 ,中国科学院遗传与发育生物学研究所,分子系统生物学中心,北京 100101
,中国科学院遗传与发育生物学研究所,分子系统生物学中心,北京 100101Advances of the technologies in large-scale membrane proteome identification
Dandan Lv, Yuanya Zhang, Haitao Ge, Xiahe Huang, Yingchun Wang,State Key Laboratory of Molecular Developmental Biology, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing 100101, China通讯作者:
编委: 陈德富
收稿日期:2019-09-8修回日期:2019-09-14网络出版日期:2019-09-20
| 基金资助: |
Editorial board:
Received:2019-09-8Revised:2019-09-14Online:2019-09-20
| Fund supported: |
作者简介 About authors
吕丹丹,博士,工程师,研究方向:蛋白质组学E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (607KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
吕丹丹, 张媛雅, 葛海涛, 黄夏禾, 汪迎春. 大规模膜蛋白质组鉴定技术进展[J]. 遗传, 2019, 41(9): 863-874 doi:10.16288/j.yczz.19-275
Dandan Lv, Yuanya Zhang, Haitao Ge, Xiahe Huang, Yingchun Wang.
生物膜主要由磷脂双分子层和镶嵌或附着在其上的蛋白质构成。动物、植物和微生物通常都有一种或多种膜系统。细胞质膜(质膜)是最为常见的一种膜系统,负责把细胞质和其他内溶物与周围环境分隔开,并负责与环境交换物质和感受信号。除质膜外,细胞内还有多种内膜系统,如高尔基体、线粒体、叶绿体等都有一层或多层膜状结构。叶绿体的膜系统分为被膜和类囊体膜,由外膜和内膜构成的被膜主要的作用是起到将叶绿体的内溶物与细胞质分开,并负责细胞质与叶绿体之间的物质和信息交换。类囊体膜则是光合作用的主要场所,光合电子传递链中的所有蛋白质复合体都定位于类囊体膜上,包括光合系统Ⅰ、光合系统Ⅱ、细胞色素Cyt b6f复合物以及ATP合酶复合物(图1)。
膜蛋白质是生物膜的重要组成成分,在物质运输和信号转导的过程中发挥着关键作用。据预测,一个物种的基因组所编码的蛋白有30%是膜蛋白[1,2]。由于膜蛋白具有一些特殊的氨基酸组成并由此产生强疏水性,从而导致对膜蛋白的蛋白质组学鉴定要远远难于可溶性蛋白,这也使得对膜蛋白的结构与功能的研究进展相对较为缓慢。本文以一种光合作用模式蓝藻集胞藻(Synechocystis sp. PCC6803)为例,介绍了膜蛋白的生物化学特征以及膜蛋白质组学相关技术,综述了过去20余年膜蛋白质组的研究进展,着重阐明了技术的进步如何在提高膜蛋白质的鉴定效率及覆盖度。
1 膜蛋白生物化学特征及分离纯化方法
1.1 膜蛋白的生物化学特征
由于膜蛋白质在功能上的重要性以及在解析膜蛋白质组上存在的巨大技术挑战,从蛋白质组学学科出现开始,膜蛋白质的分离和鉴定就成为了蛋白质组学研究的一个焦点方向。膜蛋白质的研究之所以具有巨大的技术上的挑战性,最主要的原因是由于其高于一般蛋白质的疏水性。整合膜蛋白质(integral membrane protein, IMP)一般都通过一个或多个跨膜结构域(transmembrane domain, TMD)而整合在膜上,跨膜结构域大小一般在20个氨基酸残基左右,主要由疏水氨基酸组成,可以和膜的重要组成成分(磷脂双分子层)发生疏水相互作用,从而将蛋白质固定到膜上[3,4]。除了IMP,生物膜通常还含有大量的周边膜蛋白 (peripheral membrane protein, PMP)。