0 引言
【研究意义】根系作为支撑和固定植物地上部的重要器官,在植物的生长发育中起着重要作用。小麦根系构型与其抗旱、抗干热风能力有密切的关系,是抗逆性研究的热点,但根生长于地下,对其取样研究存在极大的困难。发掘根系相关的分子标记并应用到资源鉴定和育种中,将会大大降低劳动强度、提高育种效率,对提高作物的抗旱性和产量及品种改良具有重要意义。【前人研究进展】作物根系的生长状况及在土壤中的分布对作物产量有显著影响。前人通过对水稻、小麦及大豆等作物的研究表明,增加深层土壤根系分布可获得较高的产量[1,2]。作物根系在土壤中的垂直分布与抗逆性关系密切。在充分灌溉条件下,小麦根系主要分布在浅土层,在干旱条件下,深层根系的分布明显增多[3]。根的生长发育受根尖分生组织调控。PENG等[4]研究发现,DA1-related protein2(DAR2)通过影响根尖中生长素的局部分布调控根尖分生组织大小,进而建立了细胞分裂素、生长素、SHY2和PLT调控途径间的联系。YUSAKU等[5]通过对水稻深根基因DRO1(DEEPER ROOTING 1)的研究证明,DRO1位于第9染色体,它的增强表达可以减小根系夹角,增加根系向下生长的趋势。研究表明,DRO1是一个早期生长素响应基因,通过生长素起负向调节作用参与根尖细胞的伸长,可能直接受生长素信号通路ARFs的影响。根系性状是复杂的数量性状,受多基因控制[6],基因型是根系构型的决定性因素[6,7]。根系在一定程度上受控制株高的Rht的影响,与株高相关的矮杆基因Rht-B1c、Rht-D1c和Rht12显著降低了小麦苗期的根长、根表面积、根体积;部分苗期根系性状QTL和千粒重QTL在同一标记区间内,说明千粒重与苗期根系有着密切的联系[8,9]。水稻根系性状的主效基因位点分布在第1、2、3、7和9染色体上[10,11]。在小麦中,CAO等[12]通过研究178A(短根系)和178B(长根系)2种不同根系类型小麦品种杂交后代F1、F2的根系,发现qTaLRO-B1是影响普通小麦根系长度和相关性状的数量性状位点(QTL)。关联分析(association analysis)是一种以连锁不平衡(linkage disequilibrium,LD)为基础,鉴定自然群体内表型变异与遗传标记或候选基因位点变异关系的一种分析方法[13],与传统的QTL作图相比具有明显的优势,成为植物功能基因组学研究的主要方法,广泛应用于作物产量性状[14,15]、品质性状[16,17]和抗病性[18,19]等基因的发掘与功能研究。功能标记(functional markers,FMs)[20]是根据功能基因内部引起表型性状变异的多态性基序开发的分子标记。由于功能标记与目的基因之间完全连锁,因此,利用功能标记能够在不同遗传背景下有效可靠地鉴定目标等位基因[21]。【本研究切入点】在水稻中发现深根基因DRO1对增加水稻根系深度、提高干旱胁迫耐受能力具有重要作用,但在小麦中尚未见有关TaDRO的研究报道。【拟解决的关键问题】本研究拟克隆小麦TaDRO,分析TaDRO序列在小麦品种的变异情况,通过候选基因关联分析挖掘根系相关的优异单元型,并开发有效的分子标记用于育种。1 材料与方法
1.1 试验材料
从262份中国小麦微核心种质[22](包括157份地方品种、88份现代选育品种和17份国外引进种质)中选取20份根系长度差异较大的材料和中国春共21份材料(电子附表1),对其TaDRO-5A及TaDRO-5B编码区和启动子区域进行测序。在中国春幼苗期、拔节期和抽穗期不同器官中提取RNA用于目标基因的表达分析。用中国春缺体四体材料进行TaDRO-5A和TaDRO-5B的染色体定位,用最新版中国春基因组序列对其进行准确的物理定位(https://urgi.versailles.inra. fr/blast/)。用微核心种质材料和323份材料组成的自然群体(主要来自北部冬麦区和黄淮海冬麦区)材料测定幼苗期、拔节期和灌浆期根系性状。用323份自然群体测定株高、千粒重等性状,并进行关联分析。
用262份微核心种质、348份国内育成品种、384份欧洲材料、471份北美洲材料、51份澳大利亚材料、83份俄罗斯材料和53份墨西哥材料进行TaDRO-5A和TaDRO-5B的单元型鉴定,分析TaDRO-5A和TaDRO-5B单元型在中国和全球品种中的地理分布。
1.2 目标基因克隆
在NCBI数据库(根据3个同源基因之间的序列差异,利用Primer Premier 5.0软件设计基因组A、B、D的特异引物(电子附表2)。以待检材料的基因组DNA为模板,用高保真酶Trans StartTM Fast Pfu DNA Polymerase(北京全式金公司)进行PCR扩增。反应体系为2×GC Buffer 7.5 μL、dNTP(2.5 μmol·L-1)2.4 μL、TransStartTM Fast Pfu酶(5 U)0.3 μL、模板DNA(50 ng·μL-1)1.5 μL、引物F(5 μmol·L-1)0.5 μL、引物R(5 μmol·L-1)0.5 μL、LA Taq(0.75 U)0.15 μL,补充ddH2O至15 μL。PCR反应程序为96℃ 5 min;96℃ 45 s,60—64℃(根据引物调整)45 s,72℃ 2 min,36个循环;72℃ 15 min;4℃保存。PCR仪为Biometra T1 Thermolcycler。在1.5%琼脂糖凝胶中检测PCR产物,回收纯化目标片段并测序。
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图1TaDRO-5A和TaDRO-5B的染色体定位
-->Fig.1Physical mapping of TaDRO-5A and -5B on group 5 chromosomes in wheat
-->
1.3 基因定位及功能标记开发
采用DNASTAR Lasergene7.1中的SeqMan对21份材料的测序结果进行序列组装、拼接和比对分析,根据TaDRO-5A序列启动子区-2 271bp位点的多态性(A/C)开发分子标记,命名为TaDRO-5A-KASP,并设计KASP(Kompetitive allele specific PCR)标记引物;根据TaDRO-5B启动子区-300 bp位点的多态性(InDel)开发分子标记,命名为TaDRO-5B-InDel,并设计PCR标记引物(电子附表2)。TaDRO-5A-KASP标记的基因型分析采用QuantStudio? 7 Flex实时定量PCR系统进行。PCR反应体系为模板DNA(50 ng·μL-1)2.2 μL、KASP 2×master mix 2.5 μL、MgCl2(25 mmol·L-1)0.04 μL、primer Mix(10 μmol·L-1)0.056 μL,ddH2O 补充至5.0 μL。PCR程序为stageⅠ:95℃ 15 min,1个循环;stageⅡ:95℃ 20 s,65—55℃ 1 min,10个循环(每个循环降低1.0℃);stageⅢ:95℃ 20 s,57℃ 1 min,29个循环。反应完成后自动进行荧光定量检测,读取分型结果。
TaDRO-5B-InDel标记的PCR反应体系为模板DNA(50 ng·μL-1)2.0 μL、Buffer 10.0 μL、dNTP(2.5 μmol·L-1)3.2 μL、TransStartTM Fast Pfu(5 U)0.05 μL、引物F(5 μmol·L-1)0.8 μL、引物R(5 μmol·L-1)0.8 μL、LA Taq(0.75 U)0.16 μL,ddH2O补充至20 μL。反应程序为94℃ 4 min;94℃ 30 s,62℃ 30 s,72℃ 40 s;72℃ 7 min,4℃保存。38个循环。反应完成后用2.0%的琼脂糖凝胶电泳,凝胶成像仪检测分型结果。
1.4 表型数据收集
1.4.1 小麦苗期根系性状测定小麦幼苗期根系性状选用中国小麦微核心种质测定。测定方法:按土和草木炭体积比为3﹕1拌匀,装入直径6.5 cm、高15 cm的纸钵中,每纸钵播1粒种子,每份材料播5粒,播后覆土2—3 cm,放置于中国农业科学院作物科学研究所旱棚进行培养。出苗后根据墒情补充水分并注意防虫防鸟。至4叶期时,选择生长一致的3株洗净根系,用扫描仪(Epson Flatbed Scanner)进行根系扫描,然后用根系分析系统WinRHIZO ProLA2400 (
