Spatial genetic structure of Lycium ruthenicum in the Qaidam Basin
Chun-Cheng WANG1, Song-Mei MA,,2,*, Dan ZHANG1, Shao-Ming WANG11Key Laboratory of Ecological Corps for Oasis City and Mountain Basin System, College of Life Sciences, Shihezi University, Shihezi, Xinjiang 832000, China 2Key Laboratory of Ecological Corps for Oasis City and Mountain Basin System, College of Science, Shihezi University, Shihezi, Xinjiang 832000, China
Abstract Aims Based on the cpDNA sequences, we studied the genetic diversity, genetic structure and haplotype evolution of wild Lycium ruthenicum in the Qaidam Basin and provided the scientific basis for the genetic conservation of this species. Methods We used three filtered high polymorphic cpDNA fragments (psbA-trnH, psbK-psbI and trnV) to study the genetic variation pattern of L. ruthenicum in the Qaidam Basin by employing the population genetic analysis methods. The molecular diversity indices were calculated by using the software of DnaSP 6.0 and Permut 2.0. Genetic differentiation among populations and the defined groups was estimated by the AMOVA analysis. The median-joining network and principal coordinate analysis (PCoA) were used to identify the clustering relationship of haplotype. The maximum likelihood method and Bayesian method were used to reconstruct the phylogenetic tree based on cpDNA haplotypes. Important findings The combined length of psbA-trnH, psbK-psbI and trnV was 1 454 bp. 14 polymorphic sites were detected, and a total of seven haplotypes were identified. The total genetic diversity (hT) and within-population genetic diversity (hS) were 0.916 and 0.512, respectively. Results from AMOVA suggested that more than 80% of the observed variation was due to differences among groups and populations. The maximum likelihood analysis and Beast analysis revealed that seven haplotypes clustered into two clusters, corresponding to Golmud and Delingha regions and Nuomuhong region, respectively. The revealed topological structure and clusters of haplotype network and PCoA analyses were consistent with the phylogenetic trees. Results of the Mantel test (r = 0.591 1, p = 0.000 9) indicated a non-significant correlation between geographical distance and genetic distance. The L. ruthenicum populations in the Qaidam Basin have high levels of genetic diversity and significant genetic differentiation among populations. In relation to conservation management, we identified the Nuomuhong forestry station and Xinle Village of Golmud City as having a high degree of genetic diversity and these should be the areas of the greatest focus for conservation. Keywords:Lycium ruthenicum;Qaidam Basin;chloroplast fragments;genetic variation;genetic structure
Table 1 表1 表1柴达木盆地黑果枸杞自然种群的采样信息及遗传信息 Table 1Sampling information and genetic information of natural populations of Lycium ruthenicum in the Qaidam Basin
种群名称及编码 Population name and code
海拔 Altitude (m)
经纬度 Latitude and Longitude
采样数 Sample size
单倍型及个体数 Haplotype and the individual numbers
单倍型多样性 Haplotype diversity (Hd ± SD)
核苷酸多样性 Nucleotide diversity (π ± SD)
德令哈市红光村 Hongguang Village of Delingha City (DLH)
2 970
37.38° N, 97.34° E
10
H1 (2), H4 (8)
0.356 ± 0.025
0.000 73 ± 0.33
都兰县诺木洪林业站 Nuomuhong Forestry Station of Dulan County (NMH1)
2 820
36.41° N, 96.45° E
15
H5 (7), H6 (6), H7 (2)
0.604 ± 0.069
0.000 46 ± 0.09
都兰县诺木洪乡田格力村 Tiangeli Village of Nuomuhong Township of Dulan County (NMH2)
2 762
36.39° N, 96.19° E
12
H5 (5), H7 (7)
0.530 ± 0.053
0.000 37 ± 0.05
格尔木市新乐村 Xinle Village of Golmud City (GEM1)
2 787
36.39° N, 94.86° E
12
H1 (7), H2 (4), H3 (1)
0.591 ± 0.011
0.001 26 ± 0.03
格尔木市大格勒乡 Dagele Township of Golmud City (GEM2)
2 837
36.44° N, 95.75° E
11
H1 (3), H2 (8)
0.436 ± 0.018
0.000 09 ± 0.03
H1-H7表示为本研究鉴定出的7个叶绿体单倍型。 H1-H7 represents the seven chloroplast haplotypes identified in this study.
