Identification and Development of Polymorphic Genic-SSRs in Tamarix ramosissima in Alxa Region Based on Transcriptome
Yanan Zhang1, Lei Huang1, Jiabin Li1, Lei Zhang2,3, Zhenhua Dang,1,*1Ministry of Education Key Laboratory of Ecology and Resource Use of the Mongolian Plateau/Inner Mongolia Key Laboratory of Grassland Ecology, School of Ecology and Environment, Inner Mongolia University, Hohhot 010021, China 2Inner Mongolia Academy of Forestry Sciences, Hohhot 010010, China 3Daqing Mountains Forest Ecosystem Research Station, Hohhot 010010, China
Abstract Simple sequence repeats located in gene transcribed regions (Genic-SSR) can play important roles in plant adaptation to environmental changes. In this study, the transcriptomes of Tamarix ramosissima from five different locations in Alax were sequenced, assembled, and compared. By using CandiSSR software, a total of 1 185 polymorphic Genic-SSRs representing 157 motif types were identified in 1 123 transcripts. Among them, the trinucleotide repeats (596, 50.30%) were the most abundant, followed by dinucleotide repeats (486, 41.01%). Location analysis showed that 411, 239, and 163 Genic-SSRs were located in CDSs, 5′UTRs, and 3′UTRs of the relevant transcripts, respectively; 78.47% of the trinucleotide SSRs were located in CDSs, and 94.07% of the dinucleotide SSRs were located in UTRs. Among SSRs distributed in CDSs, AGC/GCT, AGG/CCT, AAG/CTT, CCG/CGG, and ATC/GAT were relatively abundant, accounting for 64.48% of all the Genic-SSRs; AG/CT and AT/AT were the most abundant repeat types in UTRs, which together account for 55.22% of all the Genic-SSRs in UTRs. Functional annotation showed that polymorphic Genic-SSRs containing genes enriched in a wide range of GO terms and KEGG pathways that highly related to stress response in T. ramosissima. Of the 15 randomly selected Genic-SSRs, 14 were successfully amplified by using polymerase chain reaction technology and 64 alleles were found in these SSR loci. Genetic polymorphism estimation showed that the mean of expected and observed heterozygosity (He, Ho), polymorphism information content (PIC) of these SSRs were 0.553, 0.421, and 0.493, respectively, demonstrating the feasibility of developing SSR markers by RNA-seq. Keywords:Genic-SSR;polymorphism;RNA-seq;Tamarix ramosissima
PDF (2244KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文 引用本文 张雅楠, 黄蕾, 李佳彬, 张雷, 党振华. 基于转录组的阿拉善地区多枝柽柳多态Genic-SSRs的识别与开发. 植物学报, 2021, 56(4): 433-442 doi:10.11983/CBB20185 Zhang Yanan, Huang Lei, Li Jiabin, Zhang Lei, Dang Zhenhua. Identification and Development of Polymorphic Genic-SSRs in Tamarix ramosissima in Alxa Region Based on Transcriptome. Chinese Bulletin of Botany, 2021, 56(4): 433-442 doi:10.11983/CBB20185
评价物种的遗传多样性和识别适应过程中的遗传变异并鉴定其功能, 是适应性进化研究的2个重要方向(Lynch and Lande, 1993)。分子标记是遗传多样性分析的有效手段, 具有数量丰富、多态性高、直接以DNA的形式表现等优势, 比形态学标记、细胞学标记和化学标记的精准度更高。SSR (simple sequence repeat)又称微卫星DNA, 是由1-6个核苷酸串联重复而成的DNA序列, 广泛分布于真核生物基因组中, 是种群遗传学、目标基因定位及农作物种质资源等研究中常用的分子标记。研究表明, 位于基因内部的SSR (Genic-SSR或EST-SSR)变异对染色质组成、基因活性调节、DNA复制及重组、细胞周期调控以及错配修复等有重要影响, 被称为生物适应外界环境的分子进化装置, 在适应性进化过程中扮演重要角色(Trifonov, 1995; Li et al., 2002)。同时, Genic-SSR在遗传多样性评估、遗传作图及分子标记辅助育种研究中也同样有效(陈雨等, 2008; Wen et al., 2010)。可见, Genic-SSR能满足适应性进化研究的多方面需要, 极具开发潜力和研究价值。
Table 2 表2 表2测序与组装结果统计 Table 2Statistics of sequencing and assembly results
Sample
CR (No.)
Q30 (%)
GC (%)
Ug (No.)
ML (bp)
N50 (bp)
TR1
47893646
93.12
41.95
37790
1053.09
1953
TR2
43861774
93.06
41.96
35818
1083.87
1983
TR3
46194046
93.04
42.33
39252
995.67
1812
TR4
44817976
93.17
42.87
32175
1099.48
1899
TR5
44190698
92.92
42.49
34364
1082.91
1930
All-unigene
81728
823.46
1364
CR代表Clean Reads; Q30代表错误识别率小于0.1%的碱基数目; GC、Ug和ML分别代表组装所得Unigene的GC含量、数目和平均长度; N50代表将Unigene从长到短依次累加, 碱基数为Unigene总碱基数的50%时Unigene的长度。 CR represents Clean Reads; Q30 represents the Clean Reads that had Phred Quality Scores at the level of an error probability of less than 0.1%; GC, Ug and ML represent the GC content, number and the mean length of the assembled unigenes, respectively; N50 represents the length of unigene when the base number reaches 50% of the total base number of unigenes by adding the unigenes from long to short in turn.
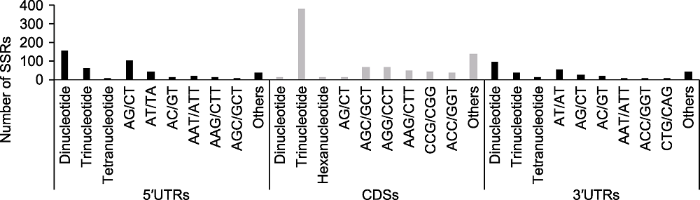
x轴表示多态性Genic-SSRs的分布和motif序列类型; y轴表示Genic-SSRs的数目。 Figure 1Distribution of the identified Genic-SSRs
The x-axis represents the distribution and motif types of polymorphic Genic-SSRs; The y-axis represents the number of Genic- SSRs.
