 ,江西中医药大学,中医基础理论分化发展研究中心,南昌330004
,江西中医药大学,中医基础理论分化发展研究中心,南昌330004DNA methylation in adipose tissue and the development of diabetes and obesity
Xin Huang, Yongqiang Chen, Guoliang Xu, Shuhong Peng,Research Center for Differentiation and Development of Basic Theory of Traditional Chinese Medicine (TCM), Jiangxi University of TCM, Nanchang 330004, China通讯作者:
编委: 陈雁
收稿日期:2018-09-10修回日期:2018-11-3网络出版日期:2019-02-25
| 基金资助: |
Editorial board:
Received:2018-09-10Revised:2018-11-3Online:2019-02-25
| Fund supported: |
作者简介 About authors
黄鑫,硕士研究生,专业方向:中医药防治代谢性疾病E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (588KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
黄鑫, 陈永强, 徐国良, 彭淑红. 脂肪组织DNA甲基化与糖尿病和肥胖的发生发展[J]. 遗传, 2019, 41(2): 98-110 doi:10.16288/j.yczz.18-206
Xin Huang, Yongqiang Chen, Guoliang Xu, Shuhong Peng.
糖尿病和肥胖作为复杂性代谢疾病,发病率日趋增加。2017年,国际糖尿病联盟(international diabetes federation, IDF)公布的数据显示,目前全球有4.25亿糖尿病患者,预计到2045年,将会有近7亿糖尿病患者,其中95%以上是2型糖尿病(type 2 diabetes, T2D)。肥胖是糖尿病发生发展的主要危险因素,流行病学调查显示60%~90%的糖尿病患者为超重或肥胖。除遗传背景影响个体对糖尿病和肥胖患病的倾向外,饮食习惯、生活方式等也与其发病密切相关[1]。这些外界因素通过改变机体表观遗传学修饰(DNA甲基化、组蛋白修饰以及microRNA等),影响相关基因表达,从而影响机体相关功能。近年来,糖尿病和肥胖的表观遗传学机制逐渐成为研究热点。作为表观遗传学的一个重要研究内容,DNA甲基化是指在DNA甲基转移酶催化下,以S-腺苷甲硫氨酸作为甲基供体使CpG岛上胞嘧啶5′端甲基化。近年来,尽管国内外已有DNA甲基化与糖尿病相关研究进展的报道[2,3],但是较少系统阐述糖尿病、肥胖与脂肪组织DNA甲基化的关系。脂肪组织是机体重要储存能量和分泌激素的器官,其分布不同和功能紊乱会直接影响T2D以及其他代谢性综合征的发生,在体重指数(body mass index,BMI)水平相同时,上半身脂肪较多的个体患T2D风险明显增加[4,5],内脏脂肪组织(visceral adipose tissue, VAT)脂肪酸水平较浅层腹部皮下脂肪高,可以导致胰岛素抵抗增加[6]。而股臀皮下脂肪沉积相对于腹部内脏脂肪沉积,能降低T2D风险性[7]。因此探讨糖尿病和肥胖受累者不同脂肪组织DNA甲基化的变化,有助于深入了解糖尿病和肥胖的发生发展机制和探寻可能的防治途径。本文从不同类型脂肪组织入手,分别阐述糖尿病患者和肥胖患者在各类型脂肪组织中DNA甲基化的变化,并对存在的问题进行思考,旨在为糖尿病和肥胖的预防治疗寻找新途径。
1 脂肪组织的分类
哺乳动物脂肪组织根据形态、功能以及发育起源的不同可分为白色脂肪组织(white adipose tissue, WAT)和棕色脂肪组织(brown adipose tissue, BAT)。WAT呈黄色,在某些哺乳动物体内呈白色(如小鼠)。主要分布在皮下组织、网膜和肠系膜等处,是体内最大的能源库,参与脂肪代谢。根据分布差异又将WAT分为皮下脂肪组织(subcutaneous adipose tissue, SAT)和VAT。SAT是指分布于全身皮肤下层的脂肪组织,VAT主要指位于内脏周围的脂肪组织。BAT呈棕色,主要分布在新生儿肩胛间、腋窝和颈后部,成人BAT主要集中在锁骨上区、颈部、腋窝等处。BAT在代谢性疾病方面有着重要意义。另外,在某些刺激下(如冷暴露、β3肾上腺能激动剂等),白色脂肪可以向棕色样脂肪转化,这种棕色样脂肪被称为米色脂肪(beige fat)[8]。2 糖尿病患者和肥胖患者SAT中DNA甲基化变化
SAT不仅是最大的脂肪储存组织,也是脂类储存最安全的位置,脂类物质在此储存对人体的危害最小。然而,其储存能力依赖于脂肪细胞肥大能力以及募集可以储存多余脂肪的新细胞的能力。当超过其储存能力时,脂肪就只能储存在皮下以外,如肝脏、骨骼肌和胰岛等,即所谓的脂肪异位沉积。脂肪的异位沉积是导致肥胖相关的胰岛素抵抗和炎症的重要因素。因此SAT储存脂肪的能力与肥胖以及糖尿病等代谢性疾病密切相关。在糖尿病或肥胖患者SAT中常常出现某些基因甲基化的异常,可能预示着脂肪组织功能的变化。2.1 糖尿病患者SAT中DNA甲基化变化
在糖尿病患者SAT中,与胰岛素抵抗、脂类运输和脂肪生成、细胞增殖以及细胞分化相关的基因会发生DNA甲基化的变化(表1)。Table 1
表1
表1 糖尿病患者SAT中DNA甲基化变化
Table 1
| 基因分类 | 基因名称 | 甲基化位置 | 甲基化变化 | 参考文献 |
|---|---|---|---|---|
| 胰岛素抵抗 | IRS1 | 第1外显子、TSS200 | ↑、↓ | [10] |
| JAZF1 | 主体、(主体)、5°UTR | ↑、(↓)、↓ | ||
| MARCH1 | 主体 | ↓ | ||
| TCF7L2 | 主体、(主体) | ↓、(↑) | ||
| 脂类运输 | ABCG1 | 尚不明确 | ↑ | [18] |
| 脂肪生成 | ST6GAL1 | 5°UTR | ↓ | [10,20] |
| PPARγ | 5°UTR、TSS1500 | ↑、↑ | ||
| HNF4A | 启动子 | ↑ | ||
| 细胞增殖 | ANK1 | 主体、3°UTR | ↓、↓ | [10,20] |
| PEPD | 第1外显子、5°UTR | ↓ | ||
| ADAMTS9 | 主体、TSS1500 | ↑、↓ | ||
| WWOX | 主体、(主体) | ↑、↓ | ||
| BARX2 | 主体 | ↓ | ||
| RND3 | TSS200 | ↑ | ||
| TP53INP1 | 5°UTR、TSS200 | ↑、↓ | ||
| CDKN2A | 启动子 | ↓ | ||
| 细胞分化 | FGF21 | 启动子 | ↑ | [10,27] |
| HHEX | 主体 | ↓ | ||
| HMG20A | 5°UTR | ↑ |
新窗口打开|下载CSV
2.1.1 糖尿病患者SAT中胰岛素抵抗相关基因甲基化变化
正常脂肪组织对于胰岛素高度敏感,胰岛素促进脂肪沉积(通过促进脂肪酸的吸收、重酯化和从头脂肪生成)并且抑制甘油三酯分解。而胰岛素抵抗的脂肪组织即使在高胰岛素水平下,也不能抑制甘油三酯分解,机体呈现葡萄糖不耐受性以及血浆游离脂肪酸水平的增加,而血浆游离脂肪酸增加会损伤肌肉胰岛素信号,促进肝脏糖异生,从而损伤葡萄糖刺激的胰岛素应答[9],导致系统胰岛素抵抗。
尽管针对糖尿病SAT胰岛素抵抗基因甲基化的报道不多,但有少数研究组从全基因组甲基化的角度分析了糖尿病群体和非糖尿病群体SAT DNA甲基化的差异,发现与胰岛素抵抗相关的部分基因出现甲基化异常。
Nilsson等[10]分析了不同遗传背景下糖尿病群体和非糖尿病群体的SAT全基因组DNA甲基化,发现两个群体有差异DNA甲基化CpG位点,经KEGG分析,差异DNA甲基化的7046个基因,其中与胰岛素抵抗相关基因有IRS1、JAZF1、MARCH1和TCF7L2等。
IRS1在胰岛素信号中发挥重要作用,作为一种重要蛋白与磷酸化胰岛素受体结合。IRS1缺失会导致胰岛素抵抗[11]。Irs1敲除小鼠模型表明,该基因在脂肪分化中也发挥重要作用[12]。在糖尿病患者SAT脂肪组织中,IRS1基因第1外显子上5个CpG位点均呈现了高甲基化,只有在转录起始位点(transcription start site, TSS)200处的一个CpG位点表现出低甲基化[10]。而且,同时发现在糖尿病患者脂肪组织中IRS1表达水平显著降低[10]。另外,Orozco等[13]也报道,代谢综合征男性糖尿病患者IRS1第1外显子的CpG位点(cg17848496、cg00727310和cg14283647)均有显著的高甲基化。一般来说,DNA启动子区域甲基化表现出与表达呈负相关,而发生在基因主体(body)和3′非翻译区(3′ untranslated region, 3′UTR)的DNA甲基化与表达呈正相关[14]。而糖尿病患者人群SAT中IRS1第1外显子区域DNA甲基化,降低了该基因表达。
JAZF1与糖异生、胰岛素抵抗、脂代谢和炎症均有关。该基因过表达可以增加胰岛素敏感性[15]。然而JAZF1在糖尿病患者SAT组织中的基因主体区域CpG位点(cg06337342和cg04493169)甲基化水平增加,而在另一个基因主体区域CpG位点(cg14491535)和5′UTR 区域CpG位点(cg23620719)甲基化水平降低。究竟哪些CpG位点甲基化与该基因的表达密切相关仍不清楚。
MARCH1是一种E3泛素化连接酶,该基因功能缺失会增强、其过表达会损伤小鼠肝脏胰岛素敏感性。MARCH1会泛素化胰岛素受体(insulin receptor, INSR)而降低细胞表面INSR水平[16]。然而糖尿病SAT MARCH1基因主体区域CpG位点的DNA甲基化水平降低。该位点DNA甲基化在脂肪组织中与基因表达水平的关系仍需进一步明确。
T2D患者SAT中TCF7L2短mRNA可变剪接体表达与禁食游离脂肪酸水平呈正相关,表明其影响了脂肪组织胰岛素作用[17]。糖尿病SAT TCF7L2在3个位于基因主体区域的CpG位点(cg24788483、cg03683087和cg05923857)甲基化水平降低,而在另一个基因主体CpG位点(cg03339956)甲基化水平升高。基因主体区域DNA甲基化除与基因表达有关,还可能与mRNA的剪接有关[14]。因此,糖尿病患者TCF7L2主体区域的甲基化位点增加或降低,预示其功能变化,然而究竟具体哪些甲基化位点如何影响到该基因功能,仍需进一步阐明。
2.1.2 糖尿病患者SAT中脂类运输和脂肪生成相关基因甲基化变化
在T2D患者SAT中的ABCG1 (cg06500161) DNA甲基化水平也有显著增加[18]。而ABCG1促进脂类(胆固醇、磷脂、鞘磷脂和氧化型胆固醇)运输,在维持脂类平衡中发挥重要作用[19]。SAT中ABCG1高甲基化预示着脂肪组织脂类运输能力降低。
在T2D患者SAT中与脂肪生成有关的ST6GAL1、PPARγ和HNF4A甲基化水平也有显著变化。ST6GAL1在5′UTR区域CpG位点甲基化水平显著降低,PPARγ5′UTR和TSS1500区域CpG位点甲基化水平显著增加,而HNF4A启动子甲基化水平也显著增加[10,20]。St6gal1在肥胖小鼠VAT显著下调,ST6GAL1对脂肪生成过程具有抑制作用[21]。其5′UTR区域甲基化水平降低,预示其表达水平增加,因此可能抑制SAT脂肪生成。而PPARγ有助于刺激脂肪细胞脂肪酸吸收和脂肪形成,敲除Pparγ小鼠即使饲喂高脂饲料(high fat diet,HFD)也不能生成脂肪组织[22]。糖尿病SAT中PPARγ高甲基化意味着SAT中摄取脂肪酸能力以及由前脂细胞向脂肪细胞分化能力下降。然而,在糖尿病小鼠(db/db)SAT中Pparγ甲基化状态有不同报道,Fujiki等[23]发现糖尿病小鼠SAT的Pparγ甲基化(-437 bp和-247 bp)降低,伴随基因mRNA水平的升高;而饮食诱导肥胖小鼠SAT中Pparγ甲基化与对照小鼠没有显著变化。可能不同的种属以及不同内在和外在环境对PPARγ影响不同。HNF4A通过与丙酮酸羧化酶(pyruvate carboxylase, PC)基因启动子相关元件结合,从而调节该基因表达。如果抑制HNF4A表达,会下调PC 60%mRNA和50%蛋白水平[24]。PC不仅在糖异生活跃组织中高表达,在白色脂肪组织中也有高水平表达[25],这可能和该酶参与脂生成有关,其产物草酰乙酸可以为脂肪酸合成提供重要的酰基基团和NADPH。因此,SAT中HNF4A发生高甲基化,提示HNF4A减少,从而PC表达降低,因此降低SAT脂生成过程。
ST6GAL1、PPARγ和HNF4A甲基化在糖尿病患者SAT中的变化提示了糖尿病SAT脂肪生成能力降低。
2.1.3 糖尿病患者SAT中细胞增殖相关基因甲基化变化
在T2D患者SAT中,与促进细胞增殖作用相关的ANK1和PEPD基因主体区域CpG位点甲基化水平显著降低;提示该基因在T2D SAT中转录水平受到抑制,因此可能降低了SAT脂肪细胞增殖作用。而与抑制细胞增殖作用相关的基因ADAMTS9、CDKN2A、WWOX、BARX2、RND3和TP53INP1在不同位点可能表现出不同甲基化变化[10,20]。ADAMTS9在基因主体区域CpG位点甲基化显著增加,但TSS1500区域CpG位点甲基化显著降低;CDKN2A基因在启动子区域甲基化降低;WWOX有2个基因主体区域CpG位点甲基化降低,1个CpG位点升高;BARX2基因主体区域CpG位点甲基化降低;RND3 TSS200区域甲基化增加;TP53INP1 5'UTR区域甲基化增加,TSS200区域甲基化降低。ADAMTS9基因主体区域CpG甲基化增加以及CDKN2A启动子DNA甲基化降低,也提示对应基因转录活性增加,因此可能具有抑制细胞增殖的作用。
总之,糖尿病SAT中ANK1、PEPD、ADAMTS9和CDKN2A等基因甲基化的变化,降低了脂肪组织细胞的增殖作用。
2.1.4 糖尿病患者SAT中细胞分化相关基因甲基化变化
FGF21在脂肪细胞中有助于葡萄糖吸收和脂肪细胞分化,并且能刺激PPARγ转录活性增强[26]。并且FGF21是DNA甲基转移酶DNMT3A的靶基因,体外研究也发现,在T2D患者SAT的脂肪细胞中,FGF21启动子区域DNA甲基化与对照比较有显著增加[27],同时其表达量显著降低。因此FGF21启动子DNA甲基化可能降低脂肪细胞分化能力。
另外,与细胞分化有关的HHEX和HMG20A在糖尿患者群中也有不同甲基化变化,其中HHEX基因主体区域DNA甲基化水平升高,而HMG20A 5′UTR甲基化水平增加[10],都提示其基因转录水平降低,从而降低脂肪细胞分化能力。
总之,糖尿病患者SAT中与胰岛素抵抗、脂肪运输、脂肪生成、细胞增殖和分化有关基因甲基化的变化,提示糖尿病SAT脂肪细胞胰岛素抵抗增加、细胞增殖能力以及脂肪生成和分化能力降低。
