 ,*中国科学院上海生命科学研究院植物生理生态研究所, 上海 200032
,*中国科学院上海生命科学研究院植物生理生态研究所, 上海 200032NRT1.1B Connects Root Microbiota and Nitrogen Use in Rice
Xiaolin Wang, Ertao Wang,*Institute of Plant Physiology and Ecology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai 200032, China通讯作者:
收稿日期:2019-03-31接受日期:2019-04-2网络出版日期:2019-07-01
Corresponding authors:
Received:2019-03-31Accepted:2019-04-2Online:2019-07-01

摘要
关键词:
Abstract
Keywords:
PDF (838KB)摘要页面多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
引用本文
王孝林, 王二涛. 根际微生物促进水稻氮利用的机制. 植物学报, 2019, 54(3): 285-287 doi:10.11983/CBB19060
Wang Xiaolin, Wang Ertao.
在土壤中, 植物根系与微生物互作。根际微生物可以促进植物在自然界的生存, 其作用机制主要包括: (1) 促进植物从环境中获取营养, 例如, 菌根真菌溶磷和根瘤菌固氮(Oldroyd et al., 2011; Mbodj et al., 2018); (2) 通过激素合成或降解调控植物的生长和环境适应性(Duan et al., 2014); (3) 通过与致病菌的作用或诱导植物产生抗性而调控免疫反应(Bulgarelli et al., 2013; Santhanam et al., 2015)。此外, 根际微生物也可以与宿主植物竞争土壤中的营养物质, 或作为致病菌攻击植物(Berendsen et al., 2012)。研究表明, 植物根系选择性地招募特定的土壤微生物, 主要包括变形杆菌(Proteobacteria)、拟杆菌(Bacteroidetes)、放线菌(Actinobacteria)和厚壁菌(Firmicutes)等, 而微生物特异富集主要取决于植物的根际区系、土壤类型、营养状况和发育阶段等因素(Bulgarelli et al., 2012; Edwards et al., 2015; Walters et al., 2018)。然而, 同一物种的不同植物群体间根际微生物的组成变化及遗传调控机理却鲜有报道。
亚洲栽培稻(Oryza sativa)主要分为籼稻(Indica)和粳稻(Japonica)。籼稻通常比粳稻具有更高的氮利用效率, 其中一个主要因素与其从土壤中吸收氮的效率相关(Hu et al., 2015)。最近, 中国科学院遗传与发育生物学研究所白洋课题组与储成才课题组合作, 揭示了土壤微生物群落调控籼稻和粳稻的氮利用效率差异的重要机制。通过对68份籼稻材料和27份粳稻材料的根际微生物进行研究, 他们发现籼稻根系特异富集的微生物类群分布在δ-变形菌纲(Deltaproteo- bacteria)、放线菌门(Actinobacteria)、酸酐菌门(Acidobacteria)和拟杆菌门(Bacteroidetes)等, 而粳稻根系富集的微生物主要集中在α-变形菌纲(Alphaproteobacteria)。对籼稻根系特异富集的微生物功能进行预测分析, 发现参与氮代谢的通路被富集, 包括氨化信号通路(nitrate ammonification and nitrite ammonification pathway)和氮呼吸信号通路(nitrate respiration, nitrite respiration and nitrogen respiration pathway) (Zhang et al., 2019)。上述结果暗示根际微生物群落的变化可能与籼粳之间氮素利用效率相关。
储成才研究团队前期对水稻氮肥利用效率的研究显示, 硝酸盐转运蛋白NRT1.1B的单个氨基酸变异是决定籼稻氮利用效率高于粳稻的主要因素之一(Hu et al., 2015)。近期, 通过比较野生型中花11与nrt1.1b突变体的根际微生物, 他们发现NRT1.1B的缺失改变了籼稻中49.6%的根际微生物群属, 表明NRT1.1B在籼稻招募特异的根际微生物中扮演着重要角色(Zhang et al., 2019)。