2.4 含多态SSR序列的功能注释
将626个Genic-SSRs对应的582个Unigenes在GO数据库中比对后得到4 947个比对结果, 涵盖3个GO分类中的1 209个子类别。在位于基因5′UTRs的Genic- SSRs中, 最丰富的GO条目是nucleus、transcription、DNA-templated、transcription factor activity, sequence-specific DNA binding和regulation of transcription, DNA-templated。在位于基因CDSs的Genic- SSRs中, 最丰富的GO条目是nucleus, 其次是transcription、DNA-templated、regulation of transcription, DNA-templated和transcription factor activity, sequence-specific DNA binding。在位于基因3′UTRs的Genic-SSRs中, nucleus、integral component of membrane和transcription factor activity, sequence- specific DNA binding最为丰富(图2A)。
(A) GO分析, 3列气泡分别代表5′UTRs、CDSs和3′UTRs含有Genic-SSRs基因的GO富集分析结果, GO条目的基因数目由气泡大小表示; 每个基因区域显示基因数目≥10的GO条目; (B) KEGG富集分析, x轴表示5′UTRs、CDSs和3′UTRs含有Genic-SSRs基因富集的代谢通路; y轴表示富集在KEGG通路中的基因数目。 Figure 2GO and KEGG enrichment of the Genic-SSR-containing sequences
(A) GO analysis, the three columns of the bubbles represent GO enrichment analysis of transcripts that containing Genic-SSRs in the 5′UTRs, CDSs and 3′UTRs, respectively, and the number of unigenes assigned to each term is indicated by the size of each bubble; GO terms that contained unigenes more than or equal to ten in one of the gene regions are shown; (B) KEGG enrichment analysis, the x-axis indicates the enriched pathways assigned to the 5°UTRs, CDSs and 3°UTRs Genic-SSR-containing sequences; the y-axis represents the number of unigenes enriched in KEGG pathways.
KEGG注释显示, 105个含有Genic-SSR的基因序列可匹配到73个KEGG通路中(图2B)。在位于基因5′UTRs、CDSs和3′UTRs的Genic-SSRs中, Metabolic pathways是最丰富的条目, 其次是plant hormone signal transduction、biosynthesis of secondary metabolites、plant-pathogen interaction和ubiquitin mediated proteolysis。
(A) TR1-TR5转录组中多态Genic-SSR 104关联基因序列的比对结果; (B) 图(A)中多态Genic-SSRs的毛细管电泳结果 Figure 3Multiple sequence alignment of polymorphic Genic-SSR and capillary electrophoresis analysis
(A) Multiple sequence alignment for five transcripts assembled in the TR1-TR5 transcriptomes that correspond to polymorphic Genic-SSR 104; (B) The polymorphism of Genic-SSRs in (A) revealed by capillary electrophoresis
Table 3 表3 表314个多态Genic-SSRs标记的基本信息及遗传学参数 Table 3Basic information and genetic parameters of the 14 polymorphic Genic-SSRs
BräutigamA, GowikU (2010). What can next generation sequencing do for you? Next generation sequencing as a valuable tool in plant research Plant Biol 12, 831-841. DOI:10.1111/plb.2010.12.issue-6URL [本文引用: 1]
CardleL, RamsayL, MilbourneD, MacaulayM, MarshallD, WaughR (2000). Computational and experimental characterization of physically clustered simple sequence repeats in plants Genetics 156, 847-854. PMID:11014830 [本文引用: 1] The type and frequency of simple sequence repeats (SSRs) in plant genomes was investigated using the expanding quantity of DNA sequence data deposited in public databases. In Arabidopsis, 306 genomic DNA sequences longer than 10 kb and 36,199 EST sequences were searched for all possible mono- to pentanucleotide repeats. The average frequency of SSRs was one every 6.04 kb in genomic DNA, decreasing to one every 14 kb in ESTs. SSR frequency and type differed between coding, intronic, and intergenic DNA. Similar frequencies were found in other plant species. On the basis of these findings, an approach is proposed and demonstrated for the targeted isolation of single or multiple, physically clustered SSRs linked to any gene that has been mapped using low-copy DNA-based markers. The approach involves sample sequencing a small number of subclones of selected randomly sheared large insert DNA clones (e.g., BACs). It is shown to be both feasible and practicable, given the probability of fortuitously sequencing through an SSR. The approach is demonstrated in barley where sample sequencing 34 subclones of a single BAC selected by hybridization to the Big1 gene revealed three SSRs. These allowed Big1 to be located at the top of barley linkage group 6HS.
