Identification and Evolution of LRR VIII-2 Subfamily Genes in Four Model Plant Species
Chenyang Yan1,2, Yingnan Chen,1,2,*1Key Laboratory of Forest Genetics and Biotechnology of Ministry of Education, Nanjing Forestry University, Nanjing 210037, China 2Co-Innovation Center for the Sustainable Forestry in Southern China, College of Forestry, Nanjing Forestry University, Nanjing 210037, China
Abstract Whole-genome duplication and tandem duplication are two important mechanisms for gene duplication, which play important roles in promoting the genomic and genetic diversity. In Arabidopsis, AtLRR-RLK encodes receptor-like kinases rich in leucine repeats, which is a multi-gene family arising from large-scale gene expansion during angiosperm evolution. It is composed of 15 subfamilies, among which, AtLRR VIII-2 is the subfamily with the highest proportion of tandem repeats. In this study, we use the genes in LRR VIII-2 as an example to analyze the gene expansion and differential retention in four model plants (Arabidopsis thaliana, Populus trichocarpa, Vitis vinifera and Carica papaya). Results showed that paralogous gene pairs were identified in the LRR VIII-2 subfamily in Arabidopsis, poplar and grape, while no such pair was found in papaya. The LRR VIII-2 subfamily expanded the most significantly in poplar and moderately expanded in Arabidopsis and grape, but some genes of the LRR VIII-2 subfamily in papaya have been lost. In addition, the paralogous and orthologous genes in the LRR VIII-2 subfamily were under strong purifying selection in the four investigated plant species, except for a pair of paralogous genes in poplar. An in-depth phylogenetic analysis of the LRR VIII-2 subfamily helps to understand the role and significance of gene duplication in plant evolution, and provides useful information for predicting the function of homologous gene among different species. This analytical pipeline is also applicable for deciphering the evolution history of other gene families. Keywords:receptor-like protein kinases;leucine-rich repeats;whole-genome duplication;tandem duplication
PDF (1184KB)摘要页面多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文 引用本文 闫晨阳, 陈赢男. 4种模式植物LRR VIII-2亚家族基因的鉴定和进化历史分析. 植物学报, 2020, 55(4): 442-456 doi:10.11983/CBB19157 Yan Chenyang, Chen Yingnan. Identification and Evolution of LRR VIII-2 Subfamily Genes in Four Model Plant Species. Chinese Bulletin of Botany, 2020, 55(4): 442-456 doi:10.11983/CBB19157
Table 2 表2 表24种模式植物中84个LRR VIII-2基因的特征 Table 2Features of 84 LRR VIII-2 genes identified in four model plant species
Code
Gene ID
Chr.
Genomic location
No. of exons
Protein length (aa)
Extracellular domain
A1
AT1G07650
At1
2359111-2366736
25
1020
LRR10
A2
AT1G16670
At1
5697216-5699870
6
390
K
A3
AT1G29720
At1
10393659-10399873
25
1019
LRR12
A4
AT1G29730
At1
10400564-10405913
24
969
LRR11
A5
AT1G29740
At1
10407220-10413119
26
1078
LRR5
A6
AT1G29750
At1
10413853-10420774
24
1021
LRR9
A7
AT1G53420
At1
19926626-19931494
23
953
LRR10
A8
AT1G53430
At1
19934987-19941185
23
1030
LRR9
A9
AT1G53440
At1
19945869-19951562
23
1035
LRR11
A10
AT1G56120
At1
20987128-20993572
23
1047
LRR11
A11
AT1G56130
At1
20994746-21001013
24
1032
LRR6
A12
AT1G56140
At1
21001269-21007855
24
1033
LRR6
A13
AT3G09010
At3
2749908-2752390
6
393
K
A14
AT3G14840
At3
4988008-4994109
24
1020
LRR11
P1
Potri.001G308600
Pt01
31227276-31234883
24
1006
LRR10
P2
Potri.001G385200
Pt01
40032658-40043384
24
1007
LRR11
P3
Potri.001G385300
Pt01
40046269-40056753
24
1021
LRR13
P4
Potri.001G385400
Pt01
40060761-40069904
24
1036
LRR10
P5
Potri.001G385900
Pt01
40135506-40145556
22
982
LRR7
P6
Potri.001G386300
Pt01
40164017-40178014
23
989
LRR5
P7
Potri.001G393200
Pt01
41232410-41235419
6
396
K
P8
Potri.001G438400
Pt01
47008564-47013154
6
396
K
P9
Potri.003G025600
Pt03
3062484-3069540
22
944
LRR3
P10
Potri.003G025800
Pt03
3088186-3100889
24
999
LRR3
P11
Potri.003G148000
Pt03
16311568-16319951
21
1041
LRR13
P12
Potri.004G040200
Pt04
3050127-3055020
6
373
K
P13
Potri.004G063200
Pt04
5205009-5216081
23
1092
LRR12
P14
Potri.004G063500
Pt04
5238500-5250295
24
1011
LRR9
P15
Potri.004G063600
Pt04
5255561-5259093
6
468
TK
P16
Potri.004G063900
Pt04
5275636-5279061
6
464
TK
P17
Potri.004G135500
Pt04
15551257-15559647
24
1029
LRR12
Code
Gene ID
Chr.
Genomic location
No. of exons
Protein length (aa)
Extracellular domain
P18
Potri.006G128500
Pt06
10474234-10477075
5
471
K
P19
Potri.007G067900
Pt07
8907797-8916407
24
1036
LRR12
P20
Potri.008G040800
Pt08
2314252-2320106
4
341
K
P21
Potri.010G221400
Pt10
20632758-20635063
6
366
K
P22
Potri.011G049600
Pt11
4238297-4243676
5
374
K
P23
Potri.011G072300
Pt11
6917401-6930365
24
1024
LRR8
P24
Potri.011G075400
Pt11
7247330-7259126
22
1003
LRR9
P25
Potri.011G106400
Pt11
12955841-12965039
22
1015
LRR11
P26
Potri.011G112000
Pt11
13684010-13689252
6
396
K
P27
Potri.011G142100
Pt11
16335090-16338863
6
393
K
P28
Potri.016G114800
Pt16
11913776-11918410
7
385
K
P29
Potri.019G005200
Pt19
554428-564520
24
801
LRR5
P30
Potri.019G005300
Pt19
582783-593615
24
1003
LRR8
P31
Potri.019G005700
Pt19
655851-664479
23
978
LRR6
P32
Potri.019G005900
Pt19
681330-690210
24
990
LRR3
P33
Potri.019G006000
Pt19
699799-708891
24
964
LRR5
P34
Potri.019G007900
Pt19
921639-930444
23
1002
LRR3
P35
Potri.019G008900
Pt19
1038688-1047049
24
972
LRR9
P36
Potri.019G009700
Pt19
1141831-1151301
13
988
LRR3
P37
Potri.019G009800
Pt19
1159163-1167756
24
989
LRR9
P38
Potri.T072700
scaffold_79
11693-18613
22
945
LRR8
V1
GSVIVT01006444001
chrUn
26044571-26050096
8
441
K
V2
GSVIVT01013608001
chrUn
1284866-1301072
24
994
LRR12
V3
GSVIVT01013612001
chrUn
1429686-1453826
21
897
LRR10
V4
GSVIVT01013621001
chrUn
1735676-1793223
24
911
LRR11
V5
GSVIVT01014113001
Vv19
501306-518217
23
1036
LRR9
V6
GSVIVT01014134001
Vv19
669185-677944
24
1021
LRR10
V7
GSVIVT01014138001
Vv19
719037-726465
23
935
LRR10
V8
GSVIVT01014145001
Vv19
793325-800964
21
904
LRR5
V9
GSVIVT01014147001
Vv19
813603-825355
24
1063
LRR5
V10
GSVIVT01014150001
Vv19
846312-864401
30
1181
LRR11
V11
GSVIVT01014221001
Vv19
1629917-1636613
6
385
K
V12
GSVIVT01020456001
Vv19
19137067-19142116
6
385
K
V13
GSVIVT01020786001
Vv12
1992495-2002600
24
1011
LRR10
V14
GSVIVT01021280001
Vv10
3340198-3390494
24
1107
LRR10
V15
GSVIVT01021285001
Vv10
3439036-3462765
29
1144
LRR12
V16
GSVIVT01021289001
Vv10
3527408-3540220
24
1008
LRR10
V17
GSVIVT01021291001
Vv10
3556246-3578182
24
1017
LRR11
V18
GSVIVT01024760001
Vv6
7166910-7169739
6
372
K
V19
GSVIVT01025676001
Vv8
13018070-13020594
7
433
K
V20
GSVIVT01033973001
Vv8
16186419-16192809
6
380
K
V21
GSVIVT01038011001
Vv10
12545889-12562513
24
1003
LRR11
C1
evm.model.supercontig_124.16
supercontig_124
342359-355054
9
651
TK
C2
evm.model.supercontig_124.19
supercontig_124
400979-417373
8
604
TK
C3
evm.model.supercontig_131.56
supercontig_131
437441-440461
6
380
K
C4
evm.model.supercontig_163.18
supercontig_163
423778-427253
6
397
K
Code
Gene ID
Chr.
Genomic location
No. of exons
Protein length (aa)
Extracellular domain
C5
evm.model.supercontig_2076.1
supercontig_2076
157-766
2
173
K
C6
evm.model.supercontig_748.4
supercontig_748
17410-21951
9
583
TK
C7
evm.model.supercontig_77.35
supercontig_77
331417-334856
10
636
LRR1
C8
evm.model.supercontig_77.40
supercontig_77
398441-404475
22
880
LRR10
C9
evm.model.supercontig_77.43
supercontig_77
453480-459449
23
829
LRR9
C10
evm.model.supercontig_864.1
supercontig_864
13501-16944
6
419
K
C11
evm.TU.contig_37590.1
contig_37590
240-2161
6
365
LRR1
LRR后的数字表示富含亮氨酸重复序列数; K表示没有LRR结构域的激酶; TK表示没有LRR结构域的跨膜激酶。 The number after LRR indicates the number of leucine-rich repeat; K indicates kinase without LRR domain; TK indicates transmembrane kinase without LRR domain.