PMP不含跨膜结构域,与膜的结合主要是通过蛋白质-蛋白质或者蛋白质与脂类的相互作用(图1)。有些蛋白质还会发生翻译后修饰,并通过修饰集团与膜的相互作用而结合到膜上。常见的化学修饰主要有磷酸肌醇修饰、粽榈酰化、豆蔻酰化等。蛋白质与膜的相互作用一般可以分为共价和非共价两种类型。共价相互作用非常稳定,磷酸肌醇修饰的蛋白与膜的结合可以看作是一种共价的结合,结合非常稳定,很难从膜上被分离[5,6]。而很多蛋白质-蛋白质之间的相互作用通常是非共价的,包括氢键、静电相互作用、疏水相互作用以及范德华力等。这些非共价的相互作用可以介导蛋白质形成复合体。通常很多PMP通过和一个或多个IMP共同形成复合体而结合在膜上。PMP与膜的结合一般是可逆的,其结合稳定度通常随细胞内微环境的变化而变化。对于通过生物化学方法分离的悬浮在缓冲液中的生物膜,如果改变缓冲液盐离子浓度或pH值,都有可能有效地将PMP从生物膜上解离下来。图1
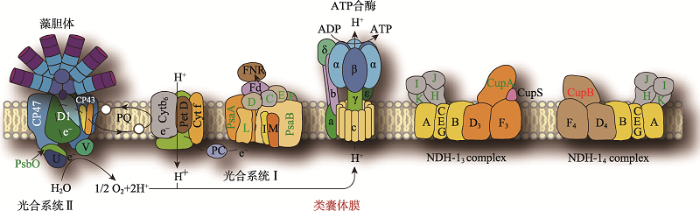 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1蓝藻类囊体膜结构示意图
类囊体膜是光合作用的主要场所,与此相关的一些主要的蛋白复合物如光合系统Ⅰ(Photosystem Ⅰ)、光合系统Ⅱ(Photosystem Ⅱ)、ATP合酶(ATP synthase)以及与CO2固定相关的NDH-13和NDH-14复合体均分布在类囊体膜上。这些膜蛋白质复合物既包含IMP, 又包含PMP。
Fig. 1Schematic diagram of cyanobacterial thylakoid membrane
疏水性是膜蛋白的一个重要生物化学特征。评价疏水性的关键指标有两个:第一个指标是分析其是否含有跨膜结构域及其数目。所有的IMP都有一个或多个跨膜结构域,通常一个膜蛋白如果所含的跨膜结构域数目越多,表明其疏水性越强,但有时也例外,因为膜蛋白整体疏水性还会受到连接跨膜结构域的膜外区域大小的影响。一个蛋白是否含有跨膜结构域及其数目的多少可以通过生物信息软件进行预测,常用的软件是基于隐马尔可夫模型的TMHMM,其预测IMP的准确度可以超过97%[7]。需要指出的是TMHMM目前有两个版本,即TMHMM1和TMHMM2。本课题组在具体的研究中发现,TMHMM2 在预测某些蛋白的跨膜结构域时反而不如TMHMM1准确。第二个指标就是蛋白质的GRAVY值。GRAVY值是从总体上评价一个蛋白质疏水性高低的参数。一般来说一个蛋白质的GRAVY值越高,表明其疏水性越强[8]。一个蛋白质的GRAVY值取决于其氨基酸序列的构成。每个不同的氨基酸残基都有一个特定的疏水值(hydropathy score),疏水性最高的为异亮氨酸(4.5),其次为缬氨酸(4.2),而亲水性最高的是精氨酸(-4.5) ,其次为赖氨酸(-3.9)。一般来说,侧链为长脂链的氨基酸其疏水性越高,而侧链为极性的尤其是带电荷的氨基酸,其亲水性越强。GRAVY值就是该蛋白质所有氨基酸疏水值的平均值,即所有氨基酸疏水值的加和除以该蛋白质的长度(所含氨基酸的数量)。
1.2 膜及膜蛋白的分离纯化
尽管一个基因组所编码的蛋白有30%被预测为膜蛋白,但总体上由于膜蛋白丰度较低,且不易在水溶液中溶解,因此在进行膜蛋白质组学分析时,经常需要先对膜组分进行分离纯化,以富集低丰度的膜蛋白。在一个全细胞裂解物中,由于膜组分的密度不同,在密度梯度离心中具有不同于可溶性组分的沉降系数,借此可以与可溶性组分分离开。对于各种细胞器如线粒体、叶绿体来说,一般都是先分离该细胞器,然后再分离其各个膜组分。以叶绿体为例,叶绿体含有3个膜组分:组成叶绿体包被的外膜、内膜和类囊体膜。叶绿体的包被可以利用一种叫做Yeda Press的装置剥离下来,并通过密度梯度离心分离外膜和内膜[9,10,11,12,13,14,15]。被剥离包被的叶绿体可以被直接裂解,并通过离心获得类囊体膜。纯化的膜组分既含有IMP, 又含有PMP。由于IMP难溶于水溶液且一般丰度较低,因此难以被现有的蛋白质组学技术鉴定。为提高IPM蛋白质组学鉴定的覆盖度,通常需要将水溶性较高的PMP与IPM分离以便富集IPM。