 ,1, 李国玲1, 莫健新1, 钟翠丽1, 李紫聪1, 顾婷1, 郑恩琴1, 刘德武1, 蔡更元1,2, 吴珍芳1,2, 张献伟,1,2
,1, 李国玲1, 莫健新1, 钟翠丽1, 李紫聪1, 顾婷1, 郑恩琴1, 刘德武1, 蔡更元1,2, 吴珍芳1,2, 张献伟,1,2Effects of RNA interference on the porcine NHEJ pathway repair factors on HR efficiency
Rong Quan,1, Guoling Li1, Jianxin Mo1, Cuili Zhong1, Zicong Li1, Ting Gu1, Enqin Zheng1, Dewu Liu1, Gengyuan Cai1,2, Zhenfang Wu1,2, Xianwei Zhang,1,2通讯作者:
编委: 张博
收稿日期:2018-04-20修回日期:2018-06-6网络出版日期:2018-09-20
| 基金资助: |
Editorial board:
Received:2018-04-20Revised:2018-06-6Online:2018-09-20
| Fund supported: |
作者简介 About authors
全绒,硕士研究生,专业方向:遗传育种E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (910KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
全绒, 李国玲, 莫健新, 钟翠丽, 李紫聪, 顾婷, 郑恩琴, 刘德武, 蔡更元, 吴珍芳, 张献伟. RNA干扰猪NHEJ通路修复因子对HR效率的影响[J]. 遗传, 2018, 40(9): 749-757 doi:10.16288/j.yczz.18-016
Rong Quan, Guoling Li, Jianxin Mo, Cuili Zhong, Zicong Li, Ting Gu, Enqin Zheng, Dewu Liu, Gengyuan Cai, Zhenfang Wu, Xianwei Zhang.
传统基因操作技术(如显微注射、转座子、慢病毒转染等)将目的基因插入基因组内的整合方式是随机的。这种随机的整合方式对后期转基因动物育种带来诸多问题,如需要多代配种繁育才能获得纯合转基因后代[1]。此外,基因治疗方面也存在未知或潜在的风险,如激活原癌基因,诱发癌症等[2]。而基因组编辑技术是一项旨在对生物基因组进行定点修饰的新技术,可以高效实现对特定基因敲除与敲入[3,4,5],其中定点敲入(定点整合)技术依赖基因组靶位点产生的DNA双链断裂(double-strand break, DSB)后,通过DNA修复机制将外源基因及其调控区整合至靶位点。定点整合效率主要取决于两个方面:一是靶位点产生DSB的效率;二是靶位点DSB与携带同源臂供体质粒(donor plasmid)发生同源重组(homologous repair, HR)的效率,同源重组机制包括同源重组定向修复(homology-directed repair, HDR)和单链退火修复(single strand annealing, SSA)两种方式,其中HDR是基因组定点整合最为依赖的修复机制。虽然锌指核酸酶(zinc finger nucleases, ZFN)、转录激活样效应因子核酸酶(transcription activator- like effector nucleases, TALEN)和规律成簇间隔短回文重复序列(clustered regularly interspaced short palidromic repeats/CRISPR-associated 9, CRISPR/Cas9)等基因组编辑技术可在基因组内高效引入DSB[3,4,5],但细胞利用HR介导的基因定点整合效率仍然十分低下(只有大约0.5%~20%),而与之竞争的非同源末端连接(nonhomologous end joining, NHEJ)发生效率却高达80%[6]。虽然有研究表明通过NHEJ的方式也能够高效实现基因组的定点整合[7,8,9],但由于NHEJ是强行将2个DNA断端连接在一起,修复过程中容易产生核苷酸片段的随机插入、缺失或突变等,对后期的研究非常不利,而利用同源重组修复能实现基因或调控序列的准确插入、缺失或突变[10, 11]。因此,如何提高HR效率对转基因经济动物品系组建和品种培育具有重要科学意义和应用价值。
经典NHEJ (classical-NHEJ, C-NHEJ) 修复通路是哺乳动物细胞最重要的修复路径[12],其过程为Ku70/Ku80蛋白识别DSB[13, 14],随后将DNA-PKcs和Artemis等修复因子招募到DSB,Ku70/80与DNA-PKcs结合形成DNA-PK复合体[15, 16],经磷酸化后与Artemis结合,产生核酸外切酶的活性,切除DSB中5°和3°的粘性末端[17, 18];接着X射线修复交叉互补蛋白4 (X-ray repair cross complementing protein 4, XRCC4)、DNA连接酶IV(DNA-ligase IV, LIG4)和XRCC4类似因子(XRCC4-like fators, XLF/ NHEJ1)结合形成XRCC4-XLF-Ligase IV复合体修复DSB[19, 20];同时,DNA 5°端募集PNKP,添加磷酸基团,以利于DNA的修复反应[21]。由于细胞内NHEJ的修复效率远大于HR的效率,可通过与HR竞争DSB位点,抑制HR效率。因而抑制NHEJ通路的关键因子是提高HR效率的重要手段。Bertolini等[22]通过RNAi敲减Ku70和Xrcc4在HCT116细胞的表达,显著降低了基因随机整合的效率,将约200 bp的插入片段的定点整合效率提高了3~4倍。Basu等[23]通过显微注射LIG4的siRNA显著增加早期胚胎中报告质粒的HDR修复效率。