拔节期和灌浆期根系性状测定材料为323份品种组成的自然群体。用PVC管培养小麦,每管播种一个品种。PVC管的内径11 cm、长度分别为120 cm(拔节期)和160 cm(灌浆期)。播种前向PVC管中装填配制的培养土(填土密度为1 200 kg·m-3),自然沉降24 h后浇水600 mL/管造墒,自然入渗12 h加适量培养土抹平表面后选饱满一致的6粒种子均匀播种于管内,覆土4—5 cm。生长至3叶期定苗3株,分别在越冬前和返青后浇水500 mL/管,至拔节期和灌浆期洗净根系后测量最大根长、根鲜重、根干重、地上部鲜重、地上部干重等指标。培养土配制按土和草木炭体积比为5﹕1掺匀,同时按每亩施有机肥100 kg、缓控释肥20 kg(美国奥绿肥,总养分含量≥32%,总氮含量≥18%,有效磷含量≥18%,有效钾含量≥18%)、磷酸二铵10 kg掺入肥料。
本研究中定义幼苗期根系生长角度(root growth angle,RGA)为小麦初生根间最大夹角。测定方法是将幼苗培养8 d后采集相片,用Adobe Photoshop CS6.0软件分析初生根最大夹角,幼苗培养参照刘秀林等[24]的凝胶室培养方法。
1.4.2 自然群体株高和千粒重等性状测定
选用323份自然群体在成熟期测定株高、千粒重等性状。参试材料于2015年、2016年种植在中国农业科学院作物科学研究所昌平和顺义试验基地,其中2015年只在顺义基地种植,共2年2点的试验结果。田间水分管理分为雨养/干旱胁迫(drought stressed,DS。小麦全生育期只浇越冬水)、灌溉(well-watered,WW。分别在越冬前、孕穗期和开花期浇1次水,每次浇水约750 m3·hm-2)、水热(hydrothermal environment,HE。小麦种植于塑料大棚中,水分管理同灌溉处理)和旱热(drythermal environment,DE。小麦种植于塑料大棚中,全生育期只浇越冬水)4种处理。每份材料种4行,行距0.3 m,行长2 m,每行均匀点播40粒种子。小麦收获前,在每小区中部随机取10株,考察株高、单株穗数、穗粒数、每穗小穗数、千粒重等指标。
1.6 关联分析
分别用基因标记TaDRO-5A-KASP和TaDRO-5B- InDel检测由323个品种构成的自然群体,进行单元型遗传效应分析。用Structure2.3.1软件进行群体遗传结构分析,估计最佳群体组群数,K值取1—11,每个K值单独运行6次。当K=2时曲线出现最大拐点,因此将323份材料分为2个类群。利用TASSEL软件[25]的混合线性模型(mixed linear model,MLM),将群体结构分析所得的Q值和亲缘关系K作为协变量,进行标记与性状的关联分析,关联信号的阈值为P<0.05。2 结果
2.1 TaDRO克隆及染色体定位
从小麦品种中国春第5部分同源群克隆到小麦TaDRO的3个同源基因TaDRO-5A、TaDRO-5B和TaDRO-5D。3个基因均由4个内含子和5个外显子组成。TaDRO-5A全长2 454 bp,包括5′-UTR 119 bp、基因区2 322 bp及3′-UTR 13 bp;TaDRO-5B全长2 319 bp,包括5′-UTR 118 bp、基因区2 168 bp及3′-UTR 33 bp;TaDRO-5D全长2 313 bp,包括5′-UTR 120 bp、基因区2 160 bp及3′-UTR 33 bp。用TaDRO-5A和TaDRO-5B基因组特异标记引物分别扩增中国春-缺体四体中的目标基因表明,TaDRO-5A-KASP和TaDRO-5B-InDel分别为5A、5B染色体特异标记(图1)。在最新组装的中国春基因组序列,TaDRO-5A、TaDRO-5B和TaDRO-5D分别定位在426 158 549—426 156 228 bp、381 044 454—381 042 287 bp和327 633 935—327 631 776 bp处,均位于染色体长臂上。2.2 TaDRO主要在根、茎和发育籽粒中表达,但不同组织差异很大
从小麦品种中国春幼苗期、拔节期和抽穗期的根、茎、叶及幼穗和种子中分别取样,用实时定量PCR分析TaDRO组织表达模式。发现TaDRO在小麦发育不同时期的根、茎、叶、及幼穗、籽粒中都有表达,但在茎中表达量最高,在籽粒中的表达量次之,在叶片中表达量最低,在根系中的表达量随生育进程推进而提高(图2),在抽穗期根中的表达量分别是幼苗期根和拔节期根中表达量的3.4倍和2.5倍。 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图2TaDRO在小麦不同发育时期的组织特异性表达
-->Fig. 2Dynamic expression of TaDRO in multi-tissue at different development stages
-->
2.3 TaDRO序列多态性分析及分子标记开发
对21份材料的测序结果进行比对分析表明,TaDRO-5A编码区及启动子区共发现3个SNP位点(图3-a),分别为-2 271位(A/C)、103位(A/C)和424位(A/T),形成2种单元型Hap-5A-A和Hap-5A-C。其中103 bp和424 bp位点位于基因的内含子区,-2 271 bp位点多态性位于基因启动子区。根据SNP-2271位点开发的标记TaDRO-5A-KASP引物对参试材料进行检测,可以区分小麦TaDRO-5A的2种单元型。TaDRO-5B编码区和启动子区检测到17个SNP和1个InDel(38 bp)(图3-b),形成2种单元型Hap-5B-Ⅰ和Hap-5B-Ⅱ。其中,启动子区有13个SNP和1个InDel,编码区有4个SNP,其中2个在外显子。用分子标记TaDRO-5B-InDel引物对参试材料进行检测,扩增出357 bp(Hap-5B-Ⅰ)和395 bp(Hap-5B-Ⅱ)2种大小片段条带(图3-b),可区分群体的2种单元型。在TaDRO-5D基因区中未发现多态性位点。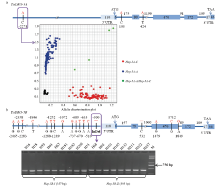 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图3TaDRO的基因结构及分子标记开发
-->Fig. 3Gene structures and development of functional markers of TaDRO-5A and -5B
-->
2.4 TaDRO变异与根长、根夹角、株高、千粒重等重要性状相关联
用标记TaDRO-5A-KASP和TaDRO-5B-InDel将323个品种组成的自然群体分别区分为2种单元型。用2年2点4种水、热处理共10种环境条件下收集的表型数据进行关联分析,TaDRO-5A与7种环境条件的株高、5种环境条件的千粒重显著相关,另外还与苗期根系生长角度显著相关;TaDRO-5B与9种环境条件的株高显著或极显著相关(表1)。Table 1
表1
表1小麦自然群体TaDRO-5A和TaDRO-5B单元型与农艺性状的关联分析
Table 1Association analysis between haplotypes at TaDRO-5A and TaDRO-5B with agronomic traits in wheat
| 年份 Year | 地点 Site | 环境 Environments | TaDRO-5A | TaDRO-5B | ||||
|---|---|---|---|---|---|---|---|---|
| 株高PH P-value | 千粒重TKW P-value | 根角度RGA P-value | 株高PH P-value | 根角度RGA P-value | ||||
| 2015 | 顺义 Shunyi | 水WW | ns | ns | 0.0017** | |||
| 旱DS | ns | 0.0431* | ns | |||||
