 ,1,*, 宋海超,2,*
,1,*, 宋海超,2,*Molecular Mechanism of the Generation of Asexual Spores of the Mango Fungal Pathogen (Colletotrichum gloeosporioides) Induced by Mechanical Injuries
Liyan Wang1, Mengyao Lu1, Yue Tong1, Xiangbin Xu1, Zhengke Zhang1, Lanhuan Meng1, Xuequn Shi,1,*, Haichao Song,2,*通讯作者: E-mail:shixuequn@163.com;songhaichao@sohu.com
责任编辑: 白羽红
收稿日期:2020-02-22接受日期:2020-05-8网络出版日期:2020-09-01
| 基金资助: |
Corresponding authors: E-mail:shixuequn@163.com;songhaichao@sohu.com
Received:2020-02-22Accepted:2020-05-8Online:2020-09-01

摘要
胶孢炭疽菌(Colletotrichum gloeosporioides)是引发芒果(Mangifera indica)炭疽病的主要病原体。室内平板培养胶孢炭疽菌不产生或产生很少分生孢子的情况时有发生, 但菌丝在机械损伤后24-48小时会产生大量分生孢子。胶孢炭疽菌应答机械损伤诱导产孢的核心基因及关键代谢通路尚未见报道。基于转录组测序(RNA-seq)技术检测了芒果胶孢炭疽菌菌丝在机械损伤处理后2小时内5个时间点的基因表达变化, 对差异表达基因进行GO富集和KEGG代谢通路富集分析, 并对菌丝响应胁迫的基因动态表达数据进行分析。基于常微分方程ODE模型结合变量选择技术, 构建了动态基因调控网络。结果表明, 有417个差异表达基因参与应答胶孢炭疽菌菌丝机械损伤, 分属12个聚类模块, 有4条通路存在显著富集, 分别是丙酮酸代谢、硫代谢、黄曲霉素合成途径和二萜合成途径。结合功能注释筛选出12个应答菌丝损伤胁迫的核心基因。研究结果为后续深入开展芒果胶孢炭疽菌产孢和致病机理研究奠定了重要基础。
关键词:
Abstract
Colletotrichum gloeosporioides is a prevalent pathogen that causes anthracnose in mango (Mangifera indica). Mycelium of C. gloeosporioides will accumulate a large number of conidia in 24-48 hours after mechanical injuries. However, it often accumulates none or few conidia during indoor culture, and the gene regulatory networks of the response to injury for a short-time (ST), or the key metabolic pathways involved in the response has not been explored. In this study, RNA-seq was carried out on RNA samples obtained at 5 time points within 2 hours after mechanical injuries. The differentially expressed genes were enriched by GO enrichment and KEGG metabolic pathway. The expression dynamics of mycelia in response to ST injury stress was analyzed. Based on a nonlinear ordinary differential equation model coupled with variable selection techniques, inter-module networks were constructed. The results showed that 417 differentially expressed genes were obtained, which belong to 12 clustered modules. KEGG enrichment analysis of differentially expressed genes was enriched in the process of pyruvate metabolism, sulfur metabolism, aflatoxin biosynthesis, diterpenoid biosynthesis. Combined with functional annotation, 12 core genes were identified that significantly correlated with ST injury-induced expression. These results provide valuable references for further research on asexual development and pathogenicity in C. gloeosporioides.
Keywords:
PDF (2141KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
引用本文
王丽妍, 卢梦瑶, 童悦, 徐祥斌, 张正科, 孟兰环, 史学群, 宋海超. 芒果胶孢炭疽病菌应答菌丝机械损伤产生无性孢子的分子机制. 植物学报, 2020, 55(5): 551-563 doi:10.11983/CBB20026
Wang Liyan, Lu Mengyao, Tong Yue, Xu Xiangbin, Zhang Zhengke, Meng Lanhuan, Shi Xuequn, Song Haichao.

芒果(Mangifera indica)属漆树科植物, 是热带和亚热带地区广泛种植的名贵水果, 被誉为“水果之王”(Jahurul et al., 2015)。但芒果在生长过程中极易发生真菌性病害(如炭疽病、蒂腐病) (吴振麟, 2011)从而引起果实腐烂。多年来, 国内外****对芒果的采后处理、贮藏保鲜及生理生化变化进行了研究(Perumal et al., 2017)。邹日娥(1995)用水杨酸或氯化钙结合全美梅氏酵母悬浮液处理采后台农芒果可有效增强多酚氧化酶(PPO)、苯丙氨酸酶(PAL)及几丁质酶(CHT)的活力, 延长芒果的保质期。也有研究表明, 对芒果施用海洋细菌嗜根寡养单胞菌可以有效降低炭疽病的发病率, 及时控制病害发生及蔓延(Shao et al., 2019)。
胶孢炭疽菌(Colletotrichum gloeosporioides)是引起芒果炭疽病的主要病原菌, 该病菌是一种丝状真菌, 其寄主范围广, 可潜伏侵染果实的多个部位, 严重影响芒果的产量和品质, 危害极大(Reyes-Perez et al., 2019)。通过无性繁殖产生分生孢子是胶孢炭疽菌的主要繁殖方式。研究表明, 丝状真菌的产孢过程涉及多个生物学过程(Adams et al., 1988), 如基因时空表达的特异性、细胞特化及细胞间相互关系。随着基因测序技术的发展以及测序成本的降低, 研究者在转录组和全基因组水平对胶孢炭疽病原菌功能基因进行了研究。黄华平(2016)利用新一代高通量测序平台Illumina RNA-seq对高致病力胶孢炭疽菌菌株的全基因组和转录组进行测序及功能注释, 筛选了疑似致病相关基因CgALS, 并通过基因敲除实验验证其致病性。张丽勍等(2017)利用Pfam数据库对草莓(Fragaria ananassa)胶孢炭疽菌全基因组进行搜索, 鉴定并分析了22个CFEM (common in fungal extracellular membrane)蛋白及其中8个分泌蛋白在不同侵染阶段的转录组数据, 为深入研究病原真菌CFEM效应子提供了理论依据。
许多丝状真菌通过分生孢子进行侵染和传播, 产生大量分生孢子是其进行侵染和扩散的前提(李伟, 2002)。在实验室培养胶孢炭疽菌时, 我们发现在培养早期, 菌丝营养生长状况良好但孢子数量少或不产孢, 如果打断菌丝, 24-48小时后会产生大量分生孢子, 说明机械损伤对该病原真菌生殖生长有诱导和促进作用。研究表明, 木霉在机械损伤后其损伤细胞附近同样会产生大量无性孢子, 利用RNA-seq结合基因敲除实验, 发现可能存在与活性氧和脂质代谢途径相关的一系列保守的损伤防御机理(Hernández- O?ate et al., 2012)。然而, 尽管有报道显示损伤会诱导产生无性孢子, 但这一动态生物学过程的反应机制及机械损伤过程中的一系列信号转导及代谢通路等科学问题尚未见报道, 有待深入研究。
本研究将芒果胶孢炭疽菌菌株于28°C液体培养基摇菌培养, 15小时后通过机械损伤打断菌丝, 并于不同时间点收集取样, 测定转录组数据。通过差异表达基因筛选、功能富集分析、聚类分析及调控网络构建, 初步揭示胶孢炭疽菌机械损伤胁迫下的基因表达模式, 筛选出响应损伤胁迫产孢的关键基因。研究结果可为阐明芒果胶孢炭疽菌产孢和致病的分子机理以及研发作用于炭疽菌具有靶标位点的新农药奠定基础。
1 材料与方法
1.1 供试菌株
实验所用菌株为芒果胶孢炭疽菌(Colletotrichum gloeosporioides (Penz.) Penz.&Sacc.), 该病菌从自然发病且具有典型芒果炭疽病症状的芒果果皮上分离得到(于海英等, 2012), 经纯化培养后进行单孢分离和鉴定, 并将菌种保存在4°C冰箱中备用。1.2 机械损伤处理菌丝转录组样品制备方法
将芒果胶孢炭疽菌菌株于马铃薯葡萄糖琼脂(PDA)平板培养6天后, 取10个直径约8 mm的菌块, 接种于含有200 mL马铃薯葡萄糖(PDB)培养基的250 mL锥形瓶中, 28°C, 每分钟180转摇床培养。用无菌的菌铲用力搅拌PDB培养基中已生长15小时的胶孢炭疽菌, 以顺时针和逆时针方向交替搅拌, 搅拌时间大于30秒, 使菌块破碎, 菌丝损伤, 严重干扰PDB培养基中胶孢炭疽菌正常的生长状态。分别在处理后0分钟、15分钟、30分钟、1小时和2小时取样。取样时用Miracloth滤膜过滤除去滤液, 收集菌丝样品。所有样品经液氮速冻2小时后置于-70°C保存备用。1.3 RNA提取及高通量测序文库构建
