDE1和BDE2(MIM:113 300)的致病基因分别为HOXD13[8]和PTHLH (parathyroid hormone like hormone)[9](表1)。
1.2 复杂短指(趾)症综合征
部分参与肢端形成的基因发生突变所引起的短指(趾)表型,可能也是某种综合征的表型之一。具有肢端缩短症状的综合征有很多,如Feingold综合征(Feingold syndrome, MIM:164 280)、Du Pan综合征(Du Pan syndrome,MIM:228900)、Robinow综合征(Robinow syndrome, MIM:268 310)、Temtamy轴前短指(趾)综合征(Temtamy preaxial brachydactyly syndrome, TPBS, MIM:605282)等(表1)。Table 1
表1
表1单纯型短指(趾)症和部分短指(趾)症综合征
Table 1
| 疾病名称 | MIM | 致病基因 | 基因定位 | 参考文献 | |
|---|---|---|---|---|---|
| 单纯型短 指(趾)症 | BDA1 | 112 500 | IHH、GDF5和BMP1RB | 2q35~q36、20q11和4q21~20 | [10~12] |
| BDA2 | 112 600 | BMPR1B、GDF5和BMP2 | 4q21~20、20q11和20p12 | [13~15] | |
| BDA3 | 112 700 | 暂无 | 暂无 | [1,2] | |
| BDA4 | 112 800 | 暂无 | 暂无 | [1,2] | |
| BDB1 | 113 000 | ROR2 | 9q22 | [5] | |
| BDB2 | 611 377 | NOG | 17q22 | [6] | |
| BDC | 113 100 | GDF5 | 20q11 | [7] | |
| BDD | 113 200 | HOXD13 | 2q31.1 | [8] | |
| BDE1 | 113 300 | HOXD13 | 2q31.1 | [8] | |
| BDE2 | 613 328 | PTHLH | 12p11.22 | [9] | |
| 部分短指 (趾)综合征 | 短指(趾)-高血压综合征 | 112 410 | PDE3A | 12p12.2 | [3] |
| Rubinstein-Taybi综合征 | 180 849 | CREBBP | 16p13.3 | [3] | |
| Adams-Olive 综合征 | 100 300 | ARHGAP31 | 3q13.32~q13.33 | [3] | |
| Catel-Manzke综合征 | 616 145 | TGDS | 13q32.1 | [3] | |
| Feingold 综合征 | 164 280 | MYCN | 2p24.3 | [3] | |
| Du Pan 综合征 | 228 900 | GDF5 | 20q11.2 | [3] | |
| Robinow 综合征 | 268 310 | ROR2 | 9q22 | [16,17] | |
| Temtamy轴前短指综合征 | 605 282 | CHSY1 | 15q26.3 | [18] |
新窗口打开|下载CSV
Robinow综合征主要表现为短指与远节指(趾)骨短缩,指甲发育不良,第五指(趾)骨弯曲。Robinow综合征有两种遗传模式,常染色体显性遗传和常染色体隐性遗传,ROR2的无义突变和移码突变可以导致该疾病[16,17]。TPBS是近年来新发现的一类短指(趾)症综合征,临床诊断多表现为先天性的骨骼发育异常,1、2、3指(趾)近节指(趾)骨部分增生,常伴有智力发育迟缓、身材矮小、小头畸形、大眼、发育不良、牙齿异常、颌骨小和骨质疏松症。2010年,本课题组通过对近亲婚配的约旦家系中的患者进行DNA序列分析,发现硫酸软骨素合成酶(chondroitin sulfate synthase1, CHSY1)基因的隐性突变是造成其先天性指(趾)骨发育异常的遗传基础,并将此疾病命名为Temtamy轴前短指(趾)综合征[18]。随着TPBS致病基因的发现,越来越多的TPBS综合征在不同地区被发现,目前在埃及、土耳其、斯里兰卡、巴基斯坦和约旦等地均有该病例的报道[19]。
2 短指(趾)症相关信号通路
2.1 Hedgehog信号通路
Hedgehog信号通路由Hedgehog配体(HH)、膜蛋白受体复合物、核内转录因子和下游靶基因等4个部分组成。在高等脊椎动物中,Hedgehog配体有3种同源蛋白IHH (indiana hedgehog)、SHH (sonic hedgehog),DHH (desert hedgehog)。IHH主要表达在肥大软骨细胞之类的软骨细胞中,可以调控软骨细胞的增殖和分化,是短指(趾)症的主要致病基因之一。在Hedgehog信号传导通路中,当IHH配体与受体PTC结合,解除PTC对SMO的抑制作用,SMO被激活,使转录因子GLI1磷酸化,从而启动下游靶基因表达。IHH蛋白在关节周围的软骨膜中可诱导PTHLH(parathyroid hormone like hormone,甲状旁腺激素样激素)表达,PTHLH可抑制IHH的表达,与IHH形成负反馈环,从而调节Hedgehog信号通路。
IHH作为Hedgehog信号通路的配体,其突变可能会导致BDA1发生。St-Jacques等[20]通过同源重组技术敲除Ihh第一个外显子,从而得到了Ihh-/-无义突变小鼠,并观察到小鼠肢端缩短并且出现各种成骨发育不良。上海交通大学贺林院士团队首先将BDA1的致病基因定位于染色体2q35~q36,并发现了3个IHH错义突变,分别为E95K、D100E和E131K[5]。随后该团队Gao等[21]发现在突变体小鼠 IhhE95K/E95K中,Ihh与受体Ptch1和拮抗蛋白Hip1的结合减弱,下游转录因子Gli1激活减弱,从而下调Hedgehog信号通路。由于指(趾)骨远端祖细胞的募集减少,指(趾)骨的生长受损致使指(趾)骨的缩短。在信号通路水平上,IHH突变(E95K)导致IHH对PTC1和HIP1的结合降低,从而增加了IHH的信号范围,但导致生长板软骨细胞中短程IHH信号降低,但导致关节周围软骨膜中的远程IHH信号升高,导致PTHLH表达增加。增加的PTHLH又阻止了远端新形成的IHH表达,最终导致远端IHH急剧下降。这一系列的信号传导及蛋白表达导致了中间指(趾)骨的缩短或缺失,即产生了BDA1的表型[21]。在对IHH的3个突变(E95K、D100E和E131K)的进一步研究发现IHH的3个突变均可使Ptc1和Gli1的激活削弱。在最适诱导浓度下,突变型IHH对Gli蛋白的诱导能力大幅减弱[22]。2011年,Ma等[23]验证了3个IHH错义突变(E95K、D100E和E131K)对IHH蛋白结构、功能和结合能力的影响,全面阐述了IHH的功能与BDA1突变之间的关系[22]。E95K改变了将其位点的表面电荷由负变为正,而不改变临近结构的表面电荷。E131K可能出现两种构象:一是与E95构象一致;二是E95与K131之间形成盐桥。这种表面电荷额度改变可能会影响IHH与PTC1的相互作用。D100E突变的表面电荷分布则没有显著改变,但在D100E结构中,氨基酸残基93~98之间的区域比正常结构具有更高的灵活性。这3个突变并没有对IHH整体结构造成显著破坏,但是对钙离子的结合区域出现了细微的变化。E95K、D100E和E131K这3种突变均通过溶酶体途径导致在胞内出现不同程度的降解。IHH蛋白的细胞内稳定性可能与钙结合槽内的细微变化有关,当钙离子增多时,突变型IHH (E95K、E131K)的蛋白正常表达水平提高。这些突变可能会影响IHH的合成或分泌,是BDA1疾病机制重要的一部分。2019年,贺林院士团队Shen等[24]通过结合野生型IHH和E95K突变体进行ChIP-chip和Microarray-based 基因表达分析,提出了BDA1的Gli1介导模型,并发现E95K突变体信号传导改变了Gli1-DNA结合模式,削弱了下游基因表达,并导致细胞增殖和迁移减弱。Runx2是一种与短指(趾)症相关的基因,并且Runx2在野生型IHH和突变型IHH之间存在差异表达。Runx2相关途径是解释E95K引发的BDA1和IHH下游信号调节变化的最相关基因之一[24]。
近年来,IHH突变引发BDA1的报道也逐渐增多。2015年,Dong等[25]在3例BDA1表型中国家系中分别发现3个IHH错义突变(E95del、D100E和D100N)。2017年,Salian等[26]发现1例手足中指(趾)骨完全缺失的BDA1儿童,并发现其携带有IHH具有错义突变(D100N)。
PTHLH是Heghehog信号通路的拮抗剂,PTHLH突变会导致BDE2的发生。Klopocki等[9]在5个无关联的家系中均发现PTHLH功能缺失会导致手部及足部的掌骨、跖骨、指(趾)骨缩短,第4和第5掌骨表现最为严重,即典型BDE2临床表型。此外BDE2患者也可能表现为身材矮小、迟发性牙萌出或少牙。Thomas-Teinturier等[27]在3个无血缘关系的患者中发现2种PTHLH的突变。第一种为c.47_ 101t73del128,该突变破坏了原先5号外显子的剪接位点。第二种突变为c.101_3delAAGT,该突变导致剪接位点出现异常。这两种突变均使转录异常,并过早出现终止密码子。3位患者均具有掌骨和跖骨缩短症状。
2.2 NOTCH信号通路
哺乳动物中有4种NOTCH受体,即NOTCH1~4。NOTCH配体在哺乳动物中分为2类,Delta-like (DLL)和Jagged。DLL有3种(DLL-1,3,4),Jagged有两种(Jagged-1,2)。当细胞膜上的NOTCH配体与跨膜的NOTCH受体结合,肿瘤坏死因子α转化酶(tumornecrosis factor convetingenzyme,TACE)在胞外第一次水解,NOTCH受体N端被配体表达细胞胞吞,C端则进入细胞在γ-secretase作用下二次水解,释放NOTCH受体活化形式(NICD),NOTCH信号通路被激活。激活后,NICD转运入核并在CSL蛋白作用下与DNA结合,通过调控HES1/5等转录因子,从而调控靶基因的转录、细胞的发育及分化。关于NOTCH通路异常引起短指(趾)症的研究不多。目前研究发现,TPBS的致病基因CHSY1可以通过调控NOTCH信号通路影响软骨的发育[18]。本课题组前期研究发现在CHSY1突变的人类成纤维表皮原代细胞中,JAG1蛋白的表达显著增高,而在胚胎发育过程中的JAG1蛋白的异常可能会引起对NOTCH信号通路的异常调控,故表明CHSY1缺失会导致NOTCH信号通路上调[18]。本课题组通过在斑马鱼胚胎中将chsy1敲低发现Notch信号通路上调,而且chsy1敲低斑马鱼表现出与人类TPBS相似的表型,骨骼和胸鳍的发育异常,同时也会引起视网膜的过度生长。Wilson 等[28]利用同源重组技术敲除Chsy1第1个外显子,从而得到Chsy1-/-小鼠。Chsy1-/-小鼠表现出软骨发育不良、骨密度降低、远节指间关节移位致使远节指(趾)骨缩短并出现分节。Filipek-Górniok等[29]通过对不同时期的斑马鱼进行阿利新兰染色,发现在早期发育阶段硫酸软骨素(chondroitin sulfate, CS)沉积于脊索鞘,通过原位杂交发现Chsy1及其他硫酸软骨素糖基转移酶基因在软骨形成部位均有表达,如咽软骨、耳囊和胸鳍。目前有研究表明CHSY1可能与HOXD13也有密切关系。通过荧光素酶报告系统和实时定量PCR发现,HOXD13的过表达会引起CHSY1上调[30]。
本课题组在对1例罕见并指(趾)症家系CLS并指(趾)症(Cenani-Lenz syndactyly syndrome, MIM: 212780)的研究中也发现,其致病基因LRP4可通过NOTCH信号通路调控肢端及骨的发育[31],进一步证明了NOTCH信号通路在指(趾)骨发育中的重要作用。
2.3 WNT信号通路
WNT信号通路在物种进化上高度保守,对早期胚胎发育具有极大影响,也参与诱导骨的增殖、分化,是调控骨代谢的重要途径之一。WNT信号通路可以分为经典WNT/β-catenin信号通路、非经典WNT/PCP信号通路以及WNT/Ca2+信号通路。经典WNT/β-catenin信号通路的传导由WNT配体与低密度脂蛋白受体5/6 (LRP5/6)、Frizzled受体结合,Dishevelled蛋白(DvL)接收信号,通过抑制APC-AXIN-GSK3β复合体,使β-catenin磷酸化水平减少,非磷酸化的β-catenin水平增加,在细胞核内与TCF/LEF结合,调控下游靶基因的转录过程。经典WNT/β-catenin信号通路通过调节间充质干细胞向成骨细胞和破骨细胞分化而使这个过程处于动态平衡。但通路的过度激活或抑制均会打破这种平衡,导致部分骨类疾病,如硬骨病和骨质疏松等[32]。
由WNT信号通路传导异常而引起的短指(趾)症目前主要为BDB1和Robinow综合征,是由基因ROR2的缺失导致[15,16]。Huang等[33]发现了一个具有BDB1表型的三代中国家系,并发现1个ROR2新的杂合子碱基缺失(c.1396-1398delaa)。这种突变导致出现一种截短的ROR2蛋白和一种具有57个氨基酸的多肽片段。Dong等[25]在1例BDB1表型中国家系发现了基因ROR2的一种新的杂合突变(S758x)。此外,基因ROR2突变导致BDB1也在土耳其、德国、巴基斯坦、中国、葡萄牙、威尔士、约旦和沙特阿拉伯等地均有报道[33]。