ChenCX, ZhouP, ChoiYA, HuangS, GmitterFG Jr (2006). Mining and characterizing microsatellites from citrus ESTs Theor Appl Genet 112, 1248-1257. DOI:10.1007/s00122-006-0226-1URL [本文引用: 1]
ChenYN, ZhouHH, ChenYP (2013). Adaptation strategies of desert riparian forest vegetation in response to drought stress Ecohydrology 6, 956-973. DOI:10.1002/eco.v6.6URL [本文引用: 1]
DangZH, HuangL, JiaYY, LockhartPJ, FongY, TianYY (2020). Identification of Genic SSRs provide a perspective for studying environmental adaptation in the endemic shrub Tetraena mongolica Genes 11, 322. DOI:10.3390/genes11030322URL [本文引用: 1]
FraserLG, HarveyCF, CrowhurstRN, SilvaHN (2004). EST-derived microsatellites from Actinidia species and their potential for mapping Theor Appl Genet 108, 1010-1016. PMID:15067386 [本文引用: 1] To increase the speed and reduce the cost of constructing a genetic map of Actinidia species (kiwifruit), for use in both breeding and functional genomics programmes, we sampled microsatellites from expressed sequence tags (ESTs) to evaluate their frequency of occurrence and level of polymorphism. Perfect dinucleotide repeats were the microsatellites selected, and these were found to be numerous in both the 5' and 3' ends of the genes represented. The microsatellites were of various lengths, the majority being repeats with the pattern (CT)(n)/(GA)(n). One hundred and fifty microsatellites, each with more than 10 dinucleotide repeat units, were chosen as possible markers, and when these were amplified, 93.5% were found to be polymorphic and segregating in a mapping population, with 22.6% amplifying more than one locus. Four marker categories were identified. Fully informative markers made up 27% of the total, 36.2% were female informative, 25.8% were male informative and 10% partly informative. The mapping population was an intraspecific cross in the diploid species Actinidia chinensis, with parents chosen for their diversity in fruit and plant characteristics, and for their geographical separation. Linkage was tested using the software 'Joinmap' and a LOD value of 3. The distribution of the EST-based markers over the linkage groups obtained appeared to be random, taking into consideration the small sample size, that the number of linkage groups (31) exceeded the chromosome number of n=29, and that a number of markers were not assigned to any group. Some microsatellite markers which amplified more than one locus mapped to separate linkage groups. According to our study in A. chinensis, EST-derived microsatellites give large numbers of possible markers very quickly and at reasonable cost. The markers are highly polymorphic, segregate in the mapping population, and increase the value of the genomic map by providing some functional information.
GaoLF, TangJF, LiHW, JiaJZ (2003). Analysis of microsatellites in major crops assessed by computational and experimental approaches Mol Breed 12, 245-261. DOI:10.1023/A:1026346121217URL [本文引用: 1]
GouXJ, ShiHR, YuSF, WangZQ, LiCX, LiuSH, MaJ, ChenGD, LiuT, LiuYX (2020). SSRMMD: a rapid and accurate algorithm for mining SSR feature loci and candidate polymorphic SSRs based on assembled sequences Front Genet 11, 706. DOI:10.3389/fgene.2020.00706URL [本文引用: 1]
HanZZ, MaXY, WeiM, ZhaoT, ZhanRT, ChenWW (2018). SSR marker development and intraspecific genetic divergence exploration of Chrysanthemum indicum based on transcriptome analysis BMC Genomics 19, 291. DOI:10.1186/s12864-018-4702-1URL [本文引用: 1]
LiYC, KorolAB, FahimaT, NevoE (2004). Microsatellites within genes: structure, function, and evolution Mol Biol Evol 21, 991-1007. DOI:10.1093/molbev/msh073URL [本文引用: 1]
LiZT, ZhongYD, YuFX, XuM (2018). Novel SSR marker development and genetic diversity analysis of Cinnamomum camphora based on transcriptome sequencing Plant Genet Res 16, 568-571. DOI:10.1017/S147926211800014XURL [本文引用: 1]
LiuKJ, MuseSV (2005). PowerMarker: an integrated analysis environment for genetic marker analysis Bioinformatics 21, 2128-2129. DOI:10.1093/bioinformatics/bti282URL [本文引用: 1]
LiuLY, FanXF, TanPH, WuJY, ZhangH, HanC, ChenC, XunLL, GuoWE, ChangZH, TengK (2021). The development of SSR markers based on RNA-sequencing and its validation between and within Carex L. species BMC Plant Biol 21, 17. DOI:10.1186/s12870-020-02792-8URL [本文引用: 1]
LynchM, LandeR (1993). Biotic Interactions and Global Change. Sunderland MA: Sinauer Assocs.Inc. pp. 234-250. [本文引用: 1]
MorganteM, HanafeyM, PowellW (2002). Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes Nat Genet 30, 194-200. PMID:11799393 [本文引用: 1] Microsatellites are a ubiquitous class of simple repetitive DNA sequence. An excess of such repetitive tracts has been described in all eukaryotes analyzed and is thought to result from the mutational effects of replication slippage. Large-scale genomic and EST sequencing provides the opportunity to evaluate the abundance and relative distribution of microsatellites between transcribed and nontranscribed regions and the relationship of these features to haploid genome size. Although this has been studied in microbial and animal genomes, information in plants is limited. We assessed microsatellite frequency in plant species with a 50-fold range in genome size that is mostly attributable to the recent amplification of repetitive DNA. Among species, the overall frequency of microsatellites was inversely related to genome size and to the proportion of repetitive DNA but remained constant in the transcribed portion of the genome. This indicates that most microsatellites reside in regions pre-dating the recent genome expansion in many plants. The microsatellite frequency was higher in transcribed regions, especially in the untranslated portions, than in genomic DNA. Contrary to previous reports suggesting a preferential mechanism for the origin of microsatellites from repetitive DNA in both animals and plants, our findings show a significant association with the low-copy fraction of plant genomes.