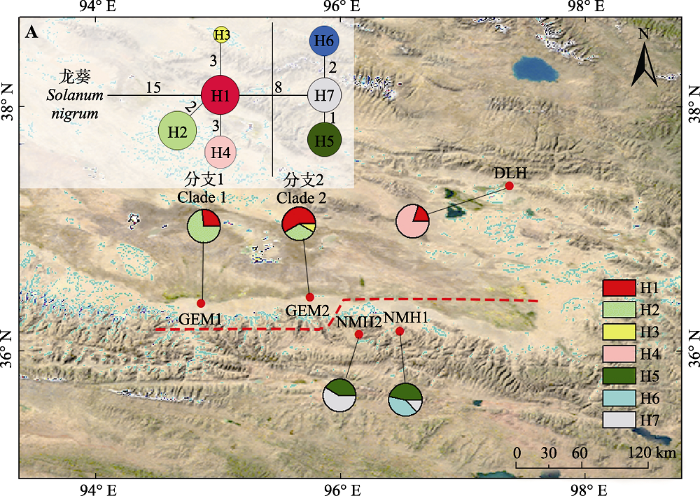
A, 单倍型网络, 图中圆圈大小与单倍型频率成正比, 节点间的分支长度大致与单倍型的突变数成正比, 相应分支附近附有步长; 龙葵作为外类群。 Fig. 1Geographical distribution and the haplotype network of seven chloroplast haplotypes (H1-H7) of Lycium ruthenicum in the Qaidam Basin. The population codes in this figure are consistent with Table 1. Pie graphs indicate the frequency of each haplotype at these locations.
A, In the median-joining haplotypes network, the sizes of the circles in the network are proportional to the haplotype frequencies. Branch lengths are roughly proportional to the number of mutation steps between haplotypes and nodes. The true number of steps is shown near the corresponding branch sections. Solanum nigrum was used as outgroup.
Table 3 表3 表3柴达木盆地黑果枸杞种群的分子方差分析(AMOVA) Table 3Analysis of molecular variance (AMOVA) of Lycium ruthenicum in the Qaidam Basin
变异来源 Source of variation
自由度 df
平方和 Sum of squares
变异组成 Variance components
变异所占比例 Percentage of variation (%)
固定指数 Fixation index
种群间 Among populations
4
105.305
2.159 37
80.01
FST= 0.800 07
种群内 Within populations
55
29.679
0.539 61
19.99
总变异 Total
59
135.953
2.698 99
组间 Among groups
1
100.383
3.254 96
82.04
FCT= 0.820 43
组内种群间 Among populations within groups
3
6.917
0.145 46
3.67
FSC= 0.204 18
种群内 Within populations
56
28.408
0.566 95
14.29
FST= 0.857 10
总变异 Total
60
135.003
3.967 37
FCT, 组间的遗传变异指数; FSC, 组内种体间的遗传变异指数; FST, 种群间的遗传变异指数。 FCT, variance among groups relative to total variance; FSC, variance among populations within groups; FST, variance among populations.
Fig. 2Plots of the first three coordinates of the principal coordinates analysis (PCoA) at the population level for Lycium ruthenicum in the Qaidam Basin.
A, 最大似然(ML)树, 分支点上方的数字为大于等于80的自展支持率。B, 贝叶斯树, 分支节点右侧的数字表示所有大于0.80的后验概率值。两系统发育树右侧的黑条表示相应的分支。 Fig. 3Phylogenetic trees of chloroplast haplotypes of Lycium ruthenicum in the Qaidam Basin.
A, Maximum likelihood (ML) tree. Bootstrap values equal to or greater than 80 are shown above the corresponding branching points. B, Bayesian tree. The values on the right of the branching points represent the posterior probability greater than 0.80. The black bars on the right of the two phylogenetic trees indicate the corresponding clades.