Table 3 表3 表34种模式植物LRR VIII-2亚家族基因旁系同源与直系同源基因对 Table 3Paralogous and orthologous gene pairs in LRR VIII-2 subfamily identified in four model plant species
Gene pair
Ka/Ks
Gene pair
Ka/Ks
Gene pair
Ka/Ks
Paralogous
A4-A5
0.2184
P9-P25
0.3214
P32-P36
0.4003
A8-A9
0.2680
P10-P17
0.1596
P32-P37
0.6808
A10-A11
0.2870
P10-P25
0.3488
P32-P38
0.3088
A10-A12
0.2910
P12-P22
0.2362
P33-P34
0.6734
A11-A12
0.2498
P13-P23
0.2871
P33-P35
0.5076
P1-P29
0.3010
P14-P15
0.2140
P33-P36
0.5615
P1-P30
0.3292
P14-P16
0.1985
P33-P37
0.6011
P1-P31
0.3020
P15-P16
1.4030
P33-P38
0.3093
P1-P32
0.3077
P15-P24
0.3642
P34-P35
0.5274
P1-P33
0.2955
P16-P24
0.3469
P34-P36
0.6011
P1-P34
0.3075
P20-P21
0.1211
P34-P37
0.7145
P1-P35
0.3188
P29-P30
0.3678
P34-P38
0.3139
P1-P36
0.2997
P29-P31
0.3321
P35-P36
0.3943
P1-P37
0.3122
P29-P32
0.3381
P35-P37
0.5829
P1-P38
0
P29-P33
0.3334
P35-P38
0.3204
P3-P4
0.3683
P29-P34
0.3317
P36-P37
0.4718
P3-P5
0.3731
P29-P35
0.3411
P36-P38
0.2992
P3-P6
0.4963
P29-P36
0.3215
P37-P38
0.3134
P3-P9
0.3791
P29-P37
0.3392
V2-V3
0.3479
P3-P10
0.4125
P29-P38
0.3169
V2-V4
0.2818
P3-P25
0.3422
P30-P31
0.5422
V2-V21
0.5377
P4-P5
0.4843
P30-P32
0.6659
V3-V4
0.3152
P4-P6
0.3907
P30-P33
0.6512
V3-V21
0.3481
P4-P9
0.7093
P30-P34
0.8013
V4-V21
0.2963
P4-P10
0.4706
P30-P35
0.5957
V6-V7
0.3152
P4-P17
0.1770
P30-P36
0.5292
V6-V8
0.2935
P4-P25
0.3206
P30-P37
0.6763
V6-V9
0.3034
P5-P6
0.3712
P30-P38
0.3368
V6-V10
0.2833
P5-P9
0.6021
P31-P32
0.4655
V7-V8
0.7300
P5-P10
0.8590
P31-P33
0.4989
V7-V9
0.5629
P5-P17
0.1715
P31-P34
0.5435
V7-V10
0.4047
P5-P25
0.3456
P31-P35
0.4251
V8-V9
0.4763
P6-P9
0.4063
P31-P36
0.3099
V8-V10
0.3629
P6-P10
0.4357
P31-P37
0.5370
V9-V10
0.3855
P6-P25
0.3556
P31-P38
0.3142
V14-V15
0.4844
P7-P26
0.5268
P32-P33
0.5711
V14-V16
0.2626
P8-P27
0.2579
P32-P34
0.6597
V15-V16
0.3233
P9-P10
0.6507
P32-P35
0.5461
V16-V17
0.2770
P9-P17
0.1859
Orthologous
P7-C3
0.2223
P28-C4
0.2070
C4-V20
0.1760
P25-V10
0.2441
C3-V11
0.2574
C9-V10
0.2076
Ka: 非同义替换率; Ks: 同义替换率 Ka: Nonsynonymous substitution rate; Ks: Synonymous substitution rate
Table 4 表4 表44种模式植物LRR VIII-2亚家族基因共线性同源基因及Ks值 Table 4Collinearity of LRR VIII-2 subfamily homologous genes and Ks value in four model plant species
Block pair No.
Code of locus 1
Code of locus 2
Block median Ks
S/WGD
Block pair within species
21
PN2
P11
0.2592
p
267
P12
P22
0.26815
p
442
P20
P21
0.26975
p
64
P8
P27
0.27455
p
61
PN1
P25
0.2765
p
61
P7
P26
0.2765
p
303
P17
PN3
0.2983
p
121
P1
P34
0.3034
p
267
P13
P23
0.3551
p
372
P18
P21
0.3625
p
515
P22
P28
0.8329
γ
284
P12
P28
0.93355
γ
26
P2
P13
1.27845
γ
274
P14
P25
1.3549
γ
361
P18
P20
1.4912
γ
503
P23
P25
1.6122
γ
57
A7
A14
0.7968
α
104
V18
V20
1.13135
γ
28
VN2
V19
1.11255
γ
Block pair between species
608
A13
P28
1.6761
γ
214
A2
P26
1.6727
γ
35
A2
P7
1.986
γ
32
A6
VN1
1.59645
γ
382
A13
V20
1.7376
γ
110
A2
V11
1.88175
γ
932
CN1
P25
1.162
γ
930
CN1
P2
1.09745
γ
153
CN2
P13
1.1864
γ
299
CN3
P28
1.2174
γ
523
CN4
P22
1.12095
γ
517
CN4
P12
1.1073
γ
203
CN5
P26
0.98095
γ
241
P14
VN1
1.0883
γ
668
P23
V10
1.52975
γ
657
P23
V13
1.4531
γ
249
P13
V13
1.5376
γ
978
PN5
V5
1.8582
γ
668
PN4
V5
1.52975
γ
81
CN6
V14
1.15
γ
108
CN5
V11
1.0547
γ
287
CN4
V1
1.0255
γ
164
CN3
V20
1.06385
γ
Block pair No.代表含有locus 1和2的成对共线性区段编号。基因编号中带字母N的表示该基因已不具备LRR VIII-2亚家族成员的特征结构域, 即非LRR VIII-2特征基因。S/WGD: 片段/全基因组重复; Ks: 同义替换率 Block pair No. indicates the number of the Block pair which contains the locus 1 and locus 2. Gene code containing a letter N represents that the gene has evolved into non-LRR VIII-2 that does not harbor the characteristic domain of LRR VIII-2 subfami-ly. S/WGD: Segmental/whole-genome duplication; Ks: Synonymous substitution rate
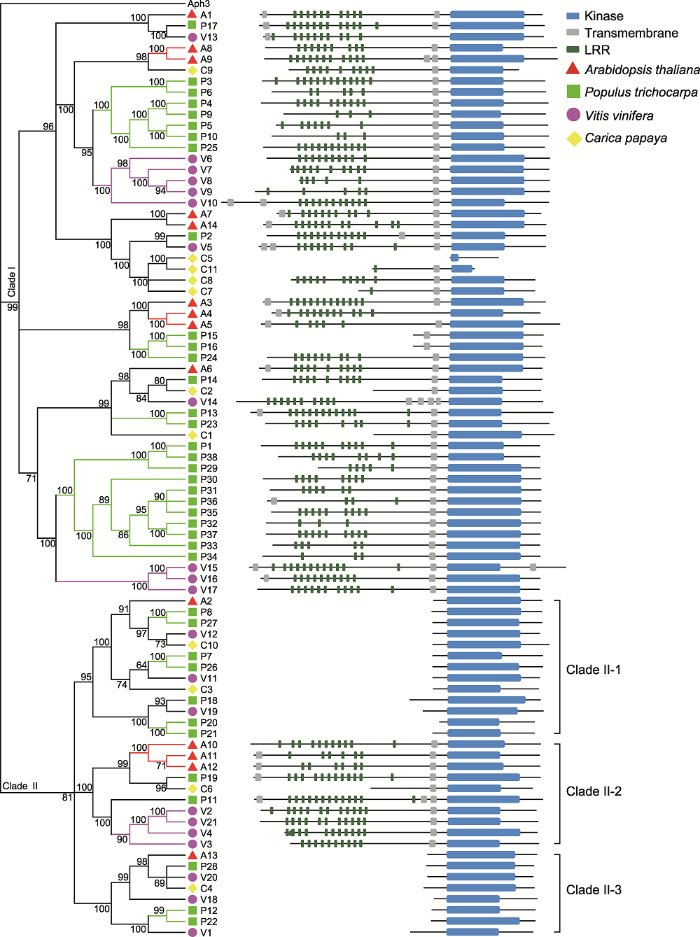
左侧第1个数字为行号。方框代表种内和种间染色体共线性区段(SCB)。方框中的编码为定位于该SCB上的LRR VIII-2基因编号; 基因编号中带字母N的表示该基因与LRR VIII-2基因存在共线性同源关系, 但该基因已不具备LRR VIII-2亚家族成员的特征结构域; 字母L表示SCB上和LRR VIII-2亚家族对应的共线性基因丢失, 但该区段被保留下来; 空白位置表示SCB完全丢失。 Figure 2Differential retention and expansion of the ancestral LRR VIII-2 gene associated with paleopolyploidy events in four model plant species
The left first digit indicates the row number. Squares in the same line represent the syntenic chromosomal blocks (SCBs) within and between species. Gene codes in the square correspond to the associated LRR VIII-2 genes; gene code containing a letter N indicates that the gene has a collinear homologous relationship with the LRR VIII-2 gene, but the gene no longer has the characteristic domain of the members of the LRR VIII-2 subfamily; the letter L represents the corresponding SCB has been retained, but the duplicated LRR VIII-2 gene on the SCB has been lost; blank regions indicate the SCBs has been completely lost.
(A) 低温胁迫下拟南芥AtLRR VIII-2亚家族基因表达热图; (B) 低温胁迫下4个山新杨LRR VIII-2亚家族基因相对表达量。*表示差异显著(t-检验, P<0.05)。 Figure 3Expression of LRR VIII-2 genes under low temperature treatment in Arabidopsis thaliana and Populus davidiana × P. bolleana
(A) Transcriptome analysis of AtLRR VIII-2 genes in Arabidopsis treated with low temperature; (B) Relative expression of four LRR VIII-2 genes in P. davidiana × P. bolleana under low temperature stress. * indicate significant differences (t-test, P<0.05).
AlbrechtC, RussinovaE, HechtV, BaaijensE, de VriesS (2005). The Arabidopsis thaliana SOMATIC EMBRYOGENESIS RECEPTOR-LIKE KINASES 1 and 2 control male sporogenesis Plant Cell 17, 3337-3349. DOI:10.1105/tpc.105.036814URLPMID:16284305 [本文引用: 1] The Arabidopsis thaliana SOMATIC EMBRYOGENESIS RECEPTOR-LIKE KINASE (SERK) family of plasma membrane receptors consists of five closely related members. The SERK1 and SERK2 genes show a complex expression pattern throughout development. Both are expressed in anther primordia up to the second parietal division. After this point, expression ceases in the sporocytes and is continued in the tapetum and middle layer precursors. Single knockout mutants of SERK1 and SERK2 show no obvious phenotypes. Double mutants of SERK1 and SERK2 are completely male sterile due to a failure in tapetum specification. Fertility can be restored by a single copy of either gene. The SERK1 and SERK2 proteins can form homodimers or heterodimers in vivo, suggesting they are interchangeable in the SERK1/SERK2 signaling complex.