使用天根(TIANGEN)植物RNA提取试剂盒(RNAsimple总RNA提取试剂盒; Cat No.DP419)提取真菌样品RNA, 具体步骤参考说明书。总RNA经质检合格后, 使用连接有Oligo (dT)的磁珠富集真核生物mRNA。用六碱基随机引物合成一链cDNA; 随后用dNTPs、RNaseH和DNA Polymerase I进行二链cDNA合成。双链产物经AMPure XP beads纯化后进行片段选择, 最后进行PCR扩增获得最终测序文库。利用Illumina Novaseq? 6000对质检合格的文库进行测序。文库的构建和测序在杭州联川生物科技有限公司完成。1.4 序列分析及注释
利用Illumina平台将测序所得的原始序列文件进行质量评估和可信度分析(Garcia-Mas et al., 2012), 去除3°端接头序列、低质量和不确定的序列(Q<20)。利用Trinity软件通过序列之间的Overlap获得Contig序列, 将Contig连接成转录本序列, 对拼接序列进行组装和拼接, 取最长的转录本为Unigene (Trapnell et al., 2010)。使用Blast2-Go软件对Unigene序列与NCBI非冗余蛋白公共数据库(Nr)、SwissProt和PFAM数据库进行比对, 取相似性大于30%、E-value小于10-5 进行注释(Conesa et al., 2005), 获得Unigene比对的E-value分布以及序列相似度分布(The Gene Ontology Consortium, 2015); 根据注释所含GO (Gene Ontology)信息, 从生物过程(BP)、分子功能(MF)以及细胞组分(CC) 3个方面对序列进行GO注释(Xie et al., 2011)。使用KOBAS软件对Unigene进行KEGG (Kyoto Encyclopedia of Genes and Genomes)注释(Langmead and Salzberg, 2012)。整理所有注释信息, 搜索胶孢炭疽菌响应损伤胁迫关键基因和代谢途径。1.5 差异基因筛选与功能富集分析
芒果胶孢炭疽菌参考基因组下载自美国国家生物技术信息中心数据库(NCBI), 将每个样品的clean reads与参考基因组进行比对(Jin et al., 2014)。使用RSEM软件对bowtie的比对结果进行统计, 获得每个样品比对到每个基因上的readcount数目。使用 Cuffdiff v2.1.1软件对其进行FPKM (fragments per kilo-base of exon per million fragments mapped)标准化转换, 从而得到基因在不同样本中的表达丰度值。比较不同处理组文库中的基因表达水平。采用R软件中edgeR包进行差异表达分析(Robinson et al., 2010)。该软件使用的负二项分布符合RNA-seq基因表达数据方差大于平均值的性质, 能够区别生物本身和技术因素所导致的基因表达的改变(McCarthy et al., 2012)。对不同时间点所有基因的表达量进行箱线图的绘制, 并利用斯皮尔曼相关系数(Spearman correlation coefficient, SCC)对所有样本的相关性进行分析, 将所有基因的FPKM值加1, 然后进行对数转换, 计算所有样本之间的SCC, 以判断测序数据的可靠性和样本间的相关性。利用GO和KEGG富集进一步分析差异表达基因, 差异基因的富集采用超几何分布检测, 对所有富集的功能类别以P-value<0.05作为筛选标准。
1.6 聚类分析
为了初步解析芒果胶孢炭疽菌在损伤胁迫下mRNA的动态表达模式, 基于实验设计对RNA-seq差异表达数据进行聚类分析(Si et al., 2013), 使具有相似表达模式的差异基因分组归类后以基因聚类图展示。图中每列代表一个实验条件, 每行代表基因表达量。1.7 调控网络构建
基因调控网络可以很好地分析胁迫中重要的分子过程或代谢通路(Wilczynski and Furlong, 2010)。损伤胁迫响应是一个动态的生物学过程, 受到许多基因及其产物组成的复杂调控网络的调节。本研究采用一种常微分方程ODE模型, 同时联合ODE参数估计和Adaptive group LASSO方法构建胶孢炭疽菌全基因组动态GRN, 通过微分方程刻画基因之间动态复杂的关系, 构建机械损伤后芒果胶孢炭疽菌全基因组动态基因调控网络。1.8 实时荧光定量PCR分析
随机选择5个基因进行实时荧光定量PCR分析。使用天根总RNA提取试剂盒提取胶孢炭疽菌菌丝RNA, 采用TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix (全式金)逆转录合成cDNA。以反转录产物为模板, Actin为内参, 采用 SuperReal PreMix Plus (SYBR Green)天根荧光定量试剂盒, 在ABI 7500 PCR仪中进行实时荧光定量检测。扩增程序如下: 95°C2分钟; 95°C10秒, 60°C1分钟, 45个循环。所有反应均进行3个生物学重复。采用2-ΔΔCT法进行相对定量分析(Livak and Schmittgen, 2001)。所用引物见表1。Table 1
表1
表1实时荧光定量PCR所用引物
Table 1
| Transcript | Upstream primer sequence (5°?3°) | Downstream primer sequence (5°?3°) |
|---|---|---|
| ELA27200 | CCTCAAACTTCGCAGATG | GGCTGTCGTGTAGAACTG |
| ELA31518 | ACTGCGAATCACATCATCTCA | TAAGAGGTCGGCGTCAGA |
| ELA26856 | CAGCAGATGGTCAAGAGG | GAGGAGACGAAGGAAGGA |
| ELA23533 | TCAGCCTTGTTGGACCTTAG | CGTAGCATCGGACCTTGT |
| ELA34567 | GAGCAGGTATCAGAAGACA | CCAGTTCGTAAGCCAGAT |
| Actin | ACGCTTCTCATCTCCAAGATCCGT | AGAGAGCCTCGTTGTCAATGCAGA |
新窗口打开|下载CSV
1.9 数据分析
实验数据均使用Excel 2016软件处理。采用Duncan多重比较法进行差异显著性分析(P<0.05)。2 结果与讨论
2.1 测序结果和统计
通过HiSeq2500测序获得210 438 160条测序读段(31.56 Gb)。对Raw reads进行质量控制后获得201 473 676个reads (30.22 Gb)。所有样本Q20值均大于99.98%, Q30值均大于97.7%, GC含量为56%。经用Trinity软件进行拼接, 获得9 958条Unigenes。箱线图(图1A)显示各样本相对于中值长度近似相等, 无明显偏斜。说明数据分布均匀, 表明测序数据质量较为可靠。通过对所有样本的相关性进行分析, 显示不同样本之间的相关系数均大于0.83 (图1B), 说明不同样本间相关性较高, 进一步表明不同样本间基因的表达在应答损伤处理这一动态机制下具有较高的稳定性。
图1
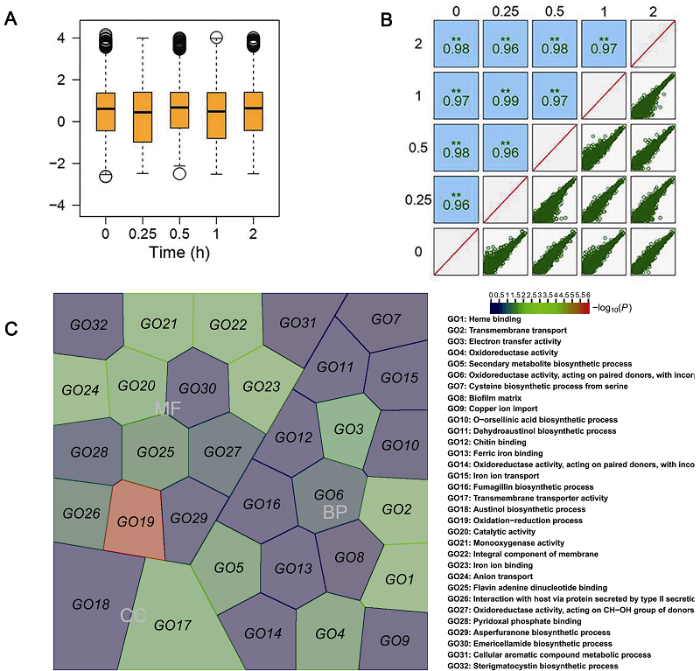 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1不同处理样品基因表达数据和差异表达基因GO富集结果
(A) 不同样本基因表达数据的分布; (B) 不同样本间的相关性; (C) 差异表达基因的GO富集分析。MF: 分子功能; BP: 生物过程; CC: 细胞组分
Figure 1Gene expression data and GO enrichment for differentially expressed genes under different treatments
(A) Gene expression in different samples; (B) Correlation between different samples; (C) GO enrichment for differentially expressed genes. MF: Molecular function; BP: Biological process; CC: Cell components
2.2 差异基因筛选与富集
使用edgeR软件对损伤处理条件下芒果胶孢炭疽菌的时间序列基因表达数据进行分析, 以1为标准(abs (logFC)>1), P-value<0.05, 获得417个差异基因。其中logFC>1的基因数量为116个, logFC<-1的基因数量为69个, 说明在机械损伤后, 许多基因上调表达。利用超几何分布统计, 计算显著差异表达基因相对全部基因的富集情况, 显示417个基因富集在127个GO 条目中。其中, 具有显著性富集的GO条目有21个(P< 0.05) (图1C)。细胞组分(CC)分类中, 跨膜转运蛋白活性(transmembrane transporter activity)富集较多的差异基因(15个)且表达水平最高; 在分子功能(MF)分类中, 大部分基因富集于氧化还原过程(oxidatIon- reduction process)及催化活性(catalytic activity); 在生物过程(BP)层面上, 显著富集的GO条目主要集中于生物过程, 富集较多的条目有跨膜转运过程(transmembrane transport)及氧化酶活性(oxidoreductase activity)等。上述结果表明, 芒果采后胶孢炭疽菌在响应损伤胁迫过程中发生了一系列生理变化。为了进一步筛选胶孢炭疽菌响应胁迫损伤期间被显著调控的代谢通路, 我们将差异表达基因进行KEGG富集分析。结果显示, 417个差异表达基因能与KEGG中的67个不同的通路匹配, 但只有4条通路存在显著富集(P<0.05) (表2)。通路主要富集到丙酮酸代谢和硫代谢(表2), 这进一步表明部分差异表达基因参与物质能量代谢, 以保证胶孢炭疽菌在响应损伤胁迫过程中的能量消耗。此外, 显著富集的代谢通路还包括黄曲霉素和二萜合成途径, 两者在真菌发育中具有重要作用。
Table 2
表2
表2差异表达基因KEGG富集结果
Table 2
| Description | Gene ratio | Bg ratio | P-value | Gene ID | Count |
|---|---|---|---|---|---|
| Aflatoxin biosynthesis | 6/113 | 46/2826 | 0.009071 | CGGC5_14023/CGGC5_13120/CGGC5_13638/ CGGC5_13456/CGGC5_10453/CGGC5_9572 | 6 |