正是基于DNA甲基化与T2D的密切关系,Orozco等[13]建立了基于SAT中DNA甲基化的生物标记物来评估T2D危险性。结果证明脂肪组织DNA甲基化对于了解代谢综合征的分子效应是一个强大工具,能很好地评估T2D发展的危险性。
2.2 肥胖患者和动物模型SAT中DNA甲基化变化
在肥胖患者或动物SAT中,常常出现与脂肪分化、形成有关的基因甲基化变化(表2),从而影响SAT功能。Table 2
表2
表2 肥胖患者及动物模型SAT中DNA甲基化的变化
Table 2
| 基因分类 | 基因名称 | 甲基化位置 | 甲基化变化 | 参考文献 |
|---|---|---|---|---|
| 脂质堆积 | CHST11 | 主体 | ↓ | [28] |
| 脂肪生成 | ZBTB16 | 主体 | ↑ | [28~31] |
| FGFRL1 | 主体、TSS1500 | ↑、↓ | ||
| SORBS3 | 主体 | ↑ | ||
| HSD17B8 | 启动子 | ↑ | ||
| FASN | 主体 | ↑ | ||
| Lipe | 尚不明确 | ↓ | ||
| Acas | 尚不明确 | ↓ | ||
| 脂肪细胞棕色化 | AXIN2 | 主体 | ↑ | [28] |
| 炎症相关 | THNSL2 | 启动子 | ↑ | [29,32] |
| HCK | 启动子 | ↓ | ||
| TNF-alpha | 启动子 | ↓ | ||
| β细胞功能 | APBB2 | 主体、TSS200 | ↓、↓ | [30] |
| 胰岛素信号 | ASAP2 | 主体 | ↓ | [28,29,31~33] |
| SERPINF1 | 3°UTR | ↑ | ||
| ATP6V1B2 | 启动子 | ↓ | ||
| LEPTIN | 启动子 | ↓ | ||
| IRS1 | 启动子 | ↑ | ||
| IGF1R | 主体 | ↓ | ||
| IRS2 | 主体 | ↑ | ||
| TGF-β信号 | RBPMS | 主体 | ↑ | [28,30] |
| SMAD3 | TSS1500 | ↓ | ||
| 线粒体功能 | MRPL23 | 主体 | ↑ | [28,30] |
| PNKD | 主体、TSS1500 | ↑、↑ | ||
| 胞内运输 | TBC1D16 | 主体 | ↑ | [28] |
| 跨膜运输 | SLC2A5 | 启动子 | ↓ | [30] |
| 细胞凋亡 | BAG6 | 启动子 | ↓ | [28] |
| 细胞增殖 | DELEC1 | TSS1500 | ↓ | [29~31] |
| RSRC2 | TSS1500 | ↓ | ||
| CDK11B | 主体、5°UTR | ↓ | ||
| SETMAR | 启动子 | ↑ | ||
| CD9 | 启动子 | ↓ | ||
| TGFB2 | 主体 | ↑ | ||
| 细胞分化 | E2F5 | 主体 | ↓ | [28,29,35,37] |
| TTLL7 | 启动子 | ↑ | ||
| RUNX1 | 启动子 | ↓ | ||
| Hoxa5 | 尚不明确 | ↓ | ||
| Igf2/H19 | 尚不明确 | ↓ | ||
| 神经元分化 | RTN1 | 启动子 | ↑ | [29] |
| C8orf46 | 启动子 | ↑ | ||
| 蛋白泛素化 | NEDD4L | 启动子 | ↑ | [29] |
| 细胞基质粘附 | EPDR1 | 启动子 | ↓ | [29] |
| 调节RNA稳定性 | EXOSC7 | 启动子 | ↓ | [29] |
| 支链氨基酸代谢 | BCKDHB | 启动子 | ↑ | [29] |
新窗口打开|下载CSV
2.2.1 肥胖患者SAT中DNA甲基化变化
Pietil?inen等[28]分析了26对BMI不一致的同卵双生双胞胎SAT中DNA甲基化和相关基因表达,发现了胖和瘦的双胞胎中有17个与肥胖相关的基因存在差异甲基化水平。这些基因主要涉及脂代谢、炎症和细胞外基质(extracellular matrix, ECM)重塑,并且与脂代谢相关基因表达下调,而炎症和ECM相关基因表达上调。Keller等[29]研究也发现,肥胖个体SAT中有46个基因与非肥胖个体甲基化显著不同,高甲基化的前10个基因为SETMAR、HSD17B8、GOLGA6L4、NEDD4L、THNSL2、RTN1、NET1、C8orf46、TTLL7和BCKDHB,低甲基化的前10个基因EPDR1、RUNX1、ATP6V1B2、GPR137B、SLC2A5、HCK、CD99L2、FAM46A、EXOSC7和CD9。Crujeiras等[30]研究发现肥胖患者SAT的差异甲基化CpG位点甲基化水平比非肥胖患者低。Dahlman等[31]分析了肥胖后(post-obese)妇女和从未肥胖(never-obese)妇女SAT的DNA甲基化,发现所有分析位点的平均甲基化程度,肥胖后妇女比从未肥胖的妇女低。此外,差异甲基化区域(differentially methylated region, DMR)主要位于CpG岛和及周边,8504个差异甲基化位点(differentially methylated sites, DMS)定位于3717个特定基因,其中27%是与脂生成有关。而Cordero等[32]研究了27位肥胖妇女SAT中LEPTIN和TNF-alpha的甲基化,他们研究发现,经8周低卡路里饮食后,体重显著减轻妇女SAT中的LEPTIN和TNF-alpha甲基化显著降低。另外,体重超重人SAT脂肪组织中的IRS1启动子甲基化水平与血浆高密度脂蛋白胆固醇呈负相关[33]。总之,在肥胖患者SAT中相关基因DNA甲基化变化可能反映了脂代谢降低、炎症发生。
2.2.2 肥胖动物模型SAT中DNA甲基化的变化
Sonne等[34]用Med-IP方法研究了饮食诱导肥胖鼠模型以及ob/ob鼠模型腹股沟白色脂肪组织(inguinal white adipose tissue, iWAT)(SAT的一种类型)的甲基化,发现在这两种模型中,SAT均出现了DNA低甲基化。饮食诱导的脂肪组织DNA甲基化变化涉及了脂生成基因表达。在iWAT中鉴定了39个基因的甲基化伴随有mRNA水平的变化,其中Lipe和Acas基因表达水平降低,这两个基因均涉及了脂肪代谢。Lipe主要编码了脂肪细胞与脂分解有关的酶,而Aacs编码了使用酮体的酶,它为脂肪酸和胆固醇提供乙酰CoA。另有研究发现高脂食物诱导肥胖小鼠SAT(iWAT)中,Hoxa5表达显著降低,伴随其在分化脂肪细胞中出现Hoxa5 DNA甲基化;因为Hoxa5具有促进脂肪细胞分化的作用,因此高脂饮食诱导肥胖小鼠SAT脂肪分化作用降低[35]。除此之外,Igf2也有促进脂肪细胞分化作用[36],高脂饮食也能诱导Igf2甲基化:Claycombe等[37]研究了出生后饮食高脂饲料的大鼠,发现其SAT中第1号染色体的Igf2/H19位点ICR/H19 差异甲基化区域,位点1~4的DNA甲基化相对于正常饮食有显著增加。总之,肥胖动物模型SAT中Lipe、Acas、Hoxa5以及Igf2甲基化变化伴随其表达量降低,表明其脂肪分解能力、脂类合成能力以及脂肪分化能力降低。
3 糖尿病患者和肥胖患者VAT中DNA甲基化变化
相比于SAT,VAT有更多血管、神经,并包含更多数目的炎症、免疫细胞和更大比例的大脂肪细胞,而且脂肪细胞代谢更加活跃,但是其前脂细胞分化能力降低,对胰岛素表现出更强的抵抗[38]。内脏脂肪型肥胖往往与死亡率增加有很强的相关性[39]。在胰岛素抵抗的糖尿病前期患者中,VAT显著增加。并且VAT与胰岛素抵抗比SAT有更强的相关性[40]。因此糖尿病患者VAT变化可能反映于糖尿病整个发展过程中,而VAT中相关基因DNA甲基化与这种VAT组织与功能的变化可能有很大关系。3.1 糖尿病患者VAT中DNA甲基化变化
Rodr??guez-Rodero等[41]分析了经过胃肠绕道术的T2D和非T2D女性VAT DNA甲基化,发现T2D和非T2D在16个基因中有24个显著不同的CpGs。其中,HOOK2基因高甲基化与T2D糖尿病显著相关。HOOK2主要参与中心体相关功能[42],但是其对于脂肪细胞相关的作用仍不清楚。Deng等[43]研究了孕期糖尿病妇女VAT中全基因组DNA甲基化和基因表达谱,发现C10orf10、FSTL1、GSTT1、HLA-DPB1、HLA-DRB5、HSPA6和MSLN有DNA甲基化变化,并且伴随基因表达的变化(表3)。Table 3
表3
表3 糖尿病患者VAT中DNA甲基化的变化
Table 3
| 基因分类 | 基因名称 | 甲基化位置 | 甲基化变化 | 参考文献 |
|---|---|---|---|---|
| 中心体相关 | HOOK2 | 尚不明确 | ↑ | [41] |
| 自噬 | C10orf10 | 尚不明确 | ↑ | [43] |
| 细胞增殖分化 | FSTL1 | 尚不明确 | ↑ | [43] |
| T细胞受体信号 | HLA-DPB1 | 尚不明确 | ↑ | [43] |
| HLA-DRB5 | ||||
| 谷胱甘肽代谢 | GSTT1 | 尚不明确 | ↑ | [43] |
| 蛋白再折叠 | HSPA6 | 尚不明确 | ↓ | [43] |
| 细胞粘附 | MSLN | 尚不明确 | ↑ | [43] |
新窗口打开|下载CSV
3.2 肥胖患者及肥胖动物模型VAT中DNA甲基化变化
有不少研究者从全基因组甲基化的角度研究肥胖患者及动物模型的VAT甲基化水平,相对于非肥胖个体其VAT甲基化差异基因主要集中在以下几个方面(表4)。Table 4
表4
表4 肥胖患者及肥胖动物模型VAT中DNA甲基化的变化
Table 4
| 基因分类 | 基因名称 | 甲基化位置 | 甲基化变化 | 参考文献 |
|---|---|---|---|---|
| 炎症 | DUSP22 | 启动子 | ↑ | [29] |
| TLE1 | 启动子 | ↓ | ||
| 细胞分化 | TBX5 | 尚不明确 | ↓ | [29,49] |
| APMAP | 启动子 | ↑ | ||
| 脂肪生成 | AQP9 | 启动子 | ↓ | [29,34,54,55,57] |
| Ankrd26 | 启动子 | ↑ | ||
| C3 | 尚不明确 | ↓ | ||
| Lipe | 尚不明确 | ↓ | ||
| Aacs | 尚不明确 | ↓ | ||
| Fasn | 启动子 | ↓ | ||
| Ndufb6 | 主体、启动子 | ↑、↓ |
新窗口打开|下载CSV
3.2.1 炎症基因甲基化变化
Keller等[29]分析了肥胖和非肥胖个体OVAT (omental visceral adipose tissue, OVAT)全基因组DNA甲基化,发现DUSP22在肥胖个体中呈高甲基化。DUSP22是一个负调控因子,通过对STAT3去磷酸化调节IL-6/LIF/STAT3介导的信号通路,IL-6可以通过激活STAT3将与肥胖相关的慢性炎症与胰岛素抵抗联系在一起。DUSP22过表达可以抑制IL-6诱导的STAT3磷酸化,而降低炎症发生[44,45],因此该基因甲基化的增加可能促进了炎症发生。然而有趣的是,TLE1同样作为炎症反向调节因子(Tle1敲除小鼠表现出慢性炎症的表型,一些炎症因子表达增加[46]),在肥胖个体中的甲基化水平降低[29]。并且在HFD诱导的肥胖小鼠附睾脂肪组织(epididymal white adipose tissue, eWAT)中Tle1表达增加[47]。DUSP22和TLE1均为炎症的负调节因子,但是其在VAT表现出完全相反的甲基化变化,具体原因仍不清楚。
3.2.2 细胞分化相关基因甲基化变化
TBX5与腹部脂肪组织的前脂细胞增殖和分化有关[48]。在IR抵抗人群中TBX5基因甲基化降低[49]。APMAP也是与脂肪细胞分化有关的蛋白,在3T3-L1细胞中沉默该基因表达会损伤前脂细胞向脂肪细胞分化[50]。在肥胖人群VAT中,该基因甲基化水平升高[29],预示其转录水平降低,从而可能损伤脂肪组织分化过程。
3.2.3 脂肪生成相关基因甲基化变化
AQP9基因在肥胖个体VAT呈现低甲基化[29]。AQP9作为甘油辅助转运体,主要在网膜和皮下脂肪表达从而促进甘油转出,在肝脏中辅助甘油转入用于糖异生;而胰岛素和瘦素能调节AQP9的活性,AQP9表达增加会促进脂肪生成[51,52]。肥胖个体VAT中,AQP9等基因低甲基化,预示着VAT在肥胖个体中脂肪生成增加。Ankrd26也是调节脂肪形成的重要因子之一,选择性降低该基因表达会促进3T3-L1细胞的脂肪形成[53]。HFD饮食会导致小鼠VAT中启动子在-436 bp和-431 bp的CpG位点出现特异性高甲基化,并损伤其表达[54]。该基因表达量降低预示其促进脂肪细胞脂肪的生成。另外,在过度肥胖人群VAT中,补体组分3(complement factor 3, C3) DNA甲基化降低,而该组分可以转化为促酰基化蛋白(acylation stimulating protein, ASP)从而刺激脂类储存[55]。
然而,也有一些报道显示与脂生成有关的基因在肥胖小鼠VAT中DNA甲基化水平降低或升高,但抑制了脂肪生成过程。如饮食诱导的肥胖模型鼠eWAT中,与脂生成有关的Lipe和Aacs甲基化水平降低,同时伴随有mRNA水平降低[34]。另外,Zfp521编码产物是脂生成抑制因子[56],其在肥胖小鼠eWAT表达增加。这些基因的变化说明,在饮食诱导肥胖小鼠eWAT中,脂肪分解水平降低以及脂类合成降低。另一研究组以HFD饲喂大鼠,发现大鼠eWAT中Fasn和Ndufb6基因表达比对照组显著减低,且伴随Fasn基因-833/-829位点甲基化水平显著降低,Ndufb6基因在+143/+158位点甲基化显著增加,而在-7/+3/+14位点甲基化降低[57]。Fasn是脂肪酸合成酶基因,参与脂肪酸合成;而Ndufb6编码了呼吸链的重要组分,参与氧化磷酸化过程。Fasn和Ndufb6基因表达水平降低,提示HFD可能抑制了VAT脂肪酸合成以及物质的氧化分解。
4 棕色脂肪组织与DNA甲基化
BAT是通过适应性产热来调节体温的脂肪组织。BAT富含线粒体,而线粒体内膜上的解偶联蛋白(uncoupling protein 1, UCP1)能使氧化磷酸化解偶联,从而使原来ATP合成过程转换为以热的形式散失。然而,除了BAT,WAT经冷暴露或β肾上腺能受体激活(β-adrenergic receptor, β-AR)处理过后,脂肪细胞能表达UCP1,因此被称为米色脂肪[58]。尽管鲜见报道糖尿病或肥胖患者BAT中DNA甲基化变化,但是由于BAT或米色脂肪组织在产热方面的能力,所以通过调节BAT或米色脂肪组织来改善肥胖或肥胖相关疾病如糖尿病引起人们的关注。