BejA, SahooBR, SwainB, BasuM, JayasankarP, SamantaM (2014). LRRsearch: an asynchronous server- based application for the prediction of leucine-rich repeat motifs and an integrative database of NOD-like receptors Comput Biol Med 53, 164-170. DOI:10.1016/j.compbiomed.2014.07.016URLPMID:25150822 [本文引用: 1] The leucine-rich repeat (LRR) motifs of the nucleotide-binding oligomerization domain like receptors (NLRs) play key roles in recognizing and binding various pathogen associated molecular patterns (PAMPs) resulting in the activation of downstream signaling and innate immunity. Therefore, identification of LRR motifs is very important to study ligand-receptor interaction. To date, available resources pose restrictions including both false negative and false positive prediction of LRR motifs from the primary protein sequence as their algorithms are relied either only on sequence based comparison or alignment techniques or are over biased for a particular LRR containing protein family. Therefore, to minimize the error ( BlancG, HokampK, WolfeKH (2003). A recent polyploidy superimposed on older large-scale duplications in the Arabidopsis genome Genome Res 13, 137-144. DOI:10.1101/gr.751803URLPMID:12566392 [本文引用: 1] The Arabidopsis genome contains numerous large duplicated chromosomal segments, but the different approaches used in previous analyses led to different interpretations regarding the number and timing of ancestral large-scale duplication events. Here, using more appropriate methodology and a more recent version of the genome sequence annotation, we investigate the scale and timing of segmental duplications in Arabidopsis. We used protein sequence similarity searches to detect duplicated blocks in the genome, used the level of synonymous substitution between duplicated genes to estimate the relative ages of the blocks containing them, and analyzed the degree of overlap between adjacent duplicated blocks. We conclude that the Arabidopsis lineage underwent at least two distinct episodes of duplication. One was a polyploidy that occurred much more recently than estimated previously, before the Arabidopsis/Brassica rapa split and probably during the early emergence of the crucifer family (24-40 Mya). An older set of duplicated blocks was formed after the monocot/dicot divergence, and the relatively low level of overlap among these blocks indicates that at least some of them are remnants of a larger duplication such as a polyploidy or aneuploidy.
BlancG, WolfeKH (2004). Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes Plant Cell 16, 1667-1678. DOI:10.1105/tpc.021345URLPMID:15208399 [本文引用: 2] It is often anticipated that many of today's diploid plant species are in fact paleopolyploids. Given that an ancient large-scale duplication will result in an excess of relatively old duplicated genes with similar ages, we analyzed the timing of duplication of pairs of paralogous genes in 14 model plant species. Using EST contigs (unigenes), we identified pairs of paralogous genes in each species and used the level of synonymous nucleotide substitution to estimate the relative ages of gene duplication. For nine of the investigated species (wheat [Triticum aestivum], maize [Zea mays], tetraploid cotton [Gossypium hirsutum], diploid cotton [G. arboretum], tomato [Lycopersicon esculentum], potato [Solanum tuberosum], soybean [Glycine max], barrel medic [Medicago truncatula], and Arabidopsis thaliana), the age distributions of duplicated genes contain peaks corresponding to short evolutionary periods during which large numbers of duplicated genes were accumulated. Large-scale duplications (polyploidy or aneuploidy) are strongly suspected to be the cause of these temporal peaks of gene duplication. However, the unusual age profile of tandem gene duplications in Arabidopsis indicates that other scenarios, such as variation in the rate at which duplicated genes are deleted, must also be considered.
ChenC, ChenH, ZhangY, ThomasHR, FrankMH, HeY, XiaR (2002). TBtools, an integrative toolkit developed for interactive analyses of big biological data Mol Plant 13, 1194-1202. URLPMID:32585190 [本文引用: 1]
ClarkSE, WilliamsRW, MeyerowitzEM (1997). The CLAVATA1 gene encodes a putative receptor kinase that controls shoot and floral meristem size in Arabidopsis Cell 89, 575-585. URLPMID:9160749 [本文引用: 1]
ColcombetJ, Boisson-DernierA, Ros-PalauR, VeraCE, SchroederJI (2005). Arabidopsis SOMATIC EMBRYOGENESIS RECEPTOR KINASES 1 and 2 are essential for tapetum development and microspore maturation Plant Cell 17, 3350-3361. URLPMID:16284306 [本文引用: 1]
ConantGC, WolfeKH (2008). Turning a hobby into a job: how duplicated genes find new functions Nat Rev Genet 9, 938-950. URLPMID:19015656 [本文引用: 1]
CummingsMP (2004). PAUP* (phylogenetic analysis using parsimony (and other methods)) Santa Monica: American Cancer Society. pp. 37-45. [本文引用: 2]
DaiXG, HuQJ, CaiQL, FengK, YeN, TuskanGA, MilneR, ChenYN, WanZB, WangZF, LuoWC, WangK, WanDS, WangMX, WangJ, LiuJQ, YinTM (2014). The willow genome and divergent evolution from poplar after the common genome duplication Cell Res 24, 1274-1277. DOI:10.1038/cr.2014.83URLPMID:24980958 [本文引用: 1]
FengC, MackeyAJ, VermuntJK, RoosDS (2007). Assessing performance of orthology detection strategies applied to eukaryotic genomes PLoS One 2, e383. URLPMID:17440619 [本文引用: 1]
FinnRD, BatemanA, ClementsJ, CoggillP, EberhardtRY, EddySR, HegerA, HetheringtonK, HolmL, MistryJ, SonnhammerELL, TateJ, PuntaM (2014). Pfam: the protein families database Nucleic Acids Res 42, D222-D230. URLPMID:24288371 [本文引用: 1]
FinnRD, ClementsJ, EddySR (2011). HMMER web server: interactive sequence similarity searching Nucleic Acids Res 39, W29-W37. DOI:10.1093/nar/gkr367URLPMID:21593126 [本文引用: 1] HMMER is a software suite for protein sequence similarity searches using probabilistic methods. Previously, HMMER has mainly been available only as a computationally intensive UNIX command-line tool, restricting its use. Recent advances in the software, HMMER3, have resulted in a 100-fold speed gain relative to previous versions. It is now feasible to make efficient profile hidden Markov model (profile HMM) searches via the web. A HMMER web server (http://hmmer.janelia.org) has been designed and implemented such that most protein database searches return within a few seconds. Methods are available for searching either a single protein sequence, multiple protein sequence alignment or profile HMM against a target sequence database, and for searching a protein sequence against Pfam. The web server is designed to cater to a range of different user expertise and accepts batch uploading of multiple queries at once. All search methods are also available as RESTful web services, thereby allowing them to be readily integrated as remotely executed tasks in locally scripted workflows. We have focused on minimizing search times and the ability to rapidly display tabular results, regardless of the number of matches found, developing graphical summaries of the search results to provide quick, intuitive appraisement of them.
FischerI, DiévartA, DrocG, DufayardJF, ChantretN (2016). Evolutionary dynamics of the leucine-rich repeat receptor-like kinase (LRR-RLK) subfamily in angiosperms Plant Physiol 170, 1595-1610. DOI:10.1104/pp.15.01470URLPMID:26773008 [本文引用: 4] Gene duplications are an important factor in plant evolution, and lineage-specific expanded (LSE) genes are of particular interest. Receptor-like kinases expanded massively in land plants, and leucine-rich repeat receptor-like kinases (LRR-RLK) constitute the largest receptor-like kinases family. Based on the phylogeny of 7,554 LRR-RLK genes from 31 fully sequenced flowering plant genomes, the complex evolutionary dynamics of this family was characterized in depth. We studied the involvement of selection during the expansion of this family among angiosperms. LRR-RLK subgroups harbor extremely contrasting rates of duplication, retention, or loss, and LSE copies are predominantly found in subgroups involved in environmental interactions. Expansion rates also differ significantly depending on the time when rounds of expansion or loss occurred on the angiosperm phylogenetic tree. Finally, using a dN/dS-based test in a phylogenetic framework, we searched for selection footprints on LSE and single-copy LRR-RLK genes. Selective constraint appeared to be globally relaxed at LSE genes, and codons under positive selection were detected in 50% of them. Moreover, the leucine-rich repeat domains, and specifically four amino acids in them, were found to be the main targets of positive selection. Here, we provide an extensive overview of the expansion and evolution of this very large gene family.
FreelingM (2009). Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition Annu Rev Plant Biol 60, 433-453. DOI:10.1146/annurev.arplant.043008.092122URLPMID:19575588 [本文引用: 2] Each mode of gene duplication (tandem, tetraploid, segmental, transpositional) retains genes in a biased manner. A reciprocal relationship exists between plant genes retained postpaleotetraploidy versus genes retained after an ancient tandem duplication. Among the models (C, neofunctionalization, balanced gene drive) and ideas that might explain this relationship, only balanced gene drive predicts reciprocity. The gene balance hypothesis explains that more
GabaldónT, DessimozC, Huxley-JonesJ, VilellaAJ, SonnhammerEL, LewisS (2009). Joining forces in the quest for orthologs Genome Biol 10, 403. DOI:10.1186/gb-2009-10-9-403URLPMID:19785718 [本文引用: 1] Better orthology-prediction resources would be beneficial for the whole biological community. A recent meeting discussed how to coordinate and leverage current efforts.
Gómez-GómezGL, BollerT (2000). FLS2: an LRR receptor-like kinase involved in the perception of the bacterial elicitor flagellin in Arabidopsis Mol Cell 5, 1003-1011. DOI:10.1016/s1097-2765(00)80265-8URLPMID:10911994 [本文引用: 1] Flagellin, the main protein of the bacterial flagella, elicits defence responses and alters growth in Arabidopsis seedlings. Previously, we identified the FLS1 locus, which confers flagellin insensitivity in Ws-0. To identify additional components involved in flagellin perception, we screened for flagellin insensitivity mutants in the flagellin-sensitive accession La-er. Here, we describe the identification of a new locus, FLS2, by a map-based strategy. The FLS2 gene is ubiquitously expressed and encodes a putative receptor kinase. FLS2 shares structural and functional homologies with known plant resistance genes and with components involved in the innate immune system of mammals and insects.