常见的分离方法有如下几种:(1)高浓度盐溶液抽提法,即利用高浓度的盐溶液(如1 mol/L NaCl)来解离与膜结合的PMP[16]。这种方法的原理是利用盐离子所带的电荷,破坏介导PMP与膜相结合的静电相互作用,从而将PMP从膜上洗脱到溶液中。经过1 mol/L NaCl抽提后的膜所含的PMP显著减少,而IMP会因此得到显著的富集;(2)高pH抽提法。细胞裂解后,膜组分由于磷脂双分子层的疏水性,在水溶液中通常是以囊泡的形式存在。这些囊泡在形成过程中会把一部分可溶性蛋白包含进去,这样在分离纯化的膜组分中就会造成可溶性蛋白的污染。用100 mmol/L Na2CO3溶液(pH11.0)抽提纯化的膜组分,可以将这些囊泡状的膜组分剪切开,形成片状,释放其中所包含的可溶性的污染蛋白[10]。同时,由于高pH值可以改变蛋白质在水溶液中的电离效率,从而改变蛋白质所带的电荷,影响蛋白质之间的静电相互作用,并因
此将PMP从膜上解离下来,达到分离PMP与IMP的效果;(3)离液剂(chaotropes)抽提法。常用的离液剂是高浓度(8 mol/L)的尿素(urea)。尿素是一种强极性的试剂,很容易与水及水溶液中的蛋白质形成氢键,从而破坏蛋白质之间的相互作用,把PMP从膜上解离出来。除尿素外,硫脲(thiourea)是一种更强的离液剂,也经常在蛋白质组学研究中用来溶解蛋白质[17,18];(4)二相分离法(two phase partition)。含有去垢剂Triton X-114在0℃时会形成均一的水溶液,当温度升高到20℃时,溶液就会分层,上层是水相,下层是去垢剂相。这时溶解在Triton X-114溶液中的膜蛋白也会随着分层,水相中主要包含可溶性蛋白,如容易从膜上解离的PMP,而去垢剂相中主要包含和膜结合紧密的PMP以及IMP[19]。需要注意的是,这些方法并不一定是单独使用,在实际研究中通常会把多种方法结合起来使用,以达到提高分离效率的目的。
2 膜蛋白质组学相关技术
2.1 双向电泳与膜蛋白质组学
双向电泳(2D electrophoresis, 2DE)是早期蛋白质组学研究最重要的技术之一,可以和MALDI-TOF (matrix-assisted laser desorption/ionization time of flight mass spectrometry)质谱技术结合(2DE-MS),进行大规模快速的蛋白质鉴定[17,18]。双向电泳本质上是一种蛋白质分离技术,可以把高复杂度的样品,如含有数万个蛋白的全细胞裂解物中的蛋白尽可能地分开[20]。一般来说,双向电泳的第一向为等电聚焦(isoelectric focusing, IEF),即根据蛋白质的等电点进行分离。蛋白质由氨基酸组成,在不同的酸碱环境下会发生不同程度的电离,因此蛋白质所处环境的pH值不同,其电离程度以及所带的净电荷也不同。在一个特定的pH值下,该蛋白质发生电离使得其所带的净电荷为0,该pH值即为该蛋白质的等电点。虽然所有的蛋白质都有一个特定的等电点,但一旦该蛋白质发生翻译后修饰,其等电点则也有可能改变, 因此在2DE中很多蛋白质会在电泳胶上形成分子量相等的多个点[17,18]。等电聚焦的本质就是让蛋白质在高电压的作用下在一个pH梯度中进行迁移,当蛋白迁移到pH梯度中与其等电点相等的位置时,由于其所带的净电荷为0,不再受到电场力的作用,从而停留在该位置。一般商业化的IEF胶条都是带有固定化的pH梯度,可以最大限度地提高实验结果的重复性。对于多种蛋白质来说,由于其等电点有所不同,因此其停留的位置也有所不同。等电聚焦就是通过这个原理对蛋白质进行分离的。需要指出的是,等电聚焦要想达到很好的分离分辨率,必须要排除环境中的带电离子对蛋白质迁移的影响。因此,等电聚焦的缓冲液不能含有盐离子,最理想的迁移环境就是电场中所有的电荷都来源于蛋白质自身。双向电泳的第二向是SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis)。SDS-PAGE主要是根据分子量的大小对蛋白质进行分离。等电聚焦完成以后,可以把含有蛋白质的IEF胶条水平地转移到SDS-PAGE凝胶顶端并用琼脂糖凝胶固定住。在电场力作用下,蛋白质从IEF胶条中垂直进入到SDS-PAGE胶中,进行第二向分离。