| 水热HE | 0.0310* | 0.0252* | 0.0011** | |||||
| 旱热DE | 0.0165* | 0.0209* | 0.0223* | |||||
| 2016 | 昌平 Changping | 水WW | 0.0223* | ns | 0.0364* | |||
| 旱DS | ns | ns | 0.0017** | |||||
| 顺义 Shunyi | 水WW | 0.0160* | 0.0342* | 0.0133* | ||||
| 旱DS | 0.0148* | 0.0198* | 0.0327* | |||||
| 水热HE | 0.0108* | ns | 0.0095** | |||||
| 旱热DE | 0.0243* | ns | 0.0122* | |||||
| 2016 | 实验室 Lab | 0.04984* | ns | |||||
新窗口打开
采集262份中国小麦微核心种质幼苗期的根总长、孕穗期和灌浆期最大根长、根系生长角度、孕穗期、灌浆期和成熟期株高、千粒重(表2、图4)。统计分析表明,单元型Hap-5A-C和Hap-5B-Ⅰ的根长、株高均高于单元型Hap-5A-A和Hap-5B-Ⅱ,而千粒重的表现刚好相反,前者低于后者。在幼苗期,携带Hap-5A-C的品种平均根总长较携带Hap-5A-A的品种长12.14 cm,株高高9.45 cm;在孕穗期,Hap-5A-C的平均最大根长较Hap-5A-A长1.46 cm,株高平均高1.47 cm;在灌浆期,Hap-5A-C的平均最大根长较Hap-5A-A长4.58 cm,株高平均高5.43 cm;千粒重前者较后者低4.73 g。
Table 2
表2
表2小麦自然群体TaDRO-5A和TaDRO-5B单元型部分性状比较
Table 2Association analysis between TaDRO-5A and TaDRO-5B haplotypes RL, PH, TKW and RGA in wheat
| 性状 Trait | TaDRO-5A | TaDRO-5B | ||
|---|---|---|---|---|
| Hap-5A-A | Hap-5A-C | Hap-5B-Ⅰ | Hap-5B-Ⅱ | |
| 根长RL | 391.07±111.56 | 403.21±95.13 | 410.91±98.80a | 383.97±112.15b |
| 株高PH | 116.85±23.22B | 126.30±18.53A | 130.20±13.34A | 111.69±24.64B |
| 千粒重TKW | 41.50±7.49A | 36.77±7.66B | 35.99±6.58B | 43.32±7.56A |
| 根夹角RGA | 112.87±1.39a | 102.61±4.66b | 112.10±3.64 | 111.73±1.46 |
新窗口打开
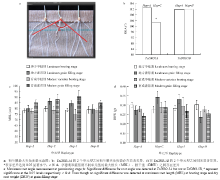 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图4TaDRO-5A与TaDRO-5B单元型与根系性状的关联结果
-->Fig. 4Association analysis of haplotype effects of TaDRO-5A and TaDRO-5B for root architecture
-->
在幼苗期,携带Hap-5B-Ⅰ的平均根总长较携带Hap-5B-Ⅱ者长26.94 cm,株高平均高18.51 cm;在孕穗期,Hap-5B-Ⅰ的平均最大根长较Hap-5B-Ⅱ短0.20 cm,株高平均高6.61 cm;在灌浆期,Hap-5B-Ⅰ的平均最大根长较Hap-5B-Ⅱ长2.05 cm,株高平均高12.20 cm,但千粒重低7.33 g。上述结果说明TaDRO在调控小麦根系生长的同时,也调控株高和千粒重,与TaDRO在根、茎、种子中表达结果是吻合的。
因此,Hap-5A-C和Hap-5B-Ⅰ为根长较长(本研究中定义为深根型单元型)、株高较高、千粒重较低的深根型单元型,Hap-5A-A和Hap-5B-Ⅱ为根长较短(本研究中定义为浅根型单元型)、株高较矮、千粒重较高的浅根型单元型。单元型Hap-5A-C的根系角度较Hap-5A-A小10.26°,说明Hap-5A-C通过减小根系角度增加根系下扎深度(表2)。
2.5 中国TaDRO-5A变异的分布
在地方品种中,生态区对单元型的选择比较明显,深根型单元型Hap-5A-C的频率在Ⅲ、Ⅳ生态区较低,在其他生态区都较高(图5-a),尤其在第Ⅹ麦区频率最高,达到90.0%。第Ⅹ麦区气候干燥,由于深根型小麦能有效利用土壤深层水分而得到选择保留,因此单元型Hap-5A-C分布频率高;第Ⅴ麦区由于降雨量大,容易发生涝灾,深根型小麦抗倒伏能力较强,因此深根单元型Hap-5A-C频率也较高;而在Ⅲ、Ⅵ麦区,气候湿润,因此以浅根型单元型Hap-5A-A为主。在育成品种中,深根单元型Hap-5A-C在各生态区小麦材料中所占比例有所降低,尤以Ⅰ、Ⅱ、Ⅴ麦区降低较多,但在Ⅵ、Ⅶ、Ⅷ、Ⅹ麦区由于干旱缺水,深根单元型Hap-5A-C仍然占有较高比例。与地方品种相比,现代育成品种中浅根型单元型Hap-5A-A在各个麦区所占频率均有较大幅度提高, 区的优势单元型(Ⅹ麦除外),主要是因为深根型单元型Hap-5A-C株高较高、千粒重低受到负向选择而淘汰。但在气候干旱的Ⅵ、Ⅶ、Ⅷ、Ⅹ等麦区深根型单元型Hap-5A-C占有较大比例,尤其在新疆春冬麦区(Ⅹ)仍为优势单元型(图5-b)。TaDRO-5B单元型的分布与TaDRO-5A相似,在地方品种中深根型单元型Hap-5B-Ⅰ的频率除在Ⅵ、Ⅶ生态区较低外,在其他生态区都占绝对优势(图5-c)。但近半个多世纪的育种选择,Hap-5B-Ⅰ在各生态区所占比例有大幅下降,只在Ⅹ麦区仍占到较大比例。因为Hap-5B-Ⅰ的株高显著高于Hap-5B-Ⅱ,而千粒重又低,因此在现代品种改良过程中,由于株型矮化及千粒重提高,造成对深根型单元型Hap-5B-Ⅰ受到负向选择。与地方品种相比,深根型单元型Hap-5B-Ⅰ在各个麦区所占频率大幅降低,甚至在Ⅸ麦区等已经完全消失,但在降雨量低、气候干旱的第Ⅹ麦区仍占有较大比例(图5-d)。
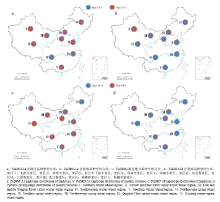 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图5TaDRO-5A和TaDRO-5B单元型在主要麦区的分布随着育种年代推进,深根型单元型Hap-5A-C、Hap-5B-Ⅰ频率呈逐年下降趋势,其频率分别从地方品种的50.3%和75.2%下降到20世纪90年代的25.0%和12.0%,而浅根型单元型Hap-5A-A、Hap-5B-Ⅱ频率呈现增加的趋势(
-->Fig. 5TaDRO-5A, TaDRO-5B haplotype distribution of landraces (a,c) and modern varieties (b,d) in ten Chinese wheat ecology regions
-->
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图6TaDRO-5A和TaDRO-5B单元型频率在中国小麦中随年代的变化
-->Fig. 6Haplotype frequencies of TaDRO-5A and TaDRO-5B with the variation tendency in different periods in China wheat
-->
2.6 全球品种中TaDRO-5A及Hap-5B-II的分布
从图7-a可知,在气候干燥的澳大利亚、墨西哥等小麦主要产区,品种以深根型单元型Hap-5A-C为主,频率分别达到60.0%和53.8%;而在中国、欧洲、北美洲和俄罗斯则以浅根型单元型Hap-5A-A为主。对TaDRO-5B而言,全球育种对浅根、矮秆大粒的Hap-5B-Ⅱ情有独钟,频率都超过90%(图7-b)。总体而言,TaDRO-5A和TaDRO-5B不同单元型在全球的分布规律是一致的。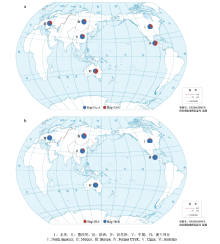 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图7TaDRO-5A(a)和TaDRO-5B(b)单元型在全球6个小麦主产区的地理分布