Chu等[24]通过转染表达短发夹RNA (short hairpin RNA, shRNA)质粒干扰人或小鼠细胞的Ku70/80的表达,使1.1 kb的插入序列定点整合效率提高4~5倍。此外,有研究报道通过小分子化合物抑制LIG4的活性,可一定程度上提高同源重组修复效率[6, 23~25]。如Maruyama等[6]使用Scr7处理哺乳动物细胞,抑制LIG4的活性,将小片段整合效率提高了19倍,而注射Scr7至小鼠受精卵使定点整合效率提高2倍以上;Chu等[24]在HEK293 细胞和NIH3T3 细胞通过Scr7和shRNA抑制LIG4活性和表达,将整合效率提高2~4倍;同时也发现使用Cas9系统共表达的腺病毒基因E1B55K和E4orf6也可将小片段整合效率提高8倍;但Zhang等[26]却发现SCR7并不能提高人类细胞的HDR效率,因此通过RNA干扰LIG4能否提高猪HR效率值得深入研究。此外,根据前人的研究[26, 27]和本实验室研究结果[28, 29]表明,以质粒为同源重组的报告系统与整合至基因组上的报告系统结果是一致的。因此,本研究利用RNAi技术敲减PFFs内NHEJ通路关键因子PNKP、LIG4、NHEJ1的表达,旨在建立一套快速且适于用其他动物细胞的检测HR效率的方法,为获得定点基因修饰动物模型制备奠定基础。
1 材料和方法
1.1 材料
报告载体SSA-GFP reporter、HDR-GFP system、ssODN- GFP system构建方法参考Li等[29];PFFs由广东温氏种猪科技有限公司提供。噻唑蓝(3-(4,5- dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide, MTT)购自美国Sigma公司;单链寡核苷酸ssODN长度设计为90nt,序列为:5°-ACATGAAGCAGCACGACTTCTTCAAGTCCGCCATGCCCGAAGGCTACGTCCAGGAGCGCACCATCTTCTTCAAGGACGACGGCAACTACA-3°,由深圳华大基因合成。1.2 siRNA Oligo合成
针对猪PNKP(NC_010448.4)、LIG4 (NC_010453.5)和NHEJ1 (NC_010457.5) 3个基因编码序列分别设计相应的siRNA,并由上海吉玛基因公司合成siRNA Oligo (表1)。阴性对照siRNA(negative control siRNA, NC siRNA)为与目的基因序列无同源性的通用阴性对照,由上海吉玛公司提供。Table 1
表1
表1 合成siRNA序列信息
Table 1
| 基因 | 编号 | 序列(5°→3°) | |
|---|---|---|---|
| 正向序列 | 反向序列 | ||
| LIG4 | siRNA-1 | GCUAAGCUUUACAUCGAAUTT | AUUCGAUGUAAAGCUUAGCTT |
| siRNA-2 | GGAAGCCCUUUCAUAGGAATT | UUCCUAUGAAAGGGCUUCCTT | |
| siRNA-3 | GCACUUUGCCCGGCAAUAUTT | AUAUUGCCGGGCAAAGUGCTT | |
| PNKP | siRNA-1 | CCUCGGGCGGGAUUUCCUUTT | AAGGAAAUCCCGCCCGAGGTT |
| siRNA-2 | GGAAGCCGGUGAUCGGCAUTT | AUGCCGAUCACCGGCUUCCTT | |
| siRNA-3 | GCUCCCAAUGCCCAAUAAATT | UUUAUUGGGCAUUGGGAGCTT | |
| NHEJ1 | siRNA-1 | GGCUACAGCUUUCUGAGAATT | UUCUCAGAAAGCUGUAGCCTT |
| siRNA-2 | GCCCGUUGUUGAAGGACAUTT | AUGUCCUUCAACAACGGGCTT | |
| siRNA-3 | GGUUGAAGACAGAGCCAUUTT | AAUGGCUCUGUCUUCAACCTT | |
新窗口打开|下载CSV
1.3 实时荧光定量PCR检测siRNA干扰效果
当复苏后的细胞汇合度达到50%~80% 时(6孔板),参考美国Thermo Fisher Scientific的RNA转染说明书Lipofectamine RNAiMAX Reagent protocol转染25 pmol siRNA至PFFs。转染后的细胞在5% CO2、37℃培养48 h后抽提总RNA,反转录后对cDNA进行实时荧光定量PCR检测LIG4、PNKP和NHEJ1的干扰效果,荧光定量PCR引物见表2。Table 2
表2
表2 实时荧光定量PCR引物序列信息
Table 2
| 基因 | 序列(5°→3°) | 产物片段 (bp) |
|---|---|---|
| LIG4 | F: GCCGCTATCGCAGACATTG | 251 |
| R: GCCATCATCTCACCATCAAGG | ||
| PNKP | F: GGACCGTGGCAGTGAAACAG | 221 |
| R: CTCTTCCTCCTCCTCGTGTGG | ||
| NHEJ1 | F: GCAGTGTTGGTGATGGAAGAC | 176 |
| R: TGGCTCCTCTGGCTGGTTC |
新窗口打开|下载CSV
1.4 细胞增殖活性检测
当复苏后的细胞汇合度达到90%时(10 cm板),用0.5%胰酶消化细胞,调整细胞密度为1000~10000个/100 μL至96孔板。24 h后转染干扰PNKP、LIG4和NHEJ1基因效果最好的的1 pmol siRNA。转染后的细胞培养48 h,使用MMT法测定细胞增殖活性:每孔加入20 μL MTT溶液(5 mg/mL),继续培养4 h。每孔加入150 μL二甲基亚砜,置摇床上低速振荡10 min。在酶联免疫检测仪OD560 nm处测量各孔的吸光值,计算细胞增殖活性。1.5 流式细胞术检测细胞周期分布