相对于WAT,BAT表达丰富的PRDM16 (co-regulator PR domain containing 16, PRDM16),该蛋白与其他转录因子PPARγ、PGC1α、C/EBPβ以及Zfp516调节BAT特异性基因如Ucp1、Dio2 (deionidase2, Dio2)以及Cidea(cell death-inducing DNA fragmentation factor alpha-like effector A, Cidea)的表达[59]。PRDM16是棕色和米色脂肪细胞中调节产热基因转录的重要因子,无论对于胚胎BAT发育以及成人BAT特异性基因的表达都是必要的[60]。PRDM16与典型的DNA转录因子如PPARγ和C/EBPβ形成转录复合物,特异性激活BAT选择性基因表达程序[61,62]。PRDM16转录起始位点含有丰富的CpG位点,可以受到DNA甲基化调控[63]。转录因子基因PPARγ[64]、PGC1α[65]、C/EBPβ[66]以及BAT组织特异性表达基因Ucp1[67]、Cidea[68]均可以受DNA甲基化调控。
因此通过调节BAT或米色脂肪组织特异性表达基因或者相关转录因子的DNA甲基化变化,从而可以促进这些脂肪组织产热,这也是许多研究者正在探寻的一种策略,用于治疗包括糖尿病在内的代谢性疾病。
5 结语与展望
糖尿病或肥胖患者在SAT和VAT中的DNA甲基化变化往往与脂肪生成、脂肪细胞分化以及炎症相关基因的变化有关(图1)。但目前有关糖尿病或肥胖患者BAT中DNA甲基化变化的报道较少。由于BAT利用脂肪的积极作用,人们将其作为有效治疗糖尿病或肥胖的一个组织靶点,正在探寻可能的治疗策略。图1
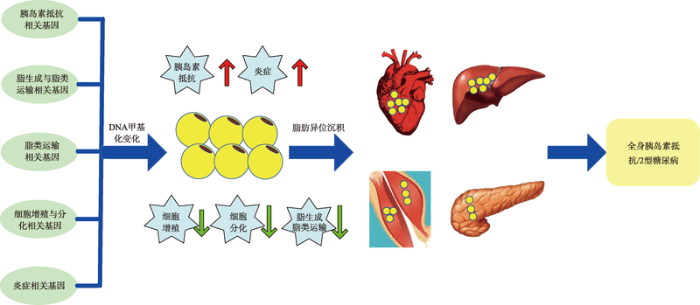 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1脂肪组织相关基因DNA甲基化与胰岛素抵抗/2型糖尿病的关联示意图
Fig. 1Schematic diagram of the association between DNA methylation of adipose tissue related genes and insulin resistance/type 2 diabetes mellitus
尽管有很多临床和实验文章报道了糖尿病和肥胖脂肪组织DNA甲基化变化,但目前仍存在一些问题尚待深入研究:(1)脂肪组织中DNA甲基化频率与肥胖、糖尿病病变程度的确切关系是怎样的?(2)脂肪组织分布广泛,SAT、VAT和BAT在机体中所承担的角色各有不同,每个组织是否有特异表达基因,肥胖和糖尿病患者这些基因的甲基化情况是否代表着组织功能的变化?(3)DNA甲基化发生在基因的不同位置(启动子、增强子、转录区、基因间隔区等)对于基因表达调控不同,肥胖和糖尿病同一脂肪组织不同基因位置甲基化与基因表达关系图谱是怎样的?(4)脂肪组织包含多种细胞类型,而DNA甲基化有细胞特异性,究竟是脂肪细胞DNA甲基化,还是其他细胞甲基化?如何有效区分将更能反映组织功能变化情况?(5)脂肪组织的DNA甲基化与其它发生胰岛素抵抗的组织(如肝脏、骨骼肌等)的DNA甲基化是否有关?两者之间有无相互影响?
总之,脂肪组织不仅是合成及储存脂肪的仓库,更是参与多种病理生理过程的内分泌器官,对于机体代谢平衡的调节具有重要影响。脂肪组织DNA甲基化与肥胖、糖尿病发生发展密切相关。由于肥胖、糖尿病发病机制的复杂性、脂肪组织分布的广泛性、类型多样性以及DNA甲基化组织细胞特异性等决定了肥胖、糖尿病与脂肪组织DNA甲基化关系仍需要进一步研究。这些具体关系的研究将有助于糖尿病发病机制的阐明,也必将积极推动基于调节DNA甲基化的治疗肥胖和糖尿病药物的开发和筛选。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:27846494 [本文引用: 1]

Abstract The rising prevalences of type 2 diabetes and obesity constitute major threats to human health globally. Powerful social and economic factors influence the distribution of these diseases among and within populations. These factors act on a substrate of individual predisposition derived from the composite effects of inherited DNA variation and a range of environmental exposures experienced throughout the life course. Although "Western" lifestyle represents a convenient catch-all culprit for such exposures, effective treatment and prevention will be informed by characterization of the most critical, causal environmental factors. In this Review, we examine how burgeoning understanding of the genetic basis of type 2 diabetes and obesity can highlight nongenetic exposures that drive development of these conditions. Copyright 2016, American Association for the Advancement of Science.
URLPMID:24779963Magsci [本文引用: 1]
To elucidate the mechanisms related to the development of type 2 diabetes (T2D) and other degenerative diseases at a molecular level, a better understanding of the changes in the chromatin structure and the corresponding functional changes in molecular pathways is still needed. For example, persons with low birth weight are at a high risk for development of T2D later in life, suggesting that the intrauterine environment contributes to the disease. One of the hypotheses is that epigenetic regulation, including changes in DNA methylation leading to modifications in chromatin structure, are behind metabolic alterations, e.g. leading to the phenomenon termed metabolic memory. Altered DNA methylation has been shown to affect healthy aging and also to promote age-related health problems. There is suggestive evidence that lifestyle changes including weight loss can have an impact on DNA methylation and consequently gene expression. In this review we provide an overview of human studies investigating DNA methylation in obesity and T2D and associated risk factors behind these diseases.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:15387477 [本文引用: 1]
The effect of fat distribution on disease risk is a subject of great interest. Central fat has been measured anthropometrically, by computed tomography, and by magnetic resonance imaging. Both cross-sectional and longitudinal studies have related central fat to type 2 diabetes mellitus and cardiovascular disease, independent of body mass index. The mechanism may relate to increased lipolysis causing the liver to increase glucose and very low density lipoprotein output, while muscle uses less. This leads to a rise in blood glucose and triglycerides, a drop in HDL cholesterol, and an increase in small, dense LDL particles. There is also an increase in blood pressure and inflammatory markers. Certain populations put on excess fat more centrally than others. These include Asian populations. It is likely that with better differentiation of abdominal fat into visceral and subcutaneous depots, clearer data will accrue on their impact on disease risk
URLPMID:1435298 [本文引用: 1]
Mass, morphology, and metabolism of total adipose tissue and its subcutaneous, visceral, and retroperitoneal subcompartments were examined in 16 men with a wide variation of total body fat. Computerized tomography (CT) scans showed that the intraabdominal fat mass comprised approximately 20% of total fat mass. Visceral and retroperitoneal fat masses were approximately 80% and 20% of total intraabdominal fat mass, respectively. Enlargement of intraabdominal fat depots was due to a parallel adipocyte enlargement only. Direct significant correlations were found between these adipose tissue masses and blood glucose and plasma insulin levels, blood pressure, and liver function tests, while glucose disposal rate during euglycemic glucose clamp measurements at submaximal insulin concentrations (GDR), plasma testosterone, and sex hormone-binding globulin concentrations correlated negatively. The correlations for glucose, insulin, and GDR were strongest with visceral fat mass. Adipose tissue lipid uptake, measured after oral administration of labeled oleic acid in triglyceride, was approximately 50% higher in omental than in subcutaneous adipose tissues. Adipocytes from omental fat also showed a higher lipolytic sensitivity and responsiveness to catecholamines. Furthermore, these adipocytes were less sensitive to the antilipolytic effects of insulin. Both lipid uptake and lipolytic sensitivity and responsiveness showed strong correlations ( r = 0.8 to 0.9) to blood glucose and plasma insulin concentrations and also to the GDR (negative), while no such correlations were found with lipid uptake in subcutaneous or retroperitoneal abdominal adipose tissues. Taken together, these results suggest a higher turnover of lipids in visceral than in the other fat depots, which is closely correlated to systemic insulin resistance and glucose metabolism in men.
