Congenital ectrodactyly caused by chromosome 10q24.31 duplication and its pathogenetic analysis
Xiuquan Zhang1,2, Jian Wang3, Fu Xiong3, Weibiao Lv1, Yuanqing Zhou1, Shaomin Yang4, Yuting Zhang2, Xiaoyan Tian2, Wei Lian2, Xiangmin Xu31. Department of General Laboratories, Reproductive Genetic Research Laboratory, Shunde Hospital, Southern Medical University, Foshan 528300, China 2. Department of Obstetrics and Gynecology, Shunde Hospital, Southern Medical University, Foshan 528300, China 3. Department of Medical Genetics, School of Basic Medical Sciences, Southern Medical University, Guangzhou 510515, China 4. Department of Radiology, Shunde Hospital, Southern Medical University, Foshan 528300, China
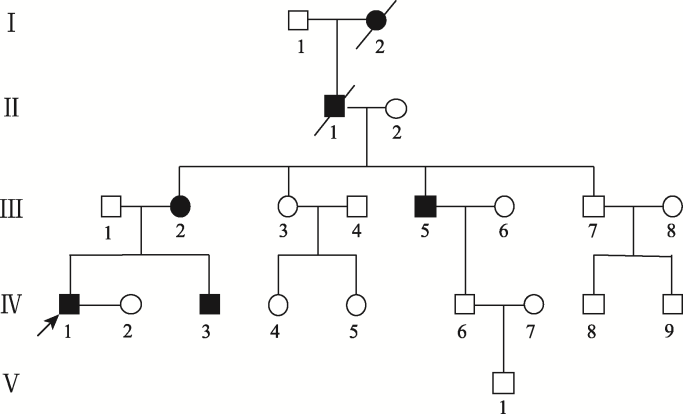
Abstract In order to investigate the genetic variations and the clinical manifestations of a range of congenital ectrodactyly family and to summarize the split hand/foot malformation (SHFM) types and their related pathogenic genes, we conducted phenotypic analyses of patient’s limbs by physical and X-ray examination. The haplotypes were analyzed by using the extracted genes from peripheral blood on D10S1709, D10S192, D10S597, D10S1693 and D10S587 loci, and the mutation duplication loci were confirmed by Array-CGH detection. The pathogenic factors and inheritance pattern of SHFM were analyzed based on family investigation and gene analysis. Results demonstrate the proband’s phenotype is typically of a congenital SHFM which is manifested by missing bilateral index and middle fingers, short bilateral thumbs, deformed left ring finger with webbing of the skin missing at the middle finger; bilateral big toe with the second and the third toe missing, fourth and fifth toe fusion leading to a deformed toe separated from the first toe by the middle of the foot. The haplotype analyses show that there is a repeat of at least 610 kb in chromosome 10q24.31-10q24.32 region. Array-CGH analysis shows 10q24.31 (102 832 650-103 511 083) ×3. Our results demonstrate that the pathogenic gene variation of ectrodactyly in this family is due to duplication of 10q24.31 (102 832 650~103 511 083). The haplotype 165-251-289-219-102 can be used as a disease marker for detecting 10q24.31~10q24.32 allele for SHFM. Keywords:congenital ectrodactyly;array-CGH;haplotype analysis;genetic counseling