2DE的分辨率和IEF胶条以及SDS-PAGE的尺寸呈正相关。尺寸越大,分辨率越高,同时蛋白质样品的上样量也越大,可以更好地鉴定低丰度蛋白。虽然大尺寸的双向电泳(如24 cm IEF胶条)理论上可以将多达1000个以上的蛋白高分辨地呈现在一块凝胶上,但一般而言2DE能分离的蛋白通常都是亲水性的可溶性蛋白,而高疏水性的尤其是含有跨膜结构域的蛋白,绝大部分都不能被双向电泳所分离。这是因为膜蛋白的溶解通常需要有去垢剂的辅助。去垢剂通常都有一个非极性的基团和一个极性的基团,非极性的基团和蛋白质发生相互作用,而极性基团和溶液中的水发生相互作用,从而帮助蛋白质溶解。不同的去垢剂辅助蛋白质溶解的能力是不同的,目前已知的助溶能力最强的去垢剂是SDS,但SDS带有负电荷,蛋白质一旦结合大量的SDS后,SDS本身的负电荷就会干扰等电聚焦,因此SDS与蛋白质的等电聚焦是不兼容的。通常与等电聚焦兼容的去垢剂都是非离子性的(如Triton X-100和NP-40),或者是双性的 (zwitterionic),即同时带一个正电荷和一个负电荷,总电荷为0,如常用的CHAPS和ASB14都是双性去垢剂 。遗憾的是,这些去垢剂的助溶能力远远低于SDS,因此绝大多数高疏水性的膜蛋白都无法被双向电泳分离[21,22,23]。
2.2 基于LC-MS的膜蛋白质组学
基于美国科学家John Yates实验室在研发基于色谱-质谱联用(liquid chromatography-mass spectrometry, LC-MS)的鸟枪法蛋白质组学(shot-gun proteomics)技术上取得的卓越成就[24,25],鸟枪法蛋白质组学技术逐渐取代2DE-MS技术,成为高通量蛋白质组学分析的主流技术。基于LC-MS的鸟枪法蛋白质组学技术的核心策略是依靠LC分离蛋白质经特异性内切酶如胰蛋白酶酶切后产生的肽段,然后经电喷雾技术离子化后进入质谱进行分析。该技术的优点是通量高、容易自动化,更为重要的是酶切后产生的肽段可以根据不同原理的利用LC进行多维分离而降低样品的复杂度,如肽段可以首先被强阳离子交换LC进行第一维分离,所收集的每个馏分可以经反相LC进行第二维分离。这种多维分离技术与MS结合可以极大限度地提高蛋白质组鉴定的覆盖度。在膜蛋白质组学方面,由于基于2DE-MS的方法很难兼容高疏水性的膜蛋白,因此该方法在膜蛋白的高通量鉴定和定量上被逐渐舍弃,而基于LC- MS的膜蛋白质组技术在不断地推陈出新。早期比较成功的技术是由John Yates实验室发表的高pH溶液抽提结合非特异性酶切的方法。该技术的关键是先利用pH 11.0的Na2CO3将提纯的囊泡状膜组分打开,形成片状膜组分。然后利用非特异性的蛋白酶Proteinase K对片状膜组分进行不完全消化,这样两个跨膜区之间的膜外区域便会被切成大小不一的肽段,可以利用LC-MS进行鉴定[14]。该方法有两个优点:第一是高pH缓冲液打开了囊泡状膜结构,使得包含在囊泡内部的膜外区域也被暴露在水溶液中,从而可以被蛋白酶消化成小片段肽段,这样就可增加膜蛋白质被分析的区域,提高氨基酸序列鉴定的覆盖度。这对小的膜蛋白尤其是膜外区域很小的膜蛋白具有重要的意义;第二个优点是使用非特异性的蛋白酶来进行消化。由于很多膜蛋白的膜外区域很小,没有合适的特异性内切酶如胰蛋白酶的识别为点,这样可能造成膜外区域不能被酶切,或酶切所产生的肽段片段过大,不能被LC-MS有效地分析。利用这种方法,454个含有预测TMD的蛋白被从脑组织裂解物中鉴定出来,虽然这些蛋白质很多都只含一个TMD,但相较基于2DE的方法来说,在膜蛋白质的鉴定上已经有了本质的提高[14]。
利用高pH结合非特异性Proteinase K部分酶切的方法虽然能显著提高膜蛋白的鉴定效率,但这种方法也有一定的弊端。首先,酶切是非特异性的,所得到的肽段的长度较难控制,并且肽段的C-端没有特异性酶切所产生的标志性氨基酸残基,如胰蛋白酶酶切后所产生的K或R,不利于后续的质谱分析。其次,由于非特异性酶切片段长短的随机性,同一个蛋白在不同的样品中,甚至在同一个样品的不同技术重复中,经过酶切所产生的肽段也是不一样的,这样样品之间就很难进行定量比较,尤其是不利于基于标记的如SILAC (stable-isotope labelling by amino acids)或iTRAQ (isobaric tags for relative and absolute quantification)的定量蛋白质组学研究。因此,该技术事实上并没有在具体的研究中得到广泛的应用。