URLPMID:29362458 [本文引用: 1]
In a series of studies of atomic bomb survivors, radiation-dose-dependent alterations in peripheral T-cell populations have been reported. For example, reduced size in na茂ve T-cell pools and impaired proliferation ability of T cells were observed. Because these alterations are also generally observed with human aging, we hypothesized that radiation exposure may accelerate the aging process of... [Show full abstract]
URLPMID:19339685 [本文引用: 1]
ABSTRACT The last several years have seen an explosion of information relating to the transcriptional control of brown fat cell development. At the same time, new data have emerged that clearly demonstrate that adult humans do indeed have substantial amounts of functioning brown adipose tissue (BAT). Together, these advances are stimulating a reassessment of the role of brown adipose tissue in human physiology and pathophysiology. These data have also opened up exciting new opportunities for the development of entirely novel classes of therapeutics for metabolic diseases like obesity and type 2 diabetes.
URLPMID:28052966 [本文引用: 1]
Abstract In the transition from normal glucose tolerance (NGT) to type 2 diabetes mellitus (T2DM), the role of β-cell dysfunction and peripheral insulin resistance (IR) is well established. However, the impact of dysfunctional adipose tissue has not been fully elucidated. The aim of the present study is to evaluate the role of resistance to the antilipolytic effect of insulin (Adipo-IR) in a large group of NGT, IGT and T2DM subjects.302 subjects with varying glucose tolerance received OGTT and euglycemic insulin clamp. We evaluated: Adipo-IR (fasting and mean OGTT plasma FFA x insulin), peripheral IR (1/(Matsuda index) and (M/I) -1 ), β-cell function (calculated as the ratio of the increment in plasma insulin to glucose (OGTT) divided by IR [ΔI/ΔG÷IR]).Fasting Adipo-IR was increased 2-folds in obese-NGT and IGT vs lean-NGT (8.0±1.1 and 9.2±0.7 vs 4.1±0.3) and 3-fold in T2DM (11.9±0.6 p<0.001). Progressive decline in ΔI/ΔG÷IR was associated with a progressive impairment in FFA suppression during OGTT, while the rise in mean plasma glucose concentration only became manifest when subjects became overtly diabetic.In conclusion, the progressive decline in β-cell function that begins in "normal" glucose tolerant individuals is associated with a progressive increase in FFA and fasting Adipo-IR. 08 2017 by the American Diabetes Association.
Magsci [本文引用: 6]
Genetics, epigenetics, and environment may together affect the susceptibility for type 2 diabetes (T2D). Our aim was to dissect molecular mechanisms underlying T2D using genome-wide expression and DNA methylation data in adipose tissue from monozygotic twin pairs discordant for T2D and independent case-control cohorts. In adipose tissue from diabetic twins, we found decreased expression of genes involved in oxidative phosphorylation; carbohydrate, amino acid, and lipid metabolism; and increased expression of genes involved in inflammation and glycan degradation. The most differentially expressed genes included ELOVL6, GYS2, FADS1, SPP1 (OPN), CCL18, and IL1RN. We replicated these results in adipose tissue from an independent case-control cohort. Several candidate genes for obesity and T2D (e.g., IRS1 and VEGFA) were differentially expressed in discordant twins. We found a heritable contribution to the genome-wide DNA methylation variability in twins. Differences in methylation between monozygotic twin pairs discordant for T2D were subsequently modest. However, 15,627 sites, representing 7,046 genes including PPARG, KCNQ1, TCF7L2, and IRS1, showed differential DNA methylation in adipose tissue from unrelated subjects with T2D compared with control subjects. A total of 1,410 of these sites also showed differential DNA methylation in the twins discordant for T2D. For the differentially methylated sites, the heritability estimate was 0.28. We also identified copy number variants (CNVs) in monozygotic twin pairs discordant for T2D. Taken together, subjects with T2D exhibit multiple transcriptional and epigenetic changes in adipose tissue relevant to the development of the disease.
URLPMID:24281010Magsci [本文引用: 1]
Abstract Insulin resistance is a major underlying mechanism responsible for the 'metabolic syndrome', which is also known as insulin resistance syndrome. The incidence of the metabolic syndrome is increasing at an alarming rate, becoming a major public and clinical problem worldwide. The metabolic syndrome is represented by a group of interrelated disorders, including obesity, hyperglycemia, hyperlipidemia, and hypertension. It is also a significant risk factor for cardiovascular disease and increased morbidity and mortality. Animal studies have demonstrated that insulin and its signaling cascade normally control cell growth, metabolism, and survival through the activation of MAPKs and activation of phosphatidylinositide-3-kinase (PI3K), in which the activation of PI3K associated with insulin receptor substrate 1 (IRS1) and IRS2 and subsequent Akt090608Foxo1 phosphorylation cascade has a central role in the control of nutrient homeostasis and organ survival. The inactivation of Akt and activation of Foxo1, through the suppression IRS1 and IRS2 in different organs following hyperinsulinemia, metabolic inflammation, and overnutrition, may act as the underlying mechanisms for the metabolic syndrome in humans. Targeting the IRS090608Akt090608Foxo1 signaling cascade will probably provide a strategy for therapeutic intervention in the treatment of type 2 diabetes and its complications. This review discusses the basis of insulin signaling, insulin resistance in different mouse models, and how a deficiency of insulin signaling components in different organs contributes to the features of the metabolic syndrome. Emphasis is placed on the role of IRS1, IRS2, and associated signaling pathways that are coupled to Akt and the forkhead/winged helix transcription factor Foxo1.
URLPMID:11259600 [本文引用: 1]
To investigate the role of insulin receptor substrate 1 (IRS-1) and IRS-2, the two ubiquitously expressed IRS proteins, in adipocyte differentiation, we established embryonic fibroblast cells with four different genotypes, i.e., wild-type, IRS-1 deficient (IRS-1(-/-)), IRS-2 deficient (IRS-2(-/-)), and IRS-1 IRS-2 double deficient (IRS-1(-/-) IRS-2(-/-)), from mouse embryos of the corresponding genotypes. The abilities of IRS-1(-/-) cells and IRS-2(-/-) cells to differentiate into adipocytes are approximately 60 and 15%, respectively, lower than that of wild-type cells, at day 8 after induction and, surprisingly, IRS-1(-/-) IRS-2(-/-) cells have no ability to differentiate into adipocytes. The expression of CCAAT/enhancer binding protein alpha (C/EBPalpha) and peroxisome proliferator-activated receptor gamma (PPARgamma) is severely decreased in IRS-1(-/-) IRS-2(-/-) cells at both the mRNA and the protein level, and the mRNAs of lipoprotein lipase and adipocyte fatty acid binding protein are severely decreased in IRS-1(-/-) IRS-2(-/-) cells. Phosphatidylinositol 3-kinase (PI 3-kinase) activity that increases during adipocyte differentiation is almost completely abolished in IRS-1(-/-) IRS-2(-/-) cells. Treatment of wild-type cells with a PI 3-kinase inhibitor, LY294002, markedly decreases the expression of C/EBPalpha and PPARgamma, a result which is associated with a complete block of adipocyte differentiation. Moreover, histologic analysis of IRS-1(-/-) IRS-2(-/-) double-knockout mice 8 h after birth reveals severe reduction in white adipose tissue mass. Our results suggest that IRS-1 and IRS-2 play a crucial role in the upregulation of the C/EBPalpha and PPARgamma expression and adipocyte differentiation.
URLPMID:29566149 [本文引用: 2]
Most epigenome-wide association studies to date have been conducted in blood. However, metabolic syndrome is mediated by a dysregulation of adiposity and therefore it is critical to study adipose tissue in order to understand the effects of this syndrome on epigenomes. Therefore, to determine if natural variation in DNA methylation was associated with metabolic syndrome traits, we profiled global methylation levels in subcutaneous abdominal adipose tissue. We measured association with 32 clinical traits related to diabetes and obesity in 201 people from the Metabolic Syndrome In Men cohort. We performed epigenome-wide association studies between DNA methylation levels and traits, and identified significant associations for 13 clinical traits in 21 loci. We prioritized candidate genes using eQTL, and identified 18 high confidence candidate genes, including known and novel genes associated with diabetes and obesity traits. We also carried out an analysis to identify which cell types may be mediating the associations, and concluded that most of the loci we identified were specific to adipocytes. We determined whether the abundance of cell types varies with metabolic traits, and found that macrophages increased in abundance with the severity of metabolic syndrome traits. Finally, we developed a DNA methylation based biomarker to assess type II diabetes risk in adipose tissue. In conclusion, our results demonstrate that profiling DNA methylation in adipose tissue is a powerful tool for understanding the molecular effects of metabolic syndrome on adipose tissue, and can be used in conjunction with traditional genetic analyses to further characterize this disorder.
URLPMID:22641018Magsci [本文引用: 2]
DNA methylation is frequently described as a 'silencing' epigenetic mark, and indeed this function of 5-methylcytosine was originally proposed in the 1970s. Now, thanks to improved genome-scale mapping of methylation, we can evaluate DNA methylation in different genomic contexts: transcriptional start sites with or without CpG islands, in gene bodies, at regulatory elements and at repeat sequences. The emerging picture is that the function of DNA methylation seems to vary with context, and the relationship between DNA methylation and transcription is more nuanced than we realized at first. Improving our understanding of the functions of DNA methylation is necessary for interpreting changes in this mark that are observed in diseases such as cancer.
URLPMID:25614086 [本文引用: 1]
International Journal of Obesity is a monthly, multi-disciplinary forum for papers describing basic, clinical and applied studies in biochemistry, genetics and nutrition, together with molecular, metabolic, psychological and epidemiological aspects of obesity and related disorders
URLPMID:27577745 [本文引用: 1]
Insulin resistance is a key driver of type 2 diabetes (T2D) and is characterized by defective insulin receptor (INSR) signalling. Although surface INSR downregulation is a well-established contributor to insulin resistance, the underlying molecular mechanisms remain obscure. Here we show that the E3 ubiquitin ligase MARCH1 impairs cellular insulin action by degrading cell surface INSR. Using a large-scale RNA interference screen, we identify MARCH1 as a negative regulator of INSR signalling.March1loss-of-function enhances, andMarch1overexpression impairs, hepatic insulin sensitivity in mice. MARCH1 ubiquitinates INSR to decrease cell surface INSR levels, but unlike other INSR ubiquitin ligases, MARCH1 acts in the basal state rather than after insulin stimulation. Thus, MARCH1 may help set the basal gain of insulin signalling.MARCH1expression is increased in white adipose tissue of obese humans, suggesting that MARCH1 contributes to the pathophysiology of T2D and could be a new therapeutic target. Insulin receptor levels at the cell surface are reduced in insulin resistance, for reasons that are not fully understood. Here, the authors identify the E3 ubiquitin ligase MARCH1 as a direct regulator of basal insulin receptor surface levels and, therefore, insulin signalling.
URLPMID:3478533Magsci [本文引用: 1]
Abstract We investigated the effects of obesity surgery-induced weight loss on transcription factor 7-like 2 gene (TCF7L2) alternative splicing in adipose tissue and liver. Furthermore, we determined the association of TCF7L2 splicing with the levels of plasma glucose and serum free fatty acids (FFAs) in three independent studies (n = 216). Expression of the short mRNA variant, lacking exons 12, 13, and 13a, decreased after weight loss in subcutaneous fat (n = 46) and liver (n = 11) and was more common in subcutaneous fat of subjects with type 2 diabetes than in subjects with normal glucose tolerance in obese individuals (n = 54) and a population-based sample (n = 49). Additionally, there was a positive correlation between this variant and the level of fasting glucose in nondiabetic individuals (n = 113). This association between TCF7L2 splicing and plasma glucose was independent of the TCF7L2 genotype. Finally, this variant was associated with high levels of serum FFAs during hyperinsulinemia, suggesting impaired insulin action in adipose tissue, whereas no association with insulin secretion or insulin-stimulated whole-body glucose uptake was observed. Our study shows that the short TCF7L2 mRNA variant in subcutaneous fat is regulated by weight loss and is associated with hyperglycemia and impaired insulin action in adipose tissue.