作者通过分离粳稻和籼稻特异富集的微生物, 进一步构建人工微生物群落(SynCom), 验证其在水稻生长及营养吸收中的作用。通过分别接种粳稻和籼稻特异富集的人工微生物群落, 在硝态氮或氨态氮充足条件下, 人工微生物群落会抑制水稻的生长; 但在以有机氮作为唯一氮源的条件下, 籼稻特异富集的人工微生物群落能促进水稻的生长, 表明水稻可以通过招募特异的根际微生物提高对有机氮源的利用效率(Zhang et al., 2019) (图1)。该研究结果为解析微生物群落调控植物氮营养, 特别是籼稻与粳稻氮利用效率的分子机制提供了重要线索。
图1
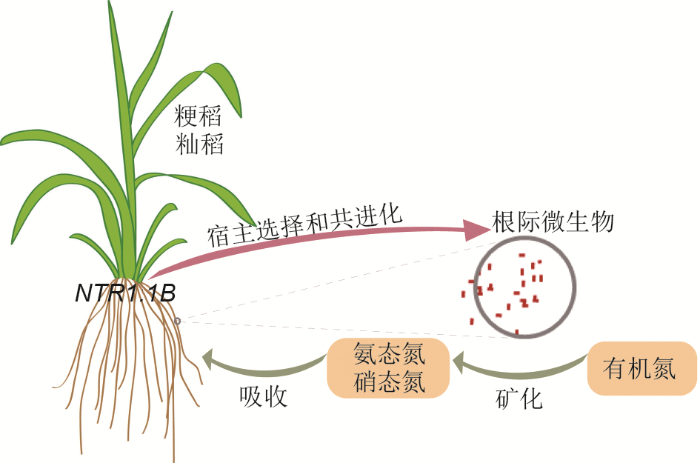 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1水稻通过NRT1.1B基因协同根系微生物利用土壤氮元素
籼稻比粳稻富集更多参与氮代谢的根系微生物。这些微生物将有机氮转化为氨态氮, 提高水稻氮元素利用效率。籼稻和粳稻特异富集的根系微生物与NRT1.1B基因相关联。
Figure 1Rice plants coordinate root microbiota to utilize soil nitrogen by NRT1.1B
Indica and japonica rice varieties recruit distinct root microb- iota. The biological progresses related to nitrogen metabolism are specifically enriched in indica-enriched bacteria. These bacteria transform organic nitrogen to ammonium, facilitating the nitrogen utilization in rice. NRT1.1B is associated with the recruitments of indica and japonica-specific bacterial taxa.
在全球生态系统退化和气候变化的背景下, 强化微生物在农业生态系统中的功能可能是未来农业可持续发展的重要方向之一(Toju et al., 2018)。然而, 如何利用微生物群落提高作物生长和环境适应性仍然面临挑战(Bender et al., 2016; Toju et al., 2018)。植物可以选择根内和根际微生物群落(Bulgarelli et al., 2013), 这一特性为通过选择特定土壤微生物促进作物营养吸收提供借鉴(Toju et al., 2018)。前人的研究表明, 对植物进行遗传修饰、改造根际微生物群落, 可提高作物的营养吸收效率, 有利于降低农业化肥污染(Chaparro et al., 2012; Beckers et al., 2016)。该研究揭示了水稻不同群体的遗传特性与根际微生物差异之间的调控机制, 为通过根际微生物管理提高作物营养吸收提供了理论支撑。
(责任编辑: 朱亚娜)
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1073/pnas.1523264113URLPMID:26755604 [本文引用: 1]

Cinnamoyl-CoA reductase (CCR), an enzyme central to the lignin biosynthetic pathway, represents a promising biotechnological target to reduce lignin levels and to improve the commercial viability of lignocellulosic biomass. However, silencing of the CCR gene results in considerable flux changes of the general and monolignol-specific lignin pathways, ultimately leading to the accumulation of var...