上述几种方法均不需要将膜蛋白真正溶解在溶液中,而是利用非特异性的酶部分消化暴露在膜外的区域。在膜蛋白质组学研究方面,研究的主流思路还是希望能把膜蛋白溶解在溶液中,然后再进行后续的分析。和基于2DE的技术一样,使用溶解能力强的去垢剂是溶解膜蛋白的最为直接也是最重要的手段。目前已知的溶解能力最强的去垢剂是SDS,但由于SDS自身带有电荷且和蛋白质结合紧密,不易去除,因此不能兼容于后续的LC-MS。如何去除蛋白质样品中的SDS因此成了很多蛋白质组学研究者孜孜不倦以求攻克的一个难题。本课题组曾经观察到用1% SDS溶液裂解的全细胞裂解液保存在4℃时,SDS会发生沉淀,可以通过离心予以去除。因此尝试是否能利用这个现象去除蛋白质样品中的SDS,但经过多次实验后发现,利用这种办法虽然能有效地去除SDS,但也有很多的蛋白质随着SDS一同沉淀,造成很大的样品损失。德国科学家Mann实验室在2009年发表了一种更为有效的从蛋白质样品中去除SDS的方法,称之为滤膜辅助的样品制备(filter aided sample preparation, FASP)[26]。该方法的基本原理是先利用高浓度的SDS裂解液,如2%或4% SDS裂解液,把蛋白质溶解出来。这种高浓度的SDS裂解液几乎能溶解所有的膜蛋白。含有SDS的蛋白质样品然后转移到超滤管进行溶液置换。超滤管的滤膜具有很多小孔,孔径的大小决定可以滤过的蛋白质分子的大小。通常用来做FASP实验的超滤膜其孔径大小的域值小于30 kDa,也就是小于30 kDa的蛋白质分子才能通过。域值越小,能通过的分子越小,但溶液置换所需的时间越长。其实30 kDa也是一个经验值。由于蛋白质分子已经结合了很多的SDS分子,且其空间构象也发生了变化,因此30 kDa域值的超滤膜也能有效地阻挡10 kDa以上的蛋白质通过。在超滤管中,可以向蛋白质样品加入8 mol/L的尿素来稀释样品。尿素作为一种很强的离水剂,可以把结合在蛋白质上的SDS置换出来,同时保持蛋白质的溶解状态[27,28]。通过离心,让置换出来的SDS透过滤膜而得到去除,同时样品得到浓缩。浓缩后的样品再次加入8 mol/L尿素稀释以置换和去除残余的SDS。重复数次,便能有效地去除样品中的SDS。最后将样品中的尿素稀释到2 mol/L,便能进行后续的DTT还原、碘乙酰胺烷基化以及蛋白酶消化等处理,得到的酶切肽段样品能完全兼容LC-MS分析。FASP方法不但能有效地鉴定膜蛋白,事实上对所有的蛋白都非常有效。因此,FASP方法已经成为蛋白质组学实验样品处理的一种最常用的方法。需要指出的是,利用超滤管去除SDS的思想并不是Mann实验室第一个提出,在此之前,Manza等[29]也提出了类似的方法,但由于在其他细节上有所不同,该方法去除SDS的效率似乎没有FASP方法好。两个研究团队也因为该方法的原创性有所争议[12],但就去除SDS的效率以及在该方法的推广上,Mann实验室应该是贡献更大。
FASP虽然在膜蛋白质的处理上具有极大的优势,但也并非完美无缺,一方面该方法需要在超滤离心管中进行多次的溶液交换,会增加实验的成本和时间,另一方面该方法会造成严重的样品损失。事实上,第二个方面造成的后果更严重,正如Manza等[29]所指出的,该方法在处理小于50 g的样品时,所能回收的肽段的量很少,很可能是肽段被吸附在滤膜上而造成了损失。为了克服这个问题,Ma等[13]提出了另外一种方法,即SCAD法(surfactant and chaotropic agent assisted sequential extraction/on pellet digestion)法。该方法同样也是先用SDS溶液裂解细胞,往裂解液中加入冰冷的丙酮(80%,V/V)过夜沉淀蛋白质,然后再用冰冷的80% 丙酮洗2 h。经离心去除丙酮后干燥样品,用8 mol/L尿素重新溶解蛋白质。利用SCAD方法,作者声称能获得更高的肽段回收率和媲美FASP方法的膜蛋白鉴定效率,该方法尤其适合微量样品的蛋白质组学分析。不过,本课题组在经过多次尝试后,发现SCAD方法和FASP法比起来并无明显优势,尤其在处理微量样品时,由于需要用丙酮沉淀蛋白质,会造成一定的样品损失。因此,在日常蛋白质组学实验中,FASP方法还是处理样品的首选方法。