GuoLH, ChenYN, YeN, DaiXG, YangWX, YinTM (2014). Differential retention and expansion of the ancestral genes associated with the paleopolyploidies in modern rosid plants, as revealed by analysis of the extensins super-gene family BMC Genomics 15, 612. URLPMID:25047956 [本文引用: 4]
HwangSG, KimDS, JangCS (2011). Comparative analysis of evolutionary dynamics of genes encoding leucine- rich repeat receptor-like kinase between rice and Arabidopsis Genetics 139, 1023. [本文引用: 1]
JiaYX, DingYL, ShiYT, ZhangXY, GongZZ, YangSH (2016). The cbfs triple mutants reveal the essential functions of cbfs in cold acclimation and allow the definition of CBF regulons in Arabidopsis New Phytol 212, 345-353. [本文引用: 1]
LeeTH, TangHB, WangXY, PatersonAH (2012). PGDD: a database of gene and genome duplication in plants Nucleic Acids Res 41, D1152-D1158. URLPMID:23180799 [本文引用: 1]
Lehti-ShiuMD, ShiuSH (2012). Diversity, classification and function of the plant protein kinase superfamily Philos Trans R Soc B Biol Sci 367, 2619-2639. [本文引用: 1]
LetunicI, BorkP (2018). 20 years of the SMART protein domain annotation resource Nucleic Acids Res 46, D493-D496. URLPMID:29040681 [本文引用: 1]
LiWH, YangJ, GuX (2005). Expression divergence between duplicate genes Trends Genet 21, 602-607. DOI:10.1016/j.tig.2005.08.006URLPMID:16140417 [本文引用: 1] A general picture of the role of expression divergence in the evolution of duplicate genes is emerging, thanks to the availability of completely sequenced genomes and functional genomic data, such as microarray data. It is now clear that expression divergence, regulatory-motif divergence and coding-sequence divergence all increase with the age of duplicate genes, although their exact interrelationships remain to be determined. It is also clear that gene duplication increases expression diversity and enables tissue or developmental specialization to evolve. However, the relative roles of subfunctionalization and neofunctionalization in the retention of duplicate genes remain to be clarified, especially for higher eukaryotes. In addition, the relationship between gene duplication and evolution of transcriptional regulatory networks is largely unexplored.
LibradoP, RozasJ (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data Bioinformatics 25, 1451-1452. DOI:10.1093/bioinformatics/btp187URLPMID:19346325 [本文引用: 1] MOTIVATION: DnaSP is a software package for a comprehensive analysis of DNA polymorphism data. Version 5 implements a number of new features and analytical methods allowing extensive DNA polymorphism analyses on large datasets. Among other features, the newly implemented methods allow for: (i) analyses on multiple data files; (ii) haplotype phasing; (iii) analyses on insertion/deletion polymorphism data; (iv) visualizing sliding window results integrated with available genome annotations in the UCSC browser. AVAILABILITY: Freely available to academic users from: (http://www.ub.edu/dnasp).
LiuZY, JiaYX, DingYL, ShiYT, LiZ, GuoY, GongZZ, YangSH (2017). Plasma membrane CRPK1-mediated phosphorylation of 14-3-3 proteins induces their nuclear import to fine-tune CBF signaling during cold response Mol Cell 66, 117-128. DOI:10.1016/j.molcel.2017.02.016URLPMID:28344081 [本文引用: 2] In plant cells, changes in fluidity of the plasma membrane may serve as the primary sensor of cold stress; however, the precise mechanism and how the cell transduces and fine-tunes cold signals remain elusive. Here we show that the cold-activated plasma membrane protein cold-responsive protein kinase 1 (CRPK1) phosphorylates 14-3-3 proteins. The phosphorylated 14-3-3 proteins shuttle from the cytosol to the nucleus, where they interact with and destabilize the key cold-responsive C-repeat-binding factor (CBF) proteins. Consistent with this, the crpk1 and 14-3-3kappalambda mutants show enhanced freezing tolerance, and transgenic plants overexpressing 14-3-3lambda show reduced freezing tolerance. Further study shows that CRPK1 is essential for the nuclear translocation of 14-3-3 proteins and for 14-3-3 function in freezing tolerance. Thus, our study reveals that the CRPK1-14-3-3 module transduces the cold signal from the plasma membrane to the nucleus to modulate CBF stability, which ensures a faithfully adjusted response to cold stress of plants.
Marchler-BauerA, LuSN, AndersonJB, ChitsazF, DerbyshireMK, DeWeese-ScottC, FongJH, GeerLY, GeerRC, GonzalesNR, GwadzM, HurwitzDI, JacksonJD, KeZX, LanczyckiCJ, LuF, MarchlerGH, MullokandovM, OmelchenkoMV, RobertsonCL, SongJS, ThankiN, YamashitaRA, ZhangDC, ZhangNG, ZhengCJ, BryantSH (2011). CDD: a conserved domain database for the functional annotation of proteins Nucleic Acids Res 39, D225-D229. URLPMID:21109532 [本文引用: 1]
MooreRC, PuruggananMD (2003). The early stages of duplicate gene evolution Proc Natl Acad Sci USA 100, 15682-15687. DOI:10.1073/pnas.2535513100URLPMID:14671323 [本文引用: 1] Gene duplications are one of the primary driving forces in the evolution of genomes and genetic systems. Gene duplicates account for 8-20% of the genes in eukaryotic genomes, and the rates of gene duplication are estimated at between 0.2% and 2% per gene per million years. Duplicate genes are believed to be a major mechanism for the establishment of new gene functions and the generation of evolutionary novelty, yet very little is known about the early stages of the evolution of duplicated gene pairs. It is unclear, for example, to what extent selection, rather than neutral genetic drift, drives the fixation and early evolution of duplicate loci. Analysis of recently duplicated genes in the Arabidopsis thaliana genome reveals significantly reduced species-wide levels of nucleotide polymorphisms in the progenitor and/or duplicate gene copies, suggesting that selective sweeps accompany the initial stages of the evolution of these duplicated gene pairs. Our results support recent theoretical work that indicates that fates of duplicate gene pairs may be determined in the initial phases of duplicate gene evolution and that positive selection plays a prominent role in the evolutionary dynamics of the very early histories of duplicate nuclear genes.
RameneniJJ, LeeY, DhandapaniV, YuXN, ChoiSR, OhMH, LimYP (2015). Genomic and post-translational modification analysis of leucine-rich-repeat receptor-like kinases in Brassica rapa PLoS One 10, e0142255. URLPMID:26588465 [本文引用: 1]
ShiuHS, BleeckerAB (2003). Expansion of the receptor-like kinase/Pelle gene family and receptor-like proteins in Arabidopsis Plant Physiol 132, 530-543. URLPMID:12805585 [本文引用: 3]
ShiuSH, BleeckerAB (2001 b). Receptor-like kinases from Arabidopsis form a monophyletic gene family related to animal receptor kinases Proc Natl Acad Sci USA 98, 10763-10768. URLPMID:11526204 [本文引用: 8]
Shumayla, SharmaS, KumarR, MenduV, SinghK, UpadhyaySK (2016). Genomic dissection and expression profiling revealed functional divergence in Triticum aestivum leucine rich repeat receptor like kinases (TaLRRKs) Front Plant Sci 7, 1374. URLPMID:27713749 [本文引用: 1]
SongDH, LiGJ, SongFM, ZhengZ (2008). Molecular characterization and expression analysis of OsBISERK1, a gene encoding a leucine-rich repeat receptor-like kinase, during disease resistance responses in rice Mol Biol Rep 35, 275-283. DOI:10.1007/s11033-007-9080-8URLPMID:17520342 [本文引用: 1] A rice gene, OsBISERK1, encoding a protein belonging to SOMATIC EMBRYOGENESIS RECEPTOR KINASE (SERK) type of leucine-rich repeat receptor-like kinases (LRR-RLKs) was identified. The OsBISERK1 encodes a 624 aa protein with high level of identity to known plant SERKs. OsBISERK1 contains a hydrophobic signal peptide, a leucine zipper, and five leucine-rich repeat motifs in the extracellular domain; the cytoplasmic region carries a proline-rich region and a single transmembrane domain, as well as a conserved intracellular serine/threonine protein kinase domain. OsBISERK1 has a low level of basal expression in leaf tissue. However, expression of OsBISERK1 was induced by treatment with benzothiadiazole (BTH), which is capable of inducing disease resistance in rice, and also up-regulated after inoculation with Magnaporthe grisea in BTH-treated rice seedlings and during incompatible interaction between a blast-resistant rice genotype and M. grisea. The results suggest that OsBISERK1 may be involved in disease resistance responses in rice.
StoneJM, WalkerJC (1995). Plant protein kinase families and signal transduction Plant Physiol 108, 451-457. URLPMID:7610156 [本文引用: 1]
SunXL, WangGL (2011). Genome-wide identification, characterization and phylogenetic analysis of the rice LRR-kinases PLoS One 6, e16079. URLPMID:21408199 [本文引用: 2]
TangHB, BowersJE, WangXY, MingR, AlamM, PatersonAH (2008a). Synteny and collinearity in plant genomes Science 320, 486-488. DOI:10.1126/science.1153917URLPMID:18436778 [本文引用: 1] Correlated gene arrangements among taxa provide a valuable framework for inference of shared ancestry of genes and for the utilization of findings from model organisms to study less-well-understood systems. In angiosperms, comparisons of gene arrangements are complicated by recurring polyploidy and extensive genome rearrangement. New genome sequences and improved analytical approaches are clarifying angiosperm evolution and revealing patterns of differential gene loss after genome duplication and differential gene retention associated with evolution of some morphological complexity. Because of variability in DNA substitution rates among taxa and genes, deviation from collinearity might be a more reliable phylogenetic character.