当复苏后的细胞汇合度达到50%~80%时(6孔板),转染25 pmol siRNA至PFFs培养48 h,消化细胞,弃上清,用约1 mL预冷PBS洗细胞两次。加入1 mL -20℃的70%乙醇,边加边振荡,于4℃固定过夜。过夜的细胞250 g离心5min,弃上清,收集细胞,以1 mL的PBS洗细胞一次,加入1 mL碘化丙啶染色液,充分振荡,4℃避光孵育30 min,用流式细胞仪进行检测。使用分析软件Modifit 5.0进行统计分析。1.6 流式细胞术检测HR效率
用筛选干扰效果最好的PNKP、LIG4和NHEJ1的siRNA分别转染PFFs 48 h后(6孔板),对细胞进行二次电转染,参考Basic Nucleofector Kit for Primary Mammalian Fibroblasts (Amaxa,德国)说明书,每孔分别转染4 μg HindⅢ限制性内切酶线性化的SSA-GFP reporter或8 μg的HDR-GFP system或ssODN-GFP system (4 μg BamHⅠ线性化的pCD3.1-deficient GFP质粒和4 μg BamHⅠ线性化的pMD-18T UNGFP质粒)。转染后的细胞培养48 h后,利用流式细胞术检测细胞荧光数的百分比,并抽提DNA,使用表3引物进行扩增和正向引物测序。Table 3
表3
表3 PCR引物序列信息
Table 3
| 名称 | 序列(5°→3°) | 产物片段 (bp) |
|---|---|---|
| SSA-GFP reporter | F: CCTACGGCAAGCTGACCCT | 571 |
| R: CATGTGATCGCGCTTCTCGTT | ||
| HDR-GFP system/ssODN-GFP system | F: CAAAATGTCGTAACAACTCCG | 1162 |
| R: CCCCAGAATAGAATGACACCT |
新窗口打开|下载CSV
2 结果与分析
2.1 RNAi对靶基因表达水平的影响
针对LIG4、PNKP和NHEJ1基因各设计3条靶向siRNA,转染PFFs,实时荧光定量PCR结果显示,所有siRNA都能够显著干扰对应基因的mRNA表达水平(P<0.05),其中对LIG4和NHEJ1干扰效果甚至达到100%,而对PNKP的干扰效果高达70% (图1)。初步筛选出干扰LIG4、PNKP和NHEJ1效果最好的siRNA,分别为LIG4- siRNA-3、NHEJ1-siRNA-2和PNKP-siRNA-2。图1
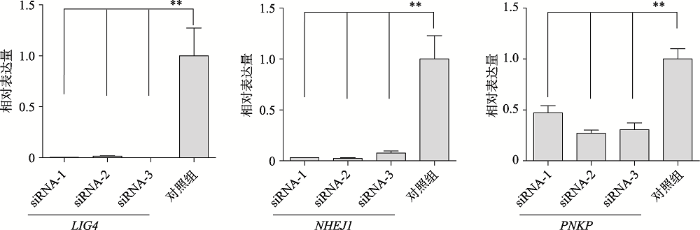 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1RNAi对基因表达水平的影响
siRNA-1,2,3为转染对应基因siRNA的编号,其中对照组为转染NC siRNA组(n=3)。**表示P<0.01。
Fig. 1Effects of RNAi on expression levels of corresponding target genes
2.2 RNAi对细胞周期和细胞增殖活性的影响
用干扰效果最好的LIG4-siRNA-3、NHEJ1- siRNA-2和PNKP-siRNA-2转染PFFs,碘化丙啶染色后利用流式细胞术检测细胞周期和MTT法检测细胞增殖活性,结果显示,LIG4-siRNA-3、NHEJ1- siRNA-2和PNKP- siRNA-2转染后,与空白组或对照组相比,细胞周期(图2A)和细胞增殖活性(图2B)均无显著影响(P>0.05)。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2RNAi对细胞周期和细胞增殖活性的影响
A:Modifit 5.0软件分析细胞周期分布(n=3);B:MTT法检测细胞活性分析结果(n=6)。LIG4、PNKP、NHEJ1组为转染效果对应最好的siRNA;对照为转染NC siRNA组;空白组为不转染组。
Fig. 2Effects of RNAi on the cell viability and cell cycle
2.3 RNAi对SSA效率的影响
用干扰效果最好的LIG4-siRNA-3、NHEJ1- siRNA-2和PNKP-siRNA-2转染PFFs后,电转染SSA-GFP reporter (图3A),测序结果显示,GFP表达框修复完整(图3B);而流式细胞术检测结果显示,PNKP-siRNA-2干扰PNKP基因表达后可显著提高SSA修复效率,为55.7% (P<0.05),而干扰LIG4和NHEJ1对SSA修复效率没有显著影响(P>0.05) (图3,C和D)。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3RNAi对SSA效率的影响
A:SSA修复的模式图。在单链退火修复下,同源的200 bp重复序列发生同源重组,GFP表达框修复完整,细胞可以检测到绿色荧光信号,而没有发生同源重组的质粒则被第一个重复序列最后的终止密码子终止表达;蓝色箭头表示引物设计位置。B:发生SSA修复前后GFP表达框的测序结果。红色框为同源重组部分序列;箭头为修复后的序列变化。C:流式细胞术检测的荧光信号的统计结果(n=3)。*表示P<0.05。D:流式细胞术检测细胞荧光信号图。对照组1为转染NC siRNA后转染SSA-GFP reporter组;对照组2为转染NC siRNA转染未线性化SSA-GFP reporter组;空白组为不转染组。
Fig. 3Effects of RNAi on SSA-mediated repair
2.4 RNAi对HDR修复效率的影响