| Pyruvate metabolism | 7/113 | 72/2826 | 0.023327 | CGGC5_14045/CGGC5_7569/CGGC5_3411/ CGGC5_10319/CGGC5_11829/CGGC5_3455/ CGGC5_5764 | 7 |
| Sulfur metabolism | 3/113 | 19/2826 | 0.037811 | CGGC5_5794/CGGC5_12200/CGGC5_15291 | 3 |
| Diterpenoid biosynthesis | 3/113 | 21/2826 | 0.048998 | CGGC5_1921/CGGC5_15293/CGGC5_11451 | 3 |
新窗口打开|下载CSV
2.3 基因表达模式解析
为了精确解析胶孢炭疽菌相关差异基因的动态表达模式, 进而筛选候选基因, 我们使用基于模型的聚类方法对具有相似表达模式的基因进行分组(Qiu et al., 2011)。结果显示, 417个损伤胁迫响应基因可分成12个聚类模块, 并获得不同基因动态表达数据的聚类结果和每个聚类内的基因数量(图2)。不同聚类模式的表达趋势不同。图2
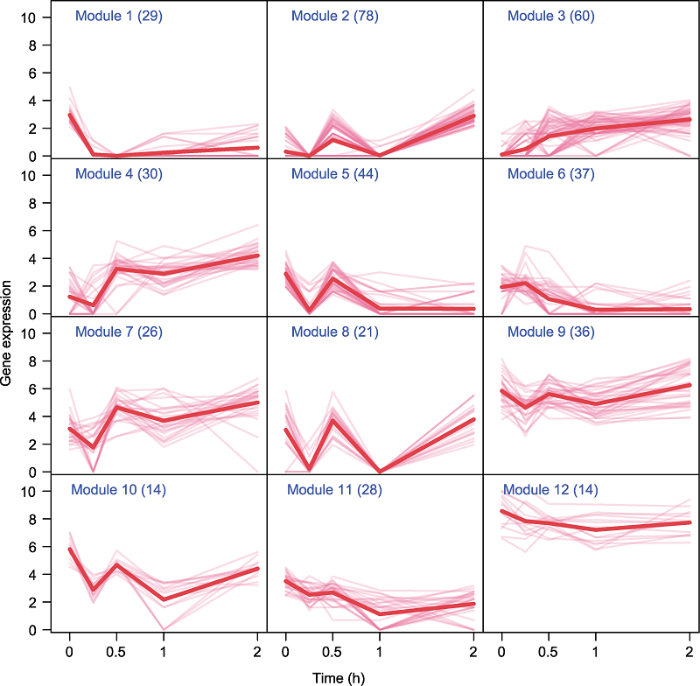 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2损伤胁迫条件下差异表达基因聚类模式
Figure 2Expression profiles for each cluster of differentially expressed genes
聚类2、4、7、8、9和10在机械损伤后0小时开始, 0到2小时整体表现先下降后上升、再下降后又上升的表达趋势, 但不同时间点变化程度差异较大。处理后0-1小时, 聚类2、5、8、9和10表现出相同的趋势, 处理后15分钟内基因表达水平迅速下降, 处理后15-30分钟基因表达呈直线上升趋势, 且在处理后1小时基因表达量与15分钟时的表达量基本一致; 聚类4和7在处理后1小时基因表达量显著高于处理15分钟时; 聚类11在处理后1小时内变化趋势相对较小且在处理后1小时的基因表达量显著低于处理15分钟时。处理后1-2小时内, 各聚类内基因表达水平变化趋势相对平缓, 聚类2、4、7、8、9、10和11基因表达水平缓慢上升; 而聚类5的基因在处理1小时后保持其表达水平。综上, 损伤处理胶孢炭疽菌后1小时内, 聚类2、4、5、7、8、9、10和11均有大量基因响应胶孢炭疽菌的瞬时应激, 这8个聚类的基因数量占总基因数量的66.43%。
聚类6与12的基因表达趋势相似, 即2小时内都呈缓慢下降趋势。但聚类6在15分钟内基因表达水平稍有上升, 后缓慢下降, 2小时后基因表达量几乎为0。聚类12基因表达水平虽然同样缓慢下降, 但是一直保持较高的水平, 且整体表达较稳定。聚类3包含60个基因, 占基因总数的14.39%, 其与其它聚类相反, 表达水平整体呈现缓慢上升趋势。聚类1在15分钟内基因表达水平迅速降低至表达量几乎为0。由上可知, 聚类3、6和12基因可能在处理后1-2小时内响应损伤应激。
2.4 聚类模块的生物学富集
分析每个聚类模块中富集的生物学功能是理解该模块在整个调控网络中所起作用的关键。为了寻找处理后不同时间不同聚类内响应的基因, 我们对每个聚类进行差异基因GO和KEGG功能富集分析(图3), 对聚类内基因功能及其调控关系进行了初步解析。图3
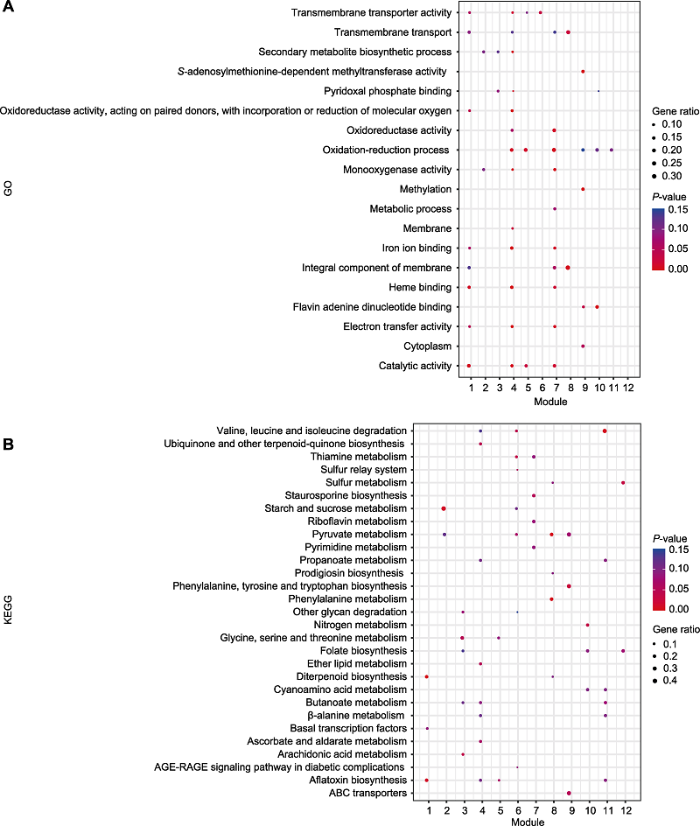 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3差异表达基因聚类模块的富集分析
(A) GO富集分析; (B) KEGG富集分析。图中横坐标表示不同聚类, 纵坐标表示富集通路的名称。不同颜色的点代表不同P-value值, 点的大小表示该通路下差异表达基因的多少。P-value取值范围为0-0.15, 数值越接近0, 表示富集越显著。
Figure 3Enrichment analysis for each gene cluster of differentially expressed genes
(A) GO enrichment analysis for each cluster; (B) KEGG enrichment analysis for each cluster. The x-axis represents the different cluster and the y-axis is the name of enrichment pathway. Different P-value are represented by dots with different colors, while the number of differentially expressed genes in each pathway are displayed for the size of the dots. P-value ranges from 0-0.15. The closer the value to 0, the more significant the enrichment is.
处理后1小时, 聚类2、4、5、7、8、9和10的基因表达趋势相似(先下降后上升)。对其中19个GO富集显著的条目进行分析, 结果(图3A)显示, 聚类4中的基因参与氧化还原过程(oxidation-reduction process), 其中包含LysM结构域蛋白相关基因。LysM结构域蛋白通过调节氨基酸和核苷酸糖代谢响应真菌病原体的防御信号转导。GO富集结果显示, 聚类7与氧化还原酶活性(oxidoreductase activity)相关, 在此聚类内发现短链脱氨酶还原酶家族(short-chain dehydrogenase reductase family)基因。聚类10的富集功能主要集中于催化活性(catalytic activity)和黄素腺嘌呤二核苷酸结合(flavin adenine dinucleotide binding)。其中, 发现具有调控转录功能的C6转录因子(C6 transcription factor)参与调控真菌子实体发育。此外, 聚类5中部分基因参与烟曲霉素的生物合成过程(fumagillin biosynthetic process)及铁离子结合(iron ion binding)等。
处理后1-2小时, 聚类3、6和12的基因总体呈缓慢下降趋势。GO富集分析显示聚类6和12的基因具有相似的功能, 如跨膜转运(transmembrane transport)和氧化还原过程(oxidation-reduction process)。GO功能富集分析显示, 聚类3与膜的组成成分(integral component of membrane)有关, 其聚类内的主要促进者超家族转运蛋白MFS (major facilitator superfamily transporter)代表了最大的二级转运家族, 为特定物质穿过疏水性脂质提供通道。
本研究对每个聚类下富集的162条通路进行了分析, 并对其中29个富集最为显著的条目进行展示。差异基因的KEGG富集散点图(图3B)显示, 基因富集最显著的代谢通路主要聚集在聚类2、8、9和11上, 分别为淀粉和蔗糖代谢(starch and sucrose metabolism)、ABC转运子(ABC transporters)及丙酮酸代谢(pyruvate metabolism)。其中富集差异基因个数最多的为淀粉和蔗糖代谢途径。聚类4富集显著的代谢通路最多, 其差异基因分布个数也最为平均。聚类模块的功能富集结果表明, 不同聚类间的生物学特性不同(图3B)。
2.5 胁迫响应基因调控网络
为了挖掘芒果胶孢炭疽菌损伤胁迫响应过程中的关键基因, 我们基于ODE模型构建基因间的动态调控网络并筛选关键的聚类模块。结合网络拓扑结构和基因注释信息, 我们发现12个胶孢炭疽菌响应损伤胁迫核心基因(表3), 分布于聚类模块1、2、6和11中, 基因间的调控网络较为稀疏(图4)。在12个聚类模块中, 调控关系主要集中在聚类模块1和2, 即核心模块。这表明核心模块中的基因在损伤胁迫的调控网络中起重要作用。Table 3
表3
表3差异表达基因12个聚类模块中的12个核心基因
Table 3
| Module | Gene numbers | Transcript | Gene description |
|---|---|---|---|
| 1 | 1 | ELA23757 | Pep1 |
| 1 | 6 | ELA25282 | Cytochrome p450 |
| 1 | 16 | ELA23240 | Nad-dependent epimerase dehydratase |
| 2 | 20 | EFCGT00000013177 | 5.8S_rRNA |
| 2 | 25 | ELA35632 | Polyketide synthase |
| 2 | 44 | ELA25574 | Integral membrane protein |
| 2 | 49 | ELA24193 | Short chain dehydrogenase reductase family |
| 2 | 57 | ELA35820 | Ankyrin repeat-containing protein |
| 2 | 189 | ELA25228 | Ankyrin repeat protein |
| 2 | 26 | ELA33919 | Laccase |
| 6 | 76 | ELA24504 | Major facilitator superfamily transporter |
| 11 | 232 | ELA37190 | Catalase |
新窗口打开|下载CSV
图4
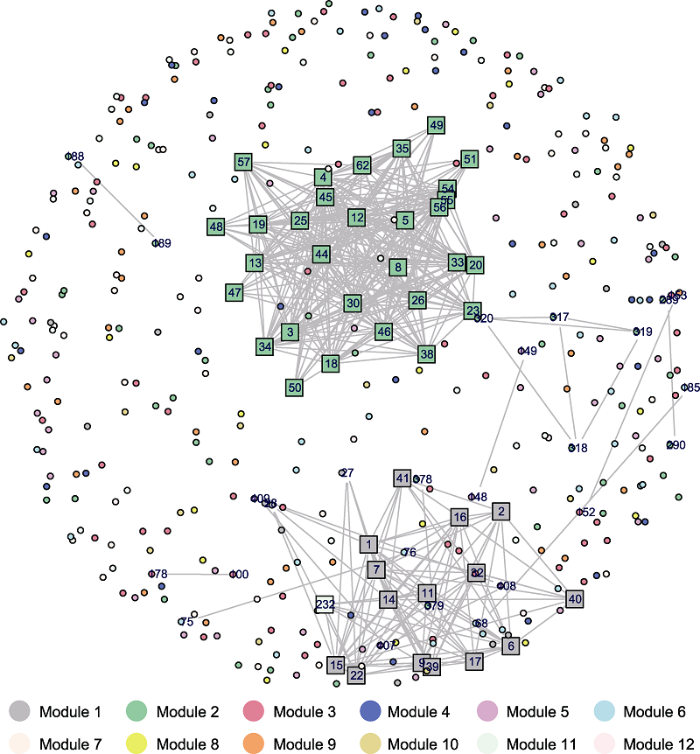 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4差异表达基因聚类模块内基因间调控网络重构建
不同颜色对应不同的聚类模块。方框中的数字代表核心模块中相互作用的基因编号, 12个核心基因包含其中。
Figure 4Inter-module regulatory network reconstruction of differentially expressed genes
Different colors correspond to different clustered modules. The numbers in the boxes represent the interacting gene numbers in core modules. The 12 core genes are included.