先证者(箭头指向)母系遗传位点为251-102-289-219-165,其弟弟母系遗传位点为251-107-289-219-165,他们的父系遗传位点均为: 251-109-291-219-167。 Fig. 4Results of the gene haplotype analysis on 5 loci
SHFLD (split hand/foot malformation with long bone deficiency)型:手/足裂畸形伴长骨发育不全,其遗传位点在17p13.3[20],为显性遗传,被证实是一个bhlha9基因重复,但有表型外显减弱的倾向,表现为手足分裂畸形合并胫/腓骨短促[10,21]。动物实验证实了这个基因在胫腓骨发育中的作用。另外,其他作者报道了19p13.11缺失导致SHFM,其EPS15L1基因变异是该疾病的根本原因[22]。
GaneBD, NatarajanP . Split-hand/feet malformation: a rare syndrome J Family Med Prim Care, 2016,5(1):168-169. [本文引用: 5]
EnriquezA, KrivanekM, FlottmannR, PetersH, WilsonM . Recurrence of split hand/foot malformation, cleft lip/palate, and severe urogenital abnormalities due to germline mosaicism for TP63 mutation Am J Med Genet A, 2016,170(9):2372-2376. [本文引用: 2]
DaiL, LiYH, DengY, ZhuJ, WangYP, LiangJ, ZhangYW, LiuZY . Prevalence of congenital split hand/split foot malformation in chinese population J Sichuan Univ(Med Sci Ed), 2010,41(2):320-323. [本文引用: 2]
BedardT, LowryRB, SibbaldB, KieferGN, MetcalfeA . Congenital limb deficiencies in Alberta-a review of 33 years (1980-2012) from the alberta congenital anomalies surveillance system (ACASS) Am J Med Genet A, 2015,167A(11):2599-2609. [本文引用: 2]
KantaputraPN, KapoorS, VermaP, IntachaiW, Ketudat CairnsJR . Split hand-foot malformation and a novel WNT10B mutation Eur J Med Genet, 2018,61(7):372-375. [本文引用: 1]
Faiyaz-Ul-HaqueM, ZaidiSH, KingLM, HaqueS, PatelM, AhmadM, SiddiqueT, AhmadW, TsuiLC, CohnDH . Fine mapping of the X-linked split-hand/split-foot malformation (SHFM2) locus to a 5.1-Mb region on Xq26.3 and analysis of candidate genes Clin Genet, 2005,67:93-97. [本文引用: 3]
FuscoC, NittisP, AlfaizAA, PellicoMT, AugelloB, MalerbaN, ZelanteL, ReymondA, MerlaG . A new split hand/foot malformation with long bone deficiency familial case J Pediatr Genet, 2016,6(2):98-102. [本文引用: 1]