基因ROR2编码酪氨酸激酶受体,在WNT信号通路中主要与WNT/Ca2+信号通路中的WNT5A结合调控经典WNT/β-catenin信号通路和WNT/Ca2+信号通路平衡与正常传导。ROR2的半胱氨酸富集区与WNT5A结合,对WNT/β-catenin信号通路起负调节作用。Oishi等[34]发现Ror2-/-小鼠身材矮小,四肢和尾部均缩短,肺部和生殖器官发育不良,并且出现室间隔缺损。Oldridge等[35]在小鼠Ror2基因中插入lacZ,导致小鼠Ror2基因功能异常,软骨无法正常生成以及中间指(趾)骨缺失。德国****Witte等[36]在Ror2W79X/W79X小鼠突变体的BDB1模型中发现间充质细胞向软骨细胞分化受阻,导致指(趾)骨无法正常延长。在指(趾)骨形成区处,BMP/pSMAD1/5/8信号减少,并且在突变体小鼠E13.5胚胎中发现远节指(趾)骨间充质细胞中Ihh表达减少,表明基因ROR2与BMP信号通路和IHH信号通路也存在密切关系。
2.4 BMP信号通路
BMP信号通路在骨的发育中占据重要的中心位置,位于指(趾)骨发育调控信号分子网络的中枢。BMP是TGF-β (转化生长因子)超家族中最大的蛋白家族,正向调节骨的增殖、分化以及形成。BMPs蛋白是BMP信号通路中的配体,包括BMP1~15。BMP受体有两种类型,Ⅰ型BMP受体(BMPR1)和Ⅱ型BMP受体(BMPR2)。Noggin(NOG)和Chordin (CHRD)是BMP信号通路上主要的抑制剂。在BMP信号通路传到中,BMPs与位于细胞膜上的BMPR1和BMPR2结合,使BMPR1和BMPR2激活。活化的BMPR1与下游SMAD1/5/8 蛋白作用并使其磷酸化。SMAD1/5/8 蛋白磷酸化后与SMAD4结合入核并在核中积累,在ID等转录因子作用下调节靶基因表达。BMP信号通路上的多数元件均与短指(趾)症密切相关。BMP2下游调控区突变的重复序列会引发BDA2。Dathe等[10]在德裔巴西家系和欧洲家系中检查了21名患者,发现BMP2基因下游110 kb处有高度保守的重复序列。Su 等[37]在一个中国BDA2家系中确定了BMP2基因下游110 kb处存在4.6 kb的杂合重复序列。Liu等[38]通过全基因组连锁分析在一个中国BDA2家族定位到了致病基因BMP2,在BMP2基因下游调控区发现一个4671 bp的重复序列。2018年,Wang等[39]在一个BDA2表型的中国家系中同样发现了BMP2基因下游具有4671 bp重复序列,但在该家系未患病的人和正常人群中并未发现,并且该家系所有成员的基因BMPR1B、GDF5和BMP2的所有外显子并无突变。这些均说明位于BMP2下游的保守重复序列对于BMP2基因功能存在远程调控作用。
GDF5蛋白是BMP信号通路中的配体,也称为BMP14。GDF5蛋白广泛表达于指(趾)间关节细胞以及关节周围的祖细胞中,参与关节形成。GDF5突变会引发BDA1、BDA2、BDB2和BDC。Byrnes等[11]在一个法国加拿大家庭中发现了3个BDA1患者,其致病基因为GDF5的纯合错义突变R399C。Degenkolbe等[40]在一个墨西哥家庭的3代人中发现Ⅱ型多发性骨性连接综合征(multiple synostoses syndrome 2, SYNS2, MIM:610017)和BDA1的患者,均为GDF5错义突变W414R,该突变位于GDF5与NOG、BMPR1的结合部位,致使BMPR1A信号缺失,而BMPR1B信号减少。
GDF5突变也可能导致BDA2。Seemann等[14]在一个具有BDA2遗传的家族中鉴定GDF5基因的错义突变L441P,他们认为BDA2的表型是由配体GDF5和受体BMPR1的结合作用受抑制引起的。Pl?ger等[41]在一个BDA2家族六代14名患者中发现GDF5的错义突变R380Q。他们认为R380Q突变导致蛋白剪切异常,致使GDF5功能降低。Kjaer等[42]通过对一个曾发现患BDA2的丹麦/挪威大型家族更新族谱并在37位患者中进行分析,发现GDF5错义突变I441P,并且预测该突变存在于与BMPR1的结合位点。2018年,Khan等[43]也在巴基斯坦BDA2家系中发现GDF5基因突变[43]。
GDF5突变也往往导致BDC型短指(趾)症。 由于GDF5突变所引发的BDC,往往会表现出分化多个关节的症状。Everman等[44]在一个BDC家族中发现12号染色体GDF5基因上插入了23 bp。Schwabe等[7]在一个大型近亲结婚的土耳其家庭中发现该家族内所有的BDC患者均为M173V杂合突变。
BMPR1和BMPR2作为BMP信号通路的受体,在软骨细胞的聚集处有较高的表达。BMPR1B 的突变也可能导致BDA1和BDA2表型。Racacho等[12]在2名类似BDA1的无亲缘关系的儿童中未发生IHH突变,但发现基因BMPR1B突变,一例为杂合错义突变K325N,另一例为杂合剪切突变。BMPR1B突变也会导致BDA2发生。Lehmann等[13]在两个无亲缘关系的患有BDA2的德国家庭中发现了BMPR1B基因的杂合突变I200K和R486W。他们通过用微团培养系统进行功能分析表明,两种突变受体都能强烈抑制软骨形成。通过逆转录病毒感染鸡胚得到过表达Bmpr1b的鸡胚,其表现出与人类指(趾)骨缩短极其相似的短指(趾)表型或会导致整个肢体的严重发育不足。
NOG蛋白是BMP信号通路的拮抗剂,基因NOG的突变会引起BDB2。Lehmann等[6]在德国、土耳其、丹麦、伊朗、英国及北美6个BDB2家系中发现致病基因为NOG,并鉴定出6种不同的杂合错义突变P35A、P35S、A36P、E48K、R167G和P187S。2015年,Ishino等[45]在一个五代日本家系中发现所有BDB2患者均为NOG的错义突变C228G,该突变破坏了NOG蛋白的FingerⅡ中半胱氨酸之间连结,致使FingerⅡ结构发生严重破坏。此外,在巴基斯坦的BDB2家系中也发现其致病基因为NOG[43]。在BDB2中,NOG的突变使NOG与BMP和GDF5的结合能力发生异常,从而破坏了BMP信号通路的稳定,使得指(趾)骨的软骨发育异常,表现出短指(趾)的表型。Brunet等[46]通过同源重组技术得到Nog突变体小鼠表现为在椎骨、肋骨和四肢上均有明显的缺陷,四肢短粗,关节出现融合,表型与人类BDB2相似。
有报道认为TPBS短指(趾)综合征的致病基因CHSY1也参与BMP信号通路的调控,但CHSY1参与BMP信号通路的具体细节仍尚未可知[47]。
3 短指(趾)症及指(趾)骨发育中的信号调控网络
Hedgehog、NOTCH、WNT、BMP信号通路均与骨骼发育密切相关,并且构成了一个复杂又庞大的信号通路网络。BMP信号通路在指(趾)骨发育中处于中枢位置,Hedgehog、NOTCH和WNT信号通路均与BMP信号通路相互作用(图1)。图1
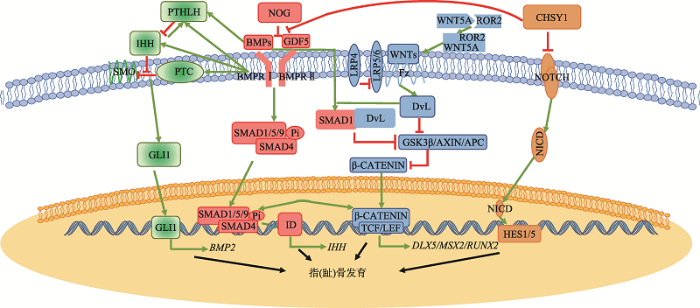 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1短指(趾)症致病基因参与信号通路调控网络
短指(趾)症致病基因参与的信号通路主要为Hedgehog、WNT、NOTCH、BMP4条信号通路。信号通路相互调控的解释见正文。大部分短指(趾)症致病基因存在于BMP信号通路中,而不直接参与BMP信号通路的短指(趾)症致病基因可通过分子网络调控将4条信号通路联系起来,最终共同影响指(趾)骨发育进程。文中对信号通路具有抑制作用的部分用红色线条表示;对信号通路具有促进作用的用绿色箭头表示。
Fig. 1The signaling pathway network involving pathogenic genes of BDs
Hedgehog信号通路作为指(趾)骨延长以及调控软骨细胞分化和增殖的不可或缺的一部分,与BMP信号通路的紧密交流也成为了骨骼正常发育的基础。IHH在BMP信号通路中具有多种调节功能。当软骨膜中进行骨生成过程中,IHH信号通路可以通过GLI激活BMPs定向的骨-软骨生成作用[48]。IHH的缺失则会减少BMP的表达量。另一方面,BMP信号通路也参与到IHH信号通路的调节中。在成骨细胞分化过程中,BMP2也可诱导IHH的表达,并下调PTC1的表达[49]。BMP/Smad信号可以直接激活IHH启动子。在生长板的发育过程中,IHH信号通路与PTHLH的调节保持软骨细胞正常增殖,有利于长骨的延伸。Zhang等[50]发现,在Smad4-/-的t突变体小鼠中,Ihh、Ptc1和Pth1r的表达量均降低。Zhang等[51]在组成性激活突变型ALK2、组成性激活突变型BMPRIA、以及组成性激活突变型BMPRIB的过表达的鸡胚的肢芽中发现,IHH、PTC1和PTH1R的表达量上升。Minina等[52]在培养小鼠肢体的外植体中发现,Bmp2可以诱导Ihh的表达,而Noggin则会抑制Ihh的表达。
WNT信号通路除参与骨骼发育外,也参与到骨细胞的分化、增殖、凋亡以及维持骨量稳定等多个生理过程。WNT信号通路与BMP信号通路在指(趾)骨发育也有着紧密的联系。BMPs蛋白对WNT信号通路的调节具有双重作用。BMP蛋白可以通过MAPK和SMAD信号通路传导,从而抑制WNT信号通路传导,调节骨的正常生理发育,并在成骨细胞中对骨量进行负调节[53,54]。在WNT信号通路中Dvl起到接收受体传导的信号,并将其信号传导至下游引起β-catenin磷酸化水平降低。而BMP2则可诱导Smad1-Dvl1复合物形成,限制了WNT信号通路的传导[55]。当ROR2与WNT5A结合后调节WNT/β-catenin信号通路,同时在软骨形成中拮抗BMP信号传导。此外,ROR2通过抑制Smad1/5/9信号传导和激活Smad非依赖性途径来调节GDF5信号传导。因此当ROR2功能异常时,往往致使指(趾)骨发育异常,从而导致短指(趾)症BDB2或Rinbown综合征的发生。BMPs同时也可以与WNT信号协同作用,共同促进成骨细胞和软骨细胞的分化,保证骨骼,尤其是指(趾)骨的正常发育。BMP2可以通过诱导WNT蛋白的表达促进经典WNT信号通路[56]。Smad和Tcf/Lef/β-catenin复合体在核内启动子上相互作用,使基因Dlx5、Msx2和Runx2表达增高,促进成骨细胞分化[57]。
NOTCH信号通路与BMP信号通路也存在着密切的联系,但NOTCH信号通路与BMP的在骨骼发育,尤其是指(趾)骨发育中的关联研究不多。2010年,本课题组在TPBS中发现,CHSY1可以通过抑制NOTCH信号通路影响指(趾)骨的发育[18]。Li等[47]在TPBS中则发现CHSY1可以通过抑制BMP信号通路影响指(趾)骨发育。因此,CHSY1很可能在BMP信号通路和NOTCH信号通路之间,起到重要的沟通作用。
4 结语与展望
在对短指(趾)症百年的研究过程中,短指(趾)症的分类、发病机制大多已被清楚认识,并建立起了短指(趾)症相关的研究体系。在短指(趾)症的治疗方面,尚无针对所有短指(趾)症类型的标准术式[58]。在能改善患者手脚功能及外形的前提下仅可以通过整形外科手术进行治疗,但在通常情况下,该类手术并非绝对必要[58]。目前对于单纯型短指(趾)症的分类,主要为Bell分类。由于Bell分类主要是以解剖学为基础,但部分短指(趾)症的分类与分子遗传学可能不完全相符,因此,在Bell分类的基础上结合指(趾)骨发育的分子遗传背景可能进一步优化短指症的诊断和分类。短指(趾)症是由于指(趾)骨的发育异常引起的缩短畸形,因此短指(趾)症在研究指(趾)骨发育方面是一个极好的模型。短指(趾)症作为由基因突变引起的先天性遗传病,各类短指(趾)症的致病基因以BMP信号通路为核心,并通过Hedgehog、NOTCH、WNT等信号通路对指(趾)骨发育起到决定性影响。随着短指(趾)症在分子水平上的研究,指(趾)骨发育的过程有望得到近一步细致的探索,并且可以通过更多分子水平上的手段得到更多对于短指(趾)症治疗的新思路和新方法。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1186/1750-1172-3-15URLPMID:18554391 [本文引用: 5]

Brachydactyly ("short digits") is a general term that refers to disproportionately short fingers and toes, and forms part of the group of limb malformations characterized by bone dysostosis. The various types of isolated brachydactyly are rare, except for types A3 and D. Brachydactyly can occur either as an isolated malformation or as a part of a complex malformation syndrome. To date, many different forms of brachydactyly have been identified. Some forms also result in short stature. In isolated brachydactyly, subtle changes elsewhere may be present. Brachydactyly may also be accompanied by other hand malformations, such as syndactyly, polydactyly, reduction defects, or symphalangism. For the majority of isolated brachydactylies and some syndromic forms of brachydactyly, the causative gene defect has been identified. In isolated brachydactyly, the inheritance is mostly autosomal dominant with variable expressivity and penetrtance. Diagnosis is clinical, anthropometric and radiological. Prenatal diagnosis