干扰效果最好的LIG4-siRNA-3、NHEJ1-siRNA-2和PNKP-siRNA-2转染PFFs后,电转染HDR-GFP system。结果显示,PNKP-siRNA-2干扰PNKP基因和LIG4-siRNA-3干扰LIG4基因后可显著提高以双链为模板的HDR修复效率,分别为37.4%和37.5% (P<0.05),而干扰NHEJ1对HDR修复效率并没有影响(图4,A~C)。Yang等[30]研究发现,ssODN同源臂长度在30~50nt之间,对EGFP的修复效率随长度增加;长度在50~60nt之间,修复效率下降;长度在60~80nt之间,修复效率又随长度增加而增加;长度在80~90nt之间,修复效率基本相同。因此,本研究采用单链寡核苷酸ssODN长度为90 nt。结果显示电转染ssODN-GFP system后,PNKP-siRNA-2干扰PNKP基因和NHEJ1-siRNA-3干扰NHEJ1 基因后可显著提高以ssODN为模板的HDR修复 效率,分别为73.1% 和76.9% (P<0.05),而干扰 LIG4对HDR修复效率没有显著影响(P>0.05) (图4,D~F)。图4
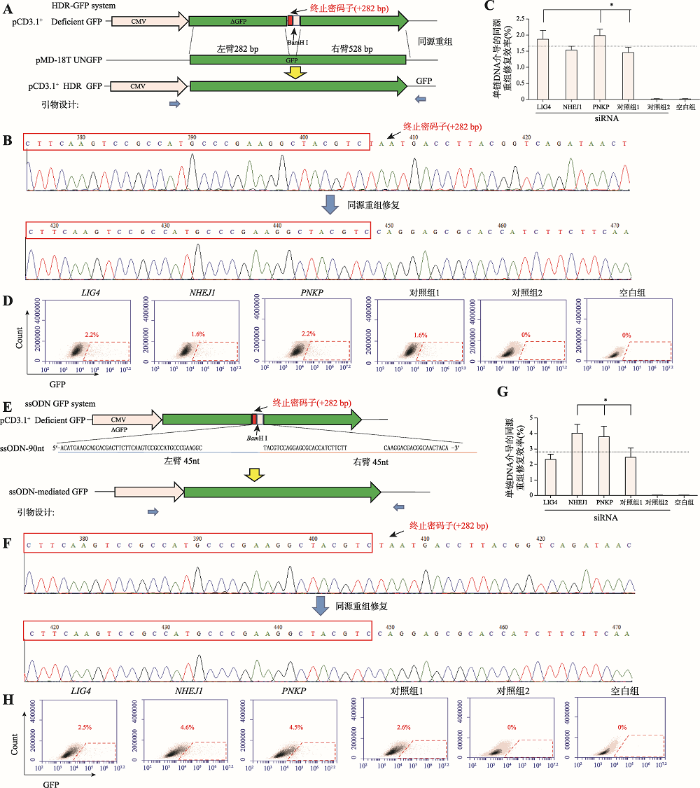 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4RNAi对HDR效率的影响
A和E:分别代表以双链DNA模板和ssODN模板的HDR修复模式图。有DSB的缺陷GFP质粒与另一个没有启动子的供体质粒(A)或左右各45nt,共90nt的ssODN (E)发生同源重组修复,GFP表达框修复完整,细胞可以检测到绿色荧光信号,而没有发生同源重组的质粒则没有荧光表达;蓝色箭头表示引物设计位置。B和F:分别为以双链DNA模板和ssODN模板修复前后的GFP表达框测序结果。红色框为同源重组5°端部分序列。C和G分别为以双链DNA模板和ssODN模板的流式细胞术检测的荧光信号统计结果(n=3)。*表示P<0.05。D和H:为对应流式细胞术检测细胞荧光信号图。对照组1为转染NC siRNA后转染HDR GFP system或ssODN GFP system组;对照组2为转染NC siRNA后仅转染pCD3.1-deficient GFP质粒组;空白组为不转染组。
Fig. 4Effects of RNAi on HDR-mediated repair efficiency
3 讨 论
RNAi技术的应用使得短暂抑制NHEJ修复的关键因子成为可能[31, 32]。以DNA依赖性蛋白激酶(DNA-dependent protein kinase, DNA-PK)为代表NHEJ修复通路则是哺乳动物细胞最重要的修复路径[12, 33, 34],在此过程中PNKP、NHEJ1和LIG4在起着至关重要的作用[19, 20]。本研究对NHEJ通路上的基因表达水平分析后发现,每个靶基因的siRNA都显著降低了相应基因的转录,特别是LIG4和NHEJ1的干扰效果甚至达到100%,而PNKP效果也可以达到70%。实验中PFFs的细胞状态,siRNA靶向位置及基因表达调控因子等因素对siRNA的干扰效果可能有一定的影响[35]。本研究采用的是Lipofectamine? RNAiMAX转染试剂,该试剂盒具有较高转染效率,基本达到对靶基因的预期抑制效果。前人研究显示,干扰LIG4,可显著提高HDR修复效率[6, 23, 24]。本研究以质粒为同源重组报告系统,也发现干扰LIG4能提高双链介导的HDR修复效率, 但并不能提高SSA介导HR效率和ssODN为模板介导的HDR修复效率。然而干扰NHEJ1基因后可显著提高以ssODN为模板的HDR修复效率,而对双链为模板的HDR修复方式却没有显著影响。导致LIG4与NHEJ1结果不一致的原因可能是两者在DNA修复过程中扮演角色的差异,其中LIG4主要负责将2个DNA断端连接在一起,而NHEJ1主要是与XRCC4和LIG4等构成复合体,起协助作 用[35, 36]。此外,虽然研究已经对NHEJ和HR通路进行一定的解析,但多以人细胞和小鼠为对象,对猪细胞内NHEJ和HR通路的研究鲜有报道[17, 19]。本研究以猪胎儿成纤维细胞为研究对象,由于实验对象的不同,干扰NHEJ修复因子对发生HDR修复的敏感性和依赖性会存在差异[37],从而导致细胞以不同的模板进行HDR修复。本研究还发现干扰PNKP可显著提高SSA和双链或单链介导的HDR修复效率。PNKP作为动物NHEJ修复通路的主要参与因子[21],通过干扰该基因的表达可以显著提高HDR修复效率,为进一步解析动物细胞内NHEJ和HR通路提供参考。
(责任编委: 张博)
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:28257857 [本文引用: 1]

AbstractThe development of transgenic technologies in the Cashmere goat (Capra hircus) has the potential to improve the quality of the meat and wool. The piggyBac (PB) transposon system is highly efficient and can be used to transpose specific target genes into the genome. Here, we developed a PB transposon system to produce transgenic Cashmere goat fetal fibroblasts (GFFs) with the enhanced green fluorescent protein (EGFP). We then used the genetically modified GFFs as nuclear donors to generate transgenic embryos by somatic cell nuclear transfer (SCNT). The embryos (n02=0240) were implanted into female goats (n02=0220). One transgenic kid that expressed EGFP throughout the surface features of its body was born. This result demonstrated the usefulness of PB transposon system in generating transgenic Cashmere goats.