-->Fig. 7Geographic distribution of haplotypes at TaDRO-5A (a) and TaDRO-5B (b) in six major wheat production regions worldwide
-->
3 讨论
3.1 根系基因TaDRO克隆及功能标记开发的意义
根系生长于地下,对其取样研究存在极大的困难。长期以来,研究者花费了大量的时间和精力,探索了不同的根系培养和取样方法[23,26-32]。这些方法各具优缺点,试验中应根据研究目标选择较为理想的研究方法。本研究探讨了根长、根系生物量与根系相关基因的相关性,幼苗期根系采用的纸钵土壤培养法,拔节期、灌浆期采用根管土柱法,小麦生长条件接近自然环境,且容易获得相对完整的根系,是能满足本研究目标要求的理想方法。虽然根系测定技术不断完善,但是现有的取样方法都存在一定的局限性,尤其在研究小麦等须根系、且扎根较深的作物时还不能提供有力的技术支撑。因此,鉴于根系改良对提高作物抗逆性和产量的重要作用,发掘与作物根系发育相关基因,开发功能标记具有十分重要的意义。LI等[33]以旱选10号为材料克隆了小麦根系相关基因TaMOR,在水稻中过量表达该基因可以显著促进根的形成和发育,最终提高穗粒数[5]。因此,借鉴在产量和抗性及水稻根系研究中的经验,加强小麦根系基因TaDRO的克隆和功能研究,发掘有效的分子标记并应用到资源鉴定和育种中,将是提高小麦产量和抗旱性研究的重要途径。
3.2 TaDRO是与小麦根深、株高和籽粒千粒重相关的一因多效基因
在谷类作物中,深根是由根系的生长角度、以及种子根和节生根决定的[34,35]。由于初生根生长角度(RGA)决定着根系的生长方向,因此决定着植物发育形成浅根或者深根。在水稻中DRO1是一个控制根系生长角度的数量性状位点。YUSAKU等[5]通过对水稻根系的研究发现DRO1过量表达可以减小根系生长角度,使其生长方向更为竖直,从而改变根系形态及在土壤中的分布,增加深层土壤根系含量而提高水稻避旱能力。因为在干旱条件下,水分在深土层的分布是不均匀的,深根系具有吸收更多深层土壤水分的能力,因此,增加深层根系的分布是作物应对干旱胁迫的有效策略[36,37,38,39],也是提升作物抗旱能力的关键[38,40-41]。又因为株高和根系成正相关关系[42],所以干旱地区的小麦一般具有较高的株高和较深的根系。本研究表明,TaDRO与OsDRO1功能相似,深根单元型Hap-5A-C可以通过减小初生根之间的夹角,使根系更加竖直生长,从而使小麦在干旱胁迫下能吸收深层土壤水分而提高耐旱能力。以中国育成品种的地理分布为例,新疆冬春麦区、西北春麦区、北部春麦区等比较干旱的地区,深根单元型Hap-5A-C和Hap-5B-I频率较高,这与当地干旱环境下,重视选择耐旱型小麦品种有关;从世界麦区分布来看,澳大利亚和墨西哥(CIMMYT)深根单元型Hap-5A-C频率较高,也与当地干旱的气候条件有关。从时间分布来看,从1940年到2000年,深根单元型Hap-5A-C和Hap-5B-Ⅰ的比例呈下降趋势,原因之一是中国农田灌溉面积比例增加(从1949年的1.60×107 hm2发展到2008年的5.87×107 hm2 [43])。灌溉条件的改善,生产上对深根型品种的需求降低,因此深根型单元型Hap-5A-C、Hap-5B-Ⅰ逐渐被淘汰。另外,根系性状是复杂的数量性状,受多基因控制。在水稻中已经报道有4个与根系性状有关的基因DRO1[44]、qSOR1[45]、DRO2[46]和DRO3[47]共同调控根系生长。因此推测在小麦中还有未被发现的根系QTL和TaDRO共同调控着根系生长角度或根系长度,有待于进一步研究。可以看出,现代育种是一个在以高产为主要目标下的千粒重、株高和根系之间相互协调的过程,在此过程中,随着生产条件的改善深根单元型的频率降低。
3.3 TaDRO-5A和TaDRO-5B单元型在全球小麦育种过程中经历了强烈的选择
受自然选择和人工选择都较强的作物品种,其基因组中的很多基因有选择的痕迹。对玉米的研究发现,其基因组中大约有1 200个基因(2%—5%)在驯化和现代育种中经受了选择[48],水稻Waxy[49]、小麦株高基因Rht-B1和Rht-D1[50]、西红柿果实大小基因fw2.2[51]等都留下了选择的痕迹。在现代小麦育种中,小麦根系基因随着株高和千粒重的改良而经受了较强的人工选择作用。一般情况下,经历过较强选择作用的基因,存在着与生态适应或与农艺性状相关且分布极不均匀的高频等位变异[52]。对中国小麦微核心种质分析表明,TaDRO 在地方品种中以深根单元型Hap-5A-C(50.32%)和Hap-5B-Ⅰ(75.16%)为主,而在现代育成品种中则以浅根单元型Hap-5A-A(65.52%)和Hap-5B-Ⅱ(81.82%)型为主。从不同年代的育成品种看,深根型单元型Hap-5A-C和Hap-5B-Ⅰ频率呈逐年下降趋势,而浅根型单元型Hap-5A-A和Hap-5B-Ⅱ呈现增长的趋势(图6)。从地理分布分析,干旱缺水的地区深根单元型Hap-5A-C和Hap-5B-Ⅰ分布频率总体较高(图5和图7),说明TaDRO-5A和TaDRO-5B单元型的选择在育种目标和种植环境的作用下经历了强烈的选择。由于根系与株高和千粒重显著相关,育种家在追求高产、稳产,防止倒伏和提高收获指数的过程中,浅根单元型Hap-5A-A和Hap-5B-Ⅱ得到正向选择并不断累积,在现代品种中被广泛选择和保留。可见,小麦根系也是一个重要的驯化和育种目标性状。从另一方面考虑,鉴于根系在植物抗旱中的重要作用和水资源日益短缺的现状,深根单元型Hap-5A-C和Hap-5B-Ⅰ在未来小麦抗逆育种中将受到选择并发挥重要作用。与地方品种相比,现代品种根长、根干重有下降的趋势(图4-c和图4-d),这一方面与水肥条件的改善相关,但另一个不可忽视的原因是地下根长与地上株高之间的相关性。一般而言,株高越高,根系越长。绿色革命在普遍降低小麦品种株高的同时,也带来根长和根体积的下降,这与半矮秆基因Rht1和Rht2的负面效应是密切相关的[53],因此,在育种中,开发利用新的半矮秆基因,减小株高降低对根系的负面影响是未来基因发掘的重要内容之一;此外,来自黑麦1RS可以显著提高根系的体积,缓解了2个半矮秆基因的负效应,从而提高水肥吸收效率,促进产量形成,这是1BL/1RS广泛存在于现代品种的另一个重要原因。如何获得携带新的抗性基因,同时消除或削弱黑麦1RS对加工品质的不良影响,是资源创新的一个重要课题。
4 结论
从小麦中克隆了水稻OsDRO的3个同源基因TaDRO-5A、TaDRO-5B和TaDRO-5D,均位于染色体长臂上;在21份多样性丰富的小麦遗传资源中检测到TaDRO-5A和TaDRO-5B序列多态性。在较大的自然群体中发现TaDRO-5A与最长根长、初生根夹角、株高及千粒重显著关联;TaDRO-5B与株高和千粒重显著关联。TaDRO-5A通过调控小麦根系生长角度调控根系在土壤中生长深度。Hap-5A-C、Hap-5B-Ⅰ为深根型单元型,但其同时引起株高的增加和千粒重的降低。TaDRO在全球小麦育种过程中经历了强烈的选择,在地方品种中以深根单元型Hap-5A-C和高杆小粒的Hap-5B-Ⅰ为主,在育成品种中以浅根型单元型Hap-5A-A和矮秆、大粒的Hap-5B-Ⅱ为主;在干旱地区以深根单元型Hap-5A-C和Hap-5B-Ⅰ为主,在水资源比较充裕的地区以浅根单元型Hap-5A-A和Hap-5B-Ⅱ为主。对大粒和矮秆的正向选择,使我国育成品种与地方品种优势单元型发生急剧转变,但不同麦区的选择强度却存在明显差异,水肥比较充裕的冬麦区明显快于水资源短缺的春麦区,深根类型能否在“双减”中发挥作用,有待进一步的研究。The authors have declared that no competing interests exist.