is usually not indicated for isolated forms of brachydactyly, but may be appropriate in syndromic forms. Molecular studies of chorionic villus samples at 11 weeks of gestation and by amniocentesis after the 14th week of gestation can provide antenatal diagnosis if the causative mutation in the family is known. The nature of genetic counseling depends both on the pattern of inheritance of the type of brachydactyly present in the family and on the presence or absence of accompanying symptoms. There is no specific management or treatment that is applicable to all forms of brachydactyly. Plastic surgery is only indicated if the brachydactyly affects hand function or for cosmetic reasons, but is typically not needed. Physical therapy and ergotherapy may ameliorate hand function. Prognosis for the brachydactylies is strongly dependent on the nature of the brachydactyly, and may vary from excellent to severely influencing hand function. If brachydactyly forms part of a syndromic entity, prognosis often depends on the nature of the associated anomalies.
URL [本文引用: 4]
DOI:10.1002/ajmg.a.37365URLPMID:26394607 [本文引用: 8]
The purpose of the nosology is to serve as a "master" list of the genetic disorders of the skeleton to facilitate diagnosis and to help delineate variant or newly recognized conditions. This is the 9th edition of the nosology and in comparison with its predecessor there are fewer conditions but many new genes. In previous editions, diagnoses that were phenotypically indistinguishable but genetically heterogenous were listed separately but we felt this was an unnecessary distinction. Thus the overall number of disorders has decreased from 456 to 436 but the number of groups has increased to 42 and the number of genes to 364. The nosology may become increasingly important today and tomorrow in the era of big data when the question for the geneticist is often whether a mutation identified by next generation sequencing technology in a particular gene can explain the clinical and radiological phenotype of their patient. This can be particularly difficult to answer conclusively in the prenatal setting. Personalized medicine emphasizes the importance of tailoring diagnosis and therapy to the individual but for our patients with rare skeletal disorders, the importance of tapping into a resource where genetic data can be centralized and made available should not be forgotten or underestimated. The nosology can also serve as a reference for the creation of locus-specific databases that are expected to help in delineating genotype-phenotype correlations and to harbor the information that will be gained by combining clinical observations and next generation sequencing results.
DOI:10.3724/SP.J.1005.2012.01522URL [本文引用: 1]
Brachydactyly (BD) is a general term that refers to shortening of the hands/feet due to small or missing metacarpals/metatarsalsand/or phalanges, and forms part of the group of limb malformations characterized by bone dy-sostosis. It may occur either as an isolated trait or as part of a syndrome. BD may also be accompanied by other hand mal-formations, such as syndactyly, polydactyly, reduction defects, and symphalangism. In isolated brachydactyly, the inheri-tance is mostly autosomal dominant with variable expressivity and penetrtance. For the majority of isolated BD and some syndromic forms of BD, the causative gene defect has been identified. These studies have shown that the bone morphoge-netic protein (BMP) pathway plays a pivotal role in the normal development of digits and joints and that the majority of brachydactyly disease genes are directly or indirectly linked to this pathway. This review summarizes the progress in the molecular genetics of BD, which will contribute to the BD pathogenic mechanism and implementation of genetic clinic.
DOI:10.3724/SP.J.1005.2012.01522URL [本文引用: 1]
Brachydactyly (BD) is a general term that refers to shortening of the hands/feet due to small or missing metacarpals/metatarsalsand/or phalanges, and forms part of the group of limb malformations characterized by bone dy-sostosis. It may occur either as an isolated trait or as part of a syndrome. BD may also be accompanied by other hand mal-formations, such as syndactyly, polydactyly, reduction defects, and symphalangism. In isolated brachydactyly, the inheri-tance is mostly autosomal dominant with variable expressivity and penetrtance. For the majority of isolated BD and some syndromic forms of BD, the causative gene defect has been identified. These studies have shown that the bone morphoge-netic protein (BMP) pathway plays a pivotal role in the normal development of digits and joints and that the majority of brachydactyly disease genes are directly or indirectly linked to this pathway. This review summarizes the progress in the molecular genetics of BD, which will contribute to the BD pathogenic mechanism and implementation of genetic clinic.
DOI:10.1002/dvdy.20993URLPMID:17061261 [本文引用: 4]
Ror2 is a receptor tyrosine kinase mutated in the human syndromes Brachydactyly type B (BDB) and recessive Robinow syndrome (RS). In this study, we used the chick as a model to investigate the role of Ror2 in skeletogenesis and to elucidate the functional consequences of Ror2 mutations. For this purpose, we cloned chicken Ror2 and analyzed its expression pattern at various embryonic stages by in situ hybridization and immunolabeling. We document expression of cRor2 in several organs, including mesonephros, heart, nervous system, intestine and cartilage. The high conservation of expression when compared with the mouse underlines the validity of the chick as a model system. Using replication-competent retroviral vector-mediated overexpression, we analyzed the functional consequences of truncating BDB and RS mutations in the developing chick limb. Overexpression of Ror2 mutants led to a disturbance of growth plate architecture and a severe block of chondrocyte differentiation, demonstrating the functional importance of Ror2 in skeletogenesis.