NataleE, ZalbaSM, OggeroA, ReinosoH (2010). Establishment of Tamarix ramosissima under different conditions of salinity and water availability: implications for its management as an invasive species J Arid Environ 74, 1399-1407. DOI:10.1016/j.jaridenv.2010.05.023URL [本文引用: 1]
QinLP, WangLQ, GuoY, LiY, ÜmütH, WangYC (2017). An ERF transcription factor from Tamarix hispida, ThCRF1, can adjust osmotic potential and reactive oxygen species scavenging capability to improve salt tolerance Plant Sci 265, 154-166. DOI:10.1016/j.plantsci.2017.10.006URL [本文引用: 1]
RanathungeC, WheelerGL, ChimahuskyME, PerkinsAD, PramodS, WelchME (2020). Transcribed microsatellite allele lengths are often correlated with gene expression in natural sunflower populations Mol Ecol 29, 1704-1716. DOI:10.1111/mec.v29.9URL [本文引用: 1]
SantiL, WangYM, StileMR, BerendzenK, WankeD, RoigC, PozziC, MüllerK, MüllerJ, RohdeW, SalaminiF (2003). The GA octodinucleotide repeat binding factor BBR participates in the transcriptional regulation of the homeobox gene Bkn3 Plant J 34, 813-826. PMID:12795701 [本文引用: 1] In the dominant mutant Hooded (K), the barley gene BKn3 is overexpressed as a result of a duplication of 305 bp in intron IV. When fused to a cauliflower mosaic virus 35S minimal promoter, the 305 bp element activates gene expression in tobacco, as does a 655 bp BKn3 promoter sequence. Both DNA fragments contain a (GA)8 repeat (GA/TC)8. A one-hybrid screen using the 305 bp element as the DNA target led to the cloning of the barley b recombinant (BBR) protein, which binds specifically to the (GA/TC)8 repeat. BBR is nuclear targeted and is a characterized nuclear localization signal (NLS) sequence, a DNA-binding domain extended up to 90 aa at the C-terminus and a putative N-terminal activation domain. The corresponding gene has no introns and is ubiquitously expressed in barley tissues. In co-transfection experiments, BBR activates (GA/TC)8-containing promoters, and its overexpression in tobacco leads to a pronounced leaf shape modification. BBR has properties of a GAGA-binding factor, but the corresponding gene has no sequence homology to Trl and Psq of Drosophila, which encode functionally analogous proteins. In Arabidopsis, (GA/TC)8 repeats occur particularly within 1500 bp upstream of gene start codons included in some homeodomain genes of different classes. The data presented suggest that expression of the barley BKn3 is regulated, at least in part, by the binding of the transcription factor BBR to GA/TC repeats.
SawayaS, BagshawA, BuschiazzoE, KumarP, ChowdhuryS, BlackMA, GemmellN (2013). Microsatellite tandem repeats are abundant in human promoters and are associated with regulatory elements PLoS One 8, e54710. DOI:10.1371/journal.pone.0054710URL [本文引用: 1]
TrifonovEN (1995). Segmented structure of protein sequences and early evolution of genome by combinatorial fusion of DNA elements J Mol Evol 40, 337-342. PMID:7723061 [本文引用: 1] A theory of an early stage of genome evolution by combinatorial fusion of circular DNA units is suggested, based on protein sequence "fossil" evidence. The evidence includes preference of protein sequence lengths for certain sizes--multiples of 123 aa for eukaryotes and multiples of 152 aa for prokaryotes. At the DNA level these sizes correspond to 350-450 base pairs--the known optimal range for DNA ring closure. The methionine residues repeatedly appear along the sequences with the same period of about 120 aa (in eukaryotes), presumably marking the sites of insertion of the early genes--rings of protein-coding DNA. No torsional constraint in this DNA results in very sharp estimate of the helical periodicity of the early DNA, indistinguishable from the experimental mean value for extant DNA. According to the combinatorial fusion theory, based on the above evidence, in the pregenomic, prerecombinational stage the genes and the noncoding sequences existed in form of autonomously replicating DNA rings of close to standard size, randomly segregating between dividing cells, like modern plasmids do. In the recombinational early genomic stage the rings started to fuse, forming larger DNA molecules consisting of several unit genes connected in various combinations and forming long protein-coding sequences (combinatorial fusion). This process, which involved, perhaps, noncoding sequences as well, eventually resulted in the formation of large genomes. The dispersed circular DNA--or, rather, evolutionarily advanced derivatives thereof--may still exist in the form of various mobile DNA elements. 药用植物柽柳内生真菌分离及其抑菌活性初步研究 1 2013
The development of SSR markers based on RNA-sequencing and its validation between and within Carex L. species 1 2021
... 高通量测序可快速获得研究对象的海量遗传信息, 使分子标记的开发步入全新时代(Bräutigam and Gowik, 2010).转录组测序是二代测序技术体系中的重要组成部分, 研究人员可从转录组数据中识别成百上千个SSR位点, 克服了传统SSR标记开发较为繁琐的不足, 推动了该标记在相关研究领域中的应用.基于转录组数据识别SSR已在植物研究中获得成功, 如芫荽(Coriandrum sativum) (Tulsani et al., 2020)、樟树(Cinnamomum camphora) (Li et al., 2018)和野菊(Chrysanthemum indicum) (Han et al., 2018).然而, 在众多候选SSR位点中开发多态性标记仍为尚未突破的瓶颈.例如, Liu等(2021)利用11个苔草(Carex sp.)个体, 对96个SSR位点进行验证, 开发出42个多态性SSR标记, 成功率为44%.Weng等(2020)验证了梨锈病病原菌(Gymnosporangium asiaticum)转录组中的72个SSR位点, 其中22个有多态性, 成功率为31%.为提高多态性标记的开发效率, 研究人员相继开发出多款生物信息学分析工具, 如CandiSSR (Xia et al., 2015, 2019; Zhang et al., 2019)和SSRMMD (Gou et al., 2020).利用这些工具可以从多个样本的高通量测序数据中识别潜在的多态性SSR位点, 方便后续标记的开发.本研究采用CandiSSR, 通过对5个多枝柽柳个体的转录组序列进行比对, 共识别出1 185个候选多态Genic-SSRs.经验证, 在随机挑选的15个SSRs中, 14个能扩增出预期PCR产物, 成功率近93%, 显示出本研究多态性SSR标记开发流程的高效性. ...
Biotic Interactions and Global Change. Sunderland MA: 1 1993
... 评价物种的遗传多样性和识别适应过程中的遗传变异并鉴定其功能, 是适应性进化研究的2个重要方向(Lynch and Lande, 1993).分子标记是遗传多样性分析的有效手段, 具有数量丰富、多态性高、直接以DNA的形式表现等优势, 比形态学标记、细胞学标记和化学标记的精准度更高.SSR (simple sequence repeat)又称微卫星DNA, 是由1-6个核苷酸串联重复而成的DNA序列, 广泛分布于真核生物基因组中, 是种群遗传学、目标基因定位及农作物种质资源等研究中常用的分子标记.研究表明, 位于基因内部的SSR (Genic-SSR或EST-SSR)变异对染色质组成、基因活性调节、DNA复制及重组、细胞周期调控以及错配修复等有重要影响, 被称为生物适应外界环境的分子进化装置, 在适应性进化过程中扮演重要角色(Trifonov, 1995; Li et al., 2002).同时, Genic-SSR在遗传多样性评估、遗传作图及分子标记辅助育种研究中也同样有效(陈雨等, 2008; Wen et al., 2010).可见, Genic-SSR能满足适应性进化研究的多方面需要, 极具开发潜力和研究价值. ...
Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes 1 2002
... 据现有Genic-SSR数据可知, 二核苷酸和三核苷酸重复SSR出现频率较高, 但不同物种的重复基元类型存在一定差别.例如, 大豆(Glycine max) (Cardle et al., 2000)和柑橘(Citrus reticulata) (Chen et al., 2006)以三核苷酸重复为主; 猕猴桃(Actinidia chinensis) (Fraser et al., 2004)、人参(Panax ginseng) (杨成君和王军, 2008)及马蹄香(Saruma henryi) (李珊等, 2010)的二核苷酸重复偏多.本研究表明, 多枝柽柳转录组中的二、三核苷酸重复分别占41.30%和50.01%, 与上述植物SSR类型的出现频率相符.此外, 基因序列中的SSRs表现出对特定核苷酸重复单元的偏好, 即三核苷酸和六核苷酸重复以较高的频率出现在CDS中, 而二核苷酸和四核苷酸重复较多出现在UTR中(Morgante et al., 2002; Gao et al., 2003; Li et al., 2004).在多枝柽柳中, 绝大多数AG/CT重复SSRs分布在基因5′UTR, 大部分AT/AT重复SSRs位于基因3′UTR, 而AGC/GCT、AGG/ CCT、AAG/CTT、CCG/CGG以及ATC/GAT等三核苷酸重复SSRs则集中分布在CDS中.上述结果在一定程度上反映了本研究结果的可靠性, 并暗示这些SSR对相关基因的影响可能存在功能相似性.通过比较应用CandiSSR在其余物种中识别多态SSRs的结果, 发现多枝柽柳的多态Genic-SSR信息较为丰富, 说明其可能具有较高的遗传多样性.在云南山茶(Camellia reticulata)及其近缘种的转录组中鉴定出835个多态Genic-SSRs (姚秋阳, 2015); 通过比较6种异质生境下的四合木(Tetraena mongolica)转录组, 鉴定出811个多态性Genic-SSR位点(Dang et al., 2020).需要指出的是, 基于此方法识别潜在多态SSR信息, 与不同研究中的样本采集距离以及种群生境间的异质性有很大关系.本研究选择地理距离相对较远的多枝柽柳个体进行测序, 以期检测到更多的变异位点, 取得了较好的效果, 可为同类研究提供借鉴.尽管Genic-SSR在进化上相对保守, 较基因组SSR多态性偏低, 但在上述验证的SSR位点中, 共检测到64个等位基因(Na), 它们的He和Ho平均值分别为0.553和0.421, PIC平均值为0.493, 均属于中、高多态性SSR标记, 表明内蒙古阿拉善地区多枝柽柳种群可能具有丰富的遗传多样性及遗传结构. ...
Establishment of Tamarix ramosissima under different conditions of salinity and water availability: implications for its management as an invasive species 1 2010
... 基因编码区和非编码区都存在大量高度可变的SSRs, 其变异可能与特定基因的活性和表达相关, 尤其是处于基因UTR区中的二核苷酸和三核苷酸重复SSRs, 可能在植物快速适应环境中发挥重要作用(Varshney et al., 2005; Sawaya et al., 2013).例如, Ranathunge等(2020)发现, 向日葵(Helianthus annuus)中479个Genic-SSRs的变异与其关联基因在不同自然种群的差异表达相关; 在这些SSR中, 70.4%位于相关基因的非翻译区.Santi等(2003)报道了植物基因调控区存在大量的(GA)n序列, 可被特定的DNA结合蛋白识别, 从而参与基因转录调控.刘林等(2010)发现, 灰色大角间座壳菌(Magnaporthe grisea)蛋白激酶基因中SSR的扩张或收缩会改变其编码产物的空间结构, 进而影响其功能.在本研究识别到的多态Genic-SSRs中, 分别有94.07%的二核苷酸和21.53%的三核苷酸重复SSR分布于相应基因的UTR区, 表明它们对相关基因具有潜在的调节作用.柽柳以其极强的耐旱和耐盐碱能力成为我国西北干旱地区的主要植被类型.据报道, 柽柳可通过根系的“提水作用”将水分从深层土壤输送到地表, 水分和养分富集形成“沃岛效应”, 间接为栖息地中的动植物提供适宜环境(Zhang et al., 2018).在潮湿河岸地带, 多枝柽柳凭借其有效的水分竞争能力和泌盐能力限制草本植物的生存, 形成单物种优势植物群落(Natale et al., 2010); 干旱时, 柽柳内的脱落酸(ABA)、吲哚乙酸(IAA)及赤霉素(GA)含量均显著增加(Chen et al., 2013).对上述逆境适应的分子机理研究表明, 植物内源激素调节和次生代谢物积累在柽柳属植物抗逆性形成过程中发挥重要作用.Zheng等(2013)发现, ThWRKY4转录因子能提高柽柳对盐、渗透和脱落酸胁迫的抗性.刚毛柽柳(T. hispida)中乙烯应答转录因子ThCRF1可调节海藻糖和脯氨酸的生物合成, 从而增强其耐盐性(Qin et al., 2017).本研究中, 对含有多态Genic-SSR基因的功能富集分析显示, 在1 123个基因中, 767个获得了注释信息.有意思的是, 其中大量基因富集分布在与柽柳植物适应生境和逆境应答密切相关的细胞过程和代谢通路中, GO条目如transcription、transcription factor activity, sequence- specific DNA binding、response to water deprivation和response to salt stress, KEGG代谢通路如plant hormone signal transduction、biosynthesis of secondary metabolites和diterpenoid biosynthesis (图2), 暗示这些SSR可能与植物的抗逆适应性相关.然而, Genic-SSR的多态性是否对相关基因的表达或功能有影响? 它们是否能驱动植物对特定环境做出响应? 这些问题仍需要进一步研究. ...