van SilfhoutAT, van den AkkerPC, DijkhuizenT, VerheijJB, Olderode-BerendsMJ, KokK, Sikkema-RaddatzB, van Ravenswaaij-ArtsCM . Split hand/foot malformation due to chromosome 7q aberrations(SHFM1): additional support for functional haploinsufficiency as the causative mechanism Eur J Hum Genet, 2009,17(11):1432-1438. [本文引用: 1]
HaberlandtE, LöfflerJ, Hirst-StadlmannA, StöcklB, JudmaierW, FischerH, Heinz-ErianP, MüllerT, UtermannG, SmithRJ, JaneckeAR . Split hand/split foot malformation associated with sensorineural deafness, inner and middle ear malformation, hypodontia, congenital vertical talus, and deletion of eight microsatellite markers in 7q21.1-q21.3 J Med Genet, 2001,38(6):405-409. [本文引用: 2]
DaiL, LiNN, DengY, MaoM, WangH, ZhuJ . Genotype-phenotype analysis of a Chinese family with split hand/split foot and syndactyly Chin J Med Gene, 2011,28(4):379-382. [本文引用: 1]
SharmaD, KumarC, BhaleraoS, PanditaA, ShastriS, SharmaP . Ectrodactyly, ectodermal dysplasia, cleft lip, and palate (EEC syndrome) with tetralogy of fallot: a very rare combination Front Pediatr, 2015,3:51. [本文引用: 1]
FilhoAB, SouzaJ, FauczFR, SotomaiorVS, DupontB, BartelF, RodriguezR, SchwartzCE, SkinnerC, AllimanS, RaskinS . Somatic/gonadal mosaicism in a syndromic form of ectrodactyly, including eye abnormalities, documented through array-based comparative genomic hybridization Am J Med Genet A, 2011,155A(5):1152-1156. [本文引用: 1]
GoodmanFR, MajewskiF, CollinsAL, ScamblerPJ . A 117-kb microdeletion removing HOXD9-HOXD13 and EVX2 causes synpolydactyly Am J Hum Genet, 2002,70(2):547-555. [本文引用: 1]
AzizA, Irfanullah, KhanS, ZimriFK, MuhammadN, RashidS, AhmadW . Novel homozygous mutations in the WNT10B gene underlying autosomal recessive split hand/foot malformation in three consanguineous families Gene, 2013,534(2):265-271. [本文引用: 1]
UgurSA, TolunA . Homozygous WNT10b mutation and complex inheritance in Split-Hand/Foot malformation Hum Mol Genet, 2008,17(17):2644-2653. [本文引用: 1]