DOI:10.1086/519697URL [本文引用: 4]
Brachydactyly type B (BDB) is characterized by terminal deficiency of fingers and toes, which is caused by heterozygous truncating mutations in the receptor tyrosine kinase–like orphan receptor 2 (ROR2) in the majority of patients. In a subset of ROR2-negative patients with BDB, clinically defined by the additional occurrence of proximal symphalangism and carpal synostosis, we identified six different point mutations (P35A, P35S, A36P, E48K, R167G, and P187S) in the bone morphogenetic protein (BMP) antagonist NOGGIN (NOG). In contrast to previously described loss-of-function mutations in NOG, which are known to cause a range of conditions associated with abnormal joint formation but without BDB, the newly identified BDB mutations do not indicate a major loss of function, as suggested by calculation of free-binding energy of the modeled NOG-GDF5 complex and functional analysis of the micromass culture system. Rather, they presumably alter NOG’s ability to bind to BMPs and growth-differentiation factors (GDFs) in a subtle way, thus disturbing the intricate balance of BMP signaling. The combined features observed in this phenotypic subtype of BDB argue for a functional connection between BMP and ROR2 signaling and support previous findings of a modulating effect of ROR2 on the BMP-receptor pathway through the formation of a heteromeric complex of the receptors at the cell surface.
DOI:10.1002/ajmg.a.20349URLPMID:14735582 [本文引用: 4]
Brachydactyly type C (BDC) is characterized by shortening of the middle phalanges of the index, middle, and little finger with hyperphalangy, usually of the index and middle finger. Heterozygous mutations of the cartilage derived morphogenetic protein-1 (CDMP1) resulting in a loss of function have been reported in BDC. We here describe a large kindred with a semi-dominant form of BDC and pronounced ulnar deviation of the second and third digits. In this family a novel homozygous missense mutation was identified (517A > G) changing methionine to valine at amino acid position 173. The mutation is located within a highly conserved seven amino acid region of the prodomain of CDMP1. Hand radiographs of heterozygous mutation carriers showed mild shortening of the metacarpals IV and V; a finding confirmed by the analysis of their metacarpophalangeal profiles (MCPPs). The mutation described here points toward an important function of the prodomain for the folding, secretion, and availability of biologically active CDMP1.
DOI:10.1086/374721URLPMID:12649808 [本文引用: 6]
HOXD13, the most 5' gene of the HOXD cluster, encodes a homeodomain transcription factor with important functions in limb patterning and growth. Heterozygous mutations of human HOXD13, encoding polyalanine expansions or frameshifts, are believed to act by dominant negative or haploinsufficiency mechanisms and are predominantly associated with synpolydactyly phenotypes. Here, we describe two mutations of HOXD13 (923C-->G encoding Ser308Cys and 940A-->C encoding Ile314Leu) that cause missense substitutions within the homeodomain. Both are associated with distinctive limb phenotypes in which brachydactyly of specific metacarpals, metatarsals, and phalangeal bones is the most constant feature, exhibiting overlap with brachydactyly types D and E. We investigated the binding of synthetic mutant proteins to double-stranded DNA targets in vitro. No consistent differences were found for the Ser308Cys mutation compared with the wild type, but the Ile314Leu mutation (which resides at the 47th position of the homeodomain) exhibited increased affinity for a target containing the core recognition sequence 5'-TTAC-3' but decreased affinity for a 5'-TTAT-3' target. Molecular modeling of the Ile314Leu mutation indicates that this mixed gain and loss of affinity may be accounted for by the relative positions of methyl groups in the amino acid side chain and target base.
DOI:10.1016/j.ajhg.2010.01.023URLPMID:20170896 [本文引用: 4]
Autosomal-dominant brachydactyly type E (BDE) is a congenital limb malformation characterized by small hands and feet predominantly as a result of shortened metacarpals and metatarsals. In a large pedigree with BDE, short stature, and learning disabilities, we detected a microdeletion of approximately 900 kb encompassing PTHLH, the gene coding for parathyroid hormone related protein (PTHRP). PTHRP is known to regulate the balance between chondrocyte proliferation and the onset of hypertrophic differentiation during endochondral bone development. Inactivation of Pthrp in mice results in short-limbed dwarfism because of premature differentiation of chondrocyte. On the basis of our initial finding, we tested further individuals with BDE and short stature for mutations in PTHLH. We identified two missense (L44P and L60P), a nonstop (X178WextX( *)54), and a nonsense (K120X) mutation. The missense mutation L60P was tested in chicken micromass culture with the replication-competent avian sarcoma leukosis virus retroviral expression system and was shown to result in a loss of function. Thus, loss-of-function mutations in PTHLH cause BDE with short stature.
DOI:10.1038/ng577URLPMID:11455389 [本文引用: 3]
Brachydactyly type A-1 (BDA-1; MIM 112500) is characterized by shortening or missing of the middle phalanges (Fig. 1a). It was first identified by Farabee in 1903 (ref. 2), is the first recorded example of a human anomaly with Mendelian autosomal-dominant inheritance and, as such, is cited in most genetic and biological textbooks. Here we show that mutations in IHH, which encodes Indian hedgehog, cause BDA-1. We have identified three heterozygous missense mutations in the region encoding the amino-terminal signaling domain in all affected members of three large, unrelated families. The three mutant amino acids, which are conserved across all vertebrates and invertebrates studied so far, are predicted to be adjacent on the surface of IHH.
DOI:10.1002/humu.21338URLPMID:20683927 [本文引用: 3]
Brachydactyly A1 (BDA1) is an autosomal dominant disorder characterized by shortness of all middle phalanges of the hands and toes, shortness of the proximal phalanges of the first digit, and short stature. Missense mutations in the Indian Hedgehog gene (IHH) are known to cause BDA1, and a second locus has been mapped to chromosome 5p. In a consanguineous French Canadian kindred with BDA1, both IHH and the 5p locus were excluded. Microsatellites flanking GDF5 on chromosome 20q were found to cosegregate with the disease. Sequencing of the GDF5 coding region revealed that a mildly affected individual in the family was heterozygous, and that all of the severely affected individuals were homozygous for a novel missense c.1195C>T mutation that predicts a p.Arg399Cys substitution at a highly conserved amino acid. Functional analysis demonstrated that although the p.Arg399Cys mutant is able to stimulate chondrogenesis, it is much less effective than wild-type GDF5. This data confirms genetic heterogeneity in BDA1, demonstrates that mutations upstream of IHH can result in BDA1, and shows that BDA1 can result from semidominant mutations in GDF5.
DOI:10.1038/ejhg.2015.38URLPMID:25758993 [本文引用: 3]
Brachydactyly type A1 is an autosomal dominant disorder primarily characterized by hypoplasia/aplasia of the middle phalanges of digits 2-5. Human and mouse genetic perturbations in the BMP-SMAD signaling pathway have been associated with many brachymesophalangies, including BDA1, as causative mutations in IHH and GDF5 have been previously identified. GDF5 interacts directly as the preferred ligand for the BMP type-1 receptor BMPR1B and is important for both chondrogenesis and digit formation. We report pathogenic variants in BMPR1B that are associated with complex BDA1. A c.975A>C (p.(Lys325Asn)) was identified in the first patient displaying absent middle phalanges and shortened distal phalanges of the toes in addition to the significant shortening of middle phalanges in digits 2, 3 and 5 of the hands. The second patient displayed a combination of brachydactyly and arachnodactyly. The sequencing of BMPR1B in this individual revealed a novel c.447-1G>A at a canonical acceptor splice site of exon 8, which is predicted to create a novel acceptor site, thus leading to a translational reading frameshift. Both mutations are most likely to act in a dominant-negative manner, similar to the effects observed in BMPR1B mutations that cause BDA2. These findings demonstrate that BMPR1B is another gene involved with the pathogenesis of BDA1 and illustrates the continuum of phenotypes between BDA1 and BDA2.
DOI:10.1073/pnas.2133476100URLPMID:14523231 [本文引用: 3]
Brachydactyly (BD) type A2 is an autosomal dominant hand malformation characterized by shortening and lateral deviation of the index fingers and, to a variable degree, shortening and deviation of the first and second toes. We performed linkage analysis in two unrelated German families and mapped a locus for BD type A2 to 4q21-q25. This interval includes the gene bone morphogenetic protein receptor 1B (BMPR1B), a type I transmembrane serinethreonine kinase. In one family, we identified a T599 --> A mutation changing an isoleucine into a lysine residue (I200K) within the glycine/serine (GS) domain of BMPR1B, a region involved in phosphorylation of the receptor. In the other family we identified a C1456 --> T mutation leading to an arginine-to-tryptophan amino acid change (R486W) in a highly conserved region C-terminal of the BMPR1B kinase domain. An in vitro kinase assay showed that the I200K mutation is kinase-deficient, whereas the R486W mutation has normal kinase activity, indicating a different pathogenic mechanism. Functional analyses with a micromass culture system revealed a strong inhibition of chondrogenesis by both mutant receptors. Overexpression of mutant chBmpR1b in vivo in chick embryos by using a retroviral system resulted either in a BD phenotype with shortening and/or missing phalanges similar to the human phenotype or in severe hypoplasia of the entire limb. These findings imply that both mutations identified in human BMPR1B affect cartilage formation in a dominant-negative manner.
DOI:10.1172/JCI25118URLPMID:16127465 [本文引用: 3]
Here we describe 2 mutations in growth and differentiation factor 5 (GDF5) that alter receptor-binding affinities. They cause brachydactyly type A2 (L441P) and symphalangism (R438L), conditions previously associated with mutations in the GDF5 receptor bone morphogenetic protein receptor type 1b (BMPR1B) and the BMP antagonist NOGGIN, respectively. We expressed the mutant proteins in limb bud micromass culture and treated ATDC5 and C2C12 cells with recombinant GDF5. Our results indicated that the L441P mutant is almost inactive. The R438L mutant, in contrast, showed increased biological activity when compared with WT GDF5. Biosensor interaction analyses revealed loss of binding to BMPR1A and BMPR1B ectodomains for the L441P mutant, whereas the R438L mutant showed normal binding to BMPR1B but increased binding to BMPR1A, the receptor normally activated by BMP2. The binding to NOGGIN was normal for both mutants. Thus, the brachydactyly type A2 phenotype (L441P) is caused by inhibition of the ligand-receptor interaction, whereas the symphalangism phenotype (R438L) is caused by a loss of receptor-binding specificity, resulting in a gain of function by the acquisition of BMP2-like properties. The presented experiments have identified some of the main determinants of GDF5 receptor-binding specificity in vivo and open new prospects for generating antagonists and superagonists of GDF5.
DOI:10.1016/j.ajhg.2009.03.001URL [本文引用: 3]
Autosomal-dominant brachydactyly type A2 (BDA2), a limb malformation characterized by hypoplastic middle phalanges of the second and fifth fingers, has been shown to be due to mutations in the Bone morphogenetic protein receptor 1B (BMPR1B) or in its ligand Growth and differentiation factor 5 (GDF5). A linkage analysis performed in a mutation-negative family identified a novel locus for BDA2 on chromosome 20p12.3 that incorporates the gene for Bone morphogenetic protein 2 (BMP2). No point mutation was identified in BMP2, so a high-density array CGH analysis covering the critical interval of ∼1.3 Mb was performed. A microduplication of ∼5.5kb in a noncoding sequence ∼110 kb downstream of BMP2 was detected. Screening of other patients by qPCR revealed a similar duplication in a second family. The duplicated region contains evolutionary highly conserved sequences suggestive of a long-range regulator. By using a transgenic mouse model we can show that this sequence is able to drive expression of a X-Gal reporter construct in the limbs. The almost complete overlap with endogenous Bmp2 expression indicates that a limb-specific enhancer of Bmp2 is located within the identified duplication. Our results reveal an additional functional mechanism for the pathogenesis of BDA2, which is duplication of a regulatory element that affects the expression of BMP2 in the developing limb.