TangHB, WangXY, BowersJE, MingR, AlamM, PatersonAH (2008b). Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps Genome Res 18, 1944-1954. DOI:10.1101/gr.080978.108URLPMID:18832442 [本文引用: 4] Large-scale (segmental or whole) genome duplication has been recurring in angiosperm evolution. Subsequent gene loss and rearrangements further affect gene copy numbers and fractionate ancestral gene linkages across multiple chromosomes. The fragmented
TangP, ZhangY, SunXQ, TianDC, YangSH, DingJ (2010). Disease resistance signature of the leucine-rich repeat receptor-like kinase genes in four plant species Plant Sci 179, 399-406. [本文引用: 1]
The Arabidopsis GenomeInitiative (2000). Analysis of the genome sequence of the flowering plant Arabidopsis thaliana Nature 408, 796-815. URLPMID:11130711 [本文引用: 1]
TianWD, SkolnickJ (2003). How well is enzyme function conserved as a function of pairwise sequence identity? J Mol Biol 333, 863-882. DOI:10.1016/j.jmb.2003.08.057URLPMID:14568541 [本文引用: 1] Enzyme function conservation has been used to derive the threshold of sequence identity necessary to transfer function from a protein of known function to an unknown protein. Using pairwise sequence comparison, several studies suggested that when the sequence identity is above 40%, enzyme function is well conserved. In contrast, Rost argued that because of database bias, the results from such simple pairwise comparisons might be misleading. Thus, by grouping enzyme sequences into families based on sequence similarity and selecting representative sequences for comparison, he showed that enzyme function starts to diverge quickly when the sequence identity is below 70%. Here, we employ a strategy similar to Rost's to reduce the database bias; however, we classify enzyme families based not only on sequence similarity, but also on functional similarity, i.e. sequences in each family must have the same four digits or the same first three digits of the enzyme commission (EC) number. Furthermore, instead of selecting representative sequences for comparison, we calculate the function conservation of each enzyme family and then average the degree of enzyme function conservation across all enzyme families. Our analysis suggests that for functional transferability, 40% sequence identity can still be used as a confident threshold to transfer the first three digits of an EC number; however, to transfer all four digits of an EC number, above 60% sequence identity is needed to have at least 90% accuracy. Moreover, when PSI-BLAST is used, the magnitude of the E-value is found to be weakly correlated with the extent of enzyme function conservation in the third iteration of PSI-BLAST. As a result, functional annotation based on the E-values from PSI-BLAST should be used with caution. We also show that by employing an enzyme family-specific sequence identity threshold above which 100% functional conservation is required, functional inference of unknown sequences can be accurately accomplished. However, this comes at a cost: those true positive sequences below this threshold cannot be uniquely identified.
WeiZR, WangJH, YangSH, SongYJ (2015). Identification and expression analysis of the LRR-RLK gene family in tomato(Solanum lycopersicum) Heinz 1706 Genome 58, 121-134. DOI:10.1139/gen-2015-0035URL [本文引用: 1]
YangSH, ZhangXH, YueJX, TianDC, ChenJQ (2008). Recent duplications dominate NBS-encoding gene expansion in two woody species Mol Genet Genomic 280, 187-198. DOI:10.1007/s00438-008-0355-0URL [本文引用: 1]
YuY, FuscoeJC, ZhaoC, GuoC, JiaMW, TaoQ, BannonDI, LancashireL, BaoWJ, DuTT, LuoH, SuZQ, JonesWD, MolandCL, BranhamWS, QianF, NingBT, LiY, HongHX, GuoL, MeiN, ShiTL, WangKY, WolfingerRD, NikolskyY, WalkerSJ, Duerksen- HughesP, MasonCE, TongWD, Thierry-MiegJ, Thierry-MiegD, ShiLM, WangC (2014). A rat RNA-Seq transcriptomic BodyMap across 11 organs and 4 developmental stages Nat Commun 5, 3230. DOI:10.1038/ncomms4230URLPMID:24510058 [本文引用: 1] The rat has been used extensively as a model for evaluating chemical toxicities and for understanding drug mechanisms. However, its transcriptome across multiple organs, or developmental stages, has not yet been reported. Here we show, as part of the SEQC consortium efforts, a comprehensive rat transcriptomic BodyMap created by performing RNA-Seq on 320 samples from 11 organs of both sexes of juvenile, adolescent, adult and aged Fischer 344 rats. We catalogue the expression profiles of 40,064 genes, 65,167 transcripts, 31,909 alternatively spliced transcript variants and 2,367 non-coding genes/non-coding RNAs (ncRNAs) annotated in AceView. We find that organ-enriched, differentially expressed genes reflect the known organ-specific biological activities. A large number of transcripts show organ-specific, age-dependent or sex-specific differential expression patterns. We create a web-based, open-access rat BodyMap database of expression profiles with crosslinks to other widely used databases, anticipating that it will serve as a primary resource for biomedical research using the rat model.
ZanYJ, JiY, ZhangY, YangSH, SongYJ, WangJH (2013). Genome-wide identification, characterization and expression analysis of Populus leucine-rich repeat receptor-like protein kinase genes BMC Genomics 14, 318. URLPMID:23663326 [本文引用: 2]
重复基因的进化——回顾与进展 1 2010
... 基因组共线性分析揭示了拟南芥、杨树、葡萄(Vitis vinifera)和番木瓜(Carica papaya)起源于共同的古六倍体祖先.在长期的进化过程中, 拟南芥基因组经历了3次全基因组重复(α-二倍化、β-二倍化和γ-三倍化), 杨树基因组存在2次全基因组重复(γ-三倍化和p-二倍化), 在葡萄和番木瓜中仅存在1次全基因组重复事件(γ-三倍化) (The Arabidopsis Genome Initiative, 2000; Tang et al., 2008a, 2008b; Guo et al., 2014; Dai et al., 2014).由于全基因组重复事件祖先基因发生倍增, 导致基因剂量增加, 并可能经历不同的进化命运: (1) 新功能化(neofunctionalization), 倍增后的2个拷贝都保留下来, 其中1个拷贝保留了祖先基因的功能, 而另外1个拷贝获得了新功能; (2) 假基因化(non-functionalization), 1个冗余拷贝由于随机突变成为无功能的假基因或者发生DNA序列丢失; (3) 亚功能化(subfunctionalization), 2个拷贝都由于突变导致功能部分受损, 必须互补合作才能完成原来祖先基因的功能(孙红正和葛颂, 2010); (4) 剂量选择(dosage selection), 对有利剂量效应的选择使2个拷贝均保留下来并保持原有的功能(Conant and Wolfe, 2008).追溯祖先基因在现代植物基因组中的扩张和保留情况有助于深入理解倍增基因在生物体进化过程中的作用和意义.本研究选用基因组共线性关系明确的4种模式植物拟南芥、杨树、番木瓜和葡萄, 以LRR VIII-2亚家族为例, 通过在不同植物中鉴定该亚家族基因成员, 分析其在不同植物中发生基因扩张、差异保留及表达分化情况, 以期为深入揭示LRR VIII-2亚家族基因的功能及从系统发育角度阐明其它基因家族的进化历史提供参考. ...
... RLK是植物中普遍存在的、数量最多的一类蛋白激酶(郑超等, 2016), 在拟南芥(Arabidopsis thaliana)中约占所有蛋白激酶的60% (Shiu and Bleecker, 2003).典型的植物RLKs具有胞外区、跨膜区和胞内激酶区(Shiu and Bleecker, 2001a).根据胞外结构域特征和胞内激酶结构域序列的系统发生关系, 可以将拟南芥AtRLK分为45个亚家族(Shiu and Bleecker, 2001b).富含亮氨酸重复序列(leucine-rich repeats, LRR)型是其中最大的一类, 包含15个亚家族(LRR I-XV).其中LRR VIII分为LRR VIII-1和LRR VIII-2.富含亮氨酸重复类受体蛋白激酶(LRR-RLKs)胞内激酶结构域相对保守, 胞外受体结构域含有富含亮氨酸的保守基序LXXLXLXX (L代表亮氨酸残基, X代表任意氨基酸残基) (Song et al., 2008).LRR-RLKs在调控植物的生长发育、激素信号转导以及响应逆境胁迫等方面发挥重要作用.例如, 拟南芥CLV (LRR XI亚家族)信号途径能够调控茎顶端分生组织干细胞数量, 维持细胞分裂与分化之间的动态平衡(Clark et al., 1997; Ogawa et al., 2008); 拟南芥BAK1 (LRR II亚家族)与BRI1 (LRR X亚家族)形成的二聚体在油菜素内酯的信号转导过程中发挥关键作用(Colcombet et al., 2005; Albrecht et al., 2005); FLS2 (LRR XII亚家族)能够通过胞外的LRR结构域识别病原菌鞭毛蛋白, 介导植物的免疫反应(Gómez-Gómez and Boller, 2000).此外, 拟南芥CRPK1 (LRR VIII-2亚家族)通过磷酸化14-3-3蛋白, 精确调控植物低温胁迫响应(Liu et al., 2017). ...
The Arabidopsis thaliana SOMATIC EMBRYOGENESIS RECEPTOR-LIKE KINASES 1 and 2 control male sporogenesis 1 2005
... RLK是植物中普遍存在的、数量最多的一类蛋白激酶(郑超等, 2016), 在拟南芥(Arabidopsis thaliana)中约占所有蛋白激酶的60% (Shiu and Bleecker, 2003).典型的植物RLKs具有胞外区、跨膜区和胞内激酶区(Shiu and Bleecker, 2001a).根据胞外结构域特征和胞内激酶结构域序列的系统发生关系, 可以将拟南芥AtRLK分为45个亚家族(Shiu and Bleecker, 2001b).富含亮氨酸重复序列(leucine-rich repeats, LRR)型是其中最大的一类, 包含15个亚家族(LRR I-XV).其中LRR VIII分为LRR VIII-1和LRR VIII-2.富含亮氨酸重复类受体蛋白激酶(LRR-RLKs)胞内激酶结构域相对保守, 胞外受体结构域含有富含亮氨酸的保守基序LXXLXLXX (L代表亮氨酸残基, X代表任意氨基酸残基) (Song et al., 2008).LRR-RLKs在调控植物的生长发育、激素信号转导以及响应逆境胁迫等方面发挥重要作用.例如, 拟南芥CLV (LRR XI亚家族)信号途径能够调控茎顶端分生组织干细胞数量, 维持细胞分裂与分化之间的动态平衡(Clark et al., 1997; Ogawa et al., 2008); 拟南芥BAK1 (LRR II亚家族)与BRI1 (LRR X亚家族)形成的二聚体在油菜素内酯的信号转导过程中发挥关键作用(Colcombet et al., 2005; Albrecht et al., 2005); FLS2 (LRR XII亚家族)能够通过胞外的LRR结构域识别病原菌鞭毛蛋白, 介导植物的免疫反应(Gómez-Gómez and Boller, 2000).此外, 拟南芥CRPK1 (LRR VIII-2亚家族)通过磷酸化14-3-3蛋白, 精确调控植物低温胁迫响应(Liu et al., 2017). ...