3 光合模式蓝藻集胞藻的膜蛋白质组学研究进展
3.1 集胞藻简介
集胞藻是一种单细胞的光合蓝藻,也是一种重要的光合作用研究的模式生物。作为模式生物,集胞藻有如下几个优点:第一,集胞藻和高等植物的叶绿体高度类似,都具有包括类囊体膜在内的多种膜结构,其光合电子传递链也和高等植物类似;第二,集胞藻的基因组在所有的蓝藻中是第一个被测定的[30],其遗传背景相较其他蓝藻更为清楚;第三,集胞藻具有天然的吸收外源DNA的能力,并能通过同源重组将外源DNA整合到自身的基因组上,非常容易进行遗传转化[31]。因此,在研究基因功能时非常容易和方便地对基因进行敲除和敲入。这些优势在进行功能蛋白质组学研究时非常重要,可以方便地对新发现的具有潜在功能的蛋白质进行遗传操作来验证功能。此外,集胞藻作为一种蓝藻,在工业上也具有重要的应用价值,可以利用其自养的特性,通过光能固定空气中的CO2来生产清洁的可再生能源和其他有用的代谢物[32,33]。目前,在合成生物学方面,集胞藻正在作为一种重要的底盘生物进行开发和利用[34]。和其他蓝藻一样,集胞藻具有多种膜结构,由外向内,包括细胞壁的外膜、质膜和多层的类囊体膜。其中质膜根据其在密度梯度离心上的浮力密度不同,又可分为低密度质膜和普通质膜[35]。各种膜结构上所含的蛋白质在很大程度上决定其功能,如外膜上有很多通透性高的孔道蛋白(porin),负责细胞与外界的物质交换。质膜上有很多转运蛋白,负责有选择性地把外界物质如各种离子运输到细胞内。类囊体上有完整的光合电子传递链,负责光合作用。需要指出的是,蓝藻类囊体上也有完整的呼吸电子传递链,且部分构成成分与光合电子传递链重合[36]。这一点与高等植物的类囊体不同,后者只具有光合电子传递链,而呼吸链则位于线粒体中。当然,各个膜组分上的蛋白质构成并非完全不同,如质膜上也有部分呼吸链和光合链的蛋白[37]。因此,鉴定这些膜组分的蛋白构成,对于理解其功能至关重要。利用膜蛋白质预测软件TMHMM预测,集胞藻的基因组编码892个含有预测TMD的蛋白,当然其中有一部分只含有一个TMD的蛋白质可能并不是真正的IMP,而是含有信号肽的可溶性蛋白。
3.2 集胞藻膜蛋白质组学研究进展
集胞藻的膜蛋白质学的研究始于1997年,日本的研究团队在完成集胞藻基因组测序后,开始进行其蛋白质组学研究。他们利用2DE结合N-端测序和MS的方法,分析了集胞藻的全细胞裂解物的蛋白质组和分离出来的类囊体蛋白组,共鉴定了不到100个蛋白质,只有少数几个蛋白含有预测的TMD[38,39]。随后,本文作者之一汪迎春研究员在做博士论文期间,利用2DE-MS方法,试图系统地解析分离的集胞藻膜组分的蛋白质组。需要指出的是,这个膜组分是经过差异离心去除可溶性组分后获得的膜组分沉淀,包含外膜、质膜和类囊体膜,只不过类囊体膜占绝大部分[17]。在经过长时间的方法优化,尝试了各种离水剂和去垢剂的组合后,他发现2DE对于溶解膜蛋白效率非常低,鉴定的蛋白只有少数几个含有预测的TMD,而这些TMD还很有可能是信号肽[17,18]。由于2DE结合MALDI-TOF的方法是早期蛋白质组学研究的主流技术,因此可以预见早期集胞藻的膜蛋白质组学研究是困难重重。但在这领域也并非没有亮点,这个时期最主要的亮点来自于对集胞藻各种膜组分的生化分离。1998年瑞典斯德哥尔摩大学的Norling等发明了一种液相二相分离法,二相分别为水溶性的多聚物Dextran-500 (5.8%)和polyethylene glycol 3350 (5.8%)组成,根据各种膜组分表面的物理化学性质不同,对不同的膜组分进行分离[40]。利用二相分离法结合密度梯度离心技术,Norling的学生黄芳博士(现为中国科学院植物研究所研究员)分离纯化了质膜和外膜,并进行了基于2DE-MS的蛋白质组学分析[41,42]。纯化的类囊体膜蛋白质组也随后由Norling实验室分析并发表[43]。