URLPMID:27148772 [本文引用: 1]
Identification of subjects with a high risk of developing type 2 diabetes (T2D) is fundamental for prevention of the disease. Consequently, it is essential to search for new biomarkers that can improve the prediction of T2D. The aim of this study was to examine whether 5 DNA methylation loci in blood DNA (ABCG1, PHOSPHO1, SOCS3, SREBF1, and TXNIP), recently reported to be associated with T2D, might predict future T2D in subjects from the Botnia prospective study. We also tested if these CpG sites exhibit altered DNA methylation in human pancreatic islets, liver, adipose tissue, and skeletal muscle from diabetic vs. non-diabetic subjects. DNA methylation at the ABCG1 locus cg06500161 in blood DNA was associated with an increased risk for future T2D (OR = 1.09, 95% CI = 1.020900091.16, P-value = 0.007, Q-value = 0.018), while DNA methylation at the PHOSPHO1 locus cg02650017 in blood DNA was associated with a decreased risk for future T2D (OR = 0.85, 95% CI = 0.750900090.95, P-value = 0.006, Q-value = 0.018) after adjustment for age, gender, fasting glucose, and family relation. Furthermore, the level of DNA methylation at the ABCG1 locus cg06500161 in blood DNA correlated positively with BMI, HbA1c, fasting insulin, and triglyceride levels, and was increased in adipose tissue and blood from the diabetic twin among monozygotic twin pairs discordant for T2D. DNA methylation at the PHOSPHO1 locus cg02650017 in blood correlated positively with HDL levels, and was decreased in skeletal muscle from diabetic vs. non-diabetic monozygotic twins. DNA methylation of cg18181703 (SOCS3), cg11024682 (SREBF1), and cg19693031 (TXNIP) was not associated with future T2D risk in subjects from the Botnia prospective study.
[本文引用: 1]
URLPMID:23251491 [本文引用: 2]
Abstract BACKGROUND: Monozygotic twins discordant for type 2 diabetes constitute an ideal model to study environmental contributions to type 2 diabetic traits. We aimed to examine whether global DNA methylation differences exist in major glucose metabolic tissues from these twins. METHODOLOGY/PRINCIPAL FINDINGS: Skeletal muscle (n = 11 pairs) and subcutaneous adipose tissue (n = 5 pairs) biopsies were collected from 53-80 year-old monozygotic twin pairs discordant for type 2 diabetes. DNA methylation was measured by microarrays at 26,850 cytosine-guanine dinucleotide (CpG) sites in the promoters of 14,279 genes. Bisulfite sequencing was applied to validate array data and to quantify methylation of intergenic repetitive DNA sequences. The overall intra-pair variation in DNA methylation was large in repetitive (LINE1, D4Z4 and NBL2) regions compared to gene promoters (standard deviation of intra-pair differences: 10% points vs. 4% points, P<0.001). Increased variation of LINE1 sequence methylation was associated with more phenotypic dissimilarity measured as body mass index (r = 0.77, P = 0.007) and 2-hour plasma glucose (r = 0.66, P = 0.03) whereas the variation in promoter methylation did not associate with phenotypic differences. Validated methylation changes were identified in the promoters of known type 2 diabetes-related genes, including PPARGC1A in muscle (13.900±6.2% vs. 9.000±4.5%, P = 0.03) and HNF4A in adipose tissue (75.200±3.8% vs. 70.500±3.7%, P<0.001) which had increased methylation in type 2 diabetic individuals. A hypothesis-free genome-wide exploration of differential methylation without correction for multiple testing identified 789 and 1,458 CpG sites in skeletal muscle and adipose tissue, respectively. These methylation changes only reached some percentage points, and few sites passed correction for multiple testing. CONCLUSIONS/SIGNIFICANCE: Our study suggests that likely acquired DNA methylation changes in skeletal muscle or adipose tissue gene promoters are quantitatively small between type 2 diabetic and non-diabetic twins. The importance of methylation changes in candidate genes such as PPARGC1A and HNF4A should be examined further by replication in larger samples.
URLPMID:5313100 [本文引用: 1]
Abstract Adipose tissue plays critical roles in obesity and related diseases such as diabetes and cardiovascular diseases. Previous reports suggest that glycans, the most common post-translational modifications, are involved in obesity-related diseases, but what type of glycan regulates adipogenesis during obesity remains unclear. In this study, we first quantified the mRNA levels of 167 genes (encoding 144 glycosyltransferases and 23 related enzymes) in visceral adipose tissues (VATs) from control mice and high fat diet (HFD)-induced obese mice. We found that a gene encoding β-galactoside α2,6-sialyltransferase-1 (St6gal1), a key enzyme responsible for the biosynthesis of α2,6-linked sialic acid in N-linked glycans, was most downregulated in VATs from obese mice. We confirmed the reduction in α2,6-sialic acid in VATs from obese mice and differentiated adipocyte model 3T3-L1 cells. Using proteomic analysis, integrin-β1 was identified as one of the target α2,6-sialylated proteins in adipose tissues, and phosphorylation of its downstream molecule focal adhesion kinase (FAK) was found to be decreased after HFD feeding. St6gal1 overexpression in differentiating 3T3-L1 cells inhibited adipogenesis with increased phosphorylation of FAK. Furthermore, St6gal1 knockout mice exhibited increased bodyweight and VAT weight after HFD feeding. The downregulation of St6gal1 during adipogenesis was canceled by treatment with a DNA methyltransferase inhibitor, suggesting an involvement of epigenetic DNA methylation in St6gal1 silencing. Our findings suggest that ST6GAL1 has an inhibitory role in adipogenesis through integrin-β1 activation, providing new insights into the roles and regulation mechanisms of glycans in adipocytes during obesity. Copyright 08 2016, The American Society for Biochemistry and Molecular Biology.
[本文引用: 1]
URLPMID:19589179 [本文引用: 1]
Background Adipose tissues serve not only as a store for energy in the form of lipid, but also as endocrine tissues that regulates metabolic activities of the organism by secreting various kinds of hormones. Peroxisome proliferator activated receptor ?? (PPAR??) is a key regulator of adipocyte differentiation that induces the expression of adipocyte-specific genes in preadipocytes and mediates their differentiation into adipocytes. Furthermore, PPAR?? has an important role to maintain the physiological function of mature adipocyte by controlling expressions of various genes properly. Therefore, any reduction in amount and activity of PPAR?? is linked to the pathogenesis of metabolic syndrome. Results In this study, we investigated the contribution of epigenetic transcriptional regulatory mechanisms, such as DNA methylation, to the expression of the PPAR?? gene, and further evaluated the contribution of such epigenetic regulatory mechanisms to the pathogenesis of metabolic syndrome. In 3T3-L1 preadipocytes, the promoter of the PPAR??2 gene was hypermethylated, but was progressively demethylated upon induction of differentiation, which was accompanied by an increase of mRNA expression. Moreover, treatment of cells with 5'-aza-cytideine, an inhibitor of DNA methylation, increased expression of the PPAR?? gene in a dose-dependent manner. Methylation in vitro of a PPAR?? promoter-driven reporter construct also repressed the transcription of a downstream reporter gene. These results suggest that the expression of the PPAR?? gene is inhibited by methylation of its promoter. We next compared the methylation status of the PPAR?? promoters in adipocytes from wild-type (WT) mice with those from two diabetic mouse models: +Leprdb/+Leprdb and diet-induced obesity mice. Interestingly, we found increased methylation of the PPAR?? promoter in visceral adipose tissues (VAT) of the mouse models of diabetes, compared to that observed in wild-type mice. We observed a concomitant decrease in the level of PPAR?? mRNA in the diabetic mice compared to the WT mice. Conclusion We conclude that the expression of PPAR?? gene is regulated by DNA methylation of its promoter region and propose that reduced expression of PPAR?? owing to DNA methylation in adipocytes of the VAT may contribute to the pathogenesis of metabolic syndrome.
[本文引用: 1]
URLPMID:1375435 [本文引用: 1]
Abstract Immunoblotting and protein microsequencing were used to identify several adipocyte proteins expressed in an obesity-related fashion in the Zucker rat. One of these was a 116-kDa particulate protein (p116). The p116 levels in adipocytes from 5- to 7-wk-old obese Zucker rats were two- to fivefold higher on a per milligram of protein basis than levels in lean animals and decreased after the induction of streptozotocin-induced diabetes mellitus. This suggests the change may be related to the actions of insulin. Hepatic levels of p116 did not change. The p116 was purified to homogeneity from obese Zucker rat adipocytes, and polyclonal antisera were prepared against the purified protein in rabbits. Microanalysis of electroblotted p116 proteolytic fragments suggested that p116 was pyruvate carboxylase (PC). Other evidence that p116 was PC included the following: 1) p116 contained biotin, 2) p116 in particulate subcellular fractions was soluble after freeze-lysis, 3) antibodies to p116 reacted with purified hepatic PC, 4) p116 and purified hepatic PC had identical pI and relative molecular weight values, and 5) similar changes were detected in adipocyte p116 and PC enzyme activity during obesity and after the induction of streptozotocin-induced diabetes mellitus. Increased adipose tissue PC probably contributes to the increased lipogenic capacity of young obese Zucker rat adipocytes.
Magsci [本文引用: 1]
Fibroblast growth factor-21 (FGF21) is a circulating hepatokine that beneficially affects carbohydrate and lipid metabolism. Here, we report that FGF21 is also an inducible, fed-state autocrine factor in adipose tissue that functions in a feed-forward loop to regulate the activity of peroxisome proliferator-activated receptor gamma (PPAR gamma), a master transcriptional regulator of adipogenesis. FGF21 knockout (KO) mice display defects in PPARg signaling including decreased body fat and attenuation of PPAR gamma-dependent gene expression. Moreover, FGF21-KO mice are refractory to both the beneficial insulin-sensitizing effects and the detrimental weight gain and edema side effects of the PPAR gamma agonist rosiglitazone. This loss of function in FGF21-KO mice is coincident with a marked increase in the sumoylation of PPAR gamma, which reduces its transcriptional activity. Adding back FGF21 prevents sumoylation and restores PPAR gamma activity. Collectively, these results reveal FGF21 as a key mediator of the physiologic and pharmacologic actions of PPAR gamma.
URLPMID:29091029 [本文引用: 1]
10.7554/eLife.30766.001Insulin resistance results from an intricate interaction between genetic make-up and environment, and thus may be orchestrated by epigenetic mechanisms like DNA methylation. Here, we demonstrate that DNA methyltransferase 3a (Dnmt3a) is both necessary and sufficient to mediate insulin resistance in cultured mouse and human adipocytes. Furthermore, adipose-specific Dnmt3a knock-out mice are protected from diet-induced insulin resistance and glucose intolerance without accompanying changes in adiposity. Unbiased gene profiling studies revealed Fgf21 as a key negatively regulated Dnmt3a target gene in adipocytes with concordant changes in DNA methylation at the Fgf21 promoter region. Consistent with this, Fgf21 can rescue Dnmt3a-mediated insulin resistance, and DNA methylation at the FGF21 locus was elevated in human subjects with diabetes and correlated negatively with expression of FGF21 in human adipose tissue. Taken together, our data demonstrate that adipose Dnmt3a is a novel epigenetic mediator of insulin resistance in vitro and in vivo.
URLPMID:26499446 [本文引用: 1]
Little is known about epigenetic alterations associated with subcutaneous adipose tissue (SAT) in . Our aim was to study genome-wide DNA and differences in SAT in monozygotic (MZ) twin pairs who are discordant for body mass index (BMI). This design completely matches lean and groups for genetic background, age, gender, and shared environment.We analyzed DNA methylome and from SAT, together with body composition (magnetic resonance imaging/spectroscopy) and tolerance test, and CRP from 26 rare BMI-discordant (intra-pair difference in BMIg/m(2)) MZ twin pairs, identified from 10 birth cohorts of young adult Finnish twins.We found 17 novel associated genes that were differentially methylated across the genome between heavy and lean co-twins. Nine of them were also differentially expressed. Pathway analyses indicated that dysregulation of SAT in includes a paradoxical downregulation of /adipogenesis, and upregulation of inflammation and remodeling. Further, CpG sites whose correlated with metabolically harmful fat depots (intra-abdominal and liver fat) also correlated with measures of resistance, and low-grade inflammation, thus suggesting that epigenetic alterations in SAT are associated with the development of unhealthy .This is the first study in BMI-discordant MZ twin pairs reporting genome-wide DNA and expression profiles in SAT. We found a number of novel genes and pathways whose and expression patterns differ within the twin pairs, suggesting that the pathological adaptation of SAT to is, at least in part, epigenetically regulated.International Journal of accepted article preview online, 26 October 2015. doi:10.1038/ijo.2015.221.
URLPMID:5220399 [本文引用: 5]
DNA methylation plays an important role in obesity and related metabolic complications. We examined genome-wide DNA promoter methylation along with mRNA profiles in paired samples of human subcutaneous adipose tissue (SAT) and omental visceral adipose tissue (OVAT) from non-obesevs.obese individuals. We identified negatively correlated methylation and expression of several obesity-associated genes in our discovery dataset andin silicoreplicatedETV6in two independent cohorts. Further, we identified six adipose tissue depot-specific genes (HAND2,HOXC6,PPARG,SORBS2,CD36, andCLDN1). The effects were further supported in additional independent cohorts. Our top hits might play a role in adipogenesis and differentiation, obesity, lipid metabolism, and adipose tissue expandability. Finally, we show thatin02vitromethylation ofSORBS2directly represses gene expression. Taken together, our data show distinct tissue specific epigenetic alterations which associate with obesity. 61Obesity-associated differences in DNA promoter methylation and transcriptome in human adipose tissue (ETV6).61Depot-specific analyses revealed novel/known genes (HAND2,HOXC6,PPARG,SORBS2,CD36,CLDN1).61EWAS revealedSSPNandCCDC125associated to BMI in SAT or OVAT, respectively.61Differentially methylated genes overlap in part with GWAS hits for obesity and fat distribution. Obesity-associated differences in DNA promoter methylation and transcriptome in human adipose tissue (ETV6). Depot-specific analyses revealed novel/known genes (HAND2,HOXC6,PPARG,SORBS2,CD36,CLDN1). EWAS revealedSSPNandCCDC125associated to BMI in SAT or OVAT, respectively. Differentially methylated genes overlap in part with GWAS hits for obesity and fat distribution.