DOI:10.1038/78107URLPMID:10932186 [本文引用: 3]
The autosomal recessive form of Robinow syndrome (RRS; MIM 268310) is a severe skeletal dysplasia with generalized limb bone shortening, segmental defects of the spine, brachydactyly and a dysmorphic facial appearance. We previously mapped the gene mutated in RRS to chromosome 9q22 (ref. 4), a region that overlaps the locus for autosomal dominant brachydactyly type B (refs 5,6). The recent identification of ROR2, encoding an orphan receptor tyrosine kinase, as the gene mutated in brachydactyly type B (BDB1; ref. 7) and the mesomelic dwarfing in mice homozygous for a lacZ and/or a neo insertion into Ror2 (refs 8,9) made this gene a candidate for RRS. Here we report homozygous missense mutations in both intracellular and extracellular domains of ROR2 in affected individuals from 3 unrelated consanguineous families, and a nonsense mutation that removes the tyrosine kinase domain and all subsequent 3' regions of the gene in 14 patients from 7 families from Oman. The nature of these mutations suggests that RRS is caused by loss of ROR2 activity. The identification of mutations in three distinct domains (containing Frizzled-like, kringle and tyrosine kinase motifs) indicates that these are all essential for ROR2 function.
DOI:10.1038/78113URLPMID:10932187 [本文引用: 2]
Robinow syndrome is a short-limbed dwarfism characterized by abnormal morphogenesis of the face and external genitalia, and vertebral segmentation. The recessive form of Robinow syndrome (RRS; OMIM 268310), particularly frequent in Turkey, has a high incidence of abnormalities of the vertebral column such as hemivertebrae and rib fusions, which is not seen in the dominant form. Some patients have cardiac malformations or facial clefting. We have mapped a gene for RRS to 9q21-q23 in 11 families. Haplotype sharing was observed between three families from Turkey, which localized the gene to a 4. 9-cM interval. The gene ROR2, which encodes an orphan membrane-bound tyrosine kinase, maps to this region. Heterozygous (presumed gain of function) mutations in ROR2 were previously shown to cause dominant brachydactyly type B (BDB; ref. 7). In contrast, Ror2-/- mice have a short-limbed phenotype that is more reminiscent of the mesomelic shortening observed in RRS. We detected several homozygous ROR2 mutations in our cohort of RRS patients that are located upstream from those previously found in BDB. The ROR2 mutations present in RRS result in premature stop codons and predict nonfunctional proteins.
DOI:10.1016/j.ajhg.2010.11.005URL [本文引用: 5]
We delineated a syndromic recessive preaxial brachydactyly with partial duplication of proximal phalanges to 16.8 Mb over 4 chromosomes. High-throughput sequencing of all 177 candidate genes detected a truncating frameshift mutation in the gene CHSY1 encoding a chondroitin synthase with a Fringe domain. CHSY1 was secreted from patients' fibroblasts and was required for synthesis of chondroitin sulfate moieties. Noticeably, its absence triggered massive production of JAG1 and subsequent NOTCH activation, which could only be reversed with a wild-type but not a Fringe catalytically dead CHSY1 construct. In vitro, depletion of CHSY1 by RNAi knockdown resulted in enhanced osteogenesis in fetal osteoblasts and remarkable upregulation of JAG2 in glioblastoma cells. In vivo, chsy1 knockdown in zebrafish embryos partially phenocopied the human disorder; it increased NOTCH output and impaired skeletal, pectoral-fin, and retinal development. We conclude that CHSY1 is a secreted FRINGE enzyme required for adjustment of NOTCH signaling throughout human and fish embryogenesis and particularly during limb patterning.
DOI:10.1097/01.MXE.0000414918.78299.94URL [本文引用: 1]
DOI:10.1101/gad.13.16.2072URLPMID:10465785 [本文引用: 1]
The mechanisms that control cell proliferation and cell differentiation during morphogenesis of the endochondral skeleton of vertebrates are poorly understood. Indian hedgehog (Ihh) signaling from prehypertrophic chondrocytes has been implicated in the control of chondrocyte maturation by way of feedback control of a second secreted factor parathyroid hormone-related peptide (PTHrP) at the articular surfaces. Analysis of an Ihh null mutant suggests a more extensive role for Ihh in skeletal development. Mutants display markedly reduced chondrocyte proliferation, maturation of chondrocytes at inappropriate position, and a failure of osteoblast development in endochondral bones. Together, the results suggest a model in which Ihh coordinates diverse aspects of skeletal morphogenesis through PTHrP-dependent and independent processes.
DOI:10.1038/nature07862URLPMID:19252479 [本文引用: 2]
Brachydactyly type A1 (BDA1) was the first recorded disorder of the autosomal dominant Mendelian trait in humans, characterized by shortened or absent middle phalanges in digits. It is associated with heterozygous missense mutations in indian hedgehog (IHH). Hedgehog proteins are important morphogens for a wide range of developmental processes. The capacity and range of signalling is thought to be regulated by its interaction with the receptor PTCH1 and antagonist HIP1. Here we show that a BDA1 mutation (E95K) in Ihh impairs the interaction of IHH with PTCH1 and HIP1. This is consistent with a recent paper showing that BDA1 mutations cluster in a calcium-binding site essential for the interaction with its receptor and cell-surface partners. Furthermore, we show that in a mouse model that recapitulates the E95K mutation, there is a change in the potency and range of signalling. The mice have digit abnormalities consistent with the human disorder.
DOI:10.2478/s11658-009-0040-2URL [本文引用: 2]
Heterozygous missense mutations in IHH result in Brachydactyly type A1 (BDA1; OMIM 112500), a condition characterized by the shortening of digits due to hypoplasia/aplasia of the middle phalanx. Indian Hedgehog signaling regulates the proliferation and differentiation of chondrocytes and is essential for endochondral bone formation. Analyses of activated IHH signaling in C3H10T1/2 cells showed that three BDA1-associated mutations (p.E95K, p.D100E and p.E131K) severely impaired the induction of targets such as Ptch1 and Gli1. However, this was not a complete loss of function, suggesting that these mutations may affect the interaction with the receptor PTCH1 or its partners, with an impact on the induction potency. From comparative microarray expression analyses and quantitative real-time PCR, we identified three additional targets, Sostdc1, Penk1 and Igfbp5, which were also severely affected. Penk1 and Igfbp5 were confirmed to be regulated by GLI1, while the induction of Sostdc1 by IHH is independent of GLI1. SOSTDC1 is a BMP antagonist, and altered BMP signaling is known to affect digit formation. The role of Penk1 and Igfbp5 in skeletogenesis is not known. However, we have shown that both Penk1 and Igfbp5 are expressed in the interzone region of the developing joint of mouse digits, providing another link for a role for IHH signaling in the formation of the distal digits.
DOI:10.1038/cr.2011.76URL [本文引用: 1]
Brachydactyly type A1 (BDA1), the first recorded Mendelian autosomal dominant disorder in humans, is characterized by a shortening or absence of the middle phalanges. Heterozygous missense mutations in the Indian Hedgehog (IHH) gene have been identified as a cause of BDA1; however, the biochemical consequences of these mutations are unclear. In this paper, we analyzed three BDA1 mutations (E95K, D100E, and E131K) in the N-terminal fragment of Indian Hedgehog (IhhN). Structural analysis showed that the E95K mutation changes a negatively charged area to a positively charged area in a calcium-binding groove, and that the D100E mutation changes the local tertiary structure. Furthermore, we showed that the E95K and D100E mutations led to a temperature-sensitive and calcium-dependent instability of IhhN, which might contribute to an enhanced intracellular degradation of the mutant proteins via the lysosome. Notably, all three mutations affected Hh binding to the receptor Patched1 (PTC1), reducing its capacity to induce cellular differentiation. We propose that these are common features of the mutations that cause BDA1, affecting the Hh tertiary structure, intracellular fate, binding to the receptor/partners, and binding to extracellular components. The combination of these features alters signaling capacity and range, but the impact is likely to be variable and mutation-dependent. The potential variation in the signaling range is characterized by an enhanced interaction with heparan sulfate for IHH with the E95K mutation, but not the E131K mutation. Taken together, our results suggest that these IHH mutations affect Hh signaling at multiple levels, causing abnormal bone development and abnormal digit formation.
DOI:10.1186/s12863-018-0697-5URLPMID:30651074 [本文引用: 2]
Brachydactyly type A1 (BDA1, OMIM 112500) is a rare inherited malformation characterized primarily by shortness or absence of middle bones of fingers and toes. It is the first recorded disorder of the autosomal dominant Mendelian trait. Indian hedgehog (IHH) gene is closely associated with BDA1, which was firstly mapped and identified in Chinese families in 2000. Previous studies have demonstrated that BDA1-related mutant IHH proteins affected interactions with its receptors and impaired IHH signaling. However, how the altered signaling pathway affects downstream transcriptional regulation remains unclear.
URLPMID:25696018 [本文引用: 2]
Autosomal dominant brachydactyly (BD) is a skeletal disorder with several subtypes, including brachydactyly type A1 (BDA1) and brachydactyly type B1 (BDB1). Mutations in Indian hedgehog (IHH) are usually associated with BDA1, whereas heterozygous mutations in receptor tyrosine kinase-like orphan receptor 2 (ROR2) are mainly responsible for BDB1. On the basis of the clinical phenotype identification, we screened IHH and ROR2 by the candidate gene approach using PCR direct sequencing. We found three known mutations of IHH (c.283_285delGAG, p.E95del; c.298 G>A, p.D100N; c.300C>G, p.D100E) in three Chinese families with BDA1, and a novel heterozygous nonsense mutation of ROR2 (c.2273C>A, p.S758X) in a BDB1 family. It was noted that c.300C>G mutation was a new nucleotide substitution compared to the reported c.300C>A, which led to the same amino acid change (p.D100E). The novel nonsense mutation p.S758X was verified by absence in the unaffected family members and the 100 randomly-selected controls. In this paper, we report three recurrent mutations with a new nucleotide substitution of IHH in three Chinese families with BDA1 and a novel nonsense mutation in BDB1 pedigree. We therefore recommend the approach of candidate gene screening as the first choice for genetic testing for BD.
DOI:10.1055/s-0037-1599201URLPMID:28794911 [本文引用: 1]
Brachydactyly type A1 (BDA1) is characterized by short middle phalanges. We report the case of a child with a severe form of BDA1 with complete absence of the middle phalanges of all extremities. He had c.298G?>?A (p.D100N) mutation in IHH gene.