LRRsearch: an asynchronous server- based application for the prediction of leucine-rich repeat motifs and an integrative database of NOD-like receptors 1 2014
Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition 2 2009
... 基因重复(gene duplication)及随后的基因功能多样性分化是基因组和遗传系统分化的重要动力, 在生物进化过程中发挥极其重要的作用(Moore and Purugganan, 2003).基因重复可以通过多种方式进行, 包括片段重复(segmental duplication)、全基因组重复(whole-genome duplication)、串联重复(tandem duplication)和转座重复(transpositional duplication) (Freeling, 2009).在众多的植物基因家族中, 对RLKs基因家族的进化历史研究比较深入, 特别是其中的LRR-RLKs (Tang et al., 2010; Fischer et al., 2016).植物基因组中存在大量的LRR-RLKs基因, 已发现在双子叶植物拟南芥、油菜(Brassica rapa)、番茄(Solanum lycopersicum)和杨树(Populus trichocarpa)基因组中分别有235、303、234和379个LRR- RLKs编码基因(Shiu and Bleecker, 2001a; Zan et al., 2013; Rameneni et al., 2015; Wei et al., 2015); 在单子叶植物水稻(Oryza sativa)中有309个, 小麦(Triticum aestivum)中多达531个(Sun and Wang, 2011; Shumayla et al., 2016).在多种植物中开展的生物信息学分析表明, 串联重复和片段或全基因组重复(segmental or whole-genome duplication, S/ WGD)是LRR-RLKs类基因发生扩张的主要机制(Shiu and Bleecker, 2001b, 2003; Sun and Wang, 2011; Zan et al., 2013).在拟南芥15个LRR-RLKs亚家族中, LRR VIII-2是通过串联重复造成基因重复比例最高的(Shiu and Bleecker, 2001b, 2003).此外, LRR VIII-2成员中既有典型的具备胞外LRR结构域+跨膜区+胞内激酶催化域的类型, 也包括只有胞内激酶域的类型, 后者可能是LRR-RLKs激酶结构域的祖先型, 在进化过程中通过基因融合将激酶催化域与胞外结构域融合形成现在的LRR-RLKs构象(Shiu and Bleecker, 2001b). ...
... 在基因组进化过程中, 基因可以通过串联重复、片段重复或全基因组重复等方式发生扩张(Freeling, 2009).串联重复基因同时满足2个条件: (1) 位于同一染色体上、基因间隔区≤100 kb (Guo et al., 2014); (2) 基因序列相似性超过阈值(相似区域覆盖较长序列的70%以上, 且Identity>70%) (Yang et al., 2008).为鉴定LRR VIII-2亚家族中通过片段/全基因组重复(S/WGD)方式扩张产生的基因, 首先从PGDD (plant genome duplication database)数据库(Lee et al., 2012)下载4种植物种内及种间基因组共线性数据, 从中筛选出含有LRR VIII-2亚家族基因的成对共线性区段(Block pair), 计算Block pair上所有共线性基因对(包括LRR VIII-2及其侧翼共线性同源基因)的Ks中值, 并根据Ks中值估算该染色体区段(Block)所经历的S/WGD事件, 即位于该区段的LRR VIII-2亚家族基因的扩张方式(Guo et al., 2014).对于既不属于串联重复也不属于S/WGD方式产生的旁系同源基因, 则认为通过其它重复方式产生(Hwang et al., 2011). ...
Joining forces in the quest for orthologs 1 2009
... 物种内旁系同源和物种间直系同源基因的识别不仅有助于比较和进化基因组学研究, 对同源基因的功能预测和注释也有重要意义.常用的同源基因鉴定方法有基于序列相似性、系统发生关系、基因组共线性分析等, 但每种方法都有其自身的局限性.例如, 根据同一物种内基因序列相似性可以鉴定旁系同源基因, 但在长期的进化过程中, 同源基因序列的相似性可能被严重侵蚀, 从而导致鉴定结果有误.本研究中, 通过基因组共线性分析表明A7-A14源于α重复事件, P2-P14源于p重复事件, 具有旁系同源关系, 但在根据序列相似性鉴定旁系同源基因的过程中未发现它们是旁系同源基因.因此, 在同源基因的鉴定过程中需要结合使用多种方法.通常认为, 直系同源基因具有相似的结构和生物学功能(Feng et al., 2007); 而由基因重复产生的旁系同源基因常伴随基因功能的分化(Sonnhammer and Koonin, 2002; Gabaldón et al., 2009).基因重复后的2个拷贝在表达上的分化是功能分化的重要一步(Li et al., 2005).本研究中, 杨树P8和P27与已有功能报道的A2直系同源, P8和P27对低温胁迫的响应情况也与A2相似.此外, P7与P26旁系同源, 但这2个基因在响应低温胁迫时的表达情况差异明显, 推测这2个基因出现了功能分化. ...
FLS2: an LRR receptor-like kinase involved in the perception of the bacterial elicitor flagellin in Arabidopsis 1 2000
... RLK是植物中普遍存在的、数量最多的一类蛋白激酶(郑超等, 2016), 在拟南芥(Arabidopsis thaliana)中约占所有蛋白激酶的60% (Shiu and Bleecker, 2003).典型的植物RLKs具有胞外区、跨膜区和胞内激酶区(Shiu and Bleecker, 2001a).根据胞外结构域特征和胞内激酶结构域序列的系统发生关系, 可以将拟南芥AtRLK分为45个亚家族(Shiu and Bleecker, 2001b).富含亮氨酸重复序列(leucine-rich repeats, LRR)型是其中最大的一类, 包含15个亚家族(LRR I-XV).其中LRR VIII分为LRR VIII-1和LRR VIII-2.富含亮氨酸重复类受体蛋白激酶(LRR-RLKs)胞内激酶结构域相对保守, 胞外受体结构域含有富含亮氨酸的保守基序LXXLXLXX (L代表亮氨酸残基, X代表任意氨基酸残基) (Song et al., 2008).LRR-RLKs在调控植物的生长发育、激素信号转导以及响应逆境胁迫等方面发挥重要作用.例如, 拟南芥CLV (LRR XI亚家族)信号途径能够调控茎顶端分生组织干细胞数量, 维持细胞分裂与分化之间的动态平衡(Clark et al., 1997; Ogawa et al., 2008); 拟南芥BAK1 (LRR II亚家族)与BRI1 (LRR X亚家族)形成的二聚体在油菜素内酯的信号转导过程中发挥关键作用(Colcombet et al., 2005; Albrecht et al., 2005); FLS2 (LRR XII亚家族)能够通过胞外的LRR结构域识别病原菌鞭毛蛋白, 介导植物的免疫反应(Gómez-Gómez and Boller, 2000).此外, 拟南芥CRPK1 (LRR VIII-2亚家族)通过磷酸化14-3-3蛋白, 精确调控植物低温胁迫响应(Liu et al., 2017). ...
Differential retention and expansion of the ancestral genes associated with the paleopolyploidies in modern rosid plants, as revealed by analysis of the extensins super-gene family 4 2014
... 基因组共线性分析揭示了拟南芥、杨树、葡萄(Vitis vinifera)和番木瓜(Carica papaya)起源于共同的古六倍体祖先.在长期的进化过程中, 拟南芥基因组经历了3次全基因组重复(α-二倍化、β-二倍化和γ-三倍化), 杨树基因组存在2次全基因组重复(γ-三倍化和p-二倍化), 在葡萄和番木瓜中仅存在1次全基因组重复事件(γ-三倍化) (The Arabidopsis Genome Initiative, 2000; Tang et al., 2008a, 2008b; Guo et al., 2014; Dai et al., 2014).由于全基因组重复事件祖先基因发生倍增, 导致基因剂量增加, 并可能经历不同的进化命运: (1) 新功能化(neofunctionalization), 倍增后的2个拷贝都保留下来, 其中1个拷贝保留了祖先基因的功能, 而另外1个拷贝获得了新功能; (2) 假基因化(non-functionalization), 1个冗余拷贝由于随机突变成为无功能的假基因或者发生DNA序列丢失; (3) 亚功能化(subfunctionalization), 2个拷贝都由于突变导致功能部分受损, 必须互补合作才能完成原来祖先基因的功能(孙红正和葛颂, 2010); (4) 剂量选择(dosage selection), 对有利剂量效应的选择使2个拷贝均保留下来并保持原有的功能(Conant and Wolfe, 2008).追溯祖先基因在现代植物基因组中的扩张和保留情况有助于深入理解倍增基因在生物体进化过程中的作用和意义.本研究选用基因组共线性关系明确的4种模式植物拟南芥、杨树、番木瓜和葡萄, 以LRR VIII-2亚家族为例, 通过在不同植物中鉴定该亚家族基因成员, 分析其在不同植物中发生基因扩张、差异保留及表达分化情况, 以期为深入揭示LRR VIII-2亚家族基因的功能及从系统发育角度阐明其它基因家族的进化历史提供参考. ...
... 在基因组进化过程中, 基因可以通过串联重复、片段重复或全基因组重复等方式发生扩张(Freeling, 2009).串联重复基因同时满足2个条件: (1) 位于同一染色体上、基因间隔区≤100 kb (Guo et al., 2014); (2) 基因序列相似性超过阈值(相似区域覆盖较长序列的70%以上, 且Identity>70%) (Yang et al., 2008).为鉴定LRR VIII-2亚家族中通过片段/全基因组重复(S/WGD)方式扩张产生的基因, 首先从PGDD (plant genome duplication database)数据库(Lee et al., 2012)下载4种植物种内及种间基因组共线性数据, 从中筛选出含有LRR VIII-2亚家族基因的成对共线性区段(Block pair), 计算Block pair上所有共线性基因对(包括LRR VIII-2及其侧翼共线性同源基因)的Ks中值, 并根据Ks中值估算该染色体区段(Block)所经历的S/WGD事件, 即位于该区段的LRR VIII-2亚家族基因的扩张方式(Guo et al., 2014).对于既不属于串联重复也不属于S/WGD方式产生的旁系同源基因, 则认为通过其它重复方式产生(Hwang et al., 2011). ...