URL [本文引用: 1]
URL [本文引用: 2]
URLPMID:24906146 [本文引用: 2]
Derived from a microbial defense system, Cas9 can be guided to specific locations within complex genomes by a short RNA. The development, applications, and future directions of the CRISPR-Cas9 system for genome engineering are discussed here.
URLPMID:25336735 [本文引用: 2]
Recent advances in the targeted modification of complex eukaryotic genomes have unlocked a new era of genome engineering. From the pioneering work using zinc-finger nucleases (ZFNs), to the advent of the versatile and specific TALEN systems, and most recently the highly accessible CRISPR/Cas9 systems, we now possess an unprecedented ability to analyze developmental processes using sophisticated designer genetic tools. In this Review, we summarize the common approaches and applications of these still-evolving tools as they are being used in the most popular model developmental systems. Excitingly, these robust and simple genomic engineering tools also promise to revolutionize developmental studies using less well established experimental organisms.
[本文引用: 4]
URL [本文引用: 1]
URLPMID:24179142 [本文引用: 1]
Sequence-specific nucleases like TALENs and the CRISPR/Cas9 system have greatly expanded the genome editing possibilities in model organisms such as zebrafish. Both systems have recently been used to create knock-out alleles with great efficiency, and TALENs have also been successfully employed in knock-in of DNA cassettes at defined loci via homologous recombination (HR). Here we report CRISPR/Cas9-mediated knock-in of DNA cassettes into the zebrafish genome at a very high rate by homology-independent double-strand break (DSB) repair pathways. After co-injection of a donor plasmid with a short guide RNA (sgRNA) and Cas9 nuclease mRNA, concurrent cleavage of donor plasmid DNA and the selected chromosomal integration site resulted in efficient targeted integration of donor DNA. We successfully employed this approach to convert eGFP into Gal4 transgenic lines, and the same plasmids and sgRNAs can be applied in any species where eGFP lines were generated as part of enhancer and gene trap screens. In addition, we show the possibility of easily targeting DNA integration at endogenous loci, thus greatly facilitating the creation of reporter and loss-of-function alleles. Due to its simplicity, flexibility, and very high efficiency, our method greatly expands the repertoire for genome editing in zebrafish and can be readily adapted to many other organisms.