An ERF transcription factor from Tamarix hispida, ThCRF1, can adjust osmotic potential and reactive oxygen species scavenging capability to improve salt tolerance 1 2017
... 基因编码区和非编码区都存在大量高度可变的SSRs, 其变异可能与特定基因的活性和表达相关, 尤其是处于基因UTR区中的二核苷酸和三核苷酸重复SSRs, 可能在植物快速适应环境中发挥重要作用(Varshney et al., 2005; Sawaya et al., 2013).例如, Ranathunge等(2020)发现, 向日葵(Helianthus annuus)中479个Genic-SSRs的变异与其关联基因在不同自然种群的差异表达相关; 在这些SSR中, 70.4%位于相关基因的非翻译区.Santi等(2003)报道了植物基因调控区存在大量的(GA)n序列, 可被特定的DNA结合蛋白识别, 从而参与基因转录调控.刘林等(2010)发现, 灰色大角间座壳菌(Magnaporthe grisea)蛋白激酶基因中SSR的扩张或收缩会改变其编码产物的空间结构, 进而影响其功能.在本研究识别到的多态Genic-SSRs中, 分别有94.07%的二核苷酸和21.53%的三核苷酸重复SSR分布于相应基因的UTR区, 表明它们对相关基因具有潜在的调节作用.柽柳以其极强的耐旱和耐盐碱能力成为我国西北干旱地区的主要植被类型.据报道, 柽柳可通过根系的“提水作用”将水分从深层土壤输送到地表, 水分和养分富集形成“沃岛效应”, 间接为栖息地中的动植物提供适宜环境(Zhang et al., 2018).在潮湿河岸地带, 多枝柽柳凭借其有效的水分竞争能力和泌盐能力限制草本植物的生存, 形成单物种优势植物群落(Natale et al., 2010); 干旱时, 柽柳内的脱落酸(ABA)、吲哚乙酸(IAA)及赤霉素(GA)含量均显著增加(Chen et al., 2013).对上述逆境适应的分子机理研究表明, 植物内源激素调节和次生代谢物积累在柽柳属植物抗逆性形成过程中发挥重要作用.Zheng等(2013)发现, ThWRKY4转录因子能提高柽柳对盐、渗透和脱落酸胁迫的抗性.刚毛柽柳(T. hispida)中乙烯应答转录因子ThCRF1可调节海藻糖和脯氨酸的生物合成, 从而增强其耐盐性(Qin et al., 2017).本研究中, 对含有多态Genic-SSR基因的功能富集分析显示, 在1 123个基因中, 767个获得了注释信息.有意思的是, 其中大量基因富集分布在与柽柳植物适应生境和逆境应答密切相关的细胞过程和代谢通路中, GO条目如transcription、transcription factor activity, sequence- specific DNA binding、response to water deprivation和response to salt stress, KEGG代谢通路如plant hormone signal transduction、biosynthesis of secondary metabolites和diterpenoid biosynthesis (图2), 暗示这些SSR可能与植物的抗逆适应性相关.然而, Genic-SSR的多态性是否对相关基因的表达或功能有影响? 它们是否能驱动植物对特定环境做出响应? 这些问题仍需要进一步研究. ...
Transcribed microsatellite allele lengths are often correlated with gene expression in natural sunflower populations 1 2020
... 基因编码区和非编码区都存在大量高度可变的SSRs, 其变异可能与特定基因的活性和表达相关, 尤其是处于基因UTR区中的二核苷酸和三核苷酸重复SSRs, 可能在植物快速适应环境中发挥重要作用(Varshney et al., 2005; Sawaya et al., 2013).例如, Ranathunge等(2020)发现, 向日葵(Helianthus annuus)中479个Genic-SSRs的变异与其关联基因在不同自然种群的差异表达相关; 在这些SSR中, 70.4%位于相关基因的非翻译区.Santi等(2003)报道了植物基因调控区存在大量的(GA)n序列, 可被特定的DNA结合蛋白识别, 从而参与基因转录调控.刘林等(2010)发现, 灰色大角间座壳菌(Magnaporthe grisea)蛋白激酶基因中SSR的扩张或收缩会改变其编码产物的空间结构, 进而影响其功能.在本研究识别到的多态Genic-SSRs中, 分别有94.07%的二核苷酸和21.53%的三核苷酸重复SSR分布于相应基因的UTR区, 表明它们对相关基因具有潜在的调节作用.柽柳以其极强的耐旱和耐盐碱能力成为我国西北干旱地区的主要植被类型.据报道, 柽柳可通过根系的“提水作用”将水分从深层土壤输送到地表, 水分和养分富集形成“沃岛效应”, 间接为栖息地中的动植物提供适宜环境(Zhang et al., 2018).在潮湿河岸地带, 多枝柽柳凭借其有效的水分竞争能力和泌盐能力限制草本植物的生存, 形成单物种优势植物群落(Natale et al., 2010); 干旱时, 柽柳内的脱落酸(ABA)、吲哚乙酸(IAA)及赤霉素(GA)含量均显著增加(Chen et al., 2013).对上述逆境适应的分子机理研究表明, 植物内源激素调节和次生代谢物积累在柽柳属植物抗逆性形成过程中发挥重要作用.Zheng等(2013)发现, ThWRKY4转录因子能提高柽柳对盐、渗透和脱落酸胁迫的抗性.刚毛柽柳(T. hispida)中乙烯应答转录因子ThCRF1可调节海藻糖和脯氨酸的生物合成, 从而增强其耐盐性(Qin et al., 2017).本研究中, 对含有多态Genic-SSR基因的功能富集分析显示, 在1 123个基因中, 767个获得了注释信息.有意思的是, 其中大量基因富集分布在与柽柳植物适应生境和逆境应答密切相关的细胞过程和代谢通路中, GO条目如transcription、transcription factor activity, sequence- specific DNA binding、response to water deprivation和response to salt stress, KEGG代谢通路如plant hormone signal transduction、biosynthesis of secondary metabolites和diterpenoid biosynthesis (图2), 暗示这些SSR可能与植物的抗逆适应性相关.然而, Genic-SSR的多态性是否对相关基因的表达或功能有影响? 它们是否能驱动植物对特定环境做出响应? 这些问题仍需要进一步研究. ...