... 亚家族基因的扩张方式(Guo et al., 2014).对于既不属于串联重复也不属于S/WGD方式产生的旁系同源基因, 则认为通过其它重复方式产生(Hwang et al., 2011). ...
b). Receptor-like kinases from Arabidopsis form a monophyletic gene family related to animal receptor kinases 8 2001
... RLK是植物中普遍存在的、数量最多的一类蛋白激酶(郑超等, 2016), 在拟南芥(Arabidopsis thaliana)中约占所有蛋白激酶的60% (Shiu and Bleecker, 2003).典型的植物RLKs具有胞外区、跨膜区和胞内激酶区(Shiu and Bleecker, 2001a).根据胞外结构域特征和胞内激酶结构域序列的系统发生关系, 可以将拟南芥AtRLK分为45个亚家族(Shiu and Bleecker, 2001b).富含亮氨酸重复序列(leucine-rich repeats, LRR)型是其中最大的一类, 包含15个亚家族(LRR I-XV).其中LRR VIII分为LRR VIII-1和LRR VIII-2.富含亮氨酸重复类受体蛋白激酶(LRR-RLKs)胞内激酶结构域相对保守, 胞外受体结构域含有富含亮氨酸的保守基序LXXLXLXX (L代表亮氨酸残基, X代表任意氨基酸残基) (Song et al., 2008).LRR-RLKs在调控植物的生长发育、激素信号转导以及响应逆境胁迫等方面发挥重要作用.例如, 拟南芥CLV (LRR XI亚家族)信号途径能够调控茎顶端分生组织干细胞数量, 维持细胞分裂与分化之间的动态平衡(Clark et al., 1997; Ogawa et al., 2008); 拟南芥BAK1 (LRR II亚家族)与BRI1 (LRR X亚家族)形成的二聚体在油菜素内酯的信号转导过程中发挥关键作用(Colcombet et al., 2005; Albrecht et al., 2005); FLS2 (LRR XII亚家族)能够通过胞外的LRR结构域识别病原菌鞭毛蛋白, 介导植物的免疫反应(Gómez-Gómez and Boller, 2000).此外, 拟南芥CRPK1 (LRR VIII-2亚家族)通过磷酸化14-3-3蛋白, 精确调控植物低温胁迫响应(Liu et al., 2017). ...
... 基因重复(gene duplication)及随后的基因功能多样性分化是基因组和遗传系统分化的重要动力, 在生物进化过程中发挥极其重要的作用(Moore and Purugganan, 2003).基因重复可以通过多种方式进行, 包括片段重复(segmental duplication)、全基因组重复(whole-genome duplication)、串联重复(tandem duplication)和转座重复(transpositional duplication) (Freeling, 2009).在众多的植物基因家族中, 对RLKs基因家族的进化历史研究比较深入, 特别是其中的LRR-RLKs (Tang et al., 2010; Fischer et al., 2016).植物基因组中存在大量的LRR-RLKs基因, 已发现在双子叶植物拟南芥、油菜(Brassica rapa)、番茄(Solanum lycopersicum)和杨树(Populus trichocarpa)基因组中分别有235、303、234和379个LRR- RLKs编码基因(Shiu and Bleecker, 2001a; Zan et al., 2013; Rameneni et al., 2015; Wei et al., 2015); 在单子叶植物水稻(Oryza sativa)中有309个, 小麦(Triticum aestivum)中多达531个(Sun and Wang, 2011; Shumayla et al., 2016).在多种植物中开展的生物信息学分析表明, 串联重复和片段或全基因组重复(segmental or whole-genome duplication, S/ WGD)是LRR-RLKs类基因发生扩张的主要机制(Shiu and Bleecker, 2001b, 2003; Sun and Wang, 2011; Zan et al., 2013).在拟南芥15个LRR-RLKs亚家族中, LRR VIII-2是通过串联重复造成基因重复比例最高的(Shiu and Bleecker, 2001b, 2003).此外, LRR VIII-2成员中既有典型的具备胞外LRR结构域+跨膜区+胞内激酶催化域的类型, 也包括只有胞内激酶域的类型, 后者可能是LRR-RLKs激酶结构域的祖先型, 在进化过程中通过基因融合将激酶催化域与胞外结构域融合形成现在的LRR-RLKs构象(Shiu and Bleecker, 2001b). ...
... 是通过串联重复造成基因重复比例最高的(Shiu and Bleecker, 2001b, 2003).此外, LRR VIII-2成员中既有典型的具备胞外LRR结构域+跨膜区+胞内激酶催化域的类型, 也包括只有胞内激酶域的类型, 后者可能是LRR-RLKs激酶结构域的祖先型, 在进化过程中通过基因融合将激酶催化域与胞外结构域融合形成现在的LRR-RLKs构象(Shiu and Bleecker, 2001b). ...
... 成员中既有典型的具备胞外LRR结构域+跨膜区+胞内激酶催化域的类型, 也包括只有胞内激酶域的类型, 后者可能是LRR-RLKs激酶结构域的祖先型, 在进化过程中通过基因融合将激酶催化域与胞外结构域融合形成现在的LRR-RLKs构象(Shiu and Bleecker, 2001b). ...
... 利用拟南芥AtLRR VIII-2亚家族14个成员的蛋白序列(Shiu and Bleecker, 2001b), 分别与毛果杨、番木瓜和葡萄的蛋白序列进行BlastP比对, 查找不同植物中LRR VIII-2亚家族的候选基因, 设置E-value≤e-20 (Shiu and Bleecker, 2001b), Identity≥40% (Tian and Skolnick, 2003).利用在线工具SMART (Letunic and Bork, 2018)、Pfam (Finn et al., 2014)和NCBI CD-search (Marchler-Bauer et al., 2011), 在比对出的候选基因中筛选出具有AtLRR VIII-2蛋白结构域的成员, 选择默认参数.同时, 应用Hmmbuild在线工具(Finn et al., 2011)构建AtLRR VIII-2亚家族的隐马尔科夫模型, 基于该模型对候选基因进行筛选.取上述2种筛选方法中共同保留下来的候选基因, 进一步利用PAUP (Phylogenetic Analysis Using Parsimony) 软件(Cummings, 2004), 基于邻接法, 最小进化、最大简约性标准(Shiu and Bleecker, 2001b), 与拟南芥所有235个LRR-RLKs成员(Shiu and Bleecker, 2001a)构建系统发育树.以葡萄球菌属(Staphylococcus)的氨基糖苷激酶APH3 (Shiu and Bleecker, 2001b)为外类群, 将其中与AtLRR VIII-2聚类在同一分支的基因鉴定为该物种的LRR VIII-2亚家族成员. ...
... 亚家族的候选基因, 设置E-value≤e-20 (Shiu and Bleecker, 2001b), Identity≥40% (Tian and Skolnick, 2003).利用在线工具SMART (Letunic and Bork, 2018)、Pfam (Finn et al., 2014)和NCBI CD-search (Marchler-Bauer et al., 2011), 在比对出的候选基因中筛选出具有AtLRR VIII-2蛋白结构域的成员, 选择默认参数.同时, 应用Hmmbuild在线工具(Finn et al., 2011)构建AtLRR VIII-2亚家族的隐马尔科夫模型, 基于该模型对候选基因进行筛选.取上述2种筛选方法中共同保留下来的候选基因, 进一步利用PAUP (Phylogenetic Analysis Using Parsimony) 软件(Cummings, 2004), 基于邻接法, 最小进化、最大简约性标准(Shiu and Bleecker, 2001b), 与拟南芥所有235个LRR-RLKs成员(Shiu and Bleecker, 2001a)构建系统发育树.以葡萄球菌属(Staphylococcus)的氨基糖苷激酶APH3 (Shiu and Bleecker, 2001b)为外类群, 将其中与AtLRR VIII-2聚类在同一分支的基因鉴定为该物种的LRR VIII-2亚家族成员. ...
... ), 基于邻接法, 最小进化、最大简约性标准(Shiu and Bleecker, 2001b), 与拟南芥所有235个LRR-RLKs成员(Shiu and Bleecker, 2001a)构建系统发育树.以葡萄球菌属(Staphylococcus)的氨基糖苷激酶APH3 (Shiu and Bleecker, 2001b)为外类群, 将其中与AtLRR VIII-2聚类在同一分支的基因鉴定为该物种的LRR VIII-2亚家族成员. ...
... (Shiu and Bleecker, 2001b)为外类群, 将其中与AtLRR VIII-2聚类在同一分支的基因鉴定为该物种的LRR VIII-2亚家族成员. ...
Genomic dissection and expression profiling revealed functional divergence in Triticum aestivum leucine rich repeat receptor like kinases (TaLRRKs) 1 2016
... 基因重复(gene duplication)及随后的基因功能多样性分化是基因组和遗传系统分化的重要动力, 在生物进化过程中发挥极其重要的作用(Moore and Purugganan, 2003).基因重复可以通过多种方式进行, 包括片段重复(segmental duplication)、全基因组重复(whole-genome duplication)、串联重复(tandem duplication)和转座重复(transpositional duplication) (Freeling, 2009).在众多的植物基因家族中, 对RLKs基因家族的进化历史研究比较深入, 特别是其中的LRR-RLKs (Tang et al., 2010; Fischer et al., 2016).植物基因组中存在大量的LRR-RLKs基因, 已发现在双子叶植物拟南芥、油菜(Brassica rapa)、番茄(Solanum lycopersicum)和杨树(Populus trichocarpa)基因组中分别有235、303、234和379个LRR- RLKs编码基因(Shiu and Bleecker, 2001a; Zan et al., 2013; Rameneni et al., 2015; Wei et al., 2015); 在单子叶植物水稻(Oryza sativa)中有309个, 小麦(Triticum aestivum)中多达531个(Sun and Wang, 2011; Shumayla et al., 2016).在多种植物中开展的生物信息学分析表明, 串联重复和片段或全基因组重复(segmental or whole-genome duplication, S/ WGD)是LRR-RLKs类基因发生扩张的主要机制(Shiu and Bleecker, 2001b, 2003; Sun and Wang, 2011; Zan et al., 2013).在拟南芥15个LRR-RLKs亚家族中, LRR VIII-2是通过串联重复造成基因重复比例最高的(Shiu and Bleecker, 2001b, 2003).此外, LRR VIII-2成员中既有典型的具备胞外LRR结构域+跨膜区+胞内激酶催化域的类型, 也包括只有胞内激酶域的类型, 后者可能是LRR-RLKs激酶结构域的祖先型, 在进化过程中通过基因融合将激酶催化域与胞外结构域融合形成现在的LRR-RLKs构象(Shiu and Bleecker, 2001b). ...