DOI:10.1002/ajmg.a.37490URLPMID:26640227 [本文引用: 1]
Autosomal-dominant brachydactyly type E is a congenital limb malformation characterized by small hands and feet as a result of shortened metacarpals and metatarsals. Alterations that predict haploinsufficiency of PTHLH, the gene coding for parathyroid hormone related protein (PTHrP), have been identified as a cause of this disorder in seven families. Here, we report three patients affected with brachydactyly type E, caused by PTHLH mutations expected to result in haploinsufficiency, and discuss our data compared to published reports.
DOI:10.1016/j.ydbio.2012.01.005URL [本文引用: 1]
Joint and skeletal development is highly regulated by extracellular matrix (ECM) proteoglycans, of which chondroitin sulfate proteoglycans (CSPGs) are a major class. Despite the requirement of joint CSPGs for skeletal flexibility and structure, relatively little is understood regarding their role in establishing joint positioning or in modulating signaling and cell behavior during joint formation. Chondroitin sulfate synthase 1 (Chsy1) is one of a family of enzymes that catalyze the extension of chondroitin and dermatan sulfate glycosaminoglycans. Recently, human syndromic brachydactylies have been described to have loss-of-function mutations at the CHSY1 locus. In concordance with these observations, we demonstrate that mice lacking Chsy1, though viable, display chondrodysplasia and decreased bone density. Notably, Chsy1(-/-) mice show a profound limb patterning defect in which orthogonally shifted ectopic joints form in the distal digits. Associated with the digit-patterning defect is a shift in cell orientation and an imbalance in chondroitin sulfation. Our results place Chsy1 as an essential regulator of joint patterning and provide a mouse model of human brachydactylies caused by mutations in CHSY1. (C) 2012 Elsevier Inc.
DOI:10.1002/dvdy.23981URLPMID:23703795 [本文引用: 1]
Chondroitin/dermatan sulfate (CS/DS) proteoglycans present in the extracellular matrix have important structural and regulatory functions.
DOI:10.7554/eLife.29329URLPMID:29205154 [本文引用: 1]
GWAS have identified hundreds of height-associated loci. However, determining causal mechanisms is challenging, especially since height-relevant tissues (e.g. growth plates) are difficult to study. To uncover mechanisms by which height GWAS variants function, we performed epigenetic profiling of murine femoral growth plates. The profiled open chromatin regions recapitulate known chondrocyte and skeletal biology, are enriched at height GWAS loci, particularly near differentially expressed growth plate genes, and enriched for binding motifs of transcription factors with roles in chondrocyte biology. At specific loci, our analyses identified compelling mechanisms for GWAS variants. For example, at CHSY1, we identified a candidate causal variant (rs9920291) overlapping an open chromatin region. Reporter assays demonstrated that rs9920291 shows allelic regulatory activity, and CRISPR/Cas9 targeting of human chondrocytes demonstrates that the region regulates CHSY1 expression. Thus, integrating biologically relevant epigenetic information (here, from growth plates) with genetic association results can identify biological mechanisms important for human growth.
DOI:10.1007/s00018-018-2928-3URLPMID:30327840 [本文引用: 1]
Low-density lipoprotein receptor-related protein 4 (LRP4) is a multi-functional protein implicated in bone, kidney and neurological diseases including Cenani-Lenz syndactyly (CLS), sclerosteosis, osteoporosis, congenital myasthenic syndrome and myasthenia gravis. Why different LRP4 mutation alleles cause distinct and even contrasting disease phenotypes remain unclear. Herein, we utilized the zebrafish model to search for pathways affected by a deficiency of LRP4. The lrp4 knockdown in zebrafish embryos exhibits cyst formations at fin structures and the caudal vein plexus, malformed pectoral fins, defective bone formation and compromised kidney morphogenesis; which partially phenocopied the human LRP4 mutations and were reminiscent of phenotypes resulting form a perturbed Notch signaling pathway. We discovered that the Lrp4-deficient zebrafish manifested increased Notch outputs in addition to enhanced Wnt signaling, with the expression of Notch ligand jagged1b being significantly elevated at the fin structures. To examine conservatism of signaling mechanisms, the effect of LRP4 missense mutations and siRNA knockdowns, including a novel missense mutation c.1117C?>?T (p.R373W) of LRP4, were tested in mammalian kidney and osteoblast cells. The results showed that LRP4 suppressed both Wnt/β-Catenin and Notch signaling pathways, and these activities were perturbed either by LRP4 missense mutations or by a knockdown of LRP4. Our finding underscore that LRP4 is required for limiting Jagged-Notch signaling throughout the fin/limb and kidney development, whose perturbation representing a novel mechanism for LRP4-related diseases. Moreover, our study reveals an evolutionarily conserved relationship between LRP4 and Jagged-Notch signaling, which may shed light on how the Notch signaling is fine-tuned during fin/limb development.
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.gene.2014.06.035URL [本文引用: 2]
Brachydactyly type B, an autosomal dominant disorder that is characterized by hypoplasia of the distal phalanges and nails, can be divided into brachydactyly type B1 (BDB1) and brachydactyly type B2 (BDB2). BDB1 is caused by mutations in the receptor tyrosine kinase gene ROR2, which maps to chromosome 9q22, whereas BDB2 is caused by point mutations in the bone morphogenetic protein antagonist NOGGIN. Here, we report a three-generation Chinese family with dominant inheritance of the BDB1 limb phenotype. Sequence analysis identified a novel heterozygous base deletion (c.1396-1398delAA) in the gene ROR2 in all affected family members. This new deletion is expected to produce a truncated Ror2 protein with a new polypeptide of 57 amino acids at the C-terminal. (C) 2014 Elsevier B.V.
DOI:10.1046/j.1365-2443.2003.00662.xURLPMID:12839624 [本文引用: 1]
Ror2 is an orphan receptor, belonging to the Ror family of receptor tyrosine kinases. Although Ror2 has been shown to play crucial roles in developmental morphogenesis, the precise signalling events that Ror2 mediates remain elusive. Since Ror2 possesses an extracellular cysteine-rich domain (CRD) that resembles the Wnt-binding sites of the Frizzled (Fz) proteins, it is conceivable that Ror2 interacts with members of the Wnt family.
DOI:10.1038/73495URLPMID:10700182 [本文引用: 1]
Inherited limb malformations provide a valuable resource for the identification of genes involved in limb development. Brachydactyly type B (BDB), an autosomal dominant disorder, is the most severe of the brachydactylies and characterized by terminal deficiency of the fingers and toes. In the typical form of BDB, the thumbs and big toes are spared, sometimes with broadening or partial duplication. The BDB1 locus was previously mapped to chromosome 9q22 within an interval of 7.5 cM (refs 9,10). Here we describe mutations in ROR2, which encodes the orphan receptor tyrosine kinase ROR2 (ref. 11), in three unrelated families with BDB1. We identified distinct heterozygous mutations (2 nonsense, 1 frameshift) within a 7-amino-acid segment of the 943-amino-acid protein, all of which predict truncation of the intracellular portion of the protein immediately after the tyrosine kinase domain. The localized nature of these mutations suggests that they confer a specific gain of function. We obtained further evidence for this by demonstrating that two patients heterozygous for 9q22 deletions including ROR2 do not exhibit BDB. Expression of the mouse mouse orthologue, Ror2, early in limb development indicates that BDB arises as a primary defect of skeletal patterning.
DOI:10.1073/pnas.1009314107URLPMID:20660756 [本文引用: 1]
Elongation of the digit rays resulting in the formation of a defined number of phalanges is a process poorly understood in mammals, whereas in the chicken distal mesenchymal bone morphogenetic protein (BMP) signaling in the so-called phalanx-forming region (PFR) or digit crescent (DC) seems to be involved. The human brachydactylies (BDs) are inheritable conditions characterized by variable degrees of digit shortening, thus providing an ideal model to analyze the development and elongation of phalanges. We used a mouse model for BDB1 (Ror2(W749X/W749X)) lacking middle phalanges and show that a signaling center corresponding to the chick PFR exists in the mouse, which is diminished in BDB1 mice. This resulted in a strongly impaired elongation of the digit condensations due to reduced chondrogenic commitment of undifferentiated distal mesenchymal cells. We further show that a similar BMP-based mechanism accounts for digit shortening in a mouse model for the closely related condition BDA1 (Ihh(E95K/E95K)), altogether indicating the functional significance of the PFR in mammals. Genetic interaction experiments as well as pathway analysis in BDB1 mice suggest that Indian hedgehog and WNT/beta-catenin signaling, which we show is inhibited by receptor tyrosine kinase-like orphan receptor 2 (ROR2) in distal limb mesenchyme, are acting upstream of BMP signaling in the PFR.
DOI:10.1136/jmg.2010.084814URL [本文引用: 1]
Background Brachydactyly type A2 (BDA2) is an autosomal dominant disorder. It was recently reported that a 5.9 kb duplication and a 5.5 kb duplication in the region 20p12.2-12.3 are associated with BDA2 in two European families.
Objective To characterise a 6-generation Chinese family with 16 members affected by BDA2 and map the gene to 20p12.2-12.3.
Methods and results A 4.6 kb duplication downstream of the bone morphogenetic protein 2 (BMP2) was identified in the family. The duplication co-segregated with the phenotype and was absent in unaffected family members and control subjects. Coding and splice-site mutations of all annotated genes in the critical region were also excluded. The duplication partially overlaps with the reported duplications but has a different breakpoint. The most conserved 2.1 kb fragment in the duplication was cloned into the pGL3-promoter vector downstream of the firefly luciferase reporter gene in the 5' to 3' orientation and transfected into osteosarcoma U-2OS and Hela cells. A reduced luciferase activity was observed.
Conclusion The smallest duplication is described, which partially overlaps the reported duplications but has a different breakpoint, and its association with BDA2 in a Chinese family is confirmed. The results also provide evidence for cis-regulatory sequences in the duplication 3' of BMP2.
DOI:10.1371/journal.pone.0094201URLPMID:24710560 [本文引用: 1]
Brachydactyly type A2 (BDA2, MIM 112600) is characterized by the deviation and shortening of the middle phalange of the index finger and the second toe. Using genome-wide linkage analysis in a Chinese BDA2 family, we mapped the maximum candidate interval of BDA2 to a ~1.5 Mb region between D20S194 and D20S115 within chromosome 20p12.3 and found that the pairwise logarithm of the odds score was highest for marker D20S156 (Zmax?=?6.09 at θ?=?0). Based on functional and positional perspectives, the bone morphogenetic protein 2 (BMP2) gene was identified as the causal gene for BDA2 in this region, even though no point mutation was detected in BMP2. Through further investigation, we identified a 4,671 bp (Chr20: 6,809,218-6,813,888) genomic duplication downstream of BMP2. This duplication was located within the linked region, co-segregated with the BDA2 phenotype in this family, and was not found in the unaffected family members and the unrelated control individuals. Compared with the previously reported duplications, the duplication in this family has a different breakpoint flanked by the microhomologous sequence GATCA and a slightly different length. Some other microhomologous nucleotides were also found in the duplicated region. In summary, our findings support the conclusions that BMP2 is the causing gene for BDA2, that the genomic location corresponding to the duplication region is prone to structural changes associated with malformation of the digits, and that this tendency is probably caused by the abundance of microhomologous sequences in the region.