AlitongQ, JinXF, YeZM, WangQF, ChenJM, YangCF (2014). Reproductive ecology research on populations of a medicinal plant Lycium ruthenicum Murr. from Xinjiang reveals factors affecting fruit production Plant Science Journal, 32, 570-576. [本文引用: 1]
DrummondAJ, NichollsGK, RodrigoAG, SolomonW (2002). Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data Genetics, 161, 1307-1320. URLPMID:12136032 [本文引用: 1] Molecular sequences obtained at different sampling times from populations of rapidly evolving pathogens and from ancient subfossil and fossil sources are increasingly available with modern sequencing technology. Here, we present a Bayesian statistical inference approach to the joint estimation of mutation rate and population size that incorporates the uncertainty in the genealogy of such temporally spaced sequences by using Markov chain Monte Carlo (MCMC) integration. The Kingman coalescent model is used to describe the time structure of the ancestral tree. We recover information about the unknown true ancestral coalescent tree, population size, and the overall mutation rate from temporally spaced data, that is, from nucleotide sequences gathered at different times, from different individuals, in an evolving haploid population. We briefly discuss the methodological implications and show what can be inferred, in various practically relevant states of prior knowledge. We develop extensions for exponentially growing population size and joint estimation of substitution model parameters. We illustrate some of the important features of this approach on a genealogy of HIV-1 envelope (env) partial sequences.
EbertD, PeakallR (2009). Chloroplast simple sequence repeats (cpSSRs): technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species Molecular Ecology Resources, 9, 673-690. DOI:10.1111/j.1755-0998.2008.02319.xURLPMID:21564725 [本文引用: 1] Chloroplast microsatellites, or simple sequence repeats (cpSSRs), are typically mononucleotide tandem repeats. When located in the noncoding regions of the chloroplast genome (cpDNA), they commonly show intraspecific variation in repeat number. Despite the growing number of studies applying cpSSRs, studies of economically important plants and their relatives remain over-represented. Thus, the potential of cpSSRs to offer unique insights into ecological and evolutionary processes in wild plant species has yet to be fully realized. This review provides an overview of the technical resources available to aid cpSSR discovery including a list of cpSSR primer sets available and cpDNA sequencing resources. Our updated analysis of 99 whole chloroplast genomes downloaded from GenBank confirms that potentially variable cpSSRs are abundant in the noncoding cpDNA of plants. Overall variation in the frequency of cpSSRs was extreme, ranging from one to 700 per genome (median = 93), while in 81 vascular plants, between 35 and 160 cpSSRs were detected per genome (median = 86). We offer five recommendations to aid wider development and application of cpSSRs: (i) When genus-specific cpSSR primers are available, cross-species amplification can often be fruitful. (ii) While potentially useful, universal cpSSR primers at best provide access to only a small number of variable markers. (iii) De novo sequencing of noncoding cpDNA is the most effective and efficient way to develop cpSSR markers in wild species. (iv) DNA sequencing of cpSSR alleles is essential, given the complex nature of the genetic variation associated with hypervariable cpDNA regions. (v) The reliability of cpSSR length based genetic assays need to be validated in all studies.
ExcoffierL, LischerHEL (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows Molecular Ecology Resources, 10, 564-567. DOI:10.1111/j.1755-0998.2010.02847.xURLPMID:21565059 [本文引用: 1] We present here a new version of the Arlequin program available under three different forms: a Windows graphical version (Winarl35), a console version of Arlequin (arlecore), and a specific console version to compute summary statistics (arlsumstat). The command-line versions run under both Linux and Windows. The main innovations of the new version include enhanced outputs in XML format, the possibility to embed graphics displaying computation results directly into output files, and the implementation of a new method to detect loci under selection from genome scans. Command-line versions are designed to handle large series of files, and arlsumstat can be used to generate summary statistics from simulated data sets within an Approximate Bayesian Computation framework.