将功能注释与基因调控网络的拓扑结构相结合, 发现聚类模块1中基因参与了转录和二萜类生物合成过程。其中核心基因包括pep1和细胞色素P450 (cytochrome p450, CYP)编码基因(表3)。研究发现, CYP参与真菌体内多种代谢过程, 并且通过参与细胞结构和激素等物质的生物合成调节真菌的生长(Rendic and Guengerich, 2015)。
聚类模块2是瞬时响应损伤胁迫的代表模块。GO富集分析表明, 其基因主要参与电子转移活性、血红素结合过程和次生代谢产物的生物合成过程, 共包含74个差异表达基因。结合基因间的调控网络和聚类富集结果, 我们筛选到7个核心基因。典型核心基因如聚酮化合物合酶PKSS (polyketide synthase)及含锚蛋白重复序列蛋白AKR2A (ankyrin repeat-containing protein)编码基因在细胞代谢中起重要作用。研究表明, PKSS参与调控真菌体内气生菌丝的生长与融合(Shimizu et al., 2016), 并在真菌体内诱导各种小分子信号过程, 从而诱导细胞的分化和再生(Bhetariya et al., 2016)。AKR2A调控由抗病性和应激反应共同参与的抗氧化作用, 是植物生长发育中不可或缺的蛋白(Yan et al., 2002)。这些模块间的调控网络表明模块内的基因协同作用响应损伤胁迫。
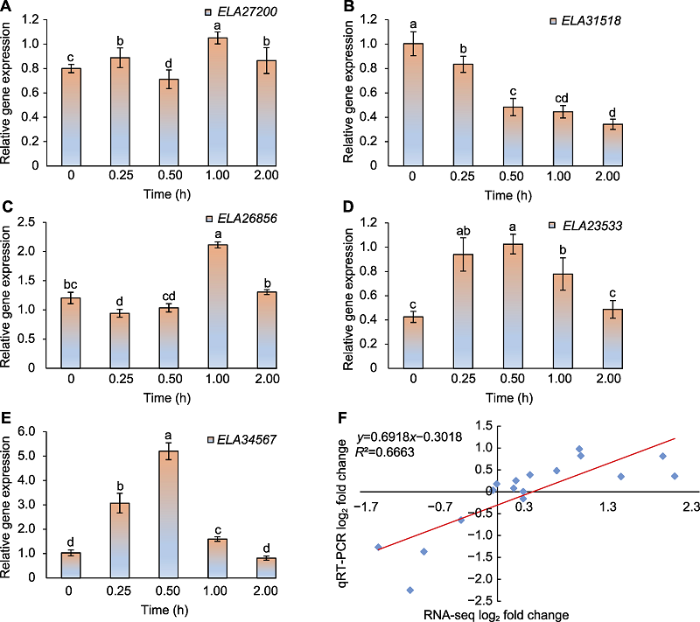
2.6 应答损伤胁迫基因的动态表达模式验证
为了验证测序结果的可靠性和应答损伤胁迫基因的动态表达模式, 我们随机选取5个mRNAs基因进行实时定量PCR检测。结果表明, 尽管基因表达水平有差异, 但从5个基因的表达趋势来看, 所选基因具有瞬时动态表达模式(图5A-E)。此外, qRT-PCR与RNA-seq数据在损伤后的时间点内线性相关系数为0.816 3 (图5F), 说明两组数据的相关性很高。本研究利用转录组测序获得的基因表达结果较为准确, 可为后续研究提供可靠的数据来源。图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图55个核心基因的动态表达模式及数据相关性
不同小写字母表示各处理间差异显著(P<0.05)。
Figure 5The dynamic expression patterns and data correlation of five core genes
Different lowercase letters indicate significant differences among treatments (P<0.05).
2.7 讨论
应答机械损伤的相关研究多与组织的再生和修复有关(Chang et al., 2009)。目前关于丝状真菌对损伤应答的认知仅限于损伤后子实体上隔膜气孔的封闭, 以防止细胞质内含物过度流失和细胞死亡(Jedd, 2011)。而真菌的损伤胁迫响应是一个动态过程, 单个时间点的静态数据很难描述这一复杂的生物学过程。目前, 对生物较长时间响应胁迫过程的研究已经较为深入(Sun et al., 2009), 而对胁迫后几分钟至数小时内生物体的瞬时生理变化正逐渐受到重视。Munns (2010)认为植物在盐胁迫下数小时已经开始产生变化, 包括细胞壁的改变、信号转导以及生长速率减慢等瞬时响应。周韬(2014)发现与长时间(60天)胁迫相比, 胁迫后6小时胡杨(Populus euphratica)叶片中SOD酶活性和根部ABA含量显著升高。Hernández-O?ate等(2012)对丝状真菌木霉菌菌丝体损伤后15、30、60分钟获得的RNA样品进行RNA-seq分析, 结果表明, 损伤会引起细胞内钙浓度升高, 激活参与氧化应激反应的基因, 其中16个编码ROS清除蛋白的基因在损伤后短时间内被强烈抑制。在信号转导方面, 范玲玲等(2010)探讨了星星草(Puccinellia tenuiflora)在响应NaHCO3胁迫中Ca2+浓度和Ca2+-ATPase活性的变化。对损伤胁迫下瞬时反应机制和生理变化的研究有助于全面理解胶孢炭疽菌产孢和生长发育中存在的通路(章树桃等, 2017)。本实验选取损伤2小时内的5个时间点进行转录组分析。聚类结果表明, 除聚类模块3和6, 其它模块基因表达量在处理后先表现为下降, 这可能是由于在胁迫开始时, 真菌中大部分生理功能会下降, 如生长受抑制或代谢活性降低, 偏离正常状态以适应多变的环境。在损伤后1-2小时, 真菌在逆境条件下继续生长, 尽管生长速度变缓慢, 但仍非常重要。网络关系拓扑图显示, 基因间的调控关系主要集中在聚类模块1和2中, 其中涉及一些与分生孢子生长、信号传递、细胞增殖分化、损伤防御及抗逆性相关的基因和代谢途径。
真菌在生长发育过程中受到逆境胁迫时, 会启动自身防御机制, 重新建立新的稳态条件并恢复生长, 这期间所发生的系列反应都是真菌生存的关键。逆境胁迫可大幅增加ROX的积累, 引起氧化应激, 从而抑制生长甚至诱导生物体死亡(Wakabayashi et al., 2012)。差异基因分析结果表明, 编码Pep1和锚蛋白重复序列蛋白的基因在激活自身抗氧化防御系统中发挥关键作用, 诱导细胞间信号转导(Sakamoto et al., 2008; Yamaguchi et al., 2010)。过氧化氢酶(catalase, CAT)是细胞防御活性氧的第一道防线。研究表明, 丝状真菌绿僵菌中cat1基因的过表达可增强过氧化氢酶活性, 加快分生孢子萌发和菌丝生长(Hernandez et al., 2010)。
富集分析结果表明, 在分子功能、细胞组分及生物学过程中占较高比重的注释有氧化还原酶活性、膜整体组分和跨膜转运蛋白活性。从这些功能注释中可以看出分生孢子生长发育过程中涉及一系列物质代谢相关途径。例如, 短链脱氢酶还原酶家族(SDR)具有功能折叠特性, 在脂质、氨基酸等物质代谢和氧化还原机制中起关键作用(Persson et al., 2008)。胶孢炭疽菌菌丝在损伤后2小时内SDR表达量呈上升趋势, 推测其可能通过传递代谢信号对损伤细胞进行修复, 从而控制代谢平衡(Kavanagh et al., 2008)。
在核心基因中也发现了很多参与真菌菌丝生长分化、细胞壁合成重塑及黑色素合成相关的基因, 包括响应胁迫过程中负责细胞壁合成和重塑的NAD脱水酶(nad-dependent epimerase dehydratase) (Zeng et al., 2014)。5.8s-rRNA突变可抑制细胞生长, 影响体外蛋白质合成(Elela and Nazar, 1997)。漆酶(LAC)在菌丝营养生长、分生孢子萌发、附着体形成和附着体的穿透性方面起重要作用(Chi et al., 2009)。内膜蛋白(integral membrane protein)负责真菌细胞壁的构建和子囊壁的组装。此外, 一些基因也调控真菌体内分子信号和大分子物质的传递。在丝状真菌中, 主要促进超家族转运蛋白MFS (major facilitator superfamily transporters)将包括糖、氨基酸、离子载体、有机物及无机阴离子在内的多种底物跨膜转运, 被称为二级活性转运蛋白(Saier et al., 2013)。
综上所述, 通过对转录组测序结果中的差异表达基因和基因间调控网络进行分析, 本研究共筛选到12个胶孢炭疽菌瞬时响应损伤胁迫核心基因, 这些核心基因不仅为芒果胶孢炭疽菌产孢和致病机理研究奠定了基础, 也为后续解析胶孢炭疽菌应答外源胁迫和诱导产孢的分子机理提供了重要参考。
(责任编辑: 白羽红)
致谢
北京林业大学姜立波老师在转录组数据分析方面给予了指导, 海南大学热带作物学院安邦老师和张贝同学在实验方案设计及方法探索上提供了帮助, 特此致谢。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/0092-8674(88)90198-5URLPMID:3293800 [本文引用: 1]

The brlA gene of A. nidulans mediates the developmental switch from the indeterminate, apical growth pattern of vegetative cells to the budding growth pattern of conidiophores. brlA encodes a 432 amino acid polypeptide containing two directly repeated motifs resembling the Zn(II) coordination sites first recognized in Xenopus TFIIIA. Misscheduled expression of brlA in vegetative cells results in transcriptional activation of developmentally regulated genes, cessation of unidirectional hyphal growth, initiation of cellular transformations resembling those that occur during normal conidiophore development, and production of viable conidiospores. We propose that BRLA is a nucleic acid-binding protein whose expression in vegetative cells is sufficient to induce sporulation through its role in regulating expression of conidiation-specific genes.