DOI:10.1016/j.tree.2016.02.016URLPMID:26993667 [本文引用: 1]
Recent evidence showed that soil biodiversity supports several ecosystem functions simultaneously, underpinning its crucial role in ecosystems worldwide. To enable the proper functioning of ecosystems, soil biodiversity has to be enhanced and maintained. Our analysis indicates that the sustainability of agricultural ecosystems can be restored by stimulating soil life and internally regulated ecosystem processes. To face the immense global problems related to a growing human population and deterioration of the global biosphere, targeted manipulations of soil organisms become necessary in addition to promoting soil biodiversity. Targeted approaches through soil ecological engineering to maximize the contribution of soil biological processes to sustainable ecosystem functioning can help to provide food security while minimizing negative environmental impacts.
DOI:10.1016/j.tplants.2012.04.001URLPMID:22564542 [本文引用: 1]
The diversity of microbes associated with plant roots is enormous, in the order of tens of thousands of species. This complex plant-associated microbial community, also referred to as the second genome of the plant, is crucial for plant health. Recent advances in plant鈥搈icrobe interactions research revealed that plants are able to shape their rhizosphere microbiome, as evidenced by the fact that different plant species host specific microbial communities when grown on the same soil. In this review, we discuss evidence that upon pathogen or insect attack, plants are able to recruit protective microorganisms, and enhance microbial activity to suppress pathogens in the rhizosphere. A comprehensive understanding of the mechanisms that govern selection and activity of microbial communities by plant roots will provide new opportunities to increase crop production.
DOI:10.1038/nature11336URLPMID:22859207 [本文引用: 1]
Abstract The plant root defines the interface between a multicellular eukaryote and soil, one of the richest microbial ecosystems on Earth. Notably, soil bacteria are able to multiply inside roots as benign endophytes and modulate plant growth and development, with implications ranging from enhanced crop productivity to phytoremediation. Endophytic colonization represents an apparent paradox of plant innate immunity because plant cells can detect an array of microbe-associated molecular patterns (also known as MAMPs) to initiate immune responses to terminate microbial multiplication. Several studies attempted to describe the structure of bacterial root endophytes; however, different sampling protocols and low-resolution profiling methods make it difficult to infer general principles. Here we describe methodology to characterize and compare soil- and root-inhabiting bacterial communities, which reveals not only a function for metabolically active plant cells but also for inert cell-wall features in the selection of soil bacteria for host colonization. We show that the roots of Arabidopsis thaliana, grown in different natural soils under controlled environmental conditions, are preferentially colonized by Proteobacteria, Bacteroidetes and Actinobacteria, and each bacterial phylum is represented by a dominating class or family. Soil type defines the composition of root-inhabiting bacterial communities and host genotype determines their ribotype profiles to a limited extent. The identification of soil-type-specific members within the root-inhabiting assemblies supports our conclusion that these represent soil-derived root endophytes. Surprisingly, plant cell-wall features of other tested plant species seem to provide a sufficient cue for the assembly of approximately 40% of the Arabidopsis bacterial root-inhabiting microbiota, with a bias for Betaproteobacteria. Thus, this root sub-community may not be Arabidopsis-specific but saprophytic bacteria that would naturally be found on any plant root or plant debris in the tested soils. By contrast, colonization of Arabidopsis roots by members of the Actinobacteria depends on other cues from metabolically active host cells.
DOI:10.1146/annurev-arplant-050312-120106URLPMID:23373698 [本文引用: 2]
Abstract Plants host distinct bacterial communities on and inside various plant organs, of which those associated with roots and the leaf surface are best characterized. The phylogenetic composition of these communities is defined by relatively few bacterial phyla, including Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria. A synthesis of available data suggests a two-step selection process by which the bacterial microbiota of roots is differentiated from the surrounding soil biome. Rhizodeposition appears to fuel an initial substrate-driven community shift in the rhizosphere, which converges with host genotype-dependent fine-tuning of microbiota profiles in the selection of root endophyte assemblages. Substrate-driven selection also underlies the establishment of phyllosphere communities but takes place solely at the immediate leaf surface. Both the leaf and root microbiota contain bacteria that provide indirect pathogen protection, but root microbiota members appear to serve additional host functions through the acquisition of nutrients from soil for plant growth. Thus, the plant microbiota emerges as a fundamental trait that includes mutualism enabled through diverse biochemical mechanisms, as revealed by studies on plant growth-promoting and plant health-promoting bacteria.