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
| [1] | . . |
| [2] | . Soil fertility and levels of applying fertilizer significantly regulated the mass root activity of wheat. With the increase of soil fertility, the mass root activity of wheat became stronger and stronger. Applying fertilizer could obviously improve the ma . Soil fertility and levels of applying fertilizer significantly regulated the mass root activity of wheat. With the increase of soil fertility, the mass root activity of wheat became stronger and stronger. Applying fertilizer could obviously improve the ma |
| [3] | . 试验在高、低两种墒情下 ,对西北干旱、半干旱地区不同年代春小麦品种水分利用率、根系干物质重、地上部生物量、根系呼吸速率、根冠比、光合作用、籽粒产量、收获指数等根系功能效率相关指标进行了研究 ,结果表明 :根系功能效率演变趋势表现为近期品种 >中期品种 >早期品种 . 试验在高、低两种墒情下 ,对西北干旱、半干旱地区不同年代春小麦品种水分利用率、根系干物质重、地上部生物量、根系呼吸速率、根冠比、光合作用、籽粒产量、收获指数等根系功能效率相关指标进行了研究 ,结果表明 :根系功能效率演变趋势表现为近期品种 >中期品种 >早期品种 |
| [4] | |
| [5] | The genetic improvement of drought resistance is essential for stable and adequate crop production in drought-prone areas(1). Here we demonstrate that alteration of root system architecture improves drought avoidance through the cloning and characterization of DEEPER ROOTING 1 (DRO1), a rice quantitative trait locus controlling root growth angle. DRO1 is negatively regulated by auxin and is involved in cell elongation in the root tip that causes asymmetric root growth and downward bending of the root in response to gravity. Higher expression of DRO1 increases the root growth angle, whereby roots grow in a more downward direction. Introducing DRO1 into a shallow-rooting rice cultivar by backcrossing enabled the resulting line to avoid drought by increasing deep rooting, which maintained high yield performance under drought conditions relative to the recipient cultivar. Our experiments suggest that control of root system architecture will contribute to drought avoidance in crops. |
| [6] | . |
| [7] | . Disparity in the root morphology of six rice (Oryza sativa L.) genotypes varying in potassium (K) efficiency was studied with three K levels: 5 mg/L (low),10 mg/L (moderate) and 40 mg/L (adequate) in hydroponic culture.Morphological parameters included root length,surface area,volume and count of lateral roots,as well as fine (diameter0.2 mm) roots.The results indicate that the root growth of all genotypes was reduced under low K,but moderate K deficiency increased the root length of the efficient genotypes.At deficient and moderate K levels,all the efficient rice genotypes developed more fine roots (diameter<0.2 mm) than the inefficient ones.Both fine root count and root surface area were found to be the best parameters to portray K stress in rice.In accordance with the root morphology,higher K concentrations were noted in shoots of the efficient genotypes when grown at moderate and deficient K levels,indicating that root morphology parameters are involved in root uptake for K and in the translocation of K up to shoots.K deficiency affected not only the root morphology,but also the root ultra-structure.The roots of high-efficient genotypes had stronger tolerance to K deficient stress for root membrane damage,and could maintain the developed root architecture to adapt to the low K growth medium. |
| [8] | . Genetic relationships between plant height and root morphology were investigated in a diverse set of wheat germplasm [199 double-haploid progeny derived from a cross between Avalon and Cadenza (Triticum aestivum L.), Rht near-isogenic lines (NILs), and accessions from the Watkins Collection] to investigate whether Rht genes controlling shoot height also control seedling root growth. A germination paper screen was developed to measure seedling root length (distinguishing seminal axes from seminal lateral roots), surface area, volume, and dry weight, and these were compared with shoot dry weight and the root to shoot ratio. Field experiments were conducted to measure mature plant height (PH) and grain characteristics for the mapping population. Forty-three quantitative trait loci (QTLs) for PH, root and seed traits were identified. Some QTLs for roots and either height or seed characteristics were coincident: chromosome 2D had co-locating root and PH QTLs; chromosomes 4D had co-locating root, PH, and seed QTLs; chromosome 5A and 6A had co-locating root and seed QTLs; and other non-co-locating root and PH QTLs were found on chromosomes 3A and 3B. Rht NILs illustrated that some known dwarfing genes reduce both PH and root proliferation. However, analysis of 25 short and 23 tall lines from the Watkins wheat germplasm collection indicated that PH and root proliferation are not simply related. |
| [9] | . |
| [10] | . |
| [11] | . in maize which encodes a conserved lateral organ boundary domain transcription factor involved in crown root initiation and development in response to auxin. Finally, we discuss the detection and validation of root development quantitative trait loci. |
| [12] | . Roots are essential for normal growth, development, and reproduction of higher plants. Consequently, improvement of root system architecture and functionality is of fundamental importance in crop improvement. However, the genetic mechanisms controlling root morphology and function are still not well understood, especially in common wheat, which possesses a complex and unsequenced hexaploid genome. Here we report a more detailed genetic analysis of qTaLRO- B1, a major quantitative trait locus (QTL) previously detected to affect root length and related traits in common wheat. A pair of QTL isolines with different qTaLRO- B1 alleles was developed. Line 178B, carrying the longer root allele, was significantly more efficient in taking up phosphate nutrient and biomass accumulation than line 178A, with the shorter root allele. We mapped qTaLRO- B1 to a 0.9-cM interval on common wheat chromosome 2BS with seven sequence-tagged-site (STS) markers developed from the genes conserved between wheat and Brachypodium distachyon. The seven STS markers were collinearly conserved in tetraploid wheat, but they covered a much larger genetic distance (22.8 cM) in the latter species. In conclusion, we have converted qTaLRO- B1 into a major gene that affects common wheat root length in a qualitative manner, and improved understanding of the genetic location of qTaLRO- B1 and the chromosomal segment carrying this important locus. The implications of our data for further study of qTaLRO- B1 are discussed. |
| [13] | . During the last two decades, DNA-based molecular markers have been extensively utilized for a variety of studies in both plant and animal systems. One of the major uses of these markers is the construction of genome-wide molecular maps and the genetic analysis of simple and complex traits. However, these studies are generally based on linkage analysis in mapping populations, thus placing serious limitations in using molecular markers for genetic analysis in a variety of plant systems. Therefore, alternative approaches have been suggested, and one of these approaches makes use of linkage disequilibrium (LD)-based association analysis. Although this approach of association analysis has already been used for studies on genetics of complex traits (including different diseases) in humans, its use in plants has just started. In the present review, we first define and distinguish between LD and association mapping, and then briefly describe various measures of LD and the two methods of its depiction. We then give a list of different factors that affect LD without discussing them, and also discuss the current issues of LD research in plants. Later, we also describe the various uses of LD in plant genomics research and summarize the present status of LD research in different plant genomes. In the end, we discuss briefly the future prospects of LD research in plants, and give a list of softwares that are useful in LD research, which is available as electronic supplementary material (ESM) |