[本文引用: 1]
DOI:10.1387/ijdb.072556ccURL [本文引用: 1]
DOI:10.1371/journal.ppat.1000401URLPMID:19390617 [本文引用: 1]
For successful colonization and further reproduction in host plants, pathogens need to overcome the innate defenses of the plant. We demonstrate that a novel pathogenicity gene, DES1, in Magnaporthe oryzae regulates counter-defenses against host basal resistance. The DES1 gene was identified by screening for pathogenicity-defective mutants in a T-DNA insertional mutant library. Bioinformatic analysis revealed that this gene encodes a serine-rich protein that has unknown biochemical properties, and its homologs are strictly conserved in filamentous Ascomycetes. Targeted gene deletion of DES1 had no apparent effect on developmental morphogenesis, including vegetative growth, conidial germination, appressorium formation, and appressorium-mediated penetration. Conidial size of the mutant became smaller than that of the wild type, but the mutant displayed no defects on cell wall integrity. The Deltades1 mutant was hypersensitive to exogenous oxidative stress and the activity and transcription level of extracellular enzymes including peroxidases and laccases were severely decreased in the mutant. In addition, ferrous ion leakage was observed in the Deltades1 mutant. In the interaction with a susceptible rice cultivar, rice cells inoculated with the Deltades1 mutant exhibited strong defense responses accompanied by brown granules in primary infected cells, the accumulation of reactive oxygen species (ROS), the generation of autofluorescent materials, and PR gene induction in neighboring tissues. The Deltades1 mutant displayed a significant reduction in infectious hyphal extension, which caused a decrease in pathogenicity. Notably, the suppression of ROS generation by treatment with diphenyleneiodonium (DPI), an inhibitor of NADPH oxidases, resulted in a significant reduction in the defense responses in plant tissues challenged with the Deltades1 mutant. Furthermore, the Deltades1 mutant recovered its normal infectious growth in DPI-treated plant tissues. These results suggest that DES1 functions as a novel pathogenicity gene that regulates the activity of fungal proteins, compromising ROS-mediated plant defense.
DOI:10.1093/bioinformatics/bti610URLPMID:16081474 [本文引用: 1]
SUMMARY: We present here Blast2GO (B2G), a research tool designed with the main purpose of enabling Gene Ontology (GO) based data mining on sequence data for which no GO annotation is yet available. B2G joints in one application GO annotation based on similarity searches with statistical analysis and highlighted visualization on directed acyclic graphs. This tool offers a suitable platform for functional genomics research in non-model species. B2G is an intuitive and interactive desktop application that allows monitoring and comprehension of the whole annotation and analysis process. AVAILABILITY: Blast2GO is freely available via Java Web Start at http://www.blast2go.de. SUPPLEMENTARY MATERIAL: http://www.blast2go.de -> Evaluation.
DOI:10.1093/nar/25.9.1788URLPMID:9108162 [本文引用: 1]
Studies on the inhibition of protein synthesis by specific anti 5.8S rRNA oligonucleotides have suggested that this RNA plays an important role in eukaryotic ribosome function. Mutations in the 5. 8S rRNA can inhibit cell growth and compromise protein synthesis in vitro . Polyribosomes from cells expressing these mutant 5.8S rRNAs are elevated in size and ribosome-associated tRNA. Cell free extracts from these cells also are more sensitive to antibiotics which act on the 60S ribosomal subunit by inhibiting elongation. The extracts are especially sensitive to cycloheximide and diphtheria toxin which act specifically to inhibit translocation. Studies of ribosomal proteins show no reproducible changes in the core proteins, but reveal reduced levels of elongation factors 1 and 2 only in ribosomes which contain large amounts of mutant 5.8S rRNA. Polyribosomes from cells which are severely inhibited, but contain little mutant 5.8S rRNA, do not show the same reductions in the elongation factors, an observation which underlines the specific nature of the change. Taken together the results demonstrate a defined and critical function for the 5.8S rRNA, suggesting that this RNA plays a role in ribosome translocation.
DOI:10.1073/pnas.1205415109URLPMID:22753475 [本文引用: 1]
We report the genome sequence of melon, an important horticultural crop worldwide. We assembled 375 Mb of the double-haploid line DHL92, representing 83.3% of the estimated melon genome. We predicted 27,427 protein-coding genes, which we analyzed by reconstructing 22,218 phylogenetic trees, allowing mapping of the orthology and paralogy relationships of sequenced plant genomes. We observed the absence of recent whole-genome duplications in the melon lineage since the ancient eudicot triplication, and our data suggest that transposon amplification may in part explain the increased size of the melon genome compared with the close relative cucumber. A low number of nucleotide-binding site-leucine-rich repeat disease resistance genes were annotated, suggesting the existence of specific defense mechanisms in this species. The DHL92 genome was compared with that of its parental lines allowing the quantification of sequence variability in the species. The use of the genome sequence in future investigations will facilitate the understanding of evolution of cucurbits and the improvement of breeding strategies.
DOI:10.1007/s00253-010-2517-3URLPMID:20361327 [本文引用: 1]
Catalases and peroxidases are the most important enzymes that degrade hydrogen peroxide into water and oxygen. These enzymes and superoxide dismutase are the first lines of cell defense against reactive oxygen species. Metarhizium anisopliae displays an increase in catalase-peroxidase activity during germination and growth. To determine the importance of catalase during the invasion process of M. anisopliae, we isolated the cat1 gene. cat1 cDNA expression in Escherichia coli and the subsequent purification of the protein confirmed that the cat1 gene codes for a monofunctional catalase. Expression analysis of this gene by RT-PCR from RNA isolated from fungus grown in liquid cultures showed a decrease in the expression level of the cat1 gene during germination and an increase during mycelium growth. The expression of this gene in the fungus during the infection process of the larvae of Plutella xylostella also showed a significant increase during invasive growth. Transgenic strains overexpressing the cat1 gene had twice the catalase activity of the wild-type strain. This increase in catalase activity was accompanied by a higher level of resistance to exogenous hydrogen peroxide and a reduction in the germination time. This improvement was also observed during the infection of P. xylostella larvae. M. anisopliae transgenic strains overexpressing the cat1 gene grew and spread faster in the soft tissue of the insect, reducing the time to death of the insect by 25% and the dose required to kill 50% of the population 14-fold.
DOI:10.1073/pnas.1209396109URLPMID:22927395 [本文引用: 2]
A conserved injury-defense mechanism is present in plants and animals, in which the production of reactive oxygen species (ROS) and lipid metabolism are essential to the response. Here, we describe that in the filamentous fungus Trichoderma atroviride, injury results in the formation of asexual reproduction structures restricted to regenerating cells. High-throughput RNA-seq analyses of the response to injury in T. atroviride suggested an oxidative response and activation of calcium-signaling pathways, as well as the participation of lipid metabolism, in this phenomenon. Gene-replacement experiments demonstrated that injury triggers NADPH oxidase (Nox)-dependent ROS production and that Nox1 and NoxR are essential for asexual development in response to damage. We further provide evidence of H(2)O(2) and oxylipin production that, as in plants and animals, may act as signal molecules in response to injury in fungi, suggesting that the three kingdoms share a conserved defense-response mechanism.
DOI:10.1016/j.foodchem.2015.03.046URLPMID:25863626 [本文引用: 1]
The large amount of waste produced by the food industries causes serious environmental problems and also results in economic losses if not utilized effectively. Different research reports have revealed that food industry by-products can be good sources of potentially valuable bioactive compounds. As such, the mango juice industry uses only the edible portions of the mangoes, and a considerable amount of peels and seeds are discarded as industrial waste. These mango by-products come from the tropical or subtropical fruit processing industries. Mango by-products, especially seeds and peels, are considered to be cheap sources of valuable food and nutraceutical ingredients. The main uses of natural food ingredients derived from mango by-products are presented and discussed, and the mainstream sectors of application for these by-products, such as in the food, pharmaceutical, nutraceutical and cosmetic industries, are highlighted.
DOI:10.1016/j.tcb.2010.09.001URLPMID:20888233 [本文引用: 1]
Peroxisome-derived Woronin bodies of the Ascomycota phyla, and the endoplasmic reticulum (ER)-derived septal pore cap (SPC) of the Basidiomycota, are both fungal organelles that prevent cytoplasmic bleeding when multicellular hyphal filaments are wounded. Analysis of Woronin body constituent proteins suggests that these organelles evolved in part through gene duplication and co-opting of non-essential genes for new functions, indicating that new organelles can arise through typical evolutionary mechanisms. Interestingly, clades possessing the Woronin body and SPC also produce the largest and most complex multicellular fungal reproductive structures. Certain Woronin body and SPC mutants have defects in growth and development, suggesting functions beyond cellular wound healing. I argue that studying these specialized systems will help to reveal the basis for fungal diversity and provide general principles for co-evolution of organelles and multicellular complexity.
DOI:10.1093/nar/gkt1016URLPMID:24174544 [本文引用: 1]
With the aim to provide a resource for functional and evolutionary study of plant transcription factors (TFs), we updated the plant TF database PlantTFDB to version 3.0 (http://planttfdb.cbi.pku.edu.cn). After refining the TF classification pipeline, we systematically identified 129 288 TFs from 83 species, of which 67 species have genome sequences, covering main lineages of green plants. Besides the abundant annotation provided in the previous version, we generated more annotations for identified TFs, including expression, regulation, interaction, conserved elements, phenotype information, expert-curated descriptions derived from UniProt, TAIR and NCBI GeneRIF, as well as references to provide clues for functional studies of TFs. To help identify evolutionary relationship among identified TFs, we assigned 69 450 TFs into 3924 orthologous groups, and constructed 9217 phylogenetic trees for TFs within the same families or same orthologous groups, respectively. In addition, we set up a TF prediction server in this version for users to identify TFs from their own sequences.
DOI:10.1007/s00018-008-8588-yURLPMID:19011750 [本文引用: 1]
Short-chain dehydrogenases/reductases (SDRs) constitute a large family of NAD(P)(H)-dependent oxidoreductases, sharing sequence motifs and displaying similar mechanisms. SDR enzymes have critical roles in lipid, amino acid, carbohydrate, cofactor, hormone and xenobiotic metabolism as well as in redox sensor mechanisms. Sequence identities are low, and the most conserved feature is an alpha/beta folding pattern with a central beta sheet flanked by 2 - 3 alpha-helices from each side, thus a classical Rossmannfold motif for nucleotide binding. The conservation of this element and an active site, often with an Asn-Ser-Tyr-Lys tetrad, provides a platform for enzymatic activities encompassing several EC classes, including oxidoreductases, epimerases and lyases. The common mechanism is an underlying hydride and proton transfer involving the nicotinamide and typically an active site tyrosine residue, whereas substrate specificity is determined by a variable C-terminal segment. Relationships exist with bacterial haloalcohol dehalogenases, which lack cofactor binding but have the active site architecture, emphasizing the versatility of the basic fold in also generating hydride transfer-independent lyases. The conserved fold and nucleotide binding emphasize the role of SDRs as scaffolds for an NAD(P)(H) redox sensor system, of importance to control metabolic routes, transcription and signalling.
DOI:10.1038/nmeth.1923URLPMID:22388286 [本文引用: 1]
As the rate of sequencing increases, greater throughput is demanded from read aligners. The full-text minute index is often used to make alignment very fast and memory-efficient, but the approach is ill-suited to finding longer, gapped alignments. Bowtie 2 combines the strengths of the full-text minute index with the flexibility and speed of hardware-accelerated dynamic programming algorithms to achieve a combination of high speed, sensitivity and accuracy.