DOI:10.1007/s00374-012-0691-4URL [本文引用: 1]
AbstractA variety of soil factors are known to increase nutrient availability and plant productivity. The most influential might be the organisms comprising the soil microbial community of the rhizosphere, which is the soil surrounding the roots of plants where complex interactions occur between the roots, soil, and microorganisms. Root exudates act as substrates and signaling molecules for microbes creating a complex and interwoven relationship between plants and the microbiome. While individual microorganisms such as endophytes, symbionts, pathogens, and plant growth promoting rhizobacteria are increasingly featured in the literature, the larger community of soil microorganisms, or soil microbiome, may have more far-reaching effects. Each microorganism functions in coordination with the overall soil microbiome to influence plant health and crop productivity. Increasing evidence indicates that plants can shape the soil microbiome through the secretion of root exudates. The molecular communication fluctuates according to the plant development stage, proximity to neighboring species, management techniques, and many other factors. This review seeks to summarize the current knowledge on this topic.
DOI:10.1111/ppl.12192URLPMID:24684436 [本文引用: 1]
Abstract Blast, caused by the fungus Magnaporthe oryzae, is one of the most devastating diseases of rice worldwide. Phenylalanine ammonia lyase (PAL) is a key enzyme in the phenylpropanoid pathway, which leads to the biosynthesis of defense-related phytohormone salicylic acid (SA) and flavonoid-type phytoalexins sakuranetin and naringenin. However, the roles and biochemical features of individual rice PALs in defense responses to pathogens remain unclear. Here, we report that rice OsPAL06, which can catalyze the formation of trans-cinnamate using l-phenylalanine, is involved in rice root-M. oryzae interaction. OsPAL06-knockout mutant showed increased susceptibility to M. oryzae invaded from roots and developed typical leaf blast symptoms, accompanied by nearly complete disappearance of sakuranetin and naringenin and a two-third reduction of the SA level in roots. This mutant also showed compensatively induced expression of chalcone synthase, which is involved in flavonoid biosynthesis, isochorismate synthase 1, which is putatively involved in SA synthesis via another pathway, reduced jasmonate content and increased ethylene content. These results suggest that OsPAL06 is a positive regulator in preventing M. oryzae infection from roots. It may regulate defense by promoting both phytoalexin accumulation and SA signaling that synergistically and antagonistically interacts with jasmonate- and ethylene-dependent signaling, respectively. 2014 Scandinavian Plant Physiology Society.
DOI:10.1073/pnas.1414592112URLPMID:25605935 [本文引用: 1]
Author(s): Edwards, J; Johnson, C; Santos-Medelln, C; Lurie, E; Podishetty, NK; Bhatnagar, S; Eisen, JA; Sundaresan, V; Jeffery, LD | Abstract: Plants depend upon beneficial interactions between roots and microbes for nutrient availability, growth promotion, and disease suppression. High-throughput sequencing approaches have provided recent insights into root microbiomes, but our current understanding is still limited relative to animal microbiomes. Here we present a detailed characterization of the root-associated microbiomes of the crop plant rice by deep sequencing, using plants grown under controlled conditions as well as field cultivation at multiple sites. The spatial resolution of the study distinguished three root-associated compartments, the endosphere (root interior), rhizoplane (root surface), and rhizosphere (soil close to the root surface), each of which was found to harbor a distinct microbiome. Under controlled greenhouse conditions, microbiome composition varied with soil source and genotype. In field conditions, geographical location and cultivation practice, namely organic vs. conventional, were factors contributing to microbiome variation. Rice cultivation is a major source of global methane emissions, and methanogenic archaea could be detected in all spatial compartments of field-grown rice. The depth and scale of this study were used to build coabundance networks that revealed potential microbial consortia, some of which were involved in methane cycling. Dynamic changes observed during microbiome acquisition, as well as steady-state compositions of spatial compartments, support a multistep model for root microbiome assembly from soil wherein the rhizoplane plays a selective gating role. Similarities in the distribution of phyla in the root microbiomes of rice and other plants suggest that conclusions derived from this study might be generally applicable to land plants.