FlanneryML, MitchellFJ, CoyneS, KavanaghTA, BurkeJI, SalaminN, DowdingP, HodkinsonTR (2006). Plastid genome characterisation in Brassica and Brassicaceae using a new set of nine SSRs Theoretical and Applied Genetics, 113, 1221-1231. DOI:10.1007/s00122-006-0377-0URLPMID:16909279 [本文引用: 1] We report a new set of nine primer pairs specifically developed for amplification of Brassica plastid SSR markers. The wide utility of these markers is demonstrated for haplotype identification and detection of polymorphism in B. napus, B. nigra, B. oleracea, B. rapa and in related genera Arabidopsis, Camelina, Raphanus and Sinapis. Eleven gene regions (ndhB-rps7 spacer, rbcL-accD spacer, rpl16 intron, rps16 intron, atpB-rbcL spacer, trnE-trnT spacer, trnL intron, trnL-trnF spacer, trnM-atpE spacer, trnR-rpoC2 spacer, ycf3-psaA spacer) were sequenced from a range of Brassica and related genera for SSR detection and primer design. Other sequences were obtained from GenBank/EMBL. Eight out of nine selected SSR loci showed polymorphism when amplified using the new primers and a combined analysis detected variation within and between Brassica species, with the number of alleles detected per locus ranging from 5 (loci MF-6, MF-1) to 11 (locus MF-7). The combined SSR data were used in a neighbour-joining analysis (SMM, D (DM) distances) to group the samples based on the presence and absence of alleles. The analysis was generally able to separate plastid types into taxon-specific groups. Multi-allelic haplotypes were plotted onto the neighbour joining tree. A total number of 28 haplotypes were detected and these differentiated 22 of the 41 accessions screened from all other accessions. None of these haplotypes was shared by more than one species and some were not characteristic of their predicted type. We interpret our results with respect to taxon differentiation, hybridisation and introgression patterns relating to the 'Triangle of U'.
FukudaT, YokoyamaJ, OhashiH (2001). Phylogeny and Biogeography of the Genus Lycium (Solanaceae): inferences from chloroplast DNA sequences Molecular Phylogenetics & Evolution, 19, 246-258. [本文引用: 1]
HaoYY, XieYW, ZhangWP, NingBS, LuXJ (2016). The research progress on desert Lycium ruthenicum. Pratacultural Science, 33, 1835-1845. [本文引用: 1]
HeWG, Nasongcaoketu, WuQE, WuCH, ZhaoJ, WangY, LiYX (2015). Natural distribution and biological characteristics of Lycium ruthenicum in Yanqi Basin of Xinjiang Chinese Wild Plant Resources, 34, 59-63. [本文引用: 2]
LiuRL, YangHW, SiJH (2011). Effect of different growth regulator on raising seedlings of Lycium ruthenicum Murr by hardwood cuttings Journal of Anhui Agricultural Sciences, 39, 11447-11448. [本文引用: 1]
LiuZG, KangHL, YueHL, MeiLJ, TaoYD, ShaoY (2018). Resources investigation of Lycium ruthenicum Murr. and analysis of fruits proanthocyanidin from different regions Lishizhen Medicine and Materia Medica Research, 29, 1713-1716. [本文引用: 1]
LiuZG, ShuQY, WangL, YuMF, HuYP, ZhangHG, TaoYD, ShaoY (2012). Genetic diversity of the endangered and medically important Lycium ruthenicum Murr. revealed by sequence-related amplified polymorphism (SRAP) markers Biochemical Systematics and Ecology, 45, 86-97. [本文引用: 2]
MaSM, NieYB, JiangXL, XuZ, JiWQ (2019). Genetic structure of the endangered, relict shrub Amygdalus mongolica(Rosaceae) in arid Northwest China Australian Journal of Botany, 67, 128-139. [本文引用: 1]
McCauleyDE (1995). The use of chloroplast DNA polymorphism in studies of gene flow in plants Trends in Ecology & Evolution, 10, 198-202. DOI:10.1016/s0169-5347(00)89052-7URLPMID:21237002 [本文引用: 1] In many species of plants, the dispersal of genes is mediated by the movement of both seeds and pollen. The relative contributions of seed and pollen movement to total gene flow can be difficult to estimate. Chloroplast DNA (cpDNA) may prove useful for resolving this problem. Over the past several years, studies of numerous species of plants have shown that intraspecific variation in cpDNA is often sufficiently abundant to serve as a marker for studies of gene flow. Recent theoretical models have shown that estimates of population structure based on cpDNA polymorphism should be especially sensitive to the impact of seed movement on gene flow, because cpDNA is often maternally inherited.