URLPMID:26678082 [本文引用: 1]
Programmable nucleases enable engineering of the genome by utilizing endogenous DNA double-strand break (DSB) repair pathways. Although homologous recombination (HR)-mediated gene knock-in is well established, it cannot necessarily be applied in every cell type and organism because of variable HR frequencies. We recently reported an alternative method of gene knock-in, named the PITCh (Precise Integration into Target Chromosome) system, assisted by microhomology-mediated end-joining (MMEJ). MMEJ harnesses independent machinery from HR, and it requires an extremely short homologous sequence (5-25 bp) for DSB repair, resulting in precise gene knock-in with a more easily constructed donor vector. Here we describe a streamlined protocol for PITCh knock-in, including the design and construction of the PITCh vectors, and their delivery to either human cell lines by transfection or to frog embryos by microinjection. The construction of the PITCh vectors requires only a few days, and the entire process takes 651.5 months to establish knocked-in cells or 651 week from injection to early genotyping in frog embryos.
URL [本文引用: 1]
Gene targeting by homologous recombination in embryonic stem cells is extensively used to generate specific mouse mutants. However, most mammalian species lack tools for targeted gene manipulation. Since double-strand breaks strongly increase the rate of homologous recombination at genomic loci, we explored whether gene targeting can be directly performed in zygotes by the use of zinc-finger nucleases. Here we report that gene targeting is achieved in 1.7 4.5% of murine one-cell embryos upon the coinjection of targeting vectors with zinc-finger nucleases, without preselection. These findings enable the manipulation of the mammalian germ line in a single step in zygotes, independent of ES cells.
[本文引用: 1]
URL [本文引用: 2]
URLPMID:23624778 [本文引用: 1]
Although the role of BRCA1 and the homologous recombination (HR) pathway in breast cancer (BC) has been extensively studied, the alternative repair pathway for DNA double-strand breaks (DSBs), non-homologous end-joining (NHEJ) remains to be defined. Ku proteins bind to DNA DSB ends and play a key role in NHEJ. In this study we aimed to assess the expression and biological significance of the KU70/KU80 heterodimer in the different molecular classes of BC. The expression of KU70/KU80 was assessed immunohistochemically in a well-characterised and annotated series of 1302 unselected invasive BC cases with a long-term follow-up together with 25 cases with known BRCA1 mutations. The results were correlated with clinicopathological parameters, other DNA repair proteins and patient outcome. The expression of KU70/KU80 protein was further evaluated in various BC cell lines using western blotting and reverse-phase protein microarray (RPPA). Nuclear KU70/KU80 expression was correlated with features of poor prognosis including higher histological grade, lymphovascular invasion, negative oestrogen receptor expression, basal-like phenotype, P53 and CHK1 positivity. KU70/KU80 was expressed in all BRCA1-associated tumours and showed an inverse correlation with nuclear BRCA1 protein and aberrant cytoplasmic RAD51 expression. RPPA confirmed these results and showed higher expression of KU70/KU80 in BRCA1-deficient cell line compared to BRCA1-proficient cell line. KU70/KU80 expression showed an association with disease-free interval; however, it was not an independent predictor of outcome. As a conclusion, KU70/KU80 may play a role in DNA DSBs repair in HR-deficient tumours. Further study of other NHEJ markers in sporadic BC is warranted.
URL [本文引用: 1]
URLPMID:8422676 [本文引用: 1]
Abstract The DNA-dependent protein kinase (DNA-PK) phosphorylates Sp1 and several other nuclear proteins. Here, we show that Sp1 and the DNA-PK must be colocalized on the same DNA molecule for efficient phosphorylation to occur. Interestingly, we find that the DNA-PK binds to and is activated by the ends of DNA molecules. Furthermore, we show that the DNA binding properties of the DNA-PK are identical to those of Ku, a well-characterized human autoimmune antigen. We demonstrate that the DNA-PK can be fractionated into two components, one of which is Ku and the other of which is a polypeptide of approximately 350 kd. DNA cross-linking and coimmunoprecipitation studies indicate that the catalytic 350 kd DNA-PK component is directed to DNA by protein-protein interactions with Ku. The implications of the unusual DNA binding mode and multicomponent nature of the DNA-PK are discussed.
URL [本文引用: 1]
URLPMID:10761921 [本文引用: 2]
Cancer susceptibility genes have been classified into two groups: gatekeepers and caretakers. Gatekeepers are genes that control cell proliferation and death, whereas caretakers are DNA repair genes whose inactivation leads to genetic instability. Abrogation of both caretaker and gatekeeper function markedly increases cancer susceptibility. Although the importance of Ku80 in DNA double-strand break repair is well established, neither Ku80 nor other components of the non-homologous end-joining pathway are known to have a caretaker role in maintaining genomic stability. Here we show that mouse cells deficient for Ku80 display a marked increase in chromosomal aberrations, including breakage, translocations and aneuploidy. Despite the observed chromosome instabilities, Ku80-/- mice have only a slightly earlier onset of cancer. Loss of p53 synergizes with Ku80 to promote tumorigenesis such that all Ku80-/- p53-/- mice succumb to disseminated pro-B-cell lymphoma before three months of age. Tumours result from a specific set of chromosomal translocations and gene amplifications involving IgH and c-Myc, reminiscent of Burkitt's lymphoma. We conclude that Ku80 is a caretaker gene that maintains the integrity of the genome by a mechanism involving the suppression of chromosomal rearrangements.