The GA octodinucleotide repeat binding factor BBR participates in the transcriptional regulation of the homeobox gene Bkn3 1 2003
... 基因编码区和非编码区都存在大量高度可变的SSRs, 其变异可能与特定基因的活性和表达相关, 尤其是处于基因UTR区中的二核苷酸和三核苷酸重复SSRs, 可能在植物快速适应环境中发挥重要作用(Varshney et al., 2005; Sawaya et al., 2013).例如, Ranathunge等(2020)发现, 向日葵(Helianthus annuus)中479个Genic-SSRs的变异与其关联基因在不同自然种群的差异表达相关; 在这些SSR中, 70.4%位于相关基因的非翻译区.Santi等(2003)报道了植物基因调控区存在大量的(GA)n序列, 可被特定的DNA结合蛋白识别, 从而参与基因转录调控.刘林等(2010)发现, 灰色大角间座壳菌(Magnaporthe grisea)蛋白激酶基因中SSR的扩张或收缩会改变其编码产物的空间结构, 进而影响其功能.在本研究识别到的多态Genic-SSRs中, 分别有94.07%的二核苷酸和21.53%的三核苷酸重复SSR分布于相应基因的UTR区, 表明它们对相关基因具有潜在的调节作用.柽柳以其极强的耐旱和耐盐碱能力成为我国西北干旱地区的主要植被类型.据报道, 柽柳可通过根系的“提水作用”将水分从深层土壤输送到地表, 水分和养分富集形成“沃岛效应”, 间接为栖息地中的动植物提供适宜环境(Zhang et al., 2018).在潮湿河岸地带, 多枝柽柳凭借其有效的水分竞争能力和泌盐能力限制草本植物的生存, 形成单物种优势植物群落(Natale et al., 2010); 干旱时, 柽柳内的脱落酸(ABA)、吲哚乙酸(IAA)及赤霉素(GA)含量均显著增加(Chen et al., 2013).对上述逆境适应的分子机理研究表明, 植物内源激素调节和次生代谢物积累在柽柳属植物抗逆性形成过程中发挥重要作用.Zheng等(2013)发现, ThWRKY4转录因子能提高柽柳对盐、渗透和脱落酸胁迫的抗性.刚毛柽柳(T. hispida)中乙烯应答转录因子ThCRF1可调节海藻糖和脯氨酸的生物合成, 从而增强其耐盐性(Qin et al., 2017).本研究中, 对含有多态Genic-SSR基因的功能富集分析显示, 在1 123个基因中, 767个获得了注释信息.有意思的是, 其中大量基因富集分布在与柽柳植物适应生境和逆境应答密切相关的细胞过程和代谢通路中, GO条目如transcription、transcription factor activity, sequence- specific DNA binding、response to water deprivation和response to salt stress, KEGG代谢通路如plant hormone signal transduction、biosynthesis of secondary metabolites和diterpenoid biosynthesis (图2), 暗示这些SSR可能与植物的抗逆适应性相关.然而, Genic-SSR的多态性是否对相关基因的表达或功能有影响? 它们是否能驱动植物对特定环境做出响应? 这些问题仍需要进一步研究. ...
Microsatellite tandem repeats are abundant in human promoters and are associated with regulatory elements 1 2013
... 基因编码区和非编码区都存在大量高度可变的SSRs, 其变异可能与特定基因的活性和表达相关, 尤其是处于基因UTR区中的二核苷酸和三核苷酸重复SSRs, 可能在植物快速适应环境中发挥重要作用(Varshney et al., 2005; Sawaya et al., 2013).例如, Ranathunge等(2020)发现, 向日葵(Helianthus annuus)中479个Genic-SSRs的变异与其关联基因在不同自然种群的差异表达相关; 在这些SSR中, 70.4%位于相关基因的非翻译区.Santi等(2003)报道了植物基因调控区存在大量的(GA)n序列, 可被特定的DNA结合蛋白识别, 从而参与基因转录调控.刘林等(2010)发现, 灰色大角间座壳菌(Magnaporthe grisea)蛋白激酶基因中SSR的扩张或收缩会改变其编码产物的空间结构, 进而影响其功能.在本研究识别到的多态Genic-SSRs中, 分别有94.07%的二核苷酸和21.53%的三核苷酸重复SSR分布于相应基因的UTR区, 表明它们对相关基因具有潜在的调节作用.柽柳以其极强的耐旱和耐盐碱能力成为我国西北干旱地区的主要植被类型.据报道, 柽柳可通过根系的“提水作用”将水分从深层土壤输送到地表, 水分和养分富集形成“沃岛效应”, 间接为栖息地中的动植物提供适宜环境(Zhang et al., 2018).在潮湿河岸地带, 多枝柽柳凭借其有效的水分竞争能力和泌盐能力限制草本植物的生存, 形成单物种优势植物群落(Natale et al., 2010); 干旱时, 柽柳内的脱落酸(ABA)、吲哚乙酸(IAA)及赤霉素(GA)含量均显著增加(Chen et al., 2013).对上述逆境适应的分子机理研究表明, 植物内源激素调节和次生代谢物积累在柽柳属植物抗逆性形成过程中发挥重要作用.Zheng等(2013)发现, ThWRKY4转录因子能提高柽柳对盐、渗透和脱落酸胁迫的抗性.刚毛柽柳(T. hispida)中乙烯应答转录因子ThCRF1可调节海藻糖和脯氨酸的生物合成, 从而增强其耐盐性(Qin et al., 2017).本研究中, 对含有多态Genic-SSR基因的功能富集分析显示, 在1 123个基因中, 767个获得了注释信息.有意思的是, 其中大量基因富集分布在与柽柳植物适应生境和逆境应答密切相关的细胞过程和代谢通路中, GO条目如transcription、transcription factor activity, sequence- specific DNA binding、response to water deprivation和response to salt stress, KEGG代谢通路如plant hormone signal transduction、biosynthesis of secondary metabolites和diterpenoid biosynthesis (图2), 暗示这些SSR可能与植物的抗逆适应性相关.然而, Genic-SSR的多态性是否对相关基因的表达或功能有影响? 它们是否能驱动植物对特定环境做出响应? 这些问题仍需要进一步研究. ...