MillerMP (2005). Alleles in space (AIS): computer software for the joint analysis of interindividual spatial and genetic information Journal of Heredity, 96, 722-724. DOI:10.1093/jhered/esi119URLPMID:16251514 [本文引用: 1]
PeakallR, SmousePE (2006). Genalex 6: genetic analysis in Excel. Population genetic software for teaching and research Molecular Ecology Notes, 6, 288-295. [本文引用: 1]
PonsO, PetitRJ (1996). Measuring and testing genetic differentiation with ordered versus unordered alleles Genetics, 144, 1237-1245. URLPMID:8913764 [本文引用: 2]
QiYY, HaoGJ, ChenJF (2018) Investigation on germplasm resources of wild Lycium ruthenicum in Qinghai Science and Technology of Qinghai Agriculture and Forestry, (3), 38-42. [本文引用: 2]
RonquistF, TeslenkoM, van der MarkP, AyresDL, DarlingA, H?hnaS, LargetB, LiuL, SuchardMA, HuelsenbeckJP (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space Systematic Biology, 61, 539-542. DOI:10.1093/sysbio/sys029URLPMID:22357727 [本文引用: 1] Since its introduction in 2001, MrBayes has grown in popularity as a software package for Bayesian phylogenetic inference using Markov chain Monte Carlo (MCMC) methods. With this note, we announce the release of version 3.2, a major upgrade to the latest official release presented in 2003. The new version provides convergence diagnostics and allows multiple analyses to be run in parallel with convergence progress monitored on the fly. The introduction of new proposals and automatic optimization of tuning parameters has improved convergence for many problems. The new version also sports significantly faster likelihood calculations through streaming single-instruction-multiple-data extensions (SSE) and support of the BEAGLE library, allowing likelihood calculations to be delegated to graphics processing units (GPUs) on compatible hardware. Speedup factors range from around 2 with SSE code to more than 50 with BEAGLE for codon problems. Checkpointing across all models allows long runs to be completed even when an analysis is prematurely terminated. New models include relaxed clocks, dating, model averaging across time-reversible substitution models, and support for hard, negative, and partial (backbone) tree constraints. Inference of species trees from gene trees is supported by full incorporation of the Bayesian estimation of species trees (BEST) algorithms. Marginal model likelihoods for Bayes factor tests can be estimated accurately across the entire model space using the stepping stone method. The new version provides more output options than previously, including samples of ancestral states, site rates, site d(N)/d(S) rations, branch rates, and node dates. A wide range of statistics on tree parameters can also be output for visualization in FigTree and compatible software.
SangT, CrawfordDJ, StuessyTF (1997). Chloroplast DNA phylogeny, reticulate evolution, and biogeography of Paeonia(Paeoniaceae) American Journal of Botany, 84, 1120-1136. DOI:10.2307/2446155URL [本文引用: 1]
SimmonsMP, OchoterenaH (2000). Gaps as characters in sequence-based phylogenetic analyses Systematic Biology, 2, 369-381. [本文引用: 1]
ShawJ, LickeyEB, SchillingEE, SmallRL (2007). Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III American Journal of Botany, 94, 275-288. DOI:10.3732/ajb.94.3.275URLPMID:21636401 [本文引用: 2] Although the chloroplast genome contains many noncoding regions, relatively few have been exploited for interspecific phylogenetic and intraspecific phylogeographic studies. In our recent evaluation of the phylogenetic utility of 21 noncoding chloroplast regions, we found the most widely used noncoding regions are among the least variable, but the more variable regions have rarely been employed. That study led us to conclude that there may be unexplored regions of the chloroplast genome that have even higher relative levels of variability. To explore the potential variability of previously unexplored regions, we compared three pairs of single-copy chloroplast genome sequences in three disparate angiosperm lineages: Atropa vs. Nicotiana (asterids); Lotus vs. Medicago (rosids); and Saccharum vs. Oryza (monocots). These three separate sequence alignments highlighted 13 mutational hotspots that may be more variable than the best regions of our former study. These 13 regions were then selected for a more detailed analysis. Here we show that nine of these newly explored regions (rpl32-trnL((UAG)), trnQ((UUG))-5'rps16, 3'trnV((UAC))-ndhC, ndhF-rpl32, psbD-trnT((GGU)), psbJ-petA, 3'rps16-5'trnK((UUU)), atpI-atpH, and petL-psbE) offer levels of variation better than the best regions identified in our earlier study and are therefore likely to be the best choices for molecular studies at low taxonomic levels.