URLPMID:28211912 [本文引用: 1]
The characterization of the epigenetic changes within the obesity-related adipose tissue will provide new insights to understand this metabolic disorder, but adipose tissue is not easy to sample in population-based studies. We aimed to evaluate the capacity of circulating leukocytes to reflect the adipose tissue-specific DNA methylation status of obesity susceptibility. DNA samples isolated from subcutaneous adipose tissue and circulating leukocytes were hybridized in the Infinium HumanMethylation 450 BeadChip. Data were compared between samples from obese (n65=6545) and non-obese (n65=658-10) patients by Wilcoxon-rank test, unadjusted for cell type distributions. A global hypomethylation of the differentially methylated CpG sites (DMCpGs) was observed in the obese subcutaneous adipose tissue and leukocytes. The overlap analysis yielded a number of genes mapped by the common DMCpGs that were identified to reflect the obesity state in the leukocytes. Specifically, the methylation levels of FGFRL1, NCAPH2, PNKD and SMAD3 exhibited excellent and statistically significant efficiencies in the discrimination of obesity from non-obesity status (AUC65>650.80; p65<650.05) and a great correlation between both tissues. Therefore, the current study provided new and valuable DNA methylation biomarkers of obesity-related adipose tissue pathogenesis through peripheral blood analysis, an easily accessible and minimally invasive biological material instead of adipose tissue.
URLPMID:25783037 [本文引用: 1]
Obese subjects have increased number of enlarged fat cells which are reduced in size but not number in post-obesity. We performed DNA methylation profiling in fat cells with the aim of identifying differentially methylated DNA sites (DMS) linked to adipose hyperplasia (many small fat cells) in post-obesity.
URLPMID:21465273Magsci [本文引用: 1]
Obesity-associated adipose tissue enlargement is characterized by an enhanced proinflammatory status and an elevated secretion of adipokines such as leptin and cytokines such as tumor necrosis factor (TNF)-alpha. Among the different mechanisms that could underlie the interindividual differences in obesity, epigenetic regulation of gene expression has emerged as a potentially important determinant. Therefore, 27 obese women (age, 32–5002years; baseline body mass index, 34.465±654.202kg/m 2 ) were prescribed an 8-week low-calorie diet, and epigenetic marks were assessed. Baseline and endpoint anthropometric parameters were measured, and blood samples were drawn. Genomic DNA and RNA from adipose tissue biopsies were isolated before and after the dietary intervention. Leptin and TNF-alpha promoter methylation were measured by MSP after bisulfite treatment, and gene expression was also analyzed. Obese women with a successful weight loss (≥5% of initial body weight, n 65=6521) improved the lipid profile and fat mass percentage (6112%, p 65<650.05). Both systolic (615%, p 65<650.05) and diastolic (618%, p 65<650.01) blood pressures significantly decreased. At baseline, women with better response to the dietary intervention showed lower promoter methylation levels of leptin (6147%, p 65<650.05) and TNF-alpha (6139%, p 65=650.071) than the non-responder group ( n 65=656), while no differences were found between responder and non-responder group in leptin and TNF-alpha gene expression analysis. These data suggest that leptin and TNF-alpha methylation levels could be used as epigenetic biomarkers concerning the response to a low-calorie diet. Indeed, methylation profile could help to predict the susceptibility to weight loss as well as some comorbidities such as hypertension or type 2 diabetes.
URLPMID:28959056 [本文引用: 1]
Abstract The SNP variant rs2943650 near IRS1 gene locus was previously associated with decreased body fat and IRS1 gene expression as well as an adverse metabolic profile in humans. Here, we hypothesize that these effects may be mediated by an interplay with epigenetic alterations. We measured IRS1 promoter DNA methylation and mRNA expression in paired human subcutaneous and omental visceral adipose tissue samples (SAT and OVAT) from 146 and 41 individuals, respectively. Genotyping of rs2943650 was performed in all individuals (N65=65146). We observed a significantly higher IRS1 promoter DNA methylation in OVAT compared to SAT (N65=65146, P65=658.065×6510 -6 ), while expression levels show the opposite effect direction (N65=6541, P65=650.011). OVAT and SAT methylation correlated negatively with IRS1 gene expression in obese subjects (N65=6516, P65=650.007 and P65=650.010). The major T-allele is related to increased DNA methylation in OVAT (N65=65146, P65=650.019). Finally, DNA methylation and gene expression in OVAT correlated with anthropometric traits (waist- circumference waist-to-hip ratio) and parameters of glucose metabolism in obese individuals. Our data suggest that the association between rs2943650 near the IRS1 gene locus with clinically relevant variables may at least be modulated by changes in DNA methylation that translates into altered IRS1 gene expression.
URLPMID:28481699 [本文引用: 2]
Abstract The present study aimed to identify genes exhibiting concomitant obesity-dependent changes in DNA methylation and gene expression in adipose tissues in the mouse using diet-induced obese (DIO) C57BL/6J and genetically obese ob/ob mice as models. Mature adipocytes were isolated from epididymal and inguinal adipose tissues of ob/ob and DIO C57BL/6J mice. DNA methylation was analyzed by MeDIP-sequencing and gene expression by microarray analysis. The majority of differentially methylated regions (DMRs) were hypomethylated in obese mice. Global methylation of long interspersed elements indicated that hypomethylation did not reflect methyl donor deficiency. In both DIO and ob/ob mice, we observed more obesity-associated methylation changes in epididymal than in inguinal adipocytes. Assignment of DMRs to promoter, exon, intron and intergenic regions demonstrated that DIO-induced changes in DNA methylation in C57BL/6J mice occurred primarily in exons, whereas inguinal adipocytes of ob/ob mice exhibited a higher enrichment of DMRs in promoter regions than in other regions of the genome, suggesting an influence of leptin on DNA methylation in inguinal adipocytes. We observed altered methylation and expression of 9 genes in epididymal adipocytes, including the known obesity-associated genes, Ehd2 and Kctd15, and a novel candidate gene, Irf8, possibly involved in immune type 1/type2 balance. The use of 2 obesity models enabled us to dissociate changes associated with high fat feeding from those associated with obesity per se. This information will be of value in future studies on the mechanisms governing the development of obesity and changes in adipocyte function associated with obesity.
URL [本文引用: 1]
Homeobox A5(Hoxa5), a member of the Hox family, plays a important role in the regulation of proliferation and apoptosis in cancer cells. The dysregulation of the adipocyte apoptosis in vivo leads to obesity and metabolic disorders. However, the effects of Hoxa5 on adipocyte apoptosis are still unknown. In this study, palmitic acid (PA) significantly increased the mRNA level of Hoxa5 and... [Show full abstract]
URLPMID:23047069Magsci [本文引用: 1]
Infantile hemangioma is a benign vascular tumor that exhibits a unique yet predictable lifecycle of rapid proliferation followed by spontaneous regression. Recent studies have identified that insulin-like growth factor-2 (IGF2), a fetal mitogen, is highly expressed during the proliferative phase of hemangioma growth. Since hemangiomas arise from CD133 + stem cells, high levels of IGF2 may regulate the activity of the stem cells and therefore, hemangioma growth. The aim of this study was to understand the functional significance of elevated IGF2 in hemangiomas. We show that IGF2 localizes to the CD133 + cells in hemangioma specimens. We, therefore, hypothesized that IGF2 may be regulating the plasticity of hemangioma stem cells. To test our hypothesis, we used CD133-selected cells from hemangiomas to knockdown the expression of IGF2. We found that IGF2 is a mitogen for hemangioma stem cells and prevents leptin induction and full terminal differentiation of hemangioma stem cells into adipocytes. We also show that IGF2 does not alter the initial commitment phase. These findings implicate an important role of IGF2 in expanding hemangioma stem cells and preventing terminal adipocyte differentiation. (C) 2012 Elsevier Inc. All rights reserved.
URLPMID:23946348 [本文引用: 1]
http://jn.nutrition.org/cgi/doi/10.3945/jn.113.178038
URLPMID:19656312 [本文引用: 1]
Obesity is a heterogeneous disorder. Obese individuals vary in their body fat distribution, their metabolic profile and degree of associated cardiovascular and metabolic risk. Abdominal obesity carries greater risk of developing diabetes and future cardiovascular events than peripheral or gluteofemoral obesity. There are differences between adipose tissue present in subcutaneous areas (SCAT) and visceral adipose tissue (VAT) present in the abdominal cavity. These include anatomical, cellular, molecular, physiological, clinical and prognostic differences. Anatomically, VAT is present mainly in the mesentery and omentum, and drains directly through the portal circulaion to the liver. VAT compared with SCAT is more cellular, vascular, innervated and contains a larger number of inflammatory and immune cells, lesser preadipocyte differentiating capacity and a greater percentage of large adipocytes. There are more glucocorticoid and androgen receptors in VAT than in SCAT. VAT adipocytes are more metabolically active, more sensitive to lipolysis and more insulin-resistant than SCAT adipocytes. VAT has a greater capacity to generate free fatty acids and to uptake glucose than SCAT and is more sensitive to adrenergic stimulation, while SCAT is more avid in absorption of circulating free fatty acids and triglycerides. VAT carries a greater prediction of mortality than SCAT.
[本文引用: 1]
URLPMID:28776439 [本文引用: 1]
To investigate the value of visceral adipose tissue (VAT) and subcutaneous adipose tissue (SAT) in a cohort of a community's residents who were diagnosed as pre-diabetes, and to evaluate the association of VAT and SAT with insulin resistance. This study was based on cross-sectional analysis of data from 107 adults. VAT and SAT were assessed by computed tomography. Insulin resistance was defined by homeostasis model assessment of insulin resistance >2.69. The relationship of VAT and SAT with insulin resistance were examined by linear regression. Logistic regression was used to analyze the association of VAT and SAT with insulin resistance. Pre-diabetic subjects with insulin resistance had elevated levels of VAT. VAT was more strongly associated with insulin resistance than SAT in Chinese subjects with pre-diabetes.
URL [本文引用: 1]
Aims/Hypothesis Failure in glucose response to insulin is a common pathology associated with obesity. In this study, we analyzed the genome wide DNA methylation profile of visceral adipose tissue (VAT) samples in a population of individuals with obesity and assessed whether differential methylation profiles are associated with the presence of type 2 diabetes (T2D). Methods More than 485,000 CpG genome sites from VAT samples from women with obesity undergoing gastric bypass (n = 18), and classified as suffering from type 2 diabetes (T2D) or not (no type 2 diabetes, NT2D), were analyzed using DNA methylation arrays. Results We found significant differential methylation between T2D and NT2D samples in 24 CpGs that map with sixteen genes, one of which, HOOK2, demonstrated a significant correlation between differentially hypermethylated regions on the gene body and the presence of type 2 diabetes. This was validated by pyrosequencing in a population of 91 samples from both males and females with obesity. Furthermore, when these results were analyzed by gender, female T2D samples were found hypermethylated at the cg04657146-region and the cg 11738485-region of HOOK2 gene, whilst, interestingly, male samples were found hypomethylated in this latter region. Conclusion The differential methylation profile of the HOOK2 gene in individuals with T2D and obesity might be related to the attendant T2D, but further studies are required to identify the potential role of HOOK2 gene in T2D disease. The finding of gender differences in T2D methylation of HOOK2 also warrants further investigation.
URLPMID:27624926 [本文引用: 1]
Polarity protein complexes function during polarized cell migration and a subset of these proteins localizes to the reoriented centrosome during this process. Despite these observations, the mechanisms behind the recruitment of these polarity complexes such as the aPKC/PAR6α complex to the centrosome are not well understood. Here we identify Hook2 as an interactor for the aPKC/PAR6α complex that functions to localize this complex at the centrosome. We first demonstrate that Hook2 is essential for the polarized Golgi re-orientation towards the migration front. Depletion of Hook2 results in a decrease of PAR6α at the centrosome during cell migration, while overexpression of Hook2 in cells induced the formation of aggresomes with the recruitment of PAR6α, aPKC and PAR3. In addition, we demonstrate that the interaction between the C-terminal domain of Hook2 and the aPKC-binding domain of PAR6α localizes PAR6α to the centrosome during cell migration. Our data suggests that Hook2, a microtubule binding protein, plays an important role in the regulation of PAR6α recruitment to the centrosome to bridge microtubules and the aPKC/PAR complex. This data reveals how some of the polarity protein complexes are recruited to the centrosome and might regulate pericentriolar and microtubule organization and potentially impact on polarized migration.