| [14] | Chinese wheat mini core collection (262 accessions) was genotyped at 531 microsatellite loci representing a mean marker density of 5.1 cM. One-thousand-kernel weights (TKW) of lines were measured in five trials (three environments in four growing seasons). Structure analysis based on 42 unlinked SSR loci indicated that the materials formed two sub-populations, viz., landraces and modern varieties. A large difference in TKW (7.08 g, P<0.001) was found between the two sub-groups. Therefore, TKW is a major yield component that was improved in the past 6 decades; it increased from a mean 31.5 g in the 1940s to 44.64 g in the 2000s, representing a 2.19 g increase in each decade. Analyses based on a mixed linear model (MLM), population structure (Q) and relative kinship (K) revealed 22 SSR loci that were significantly associated with mean TKW (MTKW) of the five trials estimated by the best linear unbiased predictor (BLUP) method. They were mainly distributed on chromosomes of homoeologous groups 1, 2, 3, 5 and 7. Six loci, cfa2234-3A, gwm156-3B, barc56-5A, gwm234-5B, wmc17-7A and cfa2257-7A individually explained more than 11.84% of the total phenotypic variation. Favored alleles for breeding at the 22 loci were inferred according to their estimated effects on MTKW based on mean difference of varieties grouped by genotypes. Statistical simulation showed that these favored alleles have additive genetic effects. Frequency changes of alleles at loci associated with TKW are much more dramatic than those at neutral loci between the sub-groups. The numbers of favored alleles in modern varieties indicate there is still considerable genetic potential for their use as markers for genome selection of TKW in wheat breeding. Alleles that can be used globally to increase TKW were inferred according to their distribution by latitude and frequency of changes between landraces and the modern varieties. |
| [15] | . Abstract Associations between markers and complex quantitative traits were investigated in a collection of 146 modern two-row spring barley cultivars, representing the current commercial germ plasm in Europe. Using 236 AFLP markers, associations between markers were found for markers as far apart as 10 cM. Subsequently, for the 146 cultivars the complex traits mean yield, adaptability (Finlay-Wilkinson slope), and stability (deviations from regression) were estimated from the analysis of variety trial data. Regression of those traits on individual marker data disclosed marker-trait associations for mean yield and yield stability. Support for identified associations was obtained from association profiles, i.e., from plots of P-values against chromosome positions. In addition, many of the associated markers were located in regions where earlier QTL were found for yield and yield components. To study the oligogenic genetic base of the traits in more detail, multiple linear regression of the traits on markers was carried out, using stepwise selection. By this procedure, 18-20 markers that accounted for 40-58% of the variation were selected. Our results indicate that association mapping approaches can be a viable alternative to classical QTL approaches based on crosses between inbred lines, especially for complex traits with costly measurements. |
| [16] | |
| [17] | Association mapping is a method for detection of gene effects based on linkage disequilibrium (LD) that complements QTL analysis in the development of tools for molecular plant breeding. In this study, association mapping was performed on a selected sample of 95 cultivars of soft winter wheat. Population structure was estimated on the basis of 36 unlinked simple-sequence repeat (SSR) markers. The extent of LD was estimated on chromosomes 2D and part of 5A, relative to the LD observed among unlinked markers. Consistent LD on chromosome 2D was <1 cM, whereas in the centromeric region of 5A, LD extended for 鈭5 cM. Association of 62 SSR loci on chromosomes 2D, 5A, and 5B with kernel morphology and milling quality was analyzed through a mixed-effects model, where subpopulation was considered as a random factor and the marker tested was considered as a fixed factor. Permutations were used to adjust the threshold of significance for multiple testing within chromosomes. In agreement with previous QTL analysis, significant markers for kernel size were detected on the three chromosomes tested, and alleles potentially useful for selection were identified. Our results demonstrated that association mapping could complement and enhance previous QTL information for marker-assisted selection. |
| [18] | Utilization of quantitative trait loci (QTL) identified in bi-parental mapping populations has had limited success for improving complex quantitative traits with low to moderate heritability. Association mapping in contemporary breeding germplasm may lead to more effective marker strategies for crop improvement. To test this approach, we conducted association mapping of two complex traits with moderate heritability; Fusarium head blight (FHB) severity and the grain concentration of mycotoxin associated with disease, deoxynivalenol (DON). To map FHB resistance in barley, 768 breeding lines were evaluated in 2006 and 2007 in four locations. All lines were genotyped with 1,536 SNP markers and QTL were mapped using a mixed model that accounts for relatedness among lines. Average linkage disequilibrium within the breeding germplasm extended beyond 4聽cM. Four QTL were identified for FHB severity and eight QTL were identified for the DON concentration in two independent sets of breeding lines. The QTL effects were small, explaining 1鈥3% of the phenotypic variation, as might be expected for complex polygenic traits. We show that using breeding germplasm to map QTL can complement bi-parental mapping studies by providing independent validation, mapping QTL with more precision, resolving questions of linkage and pleiotropy, and identifying genetic markers that can be applied immediately in crop improvement. |
| [19] | . Spot blotch, caused byCochliobolus sativus, is an important foliar disease of barley. The disease has been controlled for over 40聽years through the deployment of cultivars with durable resistance derived from the line NDB112. Pathotypes ofC.sativuswith virulence for the NDB112 resistance have been detected in Canada; thus, many commercial cultivars are vulnerable to spot blotch epidemics. To increase the diversity of spot blotch resistance in cultivated barley, we evaluated 318 diverse wild barley accessions comprising the Wild Barley Diversity Collection (WBDC) for reaction toC.sativusat the seedling stage and utilized an association mapping (AM) approach to identify and map resistance loci. A high frequency of resistance was found in the WBDC as 95% (302/318) of the accessions exhibited low infection responses. The WBDC was genotyped with 558 Diversity Array Technology (DArT庐) and 2,878 single nucleotide polymorphism (SNP) markers and subjected to structure analysis before running the AM procedure. Thirteen QTL for spot blotch resistance were identified with DArT and SNP markers. These QTL were found on chromosomes 1H, 2H, 3H, 5H, and 7H and explained from 2.3 to 3.9% of the phenotypic variance. Nearly half of the identified QTL mapped to chromosome bins where spot blotch resistance loci were previously reported, offering some validation for the AM approach. The other QTL mapped to unique genomic regions and may represent new spot blotch resistance loci. This study demonstrates that AM is an effective technique for identifying and mapping QTL for disease resistance in a wild crop progenitor. The online version of this article (doi:10.1007/s11032-010-9402-8) contains supplementary material, which is available to authorized users. |
| [20] | . |
| [21] | . Functional markers (FM) are developed from sequence polymorphisms present in allelic variants of a functional gene at a locus. FMs accurately discriminate alleles of a targeted gene, and are ideal molecular markers for marker-assisted selection in wheat breeding. In this paper, we summarize FMs developed and used in common wheat. To date, more than 30 wheat loci associated with processing quality, agronomic traits, and disease resistance, have been cloned, and 97 FMs were developed to identify 93 alleles based on the sequences of those genes. A general approach is described for isolation of wheat genes and development of FMs based on in silico cloning and comparative genomics. The divergence of DNA sequences of different alleles that affect gene function is summarized. In addition, 14 molecular markers specific for alien genes introduced from common wheat relatives were also described. This paper provides updated information on all FMs and gene-specific STS markers developed so far in wheat and should facilitate their application in wheat breeding programs. |