KhanS, BasitS, ZimriFK, AliN, AliG, AnsarM, AhmadW . A novel homozygous missense mutation in WNT10B in familial split-hand/foot malformation Clin Genet, 2012,82(1):48-55. [本文引用: 1]
KlopockiE, LohanS, DoelkenSC, StrickerS, OckeloenCW, Soares Thiele deAguiar R, LezirovitzK, Mingroni NettoRC, JamsheerA, ShahH, KurthI, HabenichtR, WarmanM, DevriendtK, KordassU, HempelM, RajabA, MäkitieO, NaveedM, RadhakrishnaU, AntonarakisSE, HornD, MundlosS . Duplications of BHLHA9 are associated with ectrodactyly and tibia hemimelia inherited in non-Mendelian fashion J Med Genet, 2012,49(2):119-125. [本文引用: 1]
Al KaissiA, GangerR, RötzerKM, KlaushoferK, GrillF . A child with split-hand/foot associated with tibial hemimelia (SHFLD syndrome) and thrombocytopenia maps to chromosome region 17p13.3 Am J Med Genet A, 2014,164A(9):2338-2343. [本文引用: 1]
UmairM, UllahA, AbbasS, AhmadF, BasitS, AhmadW . First direct evidence of involvement of a homozygous loss-of-function variant in the EPS15L1 gene underlying split-hand/split-foot malformation Clin Genet, 2018,93(3):699-702. [本文引用: 1]
DaiL, DengY, LiN, XieL, MaoM, ZhuJ . Discontinuous microduplications at chromosome 10q24.31 identified in a Chinese family with split hand and foot malformation BMC Med Genet, 2013,14:45. [本文引用: 1]
YangW, HuZJ, YuXF, LiQH, ZhangAJ, DengX, ZhangAY, GaoCS, LiuY, AoY, LoWHY, ZhangX . A DNA duplication at chromosome 10q24.3 is associated with split-hand-foot malformation in a Chinese family Nat Med J Chin, 2006,86(10):652-658. [本文引用: 1]
XiangR, DuR, GuoS, JinJY, FanLL, TangJY, ZhouZB . Microduplications of 10q24 detected in two Chinese patients with split-hand/foot malformation Type 3 Ann Clin Lab Sci, 2017,47(6):754-757. [本文引用: 1]

 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT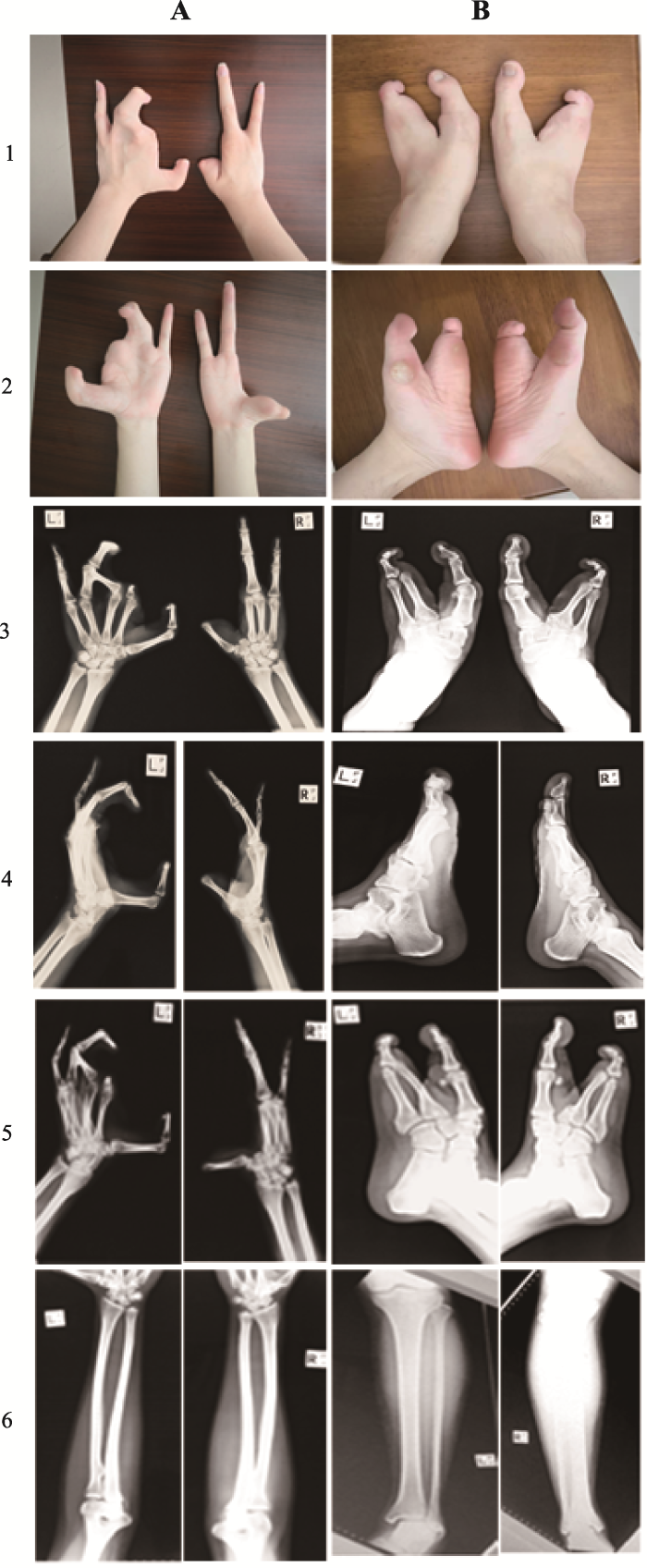 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT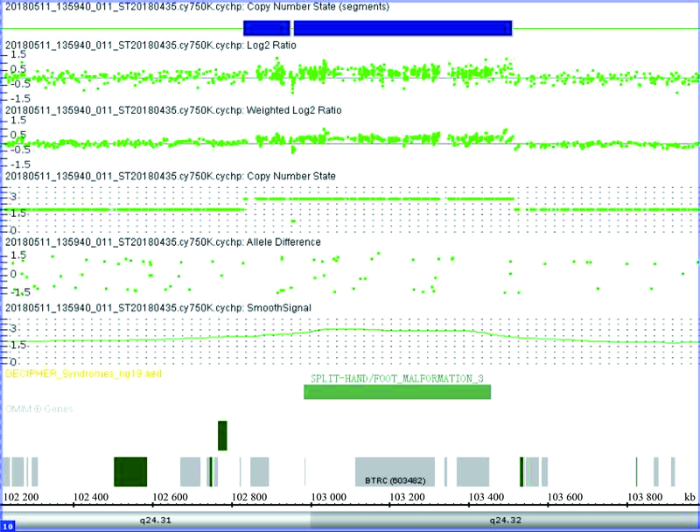 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT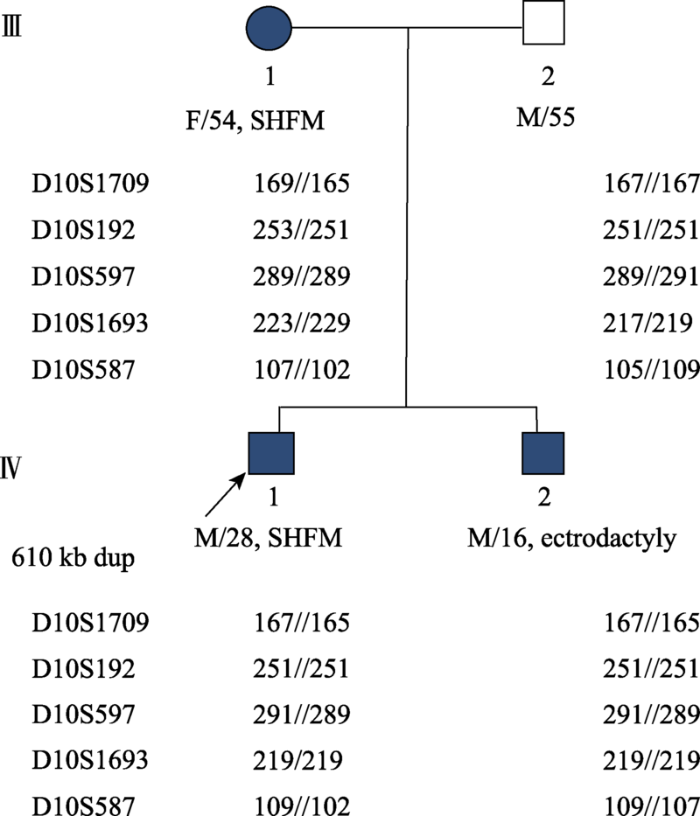 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}