Segmented structure of protein sequences and early evolution of genome by combinatorial fusion of DNA elements 1 1995
... 评价物种的遗传多样性和识别适应过程中的遗传变异并鉴定其功能, 是适应性进化研究的2个重要方向(Lynch and Lande, 1993).分子标记是遗传多样性分析的有效手段, 具有数量丰富、多态性高、直接以DNA的形式表现等优势, 比形态学标记、细胞学标记和化学标记的精准度更高.SSR (simple sequence repeat)又称微卫星DNA, 是由1-6个核苷酸串联重复而成的DNA序列, 广泛分布于真核生物基因组中, 是种群遗传学、目标基因定位及农作物种质资源等研究中常用的分子标记.研究表明, 位于基因内部的SSR (Genic-SSR或EST-SSR)变异对染色质组成、基因活性调节、DNA复制及重组、细胞周期调控以及错配修复等有重要影响, 被称为生物适应外界环境的分子进化装置, 在适应性进化过程中扮演重要角色(Trifonov, 1995; Li et al., 2002).同时, Genic-SSR在遗传多样性评估、遗传作图及分子标记辅助育种研究中也同样有效(陈雨等, 2008; Wen et al., 2010).可见, Genic-SSR能满足适应性进化研究的多方面需要, 极具开发潜力和研究价值. ...
 ,1,*
,1,*
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT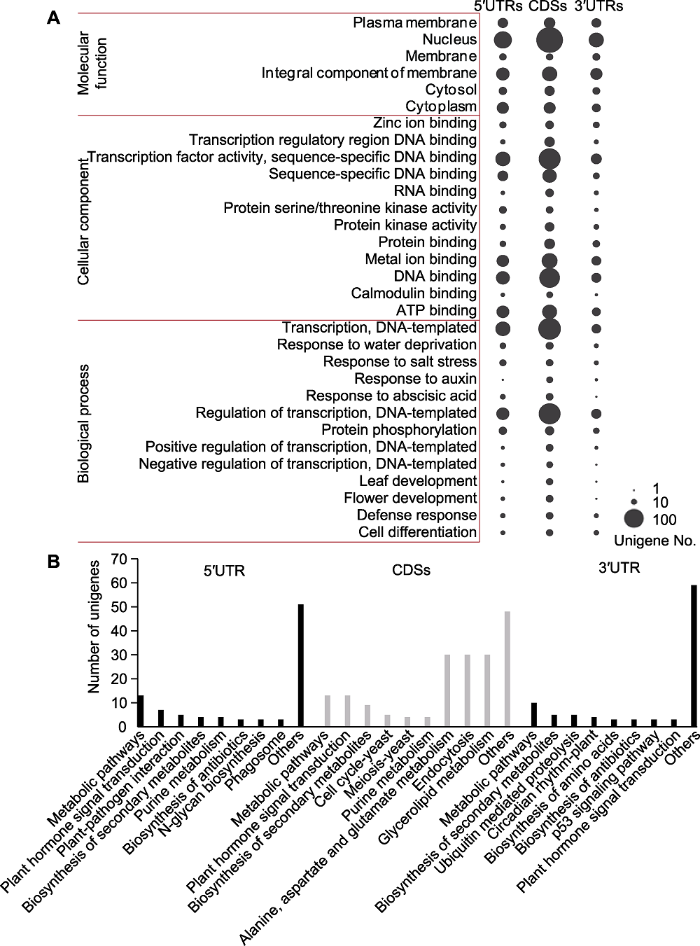 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT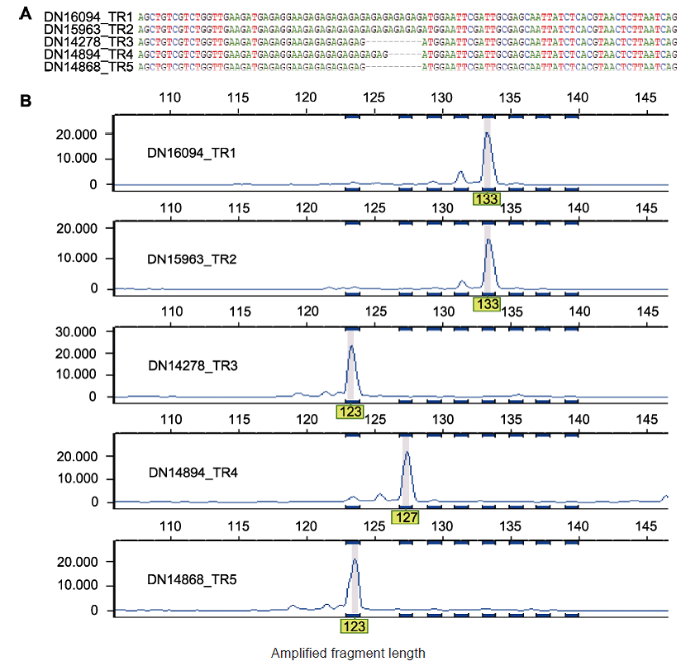 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}