Molecular characterization and expression analysis of OsBISERK1, a gene encoding a leucine-rich repeat receptor-like kinase, during disease resistance responses in rice 1 2008
... RLK是植物中普遍存在的、数量最多的一类蛋白激酶(郑超等, 2016), 在拟南芥(Arabidopsis thaliana)中约占所有蛋白激酶的60% (Shiu and Bleecker, 2003).典型的植物RLKs具有胞外区、跨膜区和胞内激酶区(Shiu and Bleecker, 2001a).根据胞外结构域特征和胞内激酶结构域序列的系统发生关系, 可以将拟南芥AtRLK分为45个亚家族(Shiu and Bleecker, 2001b).富含亮氨酸重复序列(leucine-rich repeats, LRR)型是其中最大的一类, 包含15个亚家族(LRR I-XV).其中LRR VIII分为LRR VIII-1和LRR VIII-2.富含亮氨酸重复类受体蛋白激酶(LRR-RLKs)胞内激酶结构域相对保守, 胞外受体结构域含有富含亮氨酸的保守基序LXXLXLXX (L代表亮氨酸残基, X代表任意氨基酸残基) (Song et al., 2008).LRR-RLKs在调控植物的生长发育、激素信号转导以及响应逆境胁迫等方面发挥重要作用.例如, 拟南芥CLV (LRR XI亚家族)信号途径能够调控茎顶端分生组织干细胞数量, 维持细胞分裂与分化之间的动态平衡(Clark et al., 1997; Ogawa et al., 2008); 拟南芥BAK1 (LRR II亚家族)与BRI1 (LRR X亚家族)形成的二聚体在油菜素内酯的信号转导过程中发挥关键作用(Colcombet et al., 2005; Albrecht et al., 2005); FLS2 (LRR XII亚家族)能够通过胞外的LRR结构域识别病原菌鞭毛蛋白, 介导植物的免疫反应(Gómez-Gómez and Boller, 2000).此外, 拟南芥CRPK1 (LRR VIII-2亚家族)通过磷酸化14-3-3蛋白, 精确调控植物低温胁迫响应(Liu et al., 2017). ...
Orthology, paralogy and proposed classification for paralog subtypes 1 2002
... 物种内旁系同源和物种间直系同源基因的识别不仅有助于比较和进化基因组学研究, 对同源基因的功能预测和注释也有重要意义.常用的同源基因鉴定方法有基于序列相似性、系统发生关系、基因组共线性分析等, 但每种方法都有其自身的局限性.例如, 根据同一物种内基因序列相似性可以鉴定旁系同源基因, 但在长期的进化过程中, 同源基因序列的相似性可能被严重侵蚀, 从而导致鉴定结果有误.本研究中, 通过基因组共线性分析表明A7-A14源于α重复事件, P2-P14源于p重复事件, 具有旁系同源关系, 但在根据序列相似性鉴定旁系同源基因的过程中未发现它们是旁系同源基因.因此, 在同源基因的鉴定过程中需要结合使用多种方法.通常认为, 直系同源基因具有相似的结构和生物学功能(Feng et al., 2007); 而由基因重复产生的旁系同源基因常伴随基因功能的分化(Sonnhammer and Koonin, 2002; Gabaldón et al., 2009).基因重复后的2个拷贝在表达上的分化是功能分化的重要一步(Li et al., 2005).本研究中, 杨树P8和P27与已有功能报道的A2直系同源, P8和P27对低温胁迫的响应情况也与A2相似.此外, P7与P26旁系同源, 但这2个基因在响应低温胁迫时的表达情况差异明显, 推测这2个基因出现了功能分化. ...
Plant protein kinase families and signal transduction 1 1995
Genome-wide identification, characterization and phylogenetic analysis of the rice LRR-kinases 2 2011
... 基因重复(gene duplication)及随后的基因功能多样性分化是基因组和遗传系统分化的重要动力, 在生物进化过程中发挥极其重要的作用(Moore and Purugganan, 2003).基因重复可以通过多种方式进行, 包括片段重复(segmental duplication)、全基因组重复(whole-genome duplication)、串联重复(tandem duplication)和转座重复(transpositional duplication) (Freeling, 2009).在众多的植物基因家族中, 对RLKs基因家族的进化历史研究比较深入, 特别是其中的LRR-RLKs (Tang et al., 2010; Fischer et al., 2016).植物基因组中存在大量的LRR-RLKs基因, 已发现在双子叶植物拟南芥、油菜(Brassica rapa)、番茄(Solanum lycopersicum)和杨树(Populus trichocarpa)基因组中分别有235、303、234和379个LRR- RLKs编码基因(Shiu and Bleecker, 2001a; Zan et al., 2013; Rameneni et al., 2015; Wei et al., 2015); 在单子叶植物水稻(Oryza sativa)中有309个, 小麦(Triticum aestivum)中多达531个(Sun and Wang, 2011; Shumayla et al., 2016).在多种植物中开展的生物信息学分析表明, 串联重复和片段或全基因组重复(segmental or whole-genome duplication, S/ WGD)是LRR-RLKs类基因发生扩张的主要机制(Shiu and Bleecker, 2001b, 2003; Sun and Wang, 2011; Zan et al., 2013).在拟南芥15个LRR-RLKs亚家族中, LRR VIII-2是通过串联重复造成基因重复比例最高的(Shiu and Bleecker, 2001b, 2003).此外, LRR VIII-2成员中既有典型的具备胞外LRR结构域+跨膜区+胞内激酶催化域的类型, 也包括只有胞内激酶域的类型, 后者可能是LRR-RLKs激酶结构域的祖先型, 在进化过程中通过基因融合将激酶催化域与胞外结构域融合形成现在的LRR-RLKs构象(Shiu and Bleecker, 2001b). ...
... ; Sun and Wang, 2011; Zan et al., 2013).在拟南芥15个LRR-RLKs亚家族中, LRR VIII-2是通过串联重复造成基因重复比例最高的(Shiu and Bleecker, 2001b, 2003).此外, LRR VIII-2成员中既有典型的具备胞外LRR结构域+跨膜区+胞内激酶催化域的类型, 也包括只有胞内激酶域的类型, 后者可能是LRR-RLKs激酶结构域的祖先型, 在进化过程中通过基因融合将激酶催化域与胞外结构域融合形成现在的LRR-RLKs构象(Shiu and Bleecker, 2001b). ...
Synteny and collinearity in plant genomes 1 2008
... 基因组共线性分析揭示了拟南芥、杨树、葡萄(Vitis vinifera)和番木瓜(Carica papaya)起源于共同的古六倍体祖先.在长期的进化过程中, 拟南芥基因组经历了3次全基因组重复(α-二倍化、β-二倍化和γ-三倍化), 杨树基因组存在2次全基因组重复(γ-三倍化和p-二倍化), 在葡萄和番木瓜中仅存在1次全基因组重复事件(γ-三倍化) (The Arabidopsis Genome Initiative, 2000; Tang et al., 2008a, 2008b; Guo et al., 2014; Dai et al., 2014).由于全基因组重复事件祖先基因发生倍增, 导致基因剂量增加, 并可能经历不同的进化命运: (1) 新功能化(neofunctionalization), 倍增后的2个拷贝都保留下来, 其中1个拷贝保留了祖先基因的功能, 而另外1个拷贝获得了新功能; (2) 假基因化(non-functionalization), 1个冗余拷贝由于随机突变成为无功能的假基因或者发生DNA序列丢失; (3) 亚功能化(subfunctionalization), 2个拷贝都由于突变导致功能部分受损, 必须互补合作才能完成原来祖先基因的功能(孙红正和葛颂, 2010); (4) 剂量选择(dosage selection), 对有利剂量效应的选择使2个拷贝均保留下来并保持原有的功能(Conant and Wolfe, 2008).追溯祖先基因在现代植物基因组中的扩张和保留情况有助于深入理解倍增基因在生物体进化过程中的作用和意义.本研究选用基因组共线性关系明确的4种模式植物拟南芥、杨树、番木瓜和葡萄, 以LRR VIII-2亚家族为例, 通过在不同植物中鉴定该亚家族基因成员, 分析其在不同植物中发生基因扩张、差异保留及表达分化情况, 以期为深入揭示LRR VIII-2亚家族基因的功能及从系统发育角度阐明其它基因家族的进化历史提供参考. ...
Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps 4 2008
... 基因组共线性分析揭示了拟南芥、杨树、葡萄(Vitis vinifera)和番木瓜(Carica papaya)起源于共同的古六倍体祖先.在长期的进化过程中, 拟南芥基因组经历了3次全基因组重复(α-二倍化、β-二倍化和γ-三倍化), 杨树基因组存在2次全基因组重复(γ-三倍化和p-二倍化), 在葡萄和番木瓜中仅存在1次全基因组重复事件(γ-三倍化) (The Arabidopsis Genome Initiative, 2000; Tang et al., 2008a, 2008b; Guo et al., 2014; Dai et al., 2014).由于全基因组重复事件祖先基因发生倍增, 导致基因剂量增加, 并可能经历不同的进化命运: (1) 新功能化(neofunctionalization), 倍增后的2个拷贝都保留下来, 其中1个拷贝保留了祖先基因的功能, 而另外1个拷贝获得了新功能; (2) 假基因化(non-functionalization), 1个冗余拷贝由于随机突变成为无功能的假基因或者发生DNA序列丢失; (3) 亚功能化(subfunctionalization), 2个拷贝都由于突变导致功能部分受损, 必须互补合作才能完成原来祖先基因的功能(孙红正和葛颂, 2010); (4) 剂量选择(dosage selection), 对有利剂量效应的选择使2个拷贝均保留下来并保持原有的功能(Conant and Wolfe, 2008).追溯祖先基因在现代植物基因组中的扩张和保留情况有助于深入理解倍增基因在生物体进化过程中的作用和意义.本研究选用基因组共线性关系明确的4种模式植物拟南芥、杨树、番木瓜和葡萄, 以LRR VIII-2亚家族为例, 通过在不同植物中鉴定该亚家族基因成员, 分析其在不同植物中发生基因扩张、差异保留及表达分化情况, 以期为深入揭示LRR VIII-2亚家族基因的功能及从系统发育角度阐明其它基因家族的进化历史提供参考. ...
 ,1,2,*
,1,2,*
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT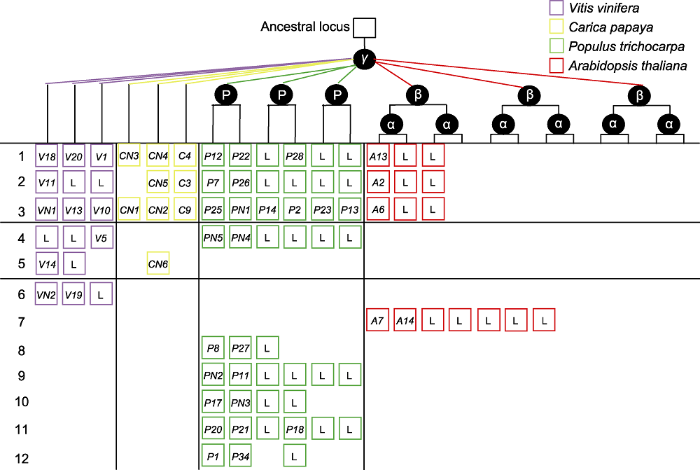 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}