URLPMID:20192759 [本文引用: 1]
Abstract Double-strand DNA breaks are common events in eukaryotic cells, and there are two major pathways for repairing them: homologous recombination (HR) and nonhomologous DNA end joining (NHEJ). The various causes of double-strand breaks (DSBs) result in a diverse chemistry of DNA ends that must be repaired. Across NHEJ evolution, the enzymes of the NHEJ pathway exhibit a remarkable degree of structural tolerance in the range of DNA end substrate configurations upon which they can act. In vertebrate cells, the nuclease, DNA polymerases, and ligase of NHEJ are the most mechanistically flexible and multifunctional enzymes in each of their classes. Unlike repair pathways for more defined lesions, NHEJ repair enzymes act iteratively, act in any order, and can function independently of one another at each of the two DNA ends being joined. NHEJ is critical not only for the repair of pathologic DSBs as in chromosomal translocations, but also for the repair of physiologic DSBs created during variable (diversity) joining [V(D)J] recombination and class switch recombination (CSR). Therefore, patients lacking normal NHEJ are not only sensitive to ionizing radiation (IR), but also severely immunodeficient.
URLPMID:5449630 [本文引用: 3]
Non-homologous end joining (NHEJ) repairs DNA double strand breaks in non-cycling eukaryotic cells. NHEJ relies on polynucleotide kinase/phosphatase (PNKP), which generates 508-phosphate/308-hydroxyl DNA termini that are critical for ligation by the NHEJ DNA ligase, LigIV. PNKP and LigIV require the NHEJ scaffolding protein, XRCC4. The PNKP FHA domain binds to the CK2-phosphorylated XRCC4 C-terminal tail, while LigIV uses its tandem BRCT repeats to bind the XRCC4 coiled-coil. Yet, the assembled PNKP-XRCC4–LigIV complex remains uncharacterized. Here, we report purification and characterization of a recombinant PNKP–XRCC4–LigIV complex. We show that the stable binding of PNKP in this complex requires XRCC4 phosphorylation and that only one PNKP protomer binds per XRCC4 dimer. Small angle X-ray scattering (SAXS) reveals a flexible multi-state complex that suggests that both the PNKP FHA and catalytic domains contact the XRCC4 coiled-coil and LigIV BRCT repeats. Hydrogen-deuterium exchange indicates protection of a surface on the PNKP phosphatase domain that may contact XRCC4–LigIV. A mutation on this surface (E326K) causes the hereditary neuro-developmental disorder, MCSZ. This mutation impairs PNKP recruitment to damaged DNA in human cells and provides a possible disease mechanism. Together, this work unveils multipoint contacts between PNKP and XRCC4–LigIV that regulate PNKP recruitment and activity within NHEJ.
URL [本文引用: 2]
URL [本文引用: 2]
URL [本文引用: 1]
[本文引用: 3]
URL [本文引用: 3]
URLPMID:5566437 [本文引用: 1]
CRISPR/Cas9 is an efficient customizable nuclease to generate double-strand breaks (DSBs) in the genome. This process results in knockout of the targeted gene or knock-in of a specific DNA fragment at the targeted locus in the genome of various species. However, efficiency of knock-in mediated by homology-directed repair (HDR) pathway is substantially lower compared with the efficiency of knockout mediated by the nonhomologous end-joining (NHEJ) pathway. Suppressing NHEJ pathway or enhancing HDR pathway has been proven to enhance the nuclease-mediated knock-in efficiency in cultured cells and model organisms. We here investigated the effect of small molecules, Scr7, L755507 and resveratrol, on promoting HDR efficiency in porcine fetal fibroblasts. Results from eGFP reporter assay showed that these small molecules could increase the HDR efficiency by 2 3-fold in porcine fetal fibroblasts. When transfecting with the homologous template DNA and CRISPR/Cas9 plasmid and treating with small molecules, the rate of knock-in porcine fetal fibroblast cell lines with large DNA fragment integration could reach more than 50% of the screened cell colonies, compared with 26.1% knock-in cell lines in the DMSO-treated group. The application of small molecules offers a beneficial approach to improve the frequency of precise genetic modifications in primary somatic cells.
URL [本文引用: 2]
URL [本文引用: 1]
URLPMID:26678082 [本文引用: 1]
Programmable nucleases enable engineering of the genome by utilizing endogenous DNA double-strand break (DSB) repair pathways. Although homologous recombination (HR)-mediated gene knock-in is well established, it cannot necessarily be applied in every cell type and organism because of variable HR frequencies. We recently reported an alternative method of gene knock-in, named the PITCh (Precise Integration into Target Chromosome) system, assisted by microhomology-mediated end-joining (MMEJ). MMEJ harnesses independent machinery from HR, and it requires an extremely short homologous sequence (5-25 bp) for DSB repair, resulting in precise gene knock-in with a more easily constructed donor vector. Here we describe a streamlined protocol for PITCh knock-in, including the design and construction of the PITCh vectors, and their delivery to either human cell lines by transfection or to frog embryos by microinjection. The construction of the PITCh vectors requires only a few days, and the entire process takes 651.5 months to establish knocked-in cells or 651 week from injection to early genotyping in frog embryos.