DOI:10.1016/j.gene.2017.11.024URLPMID:29129813 [本文引用: 1]
Brachydactyly type A2 (BDA2) is an autosomal dominant disease characterized by the deformation of the middle phalanx of the second fingers and toes. It has been reported to be associated with three genes regulating the osteogenesis, including BMPR1B, GDF5 and BMP2.
DOI:10.1371/journal.pgen.1003846URLPMID:24098149 [本文引用: 1]
Growth and Differentiation Factor 5 (GDF5) is a secreted growth factor that belongs to the Bone Morphogenetic Protein (BMP) family and plays a pivotal role during limb development. GDF5 is a susceptibility gene for osteoarthritis (OA) and mutations in GDF5 are associated with a wide variety of skeletal malformations ranging from complex syndromes such as acromesomelic chondrodysplasias to isolated forms of brachydactylies or multiple synostoses syndrome 2 (SYNS2). Here, we report on a family with an autosomal dominant inherited combination of SYNS2 and additional brachydactyly type A1 (BDA1) caused by a single point mutation in GDF5 (p.W414R). Functional studies, including chondrogenesis assays with primary mesenchymal cells, luciferase reporter gene assays and Surface Plasmon Resonance analysis, of the GDF5(W414R) variant in comparison to other GDF5 mutations associated with isolated BDA1 (p.R399C) or SYNS2 (p.E491K) revealed a dual pathomechanism characterized by a gain- and loss-of-function at the same time. On the one hand insensitivity to the main GDF5 antagonist NOGGIN (NOG) leads to a GDF5 gain of function and subsequent SYNS2 phenotype. Whereas on the other hand, a reduced signaling activity, specifically via the BMP receptor type IA (BMPR1A), is likely responsible for the BDA1 phenotype. These results demonstrate that one mutation in the overlapping interface of antagonist and receptor binding site in GDF5 can lead to a GDF5 variant with pathophysiological relevance for both, BDA1 and SYNS2 development. Consequently, our study assembles another part of the molecular puzzle of how loss and gain of function mutations in GDF5 affect bone development in hands and feet resulting in specific types of brachydactyly and SYNS2. These novel insights into the biology of GDF5 might also provide further clues on the pathophysiology of OA.
DOI:10.1093/hmg/ddn012URLPMID:18203755 [本文引用: 1]
We investigated a family with a brachydactyly type A2 and identified a heterozygous arginine to glutamine (R380Q) substitution in the growth/differentiation factor 5 (GDF5) in all affected individuals. The observed mutation is located at the processing site of the protein, at which the GDF5 precursor is thought to be cleaved releasing the mature molecule from the prodomain. In order to test the effect of the mutation, we generated the GDF5-R380Q mutant and a cleavage-resistant proGDF5 mutant (R380A/R381A) in vitro. Both mutants were secreted from chicken micromass cultures, but showed diminished biological activity. Western blot analyses showed that wt GDF5 was processed by the chicken micromass cells, whereas the mutants were not, indicating that the mutations interfere with processing and that this leads to a strong reduction of biological activity. To test the requirements for GDF5 processing in vitro we produced recombinant human (rh) proGDF5 wild-type protein in Escherichia coli. The results show that unprocessed (rh) proGDF5 is virtually inactive but can be proteolytically activated by different enzymes such as trypsin, furin, and MMP3. (rh) proGDF5 could thus be used as a locally administered depot form with retarded release of activity. In contrast to mature rhGDF5, (rh) proGDF5 shows a high solubility at physiological pH, a characteristic that might be useful for therapeutic applications.
DOI:10.1136/jmg.2005.034058URLPMID:16014698 [本文引用: 1]
Brachydactyly type A2 (OMIM 112600) is characterised by hypoplasia/aplasia of the second middle phalanx of the index finger and sometimes the little finger. BDA2 was first described by Mohr and Wriedt in a large Danish/Norwegian kindred and mutations in BMPR1B were recently demonstrated in two affected families.
DOI:10.12669/pjms.341.12885URLPMID:29643884 [本文引用: 3]
Brachdactyly a genetic disorder associated with the abnormal development of metacarpals, phalanges or both which results in the shortening of hands and feet. Mutations in the contributing genes has been recognized with the majority of the investigated syndromic form of brachdactyly. The current study was proposed to examine mutation in NOG and GDF5 genes in a Pakistani family.
DOI:10.1002/ajmg.10777URLPMID:12357473 [本文引用: 1]
Growth/differentiation factor-5 (GDF5), also known as cartilage-derived morphogenetic protein-1 (CDMP-1), is a secreted signaling molecule that participates in skeletal morphogenesis. Heterozygous mutations in GDF5, which maps to human chromosome 20, occur in individuals with autosomal dominant brachydactyly type C (BDC). Here we show that BDC is locus homogeneous by reporting a GDF5 frameshift mutation segregating with the phenotype in a family whose trait was initially thought to map to human chromosome 12. We also describe heterozygous mutations in nine additional probands/families with BDC and show nonpenetrance in a mutation carrier. Finally, we show that mutant GDF5 polypeptides containing missense mutations in their active domains do not efficiently form disulfide-linked dimers when expressed in vitro. These data support the hypothesis that BDC results from functional haploinsufficiency for GDF5.
DOI:10.1016/j.ejmg.2015.06.005URLPMID:26211601 [本文引用: 1]
Human noggin (NOG) gene mutation causes multiple bony disorders showing up as stapes ankylosis with broad thumbs and toes (SABTT), proximal symphalangism (SYM1), multiple synostoses syndrome 1 (SYNS1), tarsal-carpal coalition syndrome (TCC) and brachydactyly type B2 (BDB2). These phenotypes are defined as NOG-related syndromes with the same mutation. Some of these syndromes feature stapes ankylosis as one of the several bony symptoms. Here, we report a Japanese family with conductive hearing loss due to congenital stapes ankylosis. This family showed multiple features and was diagnosed with SABTT. We performed analysis of the NOG in the family by direct sequence analysis, and found a novel NOG mutation: c.682?T>?G (p.C228G). Our results and a review of previous cases with NOG protein conformation suggest that this mutated NOG protein lead to a change in antagonist activity in BMPs and/or a haploinsufficiency that likely impaired finger 2 structure.
DOI:10.1126/science.280.5368.1455URLPMID:9603738 [本文引用: 1]
Noggin is a bone morphogenetic protein (BMP) antagonist expressed in Spemann's organizer. Murine Noggin is expressed in condensing cartilage and immature chondrocytes, as are many BMPs. In mice lacking Noggin, cartilage condensations initiated normally but developed hyperplasia, and initiation of joint development failed as measured by the expression of growth and differentiation factor-5. The maturation of cartilage and Hoxd expression were unaffected. Excess BMP activity in the absence of Noggin antagonism may enhance the recruitment of cells into cartilage, resulting in oversized growth plates; chondrocytes are also refractory to joint-inducing positional cues.
DOI:10.1016/j.ajhg.2010.10.003URL [本文引用: 2]
Altered Bone Morphogenetic Protein (BMP) signaling leads to multiple developmental defects, including brachydactyly and deafness. Here we identify chondroitin synthase 1 (CHSY1) as a potential mediator of BMP effects. We show that loss of human CHSY1 function causes autosomal-recessive Temtamy preaxial brachydactyly syndrome (TPBS), mainly characterized by limb malformations, short stature, and hearing loss. After mapping the TPBS locus to chromosome 15q26-qterm, we identified causative mutations in five consanguineous TPBS families. In zebrafish, antisense-mediated chsy1 knockdown causes defects in multiple developmental processes, some of which are likely to also be causative in the etiology of TPBS. In the inner ears of zebrafish larvae, chsy1 is expressed similarly to the BMP inhibitor dan and in a complementary fashion to bmp2b. Furthermore, unrestricted Bmp2b signaling or loss of Dan activity leads to reduced chsy1 expression and, during epithelial morphogenesis, defects similar to those that occur upon Chsy1 inactivation, indicating that Bmp signaling affects inner-ear development by repressing chsy1. In addition, we obtained strikingly similar zebrafish phenotypes after chsy1 overexpression, which might explain why, in humans, brachydactyly can be caused by mutations leading either to loss or to gain of BMP signaling.
DOI:10.1074/jbc.M112.409342URLPMID:23423383 [本文引用: 1]
Specification of progenitors into the osteoblast lineage is an essential event for skeletogenesis. During endochondral ossification, cells in the perichondrium give rise to osteoblast precursors. Hedgehog (Hh) and bone morphogenetic protein (BMP) are suggested to regulate the commitment of these cells. However, properties of perichondrial cells and regulatory mechanisms of the specification process are still poorly understood. Here, we investigated the machineries by combining a novel organ culture system and single-cell expression analysis with mouse genetics and biochemical analyses. In a metatarsal organ culture reproducing bone collar formation, activation of BMP signaling enhanced the bone collar formation cooperatively with Hh input, whereas the signaling induced ectopic chondrocyte formation in the perichondrium without Hh input. Similar phenotypes were also observed in compound mutant mice, where signaling activities of Hh and BMP were genetically manipulated. Single-cell quantitative RT-PCR analyses showed heterogeneity of perichondrial cells in terms of natural characteristics and responsiveness to Hh input. In vitro analyses revealed that Hh signaling suppressed BMP-induced chondrogenic differentiation; Gli1 inhibited the expression of Sox5, Sox6, and Sox9 (SRY box-containing gene 9) as well as transactivation by Sox9. Indeed, ectopic expression of chondrocyte maker genes were observed in the perichondrium of metatarsals in Gli1(-/-) fetuses, and the phenotype was more severe in Gli1(-/-);Gli2(-/-) newborns. These data suggest that Hh-Gli activators alter the function of BMP to specify perichondrial cells into osteoblasts; the timing of Hh input and its target populations are critical for BMP function.
DOI:10.1242/dev.01006URLPMID:14973297 [本文引用: 1]
Indian hedgehog (Ihh) is indispensable for development of the osteoblast lineage in the endochondral skeleton. In order to determine whether Ihh is directly required for osteoblast differentiation, we have genetically manipulated smoothened (Smo), which encodes a transmembrane protein that is essential for transducing all Hedgehog (Hh) signals. Removal of Smo from perichondrial cells by the Cre-LoxP approach prevents formation of a normal bone collar and also abolishes development of the primary spongiosa. Analysis of chimeric embryos composed of wild-type and Smo(n/n) cells indicates that Smo(n/n) cells fail to contribute to osteoblasts in either the bone collar or the primary spongiosa but generate ectopic chondrocytes. In order to assess whether Ihh is sufficient to induce bone formation in vivo, we have analyzed the bone collar in the long bones of embryos in which Ihh was artificially expressed in all chondrocytes by the UAS-GAL4 bigenic system. Although ectopic Ihh does not induce overt ossification along the entire cartilage anlage, it promotes progression of the bone collar toward the epiphysis, suggesting a synergistic effect between ectopic Ihh and endogenous factors such as the bone morphogenetic proteins (BMPs). In keeping with this model, Hh signaling is further found to be required in BMP-induced osteogenesis in cultures of a limb-bud cell line. Taken together, these results demonstrate that Ihh signaling is directly required for the osteoblast lineage in the developing long bones and that Ihh functions in conjunction with other factors such as BMPs to induce osteoblast differentiation. We suggest that Ihh acts in vivo on a potential progenitor cell to promote osteoblast and prevent chondrocyte differentiation.