SunFF, NieYB, MaSM, WeiB, JiWQ (2019). Species differentiation of Haloxylon ammodendron and Haloxylon persicum based on ITS and cpDNA sequences Scientia Silvae Sinicae, 55(3), 43-53. [本文引用: 1]
TamuraK, DudleyJ, NeiM, KumarS (2007). MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0 Molecular Biology and Evolution, 24, 1596-1599. DOI:10.1093/molbev/msm092URLPMID:17488738 [本文引用: 1] We announce the release of the fourth version of MEGA software, which expands on the existing facilities for editing DNA sequence data from autosequencers, mining Web-databases, performing automatic and manual sequence alignment, analyzing sequence alignments to estimate evolutionary distances, inferring phylogenetic trees, and testing evolutionary hypotheses. Version 4 includes a unique facility to generate captions, written in figure legend format, in order to provide natural language descriptions of the models and methods used in the analyses. This facility aims to promote a better understanding of the underlying assumptions used in analyses, and of the results generated. Another new feature is the Maximum Composite Likelihood (MCL) method for estimating evolutionary distances between all pairs of sequences simultaneously, with and without incorporating rate variation among sites and substitution pattern heterogeneities among lineages. This MCL method also can be used to estimate transition/transversion bias and nucleotide substitution pattern without knowledge of the phylogenetic tree. This new version is a native 32-bit Windows application with multi-threading and multi-user supports, and it is also available to run in a Linux desktop environment (via the Wine compatibility layer) and on Intel-based Macintosh computers under the Parallels program. The current version of MEGA is available free of charge at (http://www.megasoftware.net).
The General Office of People’s Government of Qinghai Province (2017). Guidelines of the general office of the people’s government of Qinghai Province on strengthening the management of Lycium ruthenicum resources Qinghai Prataculture, 26(1), 70-71. [本文引用: 1]
WangJN, ChenJF, ChenWS, ZhouXY, XuD, LiJH, QiX (2015). Population genetic diversity of wild Lycium ruthenicum in Qaidam inferred from AFLP markers Chinese Journal of Plant Ecology, 39, 1003-1011. [本文引用: 5]
XinJP, ZhuCY (2015). Morphological structure comparison of Lycium ruthenium at different salt habitats in Qaidam Basin Journal of West China Forestry Science, 44, 73-78. [本文引用: 1]
Chloroplast simple sequence repeats (cpSSRs): technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species 1 2009
Genetic diversity of the endangered and medically important Lycium ruthenicum Murr. revealed by sequence-related amplified polymorphism (SRAP) markers 2 2012
Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III 2 2007
 ,
, ,2,*, 张丹1, 王绍明1
,2,*, 张丹1, 王绍明1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT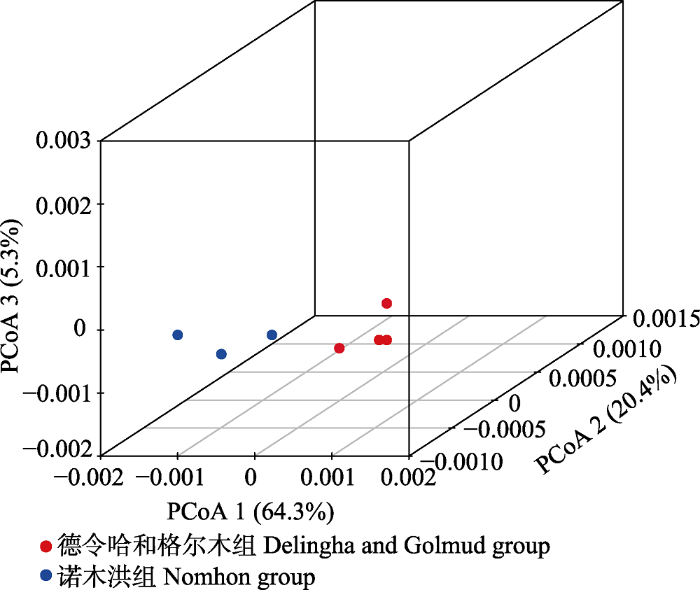 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT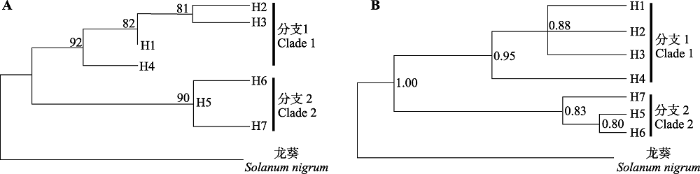 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}