DOI:10.1006/meth.2001.1262URLPMID:11846609 [本文引用: 1]
The two most commonly used methods to analyze data from real-time, quantitative PCR experiments are absolute quantification and relative quantification. Absolute quantification determines the input copy number, usually by relating the PCR signal to a standard curve. Relative quantification relates the PCR signal of the target transcript in a treatment group to that of another sample such as an untreated control. The 2(-Delta Delta C(T)) method is a convenient way to analyze the relative changes in gene expression from real-time quantitative PCR experiments. The purpose of this report is to present the derivation, assumptions, and applications of the 2(-Delta Delta C(T)) method. In addition, we present the derivation and applications of two variations of the 2(-Delta Delta C(T)) method that may be useful in the analysis of real-time, quantitative PCR data.
DOI:10.1093/nar/gks042URLPMID:22287627 [本文引用: 1]
A flexible statistical framework is developed for the analysis of read counts from RNA-Seq gene expression studies. It provides the ability to analyse complex experiments involving multiple treatment conditions and blocking variables while still taking full account of biological variation. Biological variation between RNA samples is estimated separately from the technical variation associated with sequencing technologies. Novel empirical Bayes methods allow each gene to have its own specific variability, even when there are relatively few biological replicates from which to estimate such variability. The pipeline is implemented in the edgeR package of the Bioconductor project. A case study analysis of carcinoma data demonstrates the ability of generalized linear model methods (GLMs) to detect differential expression in a paired design, and even to detect tumour-specific expression changes. The case study demonstrates the need to allow for gene-specific variability, rather than assuming a common dispersion across genes or a fixed relationship between abundance and variability. Genewise dispersions de-prioritize genes with inconsistent results and allow the main analysis to focus on changes that are consistent between biological replicates. Parallel computational approaches are developed to make non-linear model fitting faster and more reliable, making the application of GLMs to genomic data more convenient and practical. Simulations demonstrate the ability of adjusted profile likelihood estimators to return accurate estimators of biological variability in complex situations. When variation is gene-specific, empirical Bayes estimators provide an advantageous compromise between the extremes of assuming common dispersion or separate genewise dispersion. The methods developed here can also be applied to count data arising from DNA-Seq applications, including ChIP-Seq for epigenetic marks and DNA methylation analyses.
[本文引用: 1]
DOI:10.1007/s00018-008-8587-zURL [本文引用: 1]
DOI:10.1007/s11947-017-1891-6URL [本文引用: 1]
DOI:10.1093/treephys/tpr015URLPMID:21427158 [本文引用: 1]
Populus euphratica is well-adapted to extreme desert environments and is an important model species for studying the effects of abiotic stresses on trees. Here we present the first deep transcriptomic analysis of this species. To maximize representation of conditional transcripts, mRNA was obtained from living tissues of desert-grown trees and two types of callus (salt-stressed and unstressed). De novo assembly generated 86,777 Unigenes using Solexa sequence data. These sequences covered 92% of previously reported P. euphratica expressed sequence tags (ESTs) and 90% of the TIGR poplar ESTs, and a total of 58,499 high-quality unique sequences were annotated by BLAST similarity searches against public databases. We found that 27% of the total Unigenes were differentially expressed (up- or down-regulated) in response to salt stress in P. euphratica callus. These differentially expressed genes are mainly involved in transport, transcription, cellular communication and metabolism. In addition, we found that numerous putative genes involved in ABA regulation and biosynthesis were also differentially regulated. This study represents the deepest transcriptomic and gene-annotation analysis of P. euphratica to date. The genetic knowledge acquired should be very useful for future studies of the molecular adaptation of this tree species to abiotic stress and facilitate genetic manipulation of other poplar species.
DOI:10.1021/tx500444eURL [本文引用: 1]
DOI:10.1007/s13197-019-03971-8URLPMID:31741523 [本文引用: 1]
The marine bacterium Stenotrophomonas rhizophila was assessed in vitro and in vivo as biocontrol agent against anthracnose disease of mango fruit caused by Colletotrichum gloeosporioides. The results showed that in vitro inhibition of the colony diameter and spore germination of the phytopathogen was due to the production of VOCs, competition for nutrients, and lytic enzymes. When a concentration of 1 x 10(8) cells ml(-1) of the antagonist bacterium was applied to the fruit, disease incidence was reduced by 95%, and the lesion diameter of anthracnose decreased by 85%, which offered greater protection than the synthetic fungicide. This is the first report of antagonistic mechanisms of the marine bacterium S. rhizophila against anthracnose disease in mango, which in this study was found to be more effective than the synthetic fungicide.
DOI:10.1093/bioinformatics/btp616URL [本文引用: 1]
DOI:10.1093/nar/gkt1097URL [本文引用: 1]
DOI:10.1111/j.1365-313X.2008.03614.xURLPMID:18643991 [本文引用: 1]
Salt stress and abscisic acid (ABA) induce accumulation of reactive oxygen species (ROS) in plant cells. ROS not only act as second messengers for the activation of salt-stress responses, but also have deleterious effects on plant growth due to their cytotoxicity. Therefore, the timing and degree of activation of ROS-producing or ROS-scavenging enzymes must be tightly regulated under salt-stress conditions. We identified a novel locus of Arabidopsis, designated itn1 (increased tolerance to NaCl1), whose disruption leads to increased salt-stress tolerance in vegetative tissues. ITN1 encodes a transmembrane protein with an ankyrin-repeat motif that has been implicated in diverse cellular processes such as signal transduction. Comparative microarray analysis between wild-type and the itn1 mutant revealed that induction of genes encoding the ROS-producing NADPH oxidases (RBOHC and RBOHD) under salt-stress conditions was suppressed in the mutant. This suppression was accompanied by a corresponding reduction in ROS accumulation. The ABA-induced expression of RBOHC and RBOHD was also suppressed in the mutant, as was the case for RD29A, an ABA-inducible marker gene. However, the ABA-induced expression of another marker gene, RD22, was not impaired in the mutant. These results suggest that the itn1 mutation partially impairs ABA signaling pathways, possibly leading to the reduction in ROS accumulation under salt-stress conditions. We discuss the possible mechanisms underlying the salt-tolerant phenotype of the itn1 mutant.
DOI:10.1016/S2095-3119(18)62128-8URL [本文引用: 1]
DOI:10.1002/cbic.201600522URLPMID:27862822 [本文引用: 1]
Polyketide synthases (PKSs) catalyze the sequential condensation of simple acetate units to produce a large class of natural products, including pharmacologically valuable compounds. PKSs are classified into three types on the basis of their domain structures; type III PKSs have the simplest domain structure, although their products have various structures and functions. The sequence-function relationship is fundamental for predicting enzyme functions, but it has not been well investigated in type III PKSs to date. Consequently, the current methods for predicting type III PKS functions are still immature in comparison with those that target type I/II PKSs. In this review we summarize the current functional and phylogenomic knowledge about type III PKSs and propose a new classification of their enzymatic reactions. We also discuss possible directions for the development of better computational tools for functional prediction of type III PKS homologues.
DOI:10.1093/bioinformatics/btt632URLPMID:24191069 [本文引用: 1]
MOTIVATION: RNA-seq technology has been widely adopted as an attractive alternative to microarray-based methods to study global gene expression. However, robust statistical tools to analyze these complex datasets are still lacking. By grouping genes with similar expression profiles across treatments, cluster analysis provides insight into gene functions and networks, and hence is an important technique for RNA-seq data analysis. RESULTS: In this manuscript, we derive clustering algorithms based on appropriate probability models for RNA-seq data. An expectation-maximization algorithm and another two stochastic versions of expectation-maximization algorithms are described. In addition, a strategy for initialization based on likelihood is proposed to improve the clustering algorithms. Moreover, we present a model-based hybrid-hierarchical clustering method to generate a tree structure that allows visualization of relationships among clusters as well as flexibility of choosing the number of clusters. Results from both simulation studies and analysis of a maize RNA-seq dataset show that our proposed methods provide better clustering results than alternative methods such as the K-means algorithm and hierarchical clustering methods that are not based on probability models. AVAILABILITY AND IMPLEMENTATION: An R package, MBCluster.Seq, has been developed to implement our proposed algorithms. This R package provides fast computation and is publicly available at http://www.r-project.org
DOI:10.1104/pp.108.129494URLPMID:19028881 [本文引用: 1]
Using the scanning ion-selective electrode technique, fluxes of H+, Na+, and Cl- were investigated in roots and derived protoplasts of salt-tolerant Populus euphratica and salt-sensitive Populus popularis 35-44 (P. popularis). Compared to P. popularis, P. euphratica roots exhibited a higher capacity to extrude Na+ after a short-term exposure to 50 mM NaCl (24 h) and a long term in a saline environment of 100 mM NaCl (15 d). Root protoplasts, isolated from the long-term-stressed P. euphratica roots, had an enhanced Na+ efflux and a correspondingly increased H+ influx, especially at an acidic pH of 5.5. However, the NaCl-induced Na+/H+ exchange in root tissues and cells was inhibited by amiloride (a Na+/H+ antiporter inhibitor) or sodium orthovanadate (a plasma membrane H+-ATPase inhibitor). These results indicate that the Na+ extrusion in stressed P. euphratica roots is the result of an active Na+/H+ antiport across the plasma membrane. In comparison, the Na+/H+ antiport system in salt-stressed P. popularis roots was insufficient to exclude Na+ at both the tissue and cellular levels. Moreover, salt-treated P. euphratica roots retained a higher capacity for Cl- exclusion than P. popularis, especially during a long term in high salinity. The pattern of NaCl-induced fluxes of H+, Na+, and Cl- differs from that caused by isomotic mannitol in P. euphratica roots, suggesting that NaCl-induced alternations of root ion fluxes are mainly the result of ion-specific effects.
DOI:10.1093/nar/gku1179URLPMID:25428369 [本文引用: 1]
The Gene Ontology (GO; http://www.geneontology.org) is a community-based bioinformatics resource that supplies information about gene product function using ontologies to represent biological knowledge. Here we describe improvements and expansions to several branches of the ontology, as well as updates that have allowed us to more efficiently disseminate the GO and capture feedback from the research community. The Gene Ontology Consortium (GOC) has expanded areas of the ontology such as cilia-related terms, cell-cycle terms and multicellular organism processes. We have also implemented new tools for generating ontology terms based on a set of logical rules making use of templates, and we have made efforts to increase our use of logical definitions. The GOC has a new and improved web site summarizing new developments and documentation, serving as a portal to GO data. Users can perform GO enrichment analysis, and search the GO for terms, annotations to gene products, and associated metadata across multiple species using the all-new AmiGO 2 browser. We encourage and welcome the input of the research community in all biological areas in our continued effort to improve the Gene Ontology.