DOI:10.1016/j.ydbio.2005.05.036URLPMID:16023633 [本文引用: 1]
Smad4 is the central intracellular mediator of transforming growth factor-beta (TGF-beta) signals. To study the role of Smad4 in skeletal development, we introduced a conditional mutation of the gene in chondrocytes using Cre--loxP system. We showed that Smad4 was expressed strongly in prehypertrophic and hypertrophic chondrocytes. The abrogation of Smad4 in chondrocytes resulted in dwarfism with a severely disorganized growth plate characterized by expanded resting zone of chondrocytes, reduced chondrocyte proliferation, accelerated hypertrophic differentiation, increased apoptosis and ectopic bone collars in perichondrium. Meanwhile, Smad4 mutant mice exhibited decreased expression of molecules in Indian hedgehog/parathyroid hormone-related protein (Ihh/PTHrP) signaling. The cultured mutant metatarsal bones failed to response to TGF-beta1, while the hypertrophic differentiation was largely inhibited by Sonic hedgehog (Shh). This indicated that Ihh/PTHrP inhibited the hypertrophic differentiation of chondrocytes independent of the Smad4-mediated TGF-beta signals. All these data provided the first genetic evidence demonstrating that Smad4-mediated TGF-beta signals inhibit the chondrocyte hypertrophic differentiation, and are required for maintaining the normal organization of chondrocytes in the growth plate.
DOI:10.1359/jbmr.2003.18.9.1593URLPMID:12968668 [本文引用: 1]
Growth plate chondrocytes integrate multiple signals during normal development. The type I BMP receptor ALK2 is expressed in cartilage and expression of constitutively active (CA) ALK2 and other activated type I BMP receptors results in maturation-independent expression of Ihh in chondrocytes in vitro and in vivo. The findings suggest that BMP signaling modulates the Ihh/PTHrP signaling pathway that regulates the rate of chondrocyte differentiation.
URLPMID:11714677 [本文引用: 1]
During endochondral ossification, two secreted signals, Indian hedgehog (Ihh) and parathyroid hormone-related protein (PTHrP), have been shown to form a negative feedback loop regulating the onset of hypertrophic differentiation of chondrocytes. Bone morphogenetic proteins (BMPs), another family of secreted factors regulating bone formation, have been implicated as potential interactors of the Ihh/PTHrP feedback loop. To analyze the relationship between the two signaling pathways, we used an organ culture system for limb explants of mouse and chick embryos. We manipulated chondrocyte differentiation by supplementing these cultures either with BMP2, PTHrP and Sonic hedgehog as activators or with Noggin and cyclopamine as inhibitors of the BMP and Ihh/PTHrP signaling systems. Overexpression of Ihh in the cartilage elements of transgenic mice results in an upregulation of PTHrP expression and a delayed onset of hypertrophic differentiation. Noggin treatment of limbs from these mice did not antagonize the effects of Ihh overexpression. Conversely, the promotion of chondrocyte maturation induced by cyclopamine, which blocks Ihh signaling, could not be rescued with BMP2. Thus BMP signaling does not act as a secondary signal of Ihh to induce PTHrP expression or to delay the onset of hypertrophic differentiation. Similar results were obtained using cultures of chick limbs. We further investigated the role of BMP signaling in regulating proliferation and hypertrophic differentiation of chondrocytes and identified three functions of BMP signaling in this process. First we found that maintaining a normal proliferation rate requires BMP and Ihh signaling acting in parallel. We further identified a role for BMP signaling in modulating the expression of IHH: Finally, the application of Noggin to mouse limb explants resulted in advanced differentiation of terminally hypertrophic cells, implicating BMP signaling in delaying the process of hypertrophic differentiation itself. This role of BMP signaling is independent of the Ihh/PTHrP pathway.
DOI:10.1359/jbmr.090806URLPMID:19874086 [本文引用: 1]
The bone morphogenetic protein (BMP) and Wnt signaling pathways both contribute essential roles in regulating bone mass. However, the molecular interactions between these pathways in osteoblasts are poorly understood. We recently reported that osteoblast-targeted conditional knockout (cKO) of BMP receptor type IA (BMPRIA) resulted in increased bone mass during embryonic development, where diminished expression of Sost as a downstream effector of BMPRIA resulted in increased Wnt/beta-catenin signaling. Here, we report that Bmpr1a cKO mice exhibit increased bone mass during weanling stages, again with evidence of enhanced Wnt/beta-catenin signaling as assessed by Wnt reporter TOPGAL mice and TOPFLASH luciferase. Consistent with negative regulation of the Wnt pathway by BMPRIA signaling, treatment of osteoblasts with dorsomorphin, an inhibitor of Smad-dependent BMP signaling, enhanced Wnt signaling. In addition to Sost, Wnt inhibitor Dkk1 also was downregulated in cKO bone. Expression levels of Dkk1and Sost were upregulated by BMP2 treatment and downregulated by Noggin. Moreover, expression of a constitutively active Bmpr1a transgene in mice resulted in the upregulation of both Dkk1 and Sost and partially rescued the Bmpr1a cKO bone phenotype. These effectors are differentially regulated by mitogen-activated protein kinase (MAPK) p38 because pretreatment of osteoblasts with SB202190 blocked BMP2-induced Dkk1 expression but not Sost. These results demonstrate that BMPRIA in osteoblasts negatively regulates endogenous bone mass and Wnt/beta-catenin signaling and that this regulation may be mediated by the activities of Sost and Dkk1. This study highlights several interactions between BMP and Wnt signaling cascades in osteoblasts that may be amenable to therapeutic intervention for the modification of bone mass density.
DOI:10.1242/dev.025825URLPMID:18927151 [本文引用: 1]
Bone morphogenetic proteins (BMPs) are known to induce ectopic bone. However, it is largely unknown how BMP signaling in osteoblasts directly regulates endogenous bone. This study investigated the mechanism by which BMP signaling through the type IA receptor (BMPR1A) regulates endogenous bone mass using an inducible Cre-loxP system. When BMPR1A in osteoblasts was conditionally disrupted during embryonic bone development, bone mass surprisingly was increased with upregulation of canonical Wnt signaling. Although levels of bone formation markers were modestly reduced, levels of resorption markers representing osteoclastogenesis were severely reduced, resulting in a net increase in bone mass. The reduction of osteoclastogenesis was primarily caused by Bmpr1a-deficiency in osteoblasts, at least through the RANKL-OPG pathway. Sclerostin (Sost) expression was downregulated by about 90% and SOST protein was undetectable in osteoblasts and osteocytes, whereas the Wnt signaling was upregulated. Treatment of Bmpr1a-deficient calvariae with sclerostin repressed the Wnt signaling and restored normal bone morphology. By gain of Smad-dependent BMPR1A signaling in mice, Sost expression was upregulated and osteoclastogenesis was increased. Finally, the Bmpr1a-deficient bone phenotype was rescued by enhancing BMPR1A signaling, with restoration of osteoclastogenesis. These findings demonstrate that BMPR1A signaling in osteoblasts restrain endogenous bone mass directly by upregulating osteoclastogenesis through the RANKL-OPG pathway, or indirectly by downregulating canonical Wnt signaling through sclerostin, a Wnt inhibitor and a bone mass mediator.
DOI:10.1074/jbc.M513812200URLPMID:16621789 [本文引用: 1]
Genetic evidence from both humans and mice suggests that Wnt/beta-catenin and bone morphogenetic protein (BMP) signaling pathways are essential for bone marrow mesenchymal stem cells to differentiate into osteoblasts. Here we describe a mechanism through which BMPs antagonize Wnt signaling and retard bone marrow mesenchymal stem cell proliferation. Treatment with Wnt3a, but not BMP-2, stimulated Lef1-mediated transcriptional activity, whereas co-stimulation with both Wnt3a and BMP-2 markedly reduced Wnt3a-induced reporter activity. Immunoprecipitation assays in 293T cells transfected with individual Smads and Wnt pathway components revealed a specific interaction between Dvl-1 and Smad1 that was dependent on the presence of Wnt3a or BMP-2. Under unstimulated conditions, Dvl-1 and Smad1 are co-immunoprecipitated and form a complex through the linker region of Smad1. Wnt3a treatment transiently disrupted the Dvl-1/Smad1 interaction coincident with nuclear accumulation of beta-catenin. In contrast, when cells were exposed to both Wnt3a and BMP-2, there was an enhanced accumulation of the Dvl-1-Smad1 complex and a decreased nuclear accumulation of beta-catenin. Expression of a mutant Smad1 protein, which cannot be phosphorylated in response to BMP, eliminated the inhibitory effect of BMP on Wnt-inducedbeta-catenin accumulation and transcriptional activity. These results identify a potential mechanism whereby BMP-2 antagonizes Wnt signaling in osteoblast progenitors by promoting an interaction between Smad1 and Dvl-1 that restricts beta-catenin activation.
DOI:10.1359/jbmr.2003.18.10.1842URLPMID:14584895 [本文引用: 1]
Wnt/beta-catenin signaling has recently been suggested to be involved in bone biology. The precise role of this cascade in osteoblast differentiation was examined. We show that a Wnt autocrine loop mediates the induction of alkaline phosphatase and mineralization by BMP-2 in pre-osteoblastic cells.
DOI:10.1002/jbmr.260URLPMID:20878775 [本文引用: 1]
Osteoblast differentiation depends on the coordinated network of evolutionary conserved transcription factors during bone formation and homeostasis. Evidence indicates that bone morphogenetic protein (BMP) and Wnt proteins regulate several steps of skeletal development. Here, we provide a molecular description of the cooperative effects of BMP and Wnt canonical pathway on the expression of the early osteogenic genes Dlx5, Msx2, and Runx2 in C2C12 cells, primary cultures of bone marrow-mesenchymal stem cells, and organotypic calvarial cultures. Coordinated regulation of these genes leads to the cooperative activation of their downstream osteogenic target gene osterix. Induction of these genes is mediated through enhancer regions with an evolutionary conserved structure encompassing both Smad and TCF/LEF1 DNA-binding sites. Formation of a cooperative complex is mediated through DNA binding of Smads and TCF4/β-catenin to their cognate sequences, as well as protein-protein interactions between them. The formation of these cooperative transcriptional complexes results in a more efficient recruitment of coactivators such as p300. We propose that evolutionary conserved regulatory regions in specific osteogenic master genes are key integrative modules during osteogenesis.
[本文引用: 2]
[本文引用: 2]

{kind=link}
{kind=link}