URL [本文引用: 2]
URL [本文引用: 1]
URL [本文引用: 1]
URLPMID:24002223 [本文引用: 1]
Single DNA lesions such as DNA double-strand breaks (DSBs) can cause cell death or trigger genome rearrangements that have oncogenic potential, and so the pathways that mend and signal DNA damage must be highly sensitive but, at the same time, selective and reversible. When initiated, boundaries must be set to restrict the DSB response to the site of the lesion. The integration of positive and, crucially, negative control points involving post-translational modifications such as phosphorylation, ubiquitylation and acetylation is key for building fast, effective responses to DNA damage and for mitigating the impact of DNA lesions on genome integrity.
[本文引用: 1]
[本文引用: 1]
.
URL [本文引用: 1]
CRISPR/Cas系统是细菌和古生菌中抵抗外源病毒或质粒入侵的获得性免疫系统,利用CRISPR RNAs(cr RNAs)引导Cas核酸酶沉默入侵的核酸。通过分子生物学改造使Ⅱ型CRISPR/Cas系统成为一种高效的基因组定点修饰技术,并且比锌指核酸酶(Zinc-finger nucleases,ZFNs)和TALE核酸酶(Transcription activator like effector nucleases,TALENs)结构更简单,更容易设计和应用。文章主要介绍了CRISPR/Cas9系统成为高效基因组定点修饰技术的发展历程、Ⅱ型CRISPR/Cas的工作原理和改造过程以及在动物基因组定点修饰的应用,剖析了该技术存在的问题和现有改进方案,并与成功案例相结合展望了CRISPR/Cas9系统的应用前景,以期为动物性状改良和人类疾病动物模型的创立提供新思路。
URL [本文引用: 1]
CRISPR/Cas系统是细菌和古生菌中抵抗外源病毒或质粒入侵的获得性免疫系统,利用CRISPR RNAs(cr RNAs)引导Cas核酸酶沉默入侵的核酸。通过分子生物学改造使Ⅱ型CRISPR/Cas系统成为一种高效的基因组定点修饰技术,并且比锌指核酸酶(Zinc-finger nucleases,ZFNs)和TALE核酸酶(Transcription activator like effector nucleases,TALENs)结构更简单,更容易设计和应用。文章主要介绍了CRISPR/Cas9系统成为高效基因组定点修饰技术的发展历程、Ⅱ型CRISPR/Cas的工作原理和改造过程以及在动物基因组定点修饰的应用,剖析了该技术存在的问题和现有改进方案,并与成功案例相结合展望了CRISPR/Cas9系统的应用前景,以期为动物性状改良和人类疾病动物模型的创立提供新思路。
URL [本文引用: 2]
URLPMID:28512351 [本文引用: 1]
DNA double-strand breaks (DSBs) are the most dangerous type of DNA damage because they can result in the loss of large chromosomal regions. In all mammalian cells, DSBs that occur throughout the cell cycle are repaired predominantly by the non-homologous DNA end joining (NHEJ) pathway. Defects in NHEJ result in sensitivity to ionizing radiation and the ablation of lymphocytes. The NHEJ pathway utilizes proteins that recognize, resect, polymerize and ligate the DNA ends in a flexible manner. This flexibility permits NHEJ to function on a wide range of DNA-end configurations, with the resulting repaired DNA junctions often containing mutations. In this Review, we discuss the most recent findings regarding the relative involvement of the different NHEJ proteins in the repair of various DNA-end configurations. We also discuss the shunting of DNA-end repair to the auxiliary pathways of alternative end joining (a-EJ) or single-strand annealing (SSA) and the relevance of these different pathways to human disease.
URLPMID:26817820 [本文引用: 1]
Zinc-finger nuclease, transcription activator-like effector nuclease and CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 (CRISPR-associated protein 9) are becoming major tools for genome editing. Importantly, knock-in in several non-rodent species has been finally achieved thanks to these customizable nucleases; yet the rates remain to be further improved. We hypothesize that inhibiting non-homologous end joining (NHEJ) or enhancing homology-directed repair (HDR) will improve the nuclease-mediated knock-in efficiency. Here we show that thein vitroapplication of an HDR enhancer, RS-1, increases the knock-in efficiency by two- to five-fold at different loci, whereas NHEJ inhibitor SCR7 has minimal effects. We then apply RS-1 for animal production and have achieved multifold improvement on the knock-in rates as well. Our work presents tools to nuclease-mediated knock-in animal production, and sheds light on improving gene-targeting efficiencies on pluripotent stem cells. CRISPR/Cas9 and transcription activator-like effector nuclease (TALEN) are becoming major tools for genome editing. Here, Songet al. show that RS-1, a small-molecule enhancer for homology directed repair, increases the CRISPR/Cas9 and TALEN mediated knock-in efficiency bothin vitroandin vivowith rabbit.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}