URL [本文引用: 1]
We found that antigen processing and presentation pathway and immune-related genes were closely associated with gestational diabetes mellitus in the visceral omental adipose tissue of Chinese pregnant women, based on the integration analysis of expression and methylation profiles. These results may be valuable for the prognostic biomarkers and future therapeutic targets.
URLPMID:16636663 [本文引用: 1]
Abstract Signal transducer and activator of transcription 3 (STAT3), which mediates biological actions in many physiological processes, is activated by cytokines and growth factors, and has been reported to be constitutively activated in numerous cancer cells. In this study, we examined whether low molecular weight-dual specificity phosphatase two (LMW-DSP2) is involved in the regulation of the interleukin 6 (IL-6)/leukemia inhibitory factor (LIF)/STAT3-mediated signaling pathway. IL-6/LIF-induced LMW-DSP2 expression in murine testicular or hepatoma cell lines, while LMW-DSP2 overexpression in 293T cells suppressed IL-6-induced phosphorylation and activation of STAT3. Furthermore, LMW-DSP2 suppressed the expression of IL-6-induced endogenous genes. In contrast, small-interfering RNA-mediated reduction of LMW-DSP2 expression enhanced IL-6-induced STAT3-dependent transcription. In fact, LMW-DSP2 interacted with STAT3 in vivo and endogenous LMW-DSP2 bound to STAT3 in murine testicular GC-1 cells. These results strongly suggest that LMW-DSP2 acts as a negative regulator of the IL-6/LIF/STAT3-mediated signaling pathway.
Magsci [本文引用: 1]
OBJECTIVE-It has been suggested that interleukin (IL)-6 is one of the mediators linking obesity-derived chronic inflammation with insulin resistance through activation of STAT3, with subsequent upregulation of suppressor of cytokine signaling 3 (SOCS3). We evaluated whether peroxisome proliferator-activated receptor (PPAR)-beta/-delta prevented activation of the IL-6-STAT3-SOCS3 pathway and insulin resistance in adipocytes.<br/>RESEARCH DESIGN AND METHODS-Adipocytes and white adipose tissue from wild-type and PPAR-beta/-delta-null mice were used to evaluate the effect of PPAR-beta/-delta on the IL-6-STAT3-SOCS3 pathway.<br/>RESULTS-First, we observed that the PPAR-beta/-delta agonist GW501516 prevented both IL-6-dependent reduction in insulin-stimulated Akt phosphorylation and glucose uptake in adipocytes. In addition, this drug treatment abolished IL-6-induced SOCS3 expression in differentiated 3T3-L1 adipocytes. This effect was associated with the capacity of the drug to prevent IL-6-induced STAT3 phosphorylation on Tyr(705) and Ser(727) residues in vitro and in vivo. Moreover, GW501516 prevented IL-6-dependent induction of extracellular signal-related kinase (ERK)1/2, a serine-threonine-protein kinase involved in serine STAT3 phosphorylation. Furthermore, in white adipose tissue from PPAR-beta/-delta-null mice, STAT3 phosphorylation (Tyr(705) and Ser(727)), STAT3 DNA-binding activity, and SOCS3 protein levels were higher than in wild-type mice. Several steps in STAT3 activation require its association with heat shock protein 90 (Hsp90), which was prevented by GW501516 as revealed in irnmunoprecipitation studies. Consistent with this finding, the STAT3-Hsp90 association was enhanced in white adipose tissue from PPAR-beta/-delta-null mice compared with wildtype mice.<br/>CONCLUSIONS-Collectively, our findings indicate that PPAR-beta/-delta activation prevents IL-6-induced STAT3 activation by inhibiting ERK1/2 and preventing the STAT3-Hsp90 association, an effect that may contribute to the prevention of cytokine-induced insulin resistance in adipocytes. Diabetes 60:1990-1999, 2011
URLPMID:26831087 [本文引用: 1]
Abstract Tle1 (transducin-like enhancer of split 1) is a corepressor that interacts with a variety of DNA-binding transcription factors and has been implicated in many cellular functions; however, physiological studies are limited. Tle1-deficient (Tle1(Δ/Δ)) mice, although grossly normal at birth, exhibit skin defects, lung hypoplasia, severe runting, poor body condition, and early mortality. Tle1(Δ/Δ) mice display a chronic inflammatory phenotype with increased expression of inflammatory cytokines and chemokines in the skin, lung, and intestine and increased circulatory IL-6 and G-CSF, along with a hematopoietic shift toward granulocyte macrophage progenitor and myeloid cells. Tle1(Δ/Δ) macrophages produce increased inflammatory cytokines in response to Toll-like receptor (TLR) agonists and lipopolysaccharides (LPS), and Tle1(Δ/Δ) mice display an enhanced inflammatory response to ear skin 12-O-tetradecanoylphorbol-13-acetate treatment. Loss of Tle1 not only results in increased phosphorylation and activation of proinflammatory NF-κB but also results in decreased Hes1 (hairy and enhancer of split-1), a negative regulator of inflammation in macrophages. Furthermore, Tle1(Δ/Δ) mice exhibit accelerated growth of B6-F10 melanoma xenografts. Our work provides the first in vivo evidence, to our knowledge, that TLE1 is a major counterregulator of inflammation with potential roles in a variety of inflammatory diseases and in cancer progression.
URLPMID:25744306 [本文引用: 1]
The aim of the present study was to identify the genes differentially expressed in the visceral adipose tissue in a well-characterised mouse model of high-fat diet (HFD)-induced obesity. Male C57BL/6J mice (n20) were fed either HFD (189% of energy from fat) or low-fat diet (LFD, 42% of energy from fat) for 16 weeks. HFD-fed mice exhibited obesity, insulin resistance, dyslipidaemia and adipose collagen accumulation, along with higher levels of plasma leptin, resistin and plasminogen activator inhibitor type 1, although there were no significant differences in plasma cytokine levels. Energy intake was similar in the two diet groups owing to lower food intake in the HFD group; however, energy expenditure was also lower in the HFD group than in the LFD group. Microarray analysis revealed that genes related to lipolysis, fatty acid metabolism, mitochondrial energy transduction, oxidation-eduction, insulin sensitivity and skeletal system development were down-regulated in HFD-fed mice, and genes associated with extracellular matrix (ECM) components, ECM remodelling and inflammation were up-regulated. The top ten up- or down-regulated genes includeAcsm3,mt-Nd6,Fam13a,Cyp2e1,Rgs1andGpnmb, whose roles in the deterioration of obesity-associated adipose tissue are poorly understood. In conclusion, the genes identified here provide new therapeutic opportunities for prevention and treatment of diet-induced obesity.
URLPMID:24947352Magsci [本文引用: 1]
Abstract Upper- and lower-body fat depots exhibit opposing associations with obesity-related metabolic disease. We defined the relationship between DEXA-quantified fat depots and diabetes/cardiovascular risk factors in a healthy population-based cohort (n = 3,399). Gynoid fat mass correlated negatively with insulin resistance after total fat mass adjustment, whereas the opposite was seen for abdominal fat. Paired transcriptomic analysis of gluteal subcutaneous adipose tissue (GSAT) and abdominal subcutaneous adipose tissue (ASAT) was performed across the BMI spectrum (n = 49; 21.4-45.5 kg/m(2)). In both depots, energy-generating metabolic genes were negatively associated and inflammatory genes were positively associated with obesity. However, associations were significantly weaker in GSAT. At the systemic level, arteriovenous release of the proinflammatory cytokine interleukin-6 (n = 34) was lower from GSAT than ASAT. Isolated preadipocytes retained a depot-specific transcriptional "memory" of embryonic developmental genes and exhibited differential promoter DNA methylation of selected genes (HOTAIR, TBX5) between GSAT and ASAT. Short hairpin RNA-mediated silencing identified TBX5 as a regulator of preadipocyte proliferation and adipogenic differentiation in ASAT. In conclusion, intrinsic differences in the expression of developmental genes in regional adipocytes provide a mechanistic basis for diversity in adipose tissue (AT) function. The less inflammatory nature of lower-body AT offers insight into the opposing metabolic disease risk associations between upper- and lower-body obesity. 2014 by the American Diabetes Association. Readers may use this article as long as the work is properly cited, the use is educational and not for profit, and the work is not altered.
URLPMID:27477082 [本文引用: 1]
Elucidating the potential mechanisms involved in the detrimental effect of excess body weight on insulin action is an important priority in counteracting obesity-associated diseases. The present study aimed to disentangle the epigenetic basis of insulin resistance by performing a genome-wide epigenetic analysis in visceral adipose tissue (VAT) from morbidly obese patients depending on the insulin sensitivity evaluated by the clamp technique. The global human methylome screening performed in VAT from 7 insulin-resistant (IR) and 5 insulin-sensitive (IS) morbidly obese patients (discovery cohort) analyzed using the Infinium HumanMethylation450 BeadChip array identified 982 CpG sites able to perfectly separate the IR and IS samples. The identified sites represented 538 unique genes, 10% of which were diabetes-associated genes. The current work identified novel IR-related genes epigenetically regulated in VAT, such asCOL9A1, COL11A2, CD44, MUC4, ADAM2, IGF2BP1, GATA4, TET1, ZNF714, ADCY9, TBX5, andHDACM. The gene with the largest methylation fold-change and mapped by 5 differentially methylated CpG sites located in island/shore and promoter region wasZNF714. This gene presented lower methylation levels in IR than in IS patients in association with increased transcription levels, as further reflected in a validation cohort (n=24; 11 IR and 13 IS). This study reveals, for the first time, a potential epigenetic regulation involved in the dysregulation of VAT that could predispose patients to insulin resistance and future type 2 diabetes in morbid obesity, providing a potential therapeutic target and biomarkers for counteracting this process.
URLPMID:20552250Magsci [本文引用: 1]
We have developed a method for reconstructing gene association networks and have applied this method to gene profiles from 3T3-L1 cells. Priorization of the candidate genes pinpointed a transcript annotated as APMAP (adipocyte plasma membrane-associated protein). Functional studies showed that APMAP is upregulated in murine and human adipogenic cell models as well as in a genetic mouse model of obesity. Silencing APMAP in 3T3-L1 cells strongly impaired the differentiation into adipocytes. Moreover, APMAP expression was strongly induced by the PPAR ligand rosiglitazone in adipocytes in vitro and in vivo in adipose tissue. Using ChIP-qPCR and luciferase reporter assays, we show a functional PPAR binding site. In addition, we provide evidence that the extracellular C-terminal domain of APMAP is required for the function of APMAP in adipocyte differentiation. Finally, we demonstrate that APMAP translocates from the endoplasmatic reticulum to the plasma membrane during adipocyte differentiation.
URLPMID:18762715 [本文引用: 1]
The mechanism of glycerol transport by human aquaporin 9 (hAQP9), which is a liver-specific AQP water channel and can also transport glycerol, was investigated by using the Xenopus laevis oocyte expression system. It was found that specific glycerol uptake by hAQP9 was concentration-dependent (saturable) at 25C, conforming to the Michaelis-Menten kinetics with the maximum transport rate (Jmax) of 0.84 pmol/min/oocyte and the Michaelis constant (Km) of 9.2 M, and temperature-dependent, being reduced by about 70% when temperature was lowered from 25C to 4C. Such dependences on concentration and temperature are characteristic of a carrier-mediated type of mechanism rather than a channel type, which is expected not to depend on them. Furthermore, several glycerol-related compounds, such as monoacetin, were found to specifically inhibit hAQP9-mediated glycerol uptake, indicating a possibility of competition with glycerol. hAQP9-mediated glycerol uptake was, however, found not to require Na+. All these results suggest that hAQP9 functions as a facilitative carrier for glycerol, although it had been believed to function as a channel. Findings in the present study provide novel insight into its glycerol-transporting mechanism and would help exploring a possibility that hAQP9 inhibitors might help lower blood glucose level by reducing gluconeo-genesis by limiting hepatic glycerol uptake.
URLPMID:21502813Magsci [本文引用: 1]
Aquaglyceroporins (AQP3, AQP7, AQP9 and AQP10) encompass a subfamily of aquaporins that allow the movement of water and other small solutes, especially glycerol, through cell membranes. Adipose tissue constitutes a major source of glycerol via AQP7. We have recently reported that, in addition to the well-known expression of AQP7 in adipose tissue, AQP3 and AQP9 are also expressed in omental and subcutaneous fat depots. Moreover, insulin and leptin act as regulators of aquaglyceroporins through the PI3K/Akt/mTOR pathway. AQP3 and AQP7 appear to facilitate glycerol efflux from adipose tissue while reducing the glycerol influx into hepatocytes via AQP9 to prevent the excessive lipid accumulation and the subsequent aggravation of hyperglycemia in human obesity. This Extra View focuses on the control of glycerol release by aquaglyceroporins in the adipose tissue and briefly discusses the importance of glycerol as a substrate for hepatic gluconeogenesis, pancreatic insulin secretion and cardiac ATP production.