DOI:10.1038/nbt.1621URLPMID:20436464 [本文引用: 1]
High-throughput mRNA sequencing (RNA-Seq) promises simultaneous transcript discovery and abundance estimation. However, this would require algorithms that are not restricted by prior gene annotations and that account for alternative transcription and splicing. Here we introduce such algorithms in an open-source software program called Cufflinks. To test Cufflinks, we sequenced and analyzed >430 million paired 75-bp RNA-Seq reads from a mouse myoblast cell line over a differentiation time series. We detected 13,692 known transcripts and 3,724 previously unannotated ones, 62% of which are supported by independent expression data or by homologous genes in other species. Over the time series, 330 genes showed complete switches in the dominant transcription start site (TSS) or splice isoform, and we observed more subtle shifts in 1,304 other genes. These results suggest that Cufflinks can illuminate the substantial regulatory flexibility and complexity in even this well-studied model of muscle development and that it can improve transcriptome-based genome annotation.
DOI:10.1016/j.jplph.2011.10.002URLPMID:22118877 [本文引用: 1]
The relationship between the formation of cell wall-bound ferulic acid (FA) and diferulic acid (DFA) and the change in activities of phenylalanine ammonia-lyase (PAL) and cell wall-bound peroxidase (CW-PRX) was studied in rice shoots. The length and the fresh mass of shoots increased during the growth period from day 4 to 6, while coleoptiles ceased elongation growth on day 5. The amounts of FA and DFA isomers as well as cell wall polysaccharides continued to increase during the whole period. The activities of PAL and CW-PRX greatly increased in the same manner during the period. There were close correlations between the PAL activity and ferulate content or between the CW-PRX activity and DFA content. The expression levels of investigated genes for PAL and putative CW-PRX showed good accordance with the activities of these enzymes. These results suggest that increases in PAL and CW-PRX activities are cooperatively involved in the formation of ferulate network in cell walls of rice shoots and that investigated genes may be, at least in part, associated with the enzyme activities. The substantial increase in such network probably causes the maturation of cell walls and thus the cessation of elongation growth of coleoptiles.
DOI:10.1016/j.ydbio.2009.10.032URLPMID:19874814 [本文引用: 1]
Development is regulated by dynamic patterns of gene expression, which are orchestrated through the action of complex gene regulatory networks (GRNs). Substantial progress has been made in modeling transcriptional regulation in recent years, including qualitative
DOI:10.1093/nar/gkr483URLPMID:21715386 [本文引用: 1]
High-throughput experimental technologies often identify dozens to hundreds of genes related to, or changed in, a biological or pathological process. From these genes one wants to identify biological pathways that may be involved and diseases that may be implicated. Here, we report a web server, KOBAS 2.0, which annotates an input set of genes with putative pathways and disease relationships based on mapping to genes with known annotations. It allows for both ID mapping and cross-species sequence similarity mapping. It then performs statistical tests to identify statistically significantly enriched pathways and diseases. KOBAS 2.0 incorporates knowledge across 1327 species from 5 pathway databases (KEGG PATHWAY, PID, BioCyc, Reactome and Panther) and 5 human disease databases (OMIM, KEGG DISEASE, FunDO, GAD and NHGRI GWAS Catalog). KOBAS 2.0 can be accessed at http://kobas.cbi.pku.edu.cn.
DOI:10.1105/tpc.109.068874URLPMID:20179141 [本文引用: 1]
Pep1 is a 23-amino acid peptide that enhances resistance to a root pathogen, Pythium irregulare. Pep1 and its homologs (Pep2 to Pep7) are endogenous amplifiers of innate immunity of Arabidopsis thaliana that induce the transcription of defense-related genes and bind to PEPR1, a plasma membrane leucine-rich repeat (LRR) receptor kinase. Here, we identify a plasma membrane LRR receptor kinase, designated PEPR2, that has 76% amino acid similarity to PEPR1, and we characterize its role in the perception of Pep peptides and defense responses. Both PEPR1 and PEPR2 were transcriptionally induced by wounding, treatment with methyl jasmonate, Pep peptides, and pathogen-associated molecular patterns. The effects of Pep1 application on defense-related gene induction and enhancement of resistance to Pseudomonas syringae pv tomato DC3000 were partially reduced in single mutants of PEPR1 and PEPR2 and abolished completely in double mutants. Photoaffinity labeling and binding assays using transgenic tobacco (Nicotiana tabacum) cells expressing PEPR1 and PEPR2 clearly demonstrated that PEPR1 is a receptor for Pep1-6 and that PEPR2 is a receptor for Pep1 and Pep2. Our analysis demonstrates differential binding affinities of two receptors with a family of peptide ligands and the corresponding physiological effects of the specific receptor-ligand interactions. Therefore, we demonstrate that, through perception of Peps, PEPR1 and PEPR2 contribute to defense responses in Arabidopsis.
DOI:10.1046/j.0960-7412.2001.01205.xURLPMID:11862948 [本文引用: 1]
The Arabidopsis ankyrin repeat-containing protein AKR2 was identified as a GF14(lambda)-interacting protein in a yeast two-hybrid screening (GF14(lambda) is a 14-3-3 protein). Reduced expression of AKR2 by using the antisense technique results in small necrotic areas in leaves accompanied by higher production of H2O2, similar to the hypersensitive response to pathogen infection in plant disease resistance. Transcripts of genes encoding pathogen-induced protein PR-1 (pathogen-related protein 1) and stress-responsive protein GST6 (glutathione S-transferase 6) are increased in antisense plants. The resistance to a bacterial pathogen infection was also increased by at least 10-fold in antisense plants. AKR2 also interacts with another GF14(lambda)-interacting protein, the ascorbate peroxidase 3 that scavenges H2O2 in plant cells. These data suggest that AKR2 may be a negative regulator of PR-1 expression, and is probably involved in the regulation of antioxidation metabolism that is shared by both disease resistance and stress responses.
DOI:10.1007/s00425-014-2077-3URLPMID:24819712 [本文引用: 1]
MAIN CONCLUSION: Rice plants employ two strategies to cope with Cr toxicity: immobilizing Cr ions into cell walls to reduce its translocation and activating antioxidant defense to mitigate Cr-induced oxidative stress. The investigation aimed at understanding the physiological and proteomic responses of rice seedlings to hexavalent chromium (Cr(6+)) stress was conducted using two rice genotypes, which differ in Cr tolerance and accumulation. Cr toxicity (200 microM) heavily increased the accumulation of H2O2 and [Formula: see text], enhanced lipid peroxidation, decreased cell viability and consequently inhibited rice plant growth. Proteomic analyses suggest that the response of rice proteome to Cr stress is genotype- and Cr dosage-dependent and tissue specific. Sixty-four proteins, which show more than fourfold difference under either two Cr levels, have been successfully identified. They are involved in a range of cellular processes, including cell wall synthesis, energy production, primary metabolism, electron transport and detoxification. Two proteins related to cell wall structure, NAD-dependent epimerase/dehydratase and reversibly glycosylated polypeptide were greatly up-regulated by Cr stress. Their enhancements coupled with callose accumulation by Cr suggest that cell wall is an important barrier for rice plants to resist Cr stress. Some enzymes involved in antioxidant defense, such as ferredoxin-NADP reductase, NADP-isocitrate dehydrogenase, glyoxalase I (Gly I) and glutamine synthetase 1 (GS1) have also been identified in response to Cr stress. However, they were only detected in Cr-tolerant genotype, indicating the genotypic difference in the capacity of activating the defense system to fight against Cr-induced oxidative stress. Overall, two strategies in coping with Cr stress in rice plants can be hypothesized: (i) immobilizing Cr ions into cell walls to reduce its translocation and (ii) activating antioxidant defense to mitigate Cr-induced oxidative stress.
NaHCO3胁迫下星星草根中Ca 2+与Ca 2+-ATPase的超微细胞化学定位
1
2010
... 目前, 对生物较长时间响应胁迫过程的研究已经较为深入(
柱花草胶孢炭疽菌基因组、转录组测序及致病相关基因CgALS分析研究
1
2016
... 胶孢炭疽菌(Colletotrichum gloeosporioides)是引起芒果炭疽病的主要病原菌, 该病菌是一种丝状真菌, 其寄主范围广, 可潜伏侵染果实的多个部位, 严重影响芒果的产量和品质, 危害极大(
芒果胶孢炭疽病菌遗传转化体系的建立
1
2004
... 许多丝状真菌通过分生孢子进行侵染和传播, 产生大量分生孢子是其进行侵染和扩散的前提(
芒果采后生理及贮藏保鲜技术研究进展
1
2011
... 芒果(Mangifera indica)属漆树科植物, 是热带和亚热带地区广泛种植的名贵水果, 被誉为“水果之王”(
芒果胶孢炭疽菌致病性的初步研究
1
2012
... 实验所用菌株为芒果胶孢炭疽菌(Colletotrichum gloeosporioides (Penz.) Penz.&Sacc.), 该病菌从自然发病且具有典型芒果炭疽病症状的芒果果皮上分离得到(
草莓胶孢炭疽菌CFEM候选效应子的生物信息学鉴定及其侵染过程中的转录分析
1
2017
... 胶孢炭疽菌(Colletotrichum gloeosporioides)是引起芒果炭疽病的主要病原菌, 该病菌是一种丝状真菌, 其寄主范围广, 可潜伏侵染果实的多个部位, 严重影响芒果的产量和品质, 危害极大(
新蚜虫疠霉菌丝和孢子阶段的转录组学分析
1
2017
... 目前, 对生物较长时间响应胁迫过程的研究已经较为深入(
盐胁迫下胡杨的生理响应及miRNA表达动态变化
1
2014
... 目前, 对生物较长时间响应胁迫过程的研究已经较为深入(
芒果采后生理生化的变化
1
1995
... 芒果(Mangifera indica)属漆树科植物, 是热带和亚热带地区广泛种植的名贵水果, 被誉为“水果之王”(
brlA is necessary and sufficient to direct conidiophore development in Aspergillus nidulans
1
1988
... 胶孢炭疽菌(Colletotrichum gloeosporioides)是引起芒果炭疽病的主要病原菌, 该病菌是一种丝状真菌, 其寄主范围广, 可潜伏侵染果实的多个部位, 严重影响芒果的产量和品质, 危害极大(
Phylogenetic and structural analysis of polyketide synthases in Aspergilli
1
2016
... 聚类模块2是瞬时响应损伤胁迫的代表模块.GO富集分析表明, 其基因主要参与电子转移活性、血红素结合过程和次生代谢产物的生物合成过程, 共包含74个差异表达基因.结合基因间的调控网络和聚类富集结果, 我们筛选到7个核心基因.典型核心基因如聚酮化合物合酶PKSS (polyketide synthase)及含锚蛋白重复序列蛋白AKR2A (ankyrin repeat-containing protein)编码基因在细胞代谢中起重要作用.研究表明, PKSS参与调控真菌体内气生菌丝的生长与融合(
Reptile scale paradigm: evo-devo, pattern formation and regeneration
1
2009
... 应答机械损伤的相关研究多与组织的再生和修复有关(
A novel pathogenicity gene is required in the rice blast fungus to suppress the basal defenses of the host
1
2009
... 在核心基因中也发现了很多参与真菌菌丝生长分化、细胞壁合成重塑及黑色素合成相关的基因, 包括响应胁迫过程中负责细胞壁合成和重塑的NAD脱水酶(nad-dependent epimerase dehydratase) (
Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research
1
2005
... 利用Illumina平台将测序所得的原始序列文件进行质量评估和可信度分析(
Role of the 5.8S rRNA in ribosome translocation
1
1997
... 在核心基因中也发现了很多参与真菌菌丝生长分化、细胞壁合成重塑及黑色素合成相关的基因, 包括响应胁迫过程中负责细胞壁合成和重塑的NAD脱水酶(nad-dependent epimerase dehydratase) (
The genome of melon (Cucumis melo L.)