DOI:10.1038/ng.3337URLPMID:26053497 [本文引用: 2]
Asian cultivated rice (Oryza sativa L.) consists of two main subspecies, indica and japonica. Indica has higher nitrate-absorption activity than japonica, but the molecular mechanisms underlying that activity remain elusive. Here we show that variation in a nitrate-transporter gene, NRT1.1B (OsNPF6.5), may contribute to this divergence in nitrate use. Phylogenetic analysis revealed that NRT1.1B diverges between indica and japonica. NRT1.1B-indica variation was associated with enhanced nitrate uptake and root-to-shoot transport and upregulated expression of nitrate-responsive genes. The selection signature of NRT1.1B-indica suggests that nitrate-use divergence occurred during rice domestication. Notably, field tests with near-isogenic and transgenic lines confirmed that the japonica variety carrying the NRT1.1B-indica allele had significantly improved grain yield and nitrogen-use efficiency (NUE) compared to the variety without that allele. Our results show that variation in NRT1.1B largely explains nitrate-use divergence between indica and japonica and that NRT1.1B-indica can potentially improve the NUE of japonica.
DOI:10.1016/j.rhisph.2018.08.003URL [本文引用: 1]
DOI:10.1146/annurev-genet-110410-132549URLPMID:21838550 [本文引用: 1]
Abstract Rhizobial bacteria enter a symbiotic association with leguminous plants, resulting in differentiated bacteria enclosed in intracellular compartments called symbiosomes within nodules on the root. The nodules and associated symbiosomes are structured for efficient nitrogen fixation. Although the interaction is beneficial to both partners, it comes with rigid rules that are strictly enforced by the plant. Entry into root cells requires appropriate recognition of the rhizobial Nod factor signaling molecule, and this recognition activates a series of events, including polarized root-hair tip growth, invagination associated with bacterial infection, and the promotion of cell division in the cortex leading to the nodule meristem. The plant's command of the infection process has been highlighted by its enforcement of terminal differentiation upon the bacteria within nodules of some legumes, and this can result in a loss of bacterial viability while permitting effective nitrogen fixation. Here, we review the mechanisms by which the plant allows bacterial infection and promotes the formation of the nodule, as well as the details of how this intimate association plays out inside the cells of the nodule where a complex interchange of metabolites and regulatory peptides force the bacteria into a nitrogen-fixing organelle-like state.
DOI:10.1073/pnas.1505765112URLPMID:26305938 [本文引用: 1]
Plants maintain microbial associations whose functions remain largely unknown. For the past 15 y, we have planted the annual postfire tobacco Nicotiana attenuata into an experimental field plot in the plant's native habitat, and for the last 8 y the number of plants dying from a sudden wilt disease has increased, leading to crop failure. Inadvertently we had recapitulated the common agricultural dilemma of pathogen buildup associated with continuous cropping for this native plant. Plants suffered sudden tissue collapse and black roots, symptoms similar to a Fusarium-Alternaria disease complex, recently characterized in a nearby native population and developed into an in vitro pathosystem for N. attenuata. With this in vitro disease system, different protection strategies (fungicide and inoculations with native root-associated bacterial and fungal isolates), together with a biochar soil amendment, were tested further in the field. A field trial with more than 900 plants in two field plots revealed that inoculation with a mixture of native bacterial isolates significantly reduced disease incidence and mortality in the infected field plot without influencing growth, herbivore resistance, or 32 defense and signaling metabolites known to mediate resistance against native herbivores. Tests in a subsequent year revealed that a core consortium of five bacteria was essential for disease reduction. This consortium, but not individual members of the root-associated bacteria community which this plant normally recruits during germination from native seed banks, provides enduring resistance against fungal diseases, demonstrating that native plants develop opportunistic mutualisms with prokaryotes that solve context-dependent ecological problems.