| [22] | . Genetic diversity among 5029 accessions representing a proposed Chinese wheat core collection was analyzed using 78 pairs of fluorescent microsatellite (SSR) primers mapped to 21 chromosomes. A stepwise hierarchical sampling strategy with priority based on 4脳105 SSR data-points was used to construct a core collection from the 23090 initial collections. The core collection consisted of 1160 accessions, including 762 landraces, 348 modern varieties and 50 introduced varieties. The core ac-counts for 23.1% of the 5029 candidate core accessions and 5% of the 23090 initial collections, but retains 94.9% of alleles from the candidate collections and captures 91.5% of the genetic variation in the initial collections. These data indicate that it is possible to maintain genetic diversity in a core col-lection while retaining fewer accessions than the accepted standard, i.e., 10% of the initial collections captured more than 70% of their genetic diversity. Estimated genetic representation of the core con-structed by preferred sampling (91.5%) is much higher than that by random sampling (79.8%). Both mean genetic richness and genetic diversity indices of the landraces were higher than those of the modern varieties in the core. Structure and principal coordinate analysis revealed that the landraces and the modern varieties were two relatively independent subpopulations. Strong genetic differentia-tion associated with ecological environments has occurred in the landraces, but was relatively weak in the modern cultivars. In addition, a mini-core collection was constructed, which consisted of 231 ac-cessions with an estimated 70% representation of the genetic variation from the initial collections. The mini-core has been distributed to various research and breeding institutes for detailed phenotyping and breeding of genetic introgression lines. |
| [23] | . |
| [24] | . 为探讨小麦种子根结构及胚芽鞘长度的遗传基础,以小麦DH群体(旱选10号×鲁麦14)的150个株系为材料,利用凝胶室培养幼苗,测定种子根的数目和最大根长、胚芽鞘长度、根苗干重比等性状,并通过扫描仪测定幼苗种子根的总长度、根直径及角度。利用已经构建的DH群体遗传连锁图谱,采用基于混合线性模型的复合区间作图法分析上述性状的QTL。在1A、1B、2B、2D、3B、4A、4D、5A、5B、6A、7A和7B共12条染色体上检测到12个加性效应QTL和7对加性×加性互作效应QTL。QTL的加性效应值在0.02~8.45之间,对表型变异的贡献率为5.64%~12.37%。7对加性×加性互作效应QTL分布在1A–2B(2)、1A–6A、1B–2D、5B–6A、6A–7A和6A–7B等6对染色体之间,其互作效应值为0.20~7.45,对表型变异的贡献率为8.70%~15.90%。在染色体3B和7A上各检测到1个种子根结构相关性状的QTL簇。 . 为探讨小麦种子根结构及胚芽鞘长度的遗传基础,以小麦DH群体(旱选10号×鲁麦14)的150个株系为材料,利用凝胶室培养幼苗,测定种子根的数目和最大根长、胚芽鞘长度、根苗干重比等性状,并通过扫描仪测定幼苗种子根的总长度、根直径及角度。利用已经构建的DH群体遗传连锁图谱,采用基于混合线性模型的复合区间作图法分析上述性状的QTL。在1A、1B、2B、2D、3B、4A、4D、5A、5B、6A、7A和7B共12条染色体上检测到12个加性效应QTL和7对加性×加性互作效应QTL。QTL的加性效应值在0.02~8.45之间,对表型变异的贡献率为5.64%~12.37%。7对加性×加性互作效应QTL分布在1A–2B(2)、1A–6A、1B–2D、5B–6A、6A–7A和6A–7B等6对染色体之间,其互作效应值为0.20~7.45,对表型变异的贡献率为8.70%~15.90%。在染色体3B和7A上各检测到1个种子根结构相关性状的QTL簇。 |
| [25] | . Summary: Association analyses that exploit the natural diversity of a genome to map at very high resolutions are becoming increasingly important. In most studies, however, researchers must contend with the confounding effects of both population and family structure. TASSEL (Trait Analysis by aSSociation, Evolution and Linkage) implements general linear model and mixed linear model approaches for controlling population and family structure. For result interpretation, the program allows for linkage disequilibrium statistics to be calculated and visualized graphically. Database browsing and data importation is facilitated by integrated middleware. Other features include analyzing insertions/deletions, calculating diversity statistics, integration of phenotypic and genotypic data, imputing missing data and calculating principal components. Availability: The TASSEL executable, user manual, example data sets and tutorial document are freely available at http://www.maizegenetics.net/tassel. The source code for TASSEL can be found at http://sourceforge.net/projects/tassel. Contact: pjb39@cornell.edu |
| [26] | . |
| [27] | . . |
| [28] | |
| [29] | |
| [30] | . |
| [31] | . <P><FONT face=Verdana>【目的】探讨春小麦、谷子、高粱、黍子4种谷类作物根系分布的空间几何构型特点和根系生长时空分布规律。【方法】采用盆栽、根管土柱栽培、铁丝网箱栽培与田间调查相结合的方法,研究谷子、高粱、黍子、春小麦4种作物根系的生长规律。【结果】(1)4种供试作物根系的种子根数、次生根数、入土深度和根幅明显不同;根系最大入土深度为高粱>谷子>春小麦>黍子;最大根幅为高粱>黍子>谷子>春小麦。(2)随着生育时期的推进,谷子、黍子、春小麦和高粱根系的根长与根重的增长均表现为慢-快-慢的规律。(3)4种谷类作物苗期主要以根系纵向下扎为主,根长与根干重呈明显的“T”字型结构;拔节期春小麦根长分布呈现近似“8”字型,其它作物的根长和根重分布仍呈明显的“T”字型;抽穗期谷子、高粱、黍子根长在不同土层深度中的分布近“8”字型,而春小麦呈现近卵型。(4)4种谷类作物根重在不同深度土体中的垂直分布符合指数递减方程y=A·e-bx,但其垂直递减率b值大小不等。4种谷类作物的总根长在不同深度土体中的分布前期符合指数递减方程y=A·e-bx,但后期与多项式y=ax3+bx2+cx+d的拟合程度更好。【结论】4种谷类作物根系空间分布的存在相似性,该相似性可为谷类作物高产栽培的根系调控提供理论依据。</FONT></P> . <P><FONT face=Verdana>【目的】探讨春小麦、谷子、高粱、黍子4种谷类作物根系分布的空间几何构型特点和根系生长时空分布规律。【方法】采用盆栽、根管土柱栽培、铁丝网箱栽培与田间调查相结合的方法,研究谷子、高粱、黍子、春小麦4种作物根系的生长规律。【结果】(1)4种供试作物根系的种子根数、次生根数、入土深度和根幅明显不同;根系最大入土深度为高粱>谷子>春小麦>黍子;最大根幅为高粱>黍子>谷子>春小麦。(2)随着生育时期的推进,谷子、黍子、春小麦和高粱根系的根长与根重的增长均表现为慢-快-慢的规律。(3)4种谷类作物苗期主要以根系纵向下扎为主,根长与根干重呈明显的“T”字型结构;拔节期春小麦根长分布呈现近似“8”字型,其它作物的根长和根重分布仍呈明显的“T”字型;抽穗期谷子、高粱、黍子根长在不同土层深度中的分布近“8”字型,而春小麦呈现近卵型。(4)4种谷类作物根重在不同深度土体中的垂直分布符合指数递减方程y=A·e-bx,但其垂直递减率b值大小不等。4种谷类作物的总根长在不同深度土体中的分布前期符合指数递减方程y=A·e-bx,但后期与多项式y=ax3+bx2+cx+d的拟合程度更好。【结论】4种谷类作物根系空间分布的存在相似性,该相似性可为谷类作物高产栽培的根系调控提供理论依据。</FONT></P> |
| [32] | . Minirhizotrons provide a nondestructive, in situ method for directly viewing and studying fine roots. Although many insights into fine roots have been gained using minirhizotrons, a review of the literature indicates a wide variation in how minirhizotrons and minirhizotron data are used. Tube installation is critical, and steps must be taken to insure good soil/tube contact without compacting the soil. Ideally, soil adjacent to minirhizotrons will mimic bulk soil. Tube installation causes some degree of soil disturbance and has the potential to create artifacts in subsequent root data and analysis. We therefore recommend a waiting period between tube installation and image collection of 6鈥12 months to allow roots to recolonize the space around the tubes and to permit nutrients to return to pre-disturbance levels. To make repeated observations of individual roots for the purposes of quantifying their dynamic properties (e.g. root production, turnover or lifespan), tubes should be secured to prevent movement. The frequency of image collection depends upon the root parameters being measured or calculated and the time and resources available for collecting images and extracting data. However, long sampling intervals of 8 weeks or more can result in large underestimates of root dynamic properties because more fine roots will be born and die unobserved between sampling events. A sampling interval of 2 weeks or less reduces these underestimates to acceptable levels. While short sample intervals are desirable, they can lead to a potential trade-off between the number of minirhizotron tubes used and the number of frames analyzed per tube. Analyzing fewer frames per minirhizotron tube is one way to reduce costs with only minor effects on data variation. The quality of minirhizotron data should be assessed and reported; procedures for quantifying the quality of minirhizotron data are presented here. Root length is a more sensitive metric for dynamic root properties than the root number. To make minirhizotron data from separate experiments more easily comparable, idiosyncratic units should be avoided. Volumetric units compatible with aboveground plant measures make minirhizotron-based estimates of root standing crop, production and turnover more useful. Methods for calculating the volumetric root data are discussed and an example presented. Procedures for estimating fine root lifespan are discussed. |