同样,由于2DE-MS在分析膜蛋白质上存在的先天性的技术缺陷,这些蛋白质组学研究所鉴定的IMP数量也很少。鉴于这个原因,Norling实验室与韩国的Choi 团队合作,尝试用不同的技术来解析膜蛋白质组。他们开发了一种高温酸剪切结合特异性酶切的技 术[44]。该技术主要是用10 mmol/L TCEP与30%的乙腈和25%甲酸水溶液按1∶1比例混合而得到的溶液在95℃处理膜样品,膜蛋白质在溶解的同时,其天冬氨酸残基的N-端和C-端都能得到剪切,产生小于10 kDa的肽段,这些肽段经进一步胰蛋白酶和chymo胰蛋白酶酶切后,产生更小的适合LC-MS分析的肽段。利用这种方法,他们一共鉴定了155个IMP[44],和之前的报道相比,该方法所鉴定的IMP的数量有了很大的提高,但和集胞藻全基因组编码的IMP比起来尚不足20%。鉴于集胞藻在光合作用和清洁能源研究上的重要性以及在膜蛋白质组学研究上的挑战性,本课题组建立蛋白质组学实验室后又重新从事该领域的研究。2014年,本课题组利用FASP方法结合基于LC-MS的多维蛋白质组学方法,定量分析了集胞藻ORF slr0110的一个敲除突变体的蛋白质组,共鉴定了409 个IMP,占全基因组编码的IMP的50%左右,这其中有232个IMP包含2个或以上的TMD。与以往的研究相比,在IMP鉴定覆盖度方面有了巨大的提升。同时本课题组所鉴定的IMP在跨膜区数量的分布上没有表现出明显的偏好,从一个跨膜区到最多的18个跨膜区的IMP鉴定率都在50%左右[45]。这些结果也充分说明了FASP方法在膜蛋白质组学研究上的巨大优势。
尽管本课题组在集胞藻IMP鉴定的覆盖度上取得了长足的进步,但还有大约400个IMP不能在单次实验中得到鉴定。鉴于此,本课题组系统地分析了集胞藻所有从未被鉴定过蛋白的特性,发现这些蛋白主要是小蛋白和疏水性的蛋白,后者主要是IMP[46]。因此,推测这些未被鉴定的IPM太小,没有合适的特异性蛋白酶切位点,从而逃脱了基于LC-MS的鸟枪法蛋白质组学鉴定;另一个可能的原因是IMP表达量太低,目前的LC-MS技术因灵敏度不够而不能被鉴定到。对于该原因,我们推测如果将各个膜组分分离纯化,这样使得各个膜组分尤其是含量处于绝对劣势的外膜和质膜与含量上处于绝对优势的类囊体膜分开,这样就可以使外膜和质膜上的蛋白质得到富集,从而有可能使得一些低丰度的膜蛋白得到鉴定。本课题组和黄芳团队合作,从野生型的集胞藻中分离了外膜、低密度质膜、普通质膜、类囊体膜以及可溶性组分,利用基于LC-MS的无标记定量蛋白质组学技术对这5个组分进行了分析,共鉴定到2167个蛋白,其中594个蛋白为IMP,使得单次实验集胞藻IMP鉴定的数量达到了前所未有的高度(待发表)。由于此次实验所用的膜组分样品量较少,并没有进行样品消耗量较大的多维蛋白质组学分析。后续实验将大量制备各个膜组分并对每个组分进行多维蛋白质组学鉴定,预期所鉴定的IMP的数量还会有进一步提高。需要指出的是,在某个特定的条件下并非所有的编码蛋白的基因都会表达,因此在同一个实验条件下也就无法把集胞藻所有的IMP都鉴定出来。如果想要再进一步提高IMP鉴定的覆盖度,就需要在不同的条件下培养集胞藻。已有证据表明有些IMP的表达是环境诱导型的,如负责CO2转运的蛋白cmpABCD,其表达就是受低CO2浓度诱导[47,48]。利用此次无标记定量的蛋白质组学实验结果,本课题组对所鉴定的蛋白,包括IMP在各个组分上分布的相对丰度进行了定量。依此定量信息,就能获得某个蛋白在细胞内定位的信息,既可能特异性定位在某一个亚细胞结构,也可能分布在多种亚细胞结构上。本课题组一共对2027个集胞藻蛋白进行了亚细胞定位,并将该信息储存在一个网络数据库以供从事蓝藻研究的科学家检索(http://protdb.genetics.ac.cn/atlas/index.html) (图2)。
图2
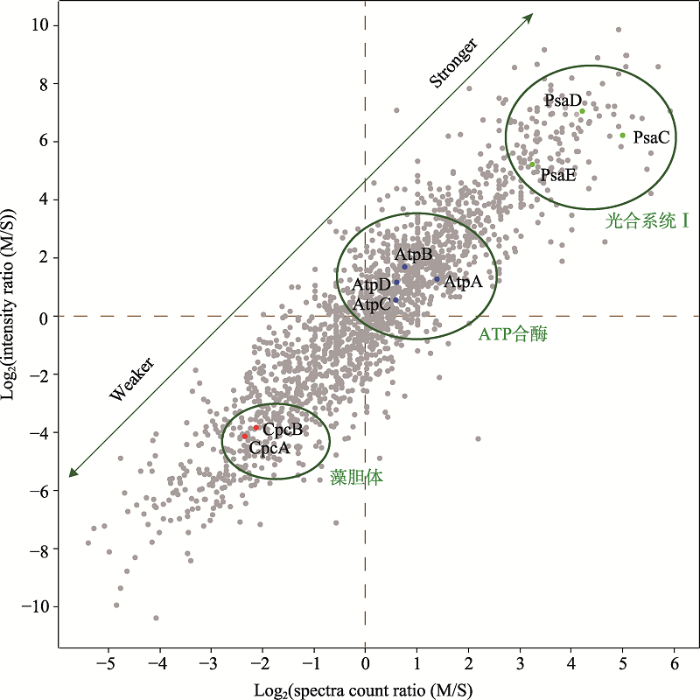 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2集胞藻可溶性蛋白与膜结合强度的评估
绿色箭头线段所示,沿绿色箭头方向,蛋白与膜结合的紧密程度依次递增或递减。图中显示光合系统Ⅰ的亚基(绿色散点)、ATP合酶的亚基(蓝色散点)以及藻胆体的亚基(红色散点)与膜结合的强度依次递减。