DOI:10.1038/s41477-018-0139-4URL [本文引用: 3]
In an era of ecosystem degradation and climate change, maximizing microbial functions in agroecosystems has become a prerequisite for the future of global agriculture. However, managing species-rich communities of plant-associated microbiomes remains a major challenge. Here, we propose interdisciplinary research strategies to optimize microbiome functions in agroecosystems. Informatics now allows us to identify members and characteristics of ‘core microbiomes’, which may be deployed to organize otherwise uncontrollable dynamics of resident microbiomes. Integration of microfluidics, robotics and machine learning provides novel ways to capitalize on core microbiomes for increasing resource-efficiency and stress-resistance of agroecosystems.
DOI:10.1073/pnas.1800918115URL [本文引用: 1]
In this very large-scale longitudinal field study of the maize rhizosphere microbiome, we identify heritable taxa. These taxa display variance in their relative abundances that can be partially explained by genetic differences between the maize lines, above and beyond the strong influences of field, plant age, and weather on the diversity of the rhizosphere microbiome. If these heritable taxa are associated with beneficial traits, they may serve as phenotypes in future breeding endeavors. Soil microbes that colonize plant roots and are responsive to differences in plant genotype remain to be ascertained for agronomically important crops. From a very large-scale longitudinal field study of 27 maize inbred lines planted in three fields, with partial replication 5 y later, we identify root-associated microbiota exhibiting reproducible associations with plant genotype. Analysis of 4,866 samples identified 143 operational taxonomic units (OTUs) whose variation in relative abundances across the samples was significantly regulated by plant genotype, and included five of seven core OTUs present in all samples. Plant genetic effects were significant amid the large effects of plant age on the rhizosphere microbiome, regardless of the specific community of each field, and despite microbiome responses to climate events. Seasonal patterns showed that the plant root microbiome is locally seeded, changes with plant growth, and responds to weather events. However, against this background of variation, specific taxa responded to differences in host genotype. If shown to have beneficial functions, microbes may be considered candidate traits for selective breeding.
[本文引用: 3]
Lignin engineering in field-grown poplar trees affects the endosphere bacterial microbiome
1
2016
... 在全球生态系统退化和气候变化的背景下, 强化微生物在农业生态系统中的功能可能是未来农业可持续发展的重要方向之一(
An underground revolution: biodiversity and soil ecological engineering for agricultural sustainability
1
2016
... 在全球生态系统退化和气候变化的背景下, 强化微生物在农业生态系统中的功能可能是未来农业可持续发展的重要方向之一(