[本文引用: 1]
URLPMID:28266632 [本文引用: 1]
Abstract Epigenetic modifications alter transcriptional activity and contribute to the effects of environment on the individual risk of obesity and Type 2 Diabetes (T2D). Here, we have estimated the in vivo effect of a fat-enriched diet (HFD) on the expression and the epigenetic regulation of the Ankyrin repeat domain 26 (Ankrd26) gene, which is associated with the onset of these disorders. In visceral adipose tissue (VAT), HFD exposure determined a specific hyper-methylation of Ankrd26 promoter at the -436 and -431 bp CpG sites (CpGs) and impaired its expression. Methylation of these 2 CpGs impaired binding of the histone acetyltransferase/transcriptional coactivator p300 to this same region, causing hypo-acetylation of histone H4 at the Ankrd26 promoter and loss of binding of RNA Pol II at the Ankrd26 Transcription Start Site (TSS). In addition, HFD increased binding of DNA methyl-transferases (DNMTs) 3a and 3b and methyl-CpG-binding domain protein 2 (MBD2) to the Ankrd26 promoter. More importantly, Ankrd26 down-regulation enhanced secretion of pro-inflammatory mediators by 3T3-L1 adipocytes as well as in human sera. Thus, in mice, the exposure to HFD induces epigenetic silencing of the Ankrd26 gene, which contributes to the adipose tissue inflammatory secretion profile induced by high-fat regimens.
URL [本文引用: 1]
Epigenetic marks, and especially DNA methylation, are becoming an important factor in obesity, which could help to explain its etiology and associated comorbidities. Adipose tissue, now considered as an important endocrine organ, produces complement system factors. Complement component 3 (C3) turns out to be an important protein in metabolic disorders, via either inflammation or the C3 subproduct acylation stimulating protein (ASP) which directly stimulates lipid storage. In this study, we analyze C3 DNA methylation in adipose tissue from subjects with a different grade of obesity. Adipose tissue samples were collected from subjects with a different degree of obesity determined by their body mass index (BMI) as: Overweight subjects (BMI ≥ 25 and <30), obese class 1/2 subjects (BMI ≥ 30 and <40) and obese class 3 subjects (BMI ≥ 40). C3 DNA methylation was measured for 7 CpGs by pyrosequencition using the Pyromark technology (Qiagen, Madrid Spain). C3 messenger RNA (mRNA) levels were analyzed by pre-designed Taqman assays (Applied biosystems, Foster City, CA, USA) and ASP/C3a was measured using a ELISA kit. The data were analyzed using the statistic package SPSS. C3 DNA methylation levels were lower in the morbid obese group. Accordingly, C3 methylation correlated negatively with BMI and leptin. However, C3 mRNA levels were more associated with insulin resistance, and positive correlations with insulin, glucose and homeostasis model assessment-estimated insulin resistance (HOMA-IR) existed. ASP correlated negatively with high density lipoprotein (HDL) cholesterol. C3 methylation levels were associated to adiposity variables, such as BMI and leptin, while the C3 mRNA levels were associated to glucose metabolism.
URLPMID:23209378 [本文引用: 1]
While there has been significant progress in determining the transcriptional cascade involved in terminal adipocyte differentiation, less is known about early events leading to lineage commitment and cell fate choice. It has been recently discovered that zinc finger protein 423 (Zfp423) is an early actor in adipose determination. Here, we show that a close paralog of Zfp423, Zfp521, acts as a key regulator of adipose commitment and differentiation in vitro and in vivo. Zfp521 exerts its actions by binding to early B cell factor 1 (Ebf1), a transcription factor required for the generation of adipocyte progenitors, and inhibiting the expression of Zfp423. Overexpression of Zfp521 in cells greatly inhibits adipogenic potential, whereas RNAi-mediated knock-down or genetic ablation of Zfp521 enhances differentiation. In addition, \(Zfp521^{\) embryos exhibit increased mass of interscapular brown adipose tissue and subcutaneous white adipocytes, a cell autonomous effect. Finally, Ebf1 participates in a negative feedback loop to repress Zfp521 as differentiation proceeds. Because Zfp521 is known to promote bone development, our results suggest that it acts as a critical switch in the commitment decision between the adipogenic and osteogenic lineages.
URLPMID:20729114 [本文引用: 1]
Experimental studies have demonstrated that dietary macronutrient distribution plays an important role in insulin regulation, a risk factor associated to obesity, diabetes and other metabolic disorders. To assess whether the macronutrient composition of the diet could be related to obesity onset by affecting the epigenetic regulation of gene expression, we investigated in rats the metabolic effects of two pair-fed isocaloric diets: control (rich in carbohydrates) and high fat diet (rich in fat; HFD). Compared to controls, HFD induced higher weight gain and adiposity and impaired glucose tolerance, which was accompanied by a slight increase in adiponectin levels and liver steatosis. Epididymal adipose tissue expression of the fatty acid synthase (FASN) gene and NADH dehydrogenase (ubiquinone) 1 subcomplex 6 (NDUFB6) were significantly reduced in HFD group. These variations in mRNA levels were accompanied by changes in the methylation patterns of several CpG islands located in the promoter region of these genes. However, no correlations were found between gene expression and the methylation status. These results suggest that high fat intake produces overweighted rats independently of total energy intake. These diets could also induce some epigenetic changes in the promoters of key genes that could influence gene expression and may be behind metabolic alterations.
[本文引用: 1]
URLPMID:27692461 [本文引用: 1]
Recent progress in our understanding of thermogenic adipose development and function has suggested a role for epigenetic mechanisms in the regulation of the thermogenic adipose program and browning of white adipose tissue. Characterizing the establishment of differential epigenetic signatures between adipose cell types may allow us to better understand thermogenic cell-fate determination and differentiation. Environmental stimuli that are known to affect brown and beige adipocyte development and function have been shown to also alter epigenetic effector activity and thus the chromatin landscape and gene regulation. A bidirectional interplay exists between metabolism and chromatin remodeling through metabolites, with epigenetic effectors playing a role in the integrative control of gene regulation for energy homeostasis.
URLPMID:24703692Magsci [本文引用: 1]
Harms etal. show that Prdm16, which can drive brown fat (BAT) differentiation, is required for BAT maintenance in adult and aged mice but dispensable for BAT development. Prdm16 suppresses white-fat-specific gene expression, and mice lacking BAT Prdm16 have reduced thermogenic capacity, though they are not prone to weight gain.
URLPMID:2754867 [本文引用: 1]
Brown adipose cells are specialized to dissipate chemical energy in the form of heat, as a physiological defense against cold and obesity1. PRDM16 (PRD1-BF1-RIZ1 homologous domain containing 16) is a 140 kDa zinc finger protein that robustly induces brown fat determination and differentiation2. Recent data suggests that brown fat cells arisein vivofrom amyf5-positive, myoblastic lineage through the action of PRDM163; however, the molecular mechanisms responsible for this developmental switch is unclear. Here we show that PRDM16 forms a transcriptional complex with the active form of C/EBP (LAP), serving as a critical molecular unit that controls the cell fate switch from myoblastic precursors to brown fat cells. Forced expression of PRDM16 and C/EBP is sufficient to induce a fully functional brown fat program in na茂ve fibroblastic cells, including skin fibroblasts from mouse and man. Transplantation of fibroblasts expressing these two factors into mice gives rise to an ectopic fat pad with the morphological and biochemical characteristics of brown fat. As with endogenous brown fat, this synthetic brown fat tissue serves as a sink for glucose uptake, as determined by18FDG-PET scanning. These data indicate that the PRDM16-C/EBP complex initiates brown fat development from myoblastic precursors, and may provide opportunities for the development of novel therapeutics for obesity and type-2 diabetes.
[本文引用: 1]
URLPMID:27641099 [本文引用: 1]
Yang et02al. show that AMPK is essential for brown adipose tissue development through the elevation of α-ketoglutarate, which facilitates TET-mediated DNA demethylation of thePrdm16promoter, committing progenitor cells to brown adipogenic differentiation. Postnatal AMPK activation by metformin rescues the obesity-induced attenuation of brown adipogenesis.
[本文引用: 1]
URLPMID:24622795Magsci [本文引用: 1]
The early environment, acting via epigenetic processes, is associated with differential risk of cardiometabolic disease (), which can be predicted by epigenetic marks in proxy tissues. However, such measurements at time points disparate from the health outcome or the environmental exposure may be confounded by intervening stochastic and environmental variation. To address this, we analyzed in the proliferator-activated receptor coactivator 1 promoter in blood from 40 children (20 boys) collected annually between 5 and 14 years of age by pyrosequencing. Body composition was measured annually by dual X-ray absorptiometry, physical activity by accelerometry, and pubertal timing by age at peak high velocity. The effect of methylation on was investigated by electrophoretic mobility shift assays. Seven (CpG) loci were identified that showed no significant temporal change or association with leukocyte populations. Modeling using generalized estimating equations showed that methylation of four loci predicted up to 14 years independent of sex, age, pubertal timing, and activity. Methylation of one predictive locus modified of the proadipogenic homeobox-1/homeobox 9 complex. These findings suggest that temporally stable CpG loci measured in childhood may have utility in predicting risk.
Magsci [本文引用: 1]
Mammalian genomes are punctuated by DNA sequences containing an atypically high frequency of CpG sites (CpG islands; CGIs) that are associated with the majority of annotated gene promoters. Methylated C bases of CpG sites inhibit the expression of downstream genes. During the differentiation of 3T3-L1 preadipocytes, the CCAAT/enhancer-binding protein (C/EBP) beta gene plays an important role. We studied the CpG island methylation status of the C/EBP beta promoter and its relationship with the GATA-2 protein. We used computer analysis to determine that the C/EBP beta promoter sequence is rich in CGIs, and observed that two of seven methylated C bases were demethylated during the preadipocyte differentiation using bisulfite sequencing PCR (BSP). This corresponded with the onset of notable C/EBP beta gene expression. Immunofluorescence and molecular docking showed that the GATA-2 protein binds the C/EBP beta promoter in front of the first demethylated CpG site. We also found that expression of GATA-2 and C/EBP beta proteins is negatively correlated. These results indicate that the methylated C bases in the C/EBP beta promoter relate to expression of the C/EBP beta gene, and that its demethylation is linked with GATA-2 protein association.
URLPMID:286056685722623238845546Magsci [本文引用: 1]
Increasing the expression of the brown adipose tissue-specific gene uncoupling protein-1 (Ucp1) is a potential target for treating obesity. We investigated the role of DNA methylation and histone modification inUcp1expression in adipose cell lines and ex vivo murine adipose tissues. Methylation state of theUcp1enhancer was studied using bisulphite mapping in murine adipose cell lines, and tissue taken from cold-stressed mice, coupled with functional assays of the effects of methylation and demethylation of theUcp1promoter on gene expression and nuclear protein binding. We show that demethylation of theUcp1promoter by 5-aza-deoxycytidine increasesUcp1expression while methylation ofUcp1promoter-eporter constructs decreases expression. Brown adipose tissue-specificUcp1expression is associated with decreased CpG dinucleotide methylation of theUcp1enhancer. The lowest CpG dinucleotide methylation state was found in two cyclic AMP response elements (CRE3, CRE2) in theUcp1promoter and methylation of the CpG in CRE2, but not CRE3 decreased nuclear protein binding. Chromatin immunoprecipitation assays revealed the presence of the silencing DiMethH3K9 modification on theUcp1enhancer in white adipose tissue and the appearance of the active TriMethH3K4 mark at theUcp1promoter in brown adipose tissue in response to a cold environment. The results demonstrate that CpG dinucleotide methylation of theUcp1enhancer exhibits tissue-specific patterns in murine tissue and cell lines and suggest that adipose tissue-specificUcp1expression involves demethylation of CpG dinucleotides found in regulatory CREs in theUcp1enhancer, as well as modification of histone tails. The online version of this article (doi:10.1007/s00125-010-1701-4) contains supplementary material, which is available to authorised users.
URLPMID:20211485 [本文引用: 1]
DNA promoter methylation is an epigenetic phenomenon for long-term gene silencing during tumorigenesis. The purpose of this study is to identify novel hypermethylated loci associated with clinicopathologic variables in endometrioid endometrial carcinomas. To find hypermethylated promoter loci, we used differential methylation hybridization coupling with microarray and further validated by combined bisulfite restriction analysis and MassARRAY assay. Methylation levels of candidate loci were corrected with clinicopathologic factors of endometrial carcinomas. Increased promoter methylation of CIDE, HAAO and RXFP3 was detected in endometrial carcinomas compared with adjacent normal tissues, and was associated with decreased gene expression of all three genes. In a clinical cohort, promoter hypermethylation on CIDEA, HAAO and RXFP3 was detected in 85, 63 and 71% of endometrial carcinomas, respectively ( n = 118, P < 0.001) compared with uninvolved normal endometrium. Methylation status of CIDEA, HAAO and RXFP3 had significant association with microsatellite instability in tumors ( P < 0.001). Furthermore, methylation levels of HAAO were further found to relate to disease-free survivals ( P = 0.034). Hypermethylation of CIDEA, HAAO and RXFP3 promoter regions appears to be a frequent event in endometrial carcinomas. Hypermethylation at these loci is strongly associated with microsatellite instability status. Moreover, HAAO methylation predicts disease-free survival in this cohort of patients with endometrioid endometrial cancer.

{kind=link}
{kind=link}