1
2012
... 利用Illumina平台将测序所得的原始序列文件进行质量评估和可信度分析(
Catalase overexpression reduces the germination time and increases the pathogenicity of the fungus Metarhizium anisopliae
1
2010
... 真菌在生长发育过程中受到逆境胁迫时, 会启动自身防御机制, 重新建立新的稳态条件并恢复生长, 这期间所发生的系列反应都是真菌生存的关键.逆境胁迫可大幅增加ROX的积累, 引起氧化应激, 从而抑制生长甚至诱导生物体死亡(
An injury-response mechanism conserved across kingdoms determines entry of the fungus Trichoderma atroviride into development
2
2012
... 许多丝状真菌通过分生孢子进行侵染和传播, 产生大量分生孢子是其进行侵染和扩散的前提(
... 目前, 对生物较长时间响应胁迫过程的研究已经较为深入(
Mango ( Mangifera indica L.) by-products and their valuable components: a review
1
2015
... 芒果(Mangifera indica)属漆树科植物, 是热带和亚热带地区广泛种植的名贵水果, 被誉为“水果之王”(
Fungal evo-devo: organelles and multicellular complexity
1
2011
... 应答机械损伤的相关研究多与组织的再生和修复有关(
PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors
1
2014
... 芒果胶孢炭疽菌参考基因组下载自美国国家生物技术信息中心数据库(NCBI), 将每个样品的clean reads与参考基因组进行比对(
Medium-and short-chain dehydrogenase/reductase gene and protein families: the SDR superfamily: functional and structural diversity within a family of metabolic and regulatory enzymes
1
2008
... 富集分析结果表明, 在分子功能、细胞组分及生物学过程中占较高比重的注释有氧化还原酶活性、膜整体组分和跨膜转运蛋白活性.从这些功能注释中可以看出分生孢子生长发育过程中涉及一系列物质代谢相关途径.例如, 短链脱氢酶还原酶家族(SDR)具有功能折叠特性, 在脂质、氨基酸等物质代谢和氧化还原机制中起关键作用(
Fast gapped-read alignment with Bowtie 2
1
2012
... 利用Illumina平台将测序所得的原始序列文件进行质量评估和可信度分析(
Analysis of relative gene expression data using real-time quantitative PCR and the ${{2}^{-\Delta \Delta \text{CT}}}$ method
1
2001
... 随机选择5个基因进行实时荧光定量PCR分析.使用天根总RNA提取试剂盒提取胶孢炭疽菌菌丝RNA, 采用TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix (全式金)逆转录合成cDNA.以反转录产物为模板, Actin为内参, 采用 SuperReal PreMix Plus (SYBR Green)天根荧光定量试剂盒, 在ABI 7500 PCR仪中进行实时荧光定量检测.扩增程序如下: 95°C2分钟; 95°C10秒, 60°C1分钟, 45个循环.所有反应均进行3个生物学重复.采用2-ΔΔCT法进行相对定量分析(
Differential expression analysis of multifactor RNA-seq experiments with respect to biological variation
1
2012
... 芒果胶孢炭疽菌参考基因组下载自美国国家生物技术信息中心数据库(NCBI), 将每个样品的clean reads与参考基因组进行比对(
1
2010
... 目前, 对生物较长时间响应胁迫过程的研究已经较为深入(
Medium-and short-chain dehydrogenase/reductase gene and protein families: the MDR superfamily
1
2008
... 富集分析结果表明, 在分子功能、细胞组分及生物学过程中占较高比重的注释有氧化还原酶活性、膜整体组分和跨膜转运蛋白活性.从这些功能注释中可以看出分生孢子生长发育过程中涉及一系列物质代谢相关途径.例如, 短链脱氢酶还原酶家族(SDR)具有功能折叠特性, 在脂质、氨基酸等物质代谢和氧化还原机制中起关键作用(
Effects of essential oil vapour treatment on the postharvest disease control and different defence responses in two mango ( Mangifera indica L.) cultivars
1
2017
... 芒果(Mangifera indica)属漆树科植物, 是热带和亚热带地区广泛种植的名贵水果, 被誉为“水果之王”(
Genome-scale transcriptome analysis of the desert poplar, Populus euphratica
1
2011
... 为了精确解析胶孢炭疽菌相关差异基因的动态表达模式, 进而筛选候选基因, 我们使用基于模型的聚类方法对具有相似表达模式的基因进行分组(
Survey of human oxidoreductases and cytochrome P450 enzymes involved in the metabolism of xenobiotic and natural chemicals
1
2015
... 将功能注释与基因调控网络的拓扑结构相结合, 发现聚类模块1中基因参与了转录和二萜类生物合成过程.其中核心基因包括pep1和细胞色素P450 (cytochrome p450, CYP)编码基因(
Postharvest biocontrol of Colletotrichum gloeosporioides on mango using the marine bacterium Stenotrophomonas rhizophila and its possible mechanisms of action
1
2019
... 胶孢炭疽菌(Colletotrichum gloeosporioides)是引起芒果炭疽病的主要病原菌, 该病菌是一种丝状真菌, 其寄主范围广, 可潜伏侵染果实的多个部位, 严重影响芒果的产量和品质, 危害极大(
edgeR: a bioconductor package for differential expression analysis of digital gene expression data
1
2010
... 芒果胶孢炭疽菌参考基因组下载自美国国家生物技术信息中心数据库(NCBI), 将每个样品的clean reads与参考基因组进行比对(
The transporter classification database
1
2014
... 在核心基因中也发现了很多参与真菌菌丝生长分化、细胞壁合成重塑及黑色素合成相关的基因, 包括响应胁迫过程中负责细胞壁合成和重塑的NAD脱水酶(nad-dependent epimerase dehydratase) (
ITN1, a novel gene encoding an ankyrin-repeat protein that affects the ABA-mediated production of reactive oxygen species and is involved in salt-stress tolerance in Arabidopsis thaliana
1
2008
... 真菌在生长发育过程中受到逆境胁迫时, 会启动自身防御机制, 重新建立新的稳态条件并恢复生长, 这期间所发生的系列反应都是真菌生存的关键.逆境胁迫可大幅增加ROX的积累, 引起氧化应激, 从而抑制生长甚至诱导生物体死亡(
The chemical treatments combined with antagonistic yeast control anthracnose and maintain the quality of postharvest mango fruit
1
2019
... 芒果(Mangifera indica)属漆树科植物, 是热带和亚热带地区广泛种植的名贵水果, 被誉为“水果之王”(
Type III polyketide synthases: functional classification and phylogenomics
1
2017
... 聚类模块2是瞬时响应损伤胁迫的代表模块.GO富集分析表明, 其基因主要参与电子转移活性、血红素结合过程和次生代谢产物的生物合成过程, 共包含74个差异表达基因.结合基因间的调控网络和聚类富集结果, 我们筛选到7个核心基因.典型核心基因如聚酮化合物合酶PKSS (polyketide synthase)及含锚蛋白重复序列蛋白AKR2A (ankyrin repeat-containing protein)编码基因在细胞代谢中起重要作用.研究表明, PKSS参与调控真菌体内气生菌丝的生长与融合(
Model-based clustering for RNA-seq data
1
2014
... 为了初步解析芒果胶孢炭疽菌在损伤胁迫下mRNA的动态表达模式, 基于实验设计对RNA-seq差异表达数据进行聚类分析(
NaCl-induced alternations of cellular and tissue ion fluxes in roots of salt-resistant and salt-sensitive poplar species
1
2009
... 目前, 对生物较长时间响应胁迫过程的研究已经较为深入(
Gene ontology consortium: going forward
1
2015
... 利用Illumina平台将测序所得的原始序列文件进行质量评估和可信度分析(
Transcript assembly and quantification by RNA- seq reveals unannotated transcripts and isoform switching during cell differentiation
1
2010
... 利用Illumina平台将测序所得的原始序列文件进行质量评估和可信度分析(
Phenylalanine ammonia-lyase and cell wall peroxidase are cooperatively involved in the extensive formation of ferulate network in cell walls of developing rice shoots
1
2012
... 真菌在生长发育过程中受到逆境胁迫时, 会启动自身防御机制, 重新建立新的稳态条件并恢复生长, 这期间所发生的系列反应都是真菌生存的关键.逆境胁迫可大幅增加ROX的积累, 引起氧化应激, 从而抑制生长甚至诱导生物体死亡(
Challenges for modeling global gene regulatory networks during development: insights from Drosophila
1
2010
... 基因调控网络可以很好地分析胁迫中重要的分子过程或代谢通路(
KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases
1
2011
... 利用Illumina平台将测序所得的原始序列文件进行质量评估和可信度分析(
PEPR2 is a second receptor for the Pep1 and Pep2 peptides and contributes to defense responses in Arabidopsis
1
2010
... 真菌在生长发育过程中受到逆境胁迫时, 会启动自身防御机制, 重新建立新的稳态条件并恢复生长, 这期间所发生的系列反应都是真菌生存的关键.逆境胁迫可大幅增加ROX的积累, 引起氧化应激, 从而抑制生长甚至诱导生物体死亡(
An ankyrin repeat-containing protein plays a role in both disease resistance and antioxidation metabolism
1
2002
... 聚类模块2是瞬时响应损伤胁迫的代表模块.GO富集分析表明, 其基因主要参与电子转移活性、血红素结合过程和次生代谢产物的生物合成过程, 共包含74个差异表达基因.结合基因间的调控网络和聚类富集结果, 我们筛选到7个核心基因.典型核心基因如聚酮化合物合酶PKSS (polyketide synthase)及含锚蛋白重复序列蛋白AKR2A (ankyrin repeat-containing protein)编码基因在细胞代谢中起重要作用.研究表明, PKSS参与调控真菌体内气生菌丝的生长与融合(
Physiological and proteomic alterations in rice ( Oryza sativa L.) seedlings under hexavalent chromium stress
1
2014
... 在核心基因中也发现了很多参与真菌菌丝生长分化、细胞壁合成重塑及黑色素合成相关的基因, 包括响应胁迫过程中负责细胞壁合成和重塑的NAD脱水酶(nad-dependent epimerase dehydratase) (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}