The rhizosphere microbiome and plant health
1
2012
... 在土壤中, 植物根系与微生物互作.根际微生物可以促进植物在自然界的生存, 其作用机制主要包括: (1) 促进植物从环境中获取营养, 例如, 菌根真菌溶磷和根瘤菌固氮(
Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota
1
2012
... 在土壤中, 植物根系与微生物互作.根际微生物可以促进植物在自然界的生存, 其作用机制主要包括: (1) 促进植物从环境中获取营养, 例如, 菌根真菌溶磷和根瘤菌固氮(
Structure and functions of the bacterial microbiota of plants
2
2013
... 在土壤中, 植物根系与微生物互作.根际微生物可以促进植物在自然界的生存, 其作用机制主要包括: (1) 促进植物从环境中获取营养, 例如, 菌根真菌溶磷和根瘤菌固氮(
... 在全球生态系统退化和气候变化的背景下, 强化微生物在农业生态系统中的功能可能是未来农业可持续发展的重要方向之一(
Manipulating the soil microbiome to increase soil health and plant fertility
1
2012
... 在全球生态系统退化和气候变化的背景下, 强化微生物在农业生态系统中的功能可能是未来农业可持续发展的重要方向之一(
Multiple phytohormones and phytoalexins are involved in disease resistance to Magnaporthe oryzae invaded from roots in rice
1
2014
... 在土壤中, 植物根系与微生物互作.根际微生物可以促进植物在自然界的生存, 其作用机制主要包括: (1) 促进植物从环境中获取营养, 例如, 菌根真菌溶磷和根瘤菌固氮(
Structure, variation, and assembly of the root- associated microbiomes of rice
1
2015
... 在土壤中, 植物根系与微生物互作.根际微生物可以促进植物在自然界的生存, 其作用机制主要包括: (1) 促进植物从环境中获取营养, 例如, 菌根真菌溶磷和根瘤菌固氮(
Variation in NRT1.1B contributes to nitrate-use divergence between rice subspecies
2
2015
... 亚洲栽培稻(Oryza sativa)主要分为籼稻(Indica)和粳稻(Japonica).籼稻通常比粳稻具有更高的氮利用效率, 其中一个主要因素与其从土壤中吸收氮的效率相关(
... 储成才研究团队前期对水稻氮肥利用效率的研究显示, 硝酸盐转运蛋白NRT1.1B的单个氨基酸变异是决定籼稻氮利用效率高于粳稻的主要因素之一(
Arbuscular mycorrhizal symbiosis in rice: establishment, environmental control and impact on plant growth and resistance to abiotic stresses
1
2018
... 在土壤中, 植物根系与微生物互作.根际微生物可以促进植物在自然界的生存, 其作用机制主要包括: (1) 促进植物从环境中获取营养, 例如, 菌根真菌溶磷和根瘤菌固氮(
The rules of engagement in the legume-rhizobial symbiosis
1
2011
... 在土壤中, 植物根系与微生物互作.根际微生物可以促进植物在自然界的生存, 其作用机制主要包括: (1) 促进植物从环境中获取营养, 例如, 菌根真菌溶磷和根瘤菌固氮(
Native root-associated bacteria rescue a plant from a sudden-wilt disease that emerged during continuous cropping
1
2015
... 在土壤中, 植物根系与微生物互作.根际微生物可以促进植物在自然界的生存, 其作用机制主要包括: (1) 促进植物从环境中获取营养, 例如, 菌根真菌溶磷和根瘤菌固氮(
Core microbiomes for sustainable agroecosystems
3
2018
... 在全球生态系统退化和气候变化的背景下, 强化微生物在农业生态系统中的功能可能是未来农业可持续发展的重要方向之一(
... ;
... ), 这一特性为通过选择特定土壤微生物促进作物营养吸收提供借鉴(
Large-scale replicated field study of maize rhizosphere identifies heritable microbes
1
2018
... 在土壤中, 植物根系与微生物互作.根际微生物可以促进植物在自然界的生存, 其作用机制主要包括: (1) 促进植物从环境中获取营养, 例如, 菌根真菌溶磷和根瘤菌固氮(
NRT1.1B contributes the association of root microbiota and nitrogen use in rice
3
2019
... 亚洲栽培稻(Oryza sativa)主要分为籼稻(Indica)和粳稻(Japonica).籼稻通常比粳稻具有更高的氮利用效率, 其中一个主要因素与其从土壤中吸收氮的效率相关(
... 储成才研究团队前期对水稻氮肥利用效率的研究显示, 硝酸盐转运蛋白NRT1.1B的单个氨基酸变异是决定籼稻氮利用效率高于粳稻的主要因素之一(
... 作者通过分离粳稻和籼稻特异富集的微生物, 进一步构建人工微生物群落(SynCom), 验证其在水稻生长及营养吸收中的作用.通过分别接种粳稻和籼稻特异富集的人工微生物群落, 在硝态氮或氨态氮充足条件下, 人工微生物群落会抑制水稻的生长; 但在以有机氮作为唯一氮源的条件下, 籼稻特异富集的人工微生物群落能促进水稻的生长, 表明水稻可以通过招募特异的根际微生物提高对有机氮源的利用效率(
备案号: 京ICP备16067583号-21
版权所有 © 2021 《植物学报》编辑部
地址:北京香山南辛村20号 邮编:100093
电话:010-62836135 010-62836131 E-mail:cbb@ibcas.ac.cn
本系统由北京玛格泰克科技发展有限公司设计开发

{kind=link}
{kind=link}