Fig. 2Evaluation of the tightness of membrane association for non-transmembrane proteins
生物膜上除了含有IMP外,还有大量的PMP。在分离膜组分的时候,和膜结合稳定的PMP一般会随膜一起分离出来,而与膜结合不稳定的有时会从膜上解离下来。此外,膜组分在分离时不可避免地会遭到一些可溶性蛋白的污染,这些污染蛋白并不是PMP,且与膜的结合是非特异性的。如何区分从膜组分中鉴定出来的PMP和污染蛋白一直是一个技术上的挑战,也是开展对PMP功能的研究需要解决的首要问题。因此,本课题组设计了一个方法用于区分真正的PMP和污染蛋白。该方法基于如下的一个假想:所有的非IMP蛋白,在细胞内都有两个池 (pool),一个是结合在膜上,一个是在可溶性组分。如果一个蛋白和膜结合得很紧密,那么它在膜上的池就较大,在可溶性组分中的池就较小,反之亦然。如果一个蛋白和膜结合得越紧密,那么它越有可能是PMP,反之则越有可能是可溶性污染蛋白。基于这个假想,本课题组定量分析了野生型集胞藻膜组分和可溶性组分的蛋白质组,并利用表征蛋白质丰度的两个质谱学参数(质谱信号强度和谱图匹配数)来计算蛋白质在膜组分和可溶性组分中丰度的比值,并依此作出二维散点图[16]。和膜结合越紧密的蛋白,其膜组分/可溶性组分的信号强度和谱图匹配数的比值就越大,其在散点图上的位置就越靠近右上角。如光合系统Ⅰ的非IMP组分PsaC、PsaD和PsaE等与膜结合非常紧密,因此位于右上角,ATP合酶的亚基AtpA和AtpB与膜的结合稍弱,处于中间,而藻胆体非常不稳地,其多个亚基如CpcA和CpcB处于左下方(图3)。污染蛋白和膜结合的紧密度一般较低,其比值较小,因此都分布于左下角[16]。该方法为判断某个蛋白是否是PMP提供了定量依据。需要指出的是,PMP与膜结合的紧密度不是一成不变的,而是随着环境的变化而变化。这也是细胞内各种生理生化反应以及信号转导的一个重要调控方式。
图3
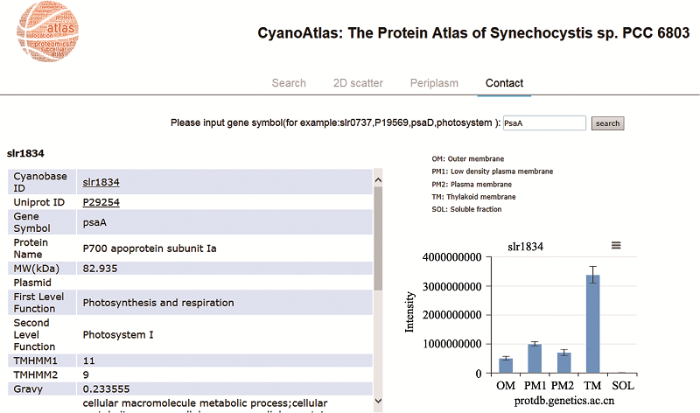 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3集胞藻蛋白质地图集网络数据库
数据库提供一个搜索引擎,用户可以输入要检索的蛋白,如图中所示的光合系统Ⅰ蛋白PsaA,该蛋白在各亚细胞结构上的相对丰度(柱状图)以及其他结构和功能信息被显示出来。
Fig. 3The Proteome Atlas of Synechocystis
4 结语与展望
现代蛋白质组学技术的快速发展使得大多数膜蛋白组研究上存在的难题正在被一一克服,在蓝藻的膜蛋白质组学研究进展也同样如此。这使得绝大数IMP都能被现代蛋白质组学技术鉴定到。但目前仍存在两个主要的问题:第一是IMP跨膜区很难被鉴定到,这主要是由于跨膜区强疏水性以及缺少合适的特异性酶切位点造成的。跨膜区一般不含胰蛋白酶特异识别的精氨酸和赖氨酸位点,因此需要选择不同的特异性内切酶才能获得可用于质谱分析的肽段;第二是很多IMP是只含一个跨膜区的小蛋白,没有合适的酶切位点,也不适合基于鸟枪法的蛋白质组学鉴定。对于这种小蛋白,目前新兴的从上而下的蛋白质组学(top-down proteomics)也许是个更好的技术选择。从上而下的蛋白质组学方法在进行质谱分析前,不需要将完整的蛋白质切成小的肽段,直接以蛋白质本身为分析检测对象,因此可以更完整、丰富和准确地提供分析对象的生物学信息。同时,从上而下的蛋白质组学方法也弥补了鸟枪法蛋白质组分析鉴定的一些不足,在小分子量或缺少蛋白酶酶切位点的蛋白质鉴定方面也许会有更大的优势[49,50,51]。因此,有理由相信在不久的将来将会实现对膜蛋白质组全覆盖度的鉴定。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 3]
[本文引用: 1]
[本文引用: 3]
[本文引用: 5]
[本文引用: 4]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}