| [33] | AP2/EREBPs play significant roles in plant growth and development. A novel, pleiotropicTaPARG(PLANT ARCHITECTURE-RELATED GENE), a member of the AP2/EREBP transcription factor gene family, and its flanking sequences were isolated in wheat (Triticum aestivumL.). TwoTaPARGgenes were identified and named asTaPARG-2AandTaPARG-2D. Their amino acid sequences were highly similar especially in the functional domains.TaPARG-2Aon chromosome 2A was flanked by markersXwmc63andXgwm372.TaPARG-2Dwas mapped to chromosome 2D. Subcellular localization revealed that TaPARG-2D was localized in the nucleus. The results of tissue expression pattern, overexpression in rice, association analysis and distinct population verification jointly revealed thatTaPARGfunctions during the entire growth cycle of wheat. Its functions include regulation of plant architecture-related and yield-related traits. Association analysis, geographic distribution and allelic frequencies suggested that favored haplotypesHap-2A-2andHap-2A-3were selected in Chinese wheat breeding programs. Both favored haplotypes might be caused by a single amino acid substitution (His/Tyr). These results suggest thatTaPARGis a regulatory factor in plant growth and development, and that the favored alleles might be useful for improving plant architecture and grain yield of wheat. |
| [34] | . The root system of a rice plant (Oryza sativa L.) consists of numerous nodal roots and their laterals. The growth direction of these nodal roots affects the spatial distribution of the root system in soil, which seems to relate to yield and lodging resistance. The growth angle of a nodal root varies with the type and timing of emergence of the nodal root. The body of a rice plant can be recognized as an integrated set of shoot units, each unit consisting of an intern ode with a leaf and several roots. Nodal roots formed at the apical part of a shoot unit often elongate horizontally, whereas those formed at the basal part of the shoot unit show various growth directions depending on both the growth stages of the plant and the environmental conditions. Moreover, nodal roots that emerge from the most basal shoot unit of a tiller are usually thick and grow downwards. External factors such as planting density and nitrogen application affect the growth direction of nodal roots, probably partly because of the changing tillering pattern of the shoot. In addition to the growth angle of nodal roots, size of nodal roots may be another important factor determining the spatial distribution of the root system in soil. |
| [35] | . |
| [36] | . |
| [37] | . Drought is a major problem for rice grown under rainfed lowland and upland conditions, but progress in breeding to improve drought resistance has been slow. This paper describes patterns of water-stress development in rice fields, reviews genetic variation in physio-morphological traits for drought resistance in rice, and suggests how knowledge of stress physiology can contribute to plant breeding programmes that aim to increase yield in water-limiting environments. To provide a basis for integrating physiological research with plant-breeding objectives we define drought resistance in terms of relative yield of genotypes. Therefore, a drought-resistant genotype will be one which has a higher grain yield than others when all genotypes are exposed to the same level of water stress. A major reason for the slow progress in breeding for drought resistance in rice is the complexity of the drought environment, which often results in the lack of clear identification of the target environment(s). There is a need to identify the relative importance of the three common drought types; early-season drought which often causes delay in transplanting, mild intermittent stress which can have a severe cumulative effect, and late stress which affects particularly late-maturing genotypes. In addition, in rainfed lowland rice, flooded and non-flooded soil conditions may alternate during the growing season, and affect nutrient availability or cause toxicity. Several drought-resistance mechanisms, and putative traits which contribute to them, have been identified for rice; important among these being drought escape via appropriate phenology, root characteristics, specific dehydration avoidance and tolerance mechanisms, and drought recovery. Some of these mechanisms/traits have been shown to confer drought resistance and others show potential to do so in rice. The most important is the appropriate phenology which matches crop growth and development with the water environment. A deep root system, with high root length density at depth is useful in extracting water thoroughly in upland conditions, but does not appear to offer much scope for improving drought resistance in rainfed lowland rice where the development of a hard pan may prevent deep root penetration. Under water-limiting environments, genotypes which maintain the highest leaf water potential generally grow best, but it is not known if genotypic variation in leaf water potential is solely caused by root factors. Osmotic adjustment is promising, because it can potentially counteract the effects of a rapid decline in leaf water potential and there is large genetic variation for this trait. There is genotypic variation in expression of green leaf retention which appears to be a useful character for prolonged droughts, but it is affected by plant size which complicates its use as a selection criterion for drought resistance. There is a general lack of drought related research for rice in rainfed lowland conditions. This needs to be rectified, particularly considering their importance relative to upland conditions in Asian countries. We suggest that focussing physiological-genetic research efforts onto clearly defined, major target environments should provide a basis for increasing the relevance of stress physiology and the efficiency of breeding programmes for development of drought-resistant genotypes. |
| [38] | . Abstract Charles Darwin founded root system architecture research in 1880 when he described a root bending with gravity. Curving, elongating, and branching are the three cellular processes in roots that underlie root architecture. Together they determine the distribution of roots through soil and time, and hence the plants' access to water and nutrients, and anchorage. Most knowledge of these cellular processes comes from seedlings of the model dicotyledon, Arabidopsis, grown in soil-less conditions with single treatments. Root systems in the field, however, face multiple stimuli that interact with the plant genetics to result in the root system architecture. Here we review how soil conditions influence root system architecture; focusing on cereals. Cereals provide half of human calories, and their root systems differ from those of dicotyledons. We find that few controlled-environment studies combine more than one soil stimulus and, those that do, highlight the complexity of responses. Most studies are conducted on seedling roots; those on adult roots generally show low correlations to seedling studies. Few field studies report root and soil conditions. Until technologies are available to track root architecture in the field, soil analyses combined with knowledge of the effects of factors on elongation and gravitropism could be ranked to better predict the interaction between genetics and environment (G脙聴E) for a given crop. Understanding how soil conditions regulate root architecture can be effectively used to design soil management and plant genetics that best exploit synergies from G脙聴E of roots. |
| [39] | . Abstract Wheat yields globally will depend increasingly on good management to conserve rainfall and new varieties that use water efficiently for grain production. Here we propose an approach for developing new varieties to make better use of deep stored water. We focus on water-limited wheat production in the summer-dominant rainfall regions of India and Australia, but the approach is generally applicable to other environments and root-based constraints. Use of stored deep water is valuable because it is more predictable than variable in-season rainfall and can be measured prior to sowing. Further, this moisture is converted into grain with twice the efficiently of in-season rainfall since it is taken up later in crop growth during the grain-filling period when the roots reach deeper layers. We propose that wheat varieties with a deeper root system, a redistribution of branch root density from the surface to depth, and with greater radial hydraulic conductivity at depth would have higher yields in rainfed systems where crops rely on deep water for grain fill. Developing selection systems for mature root system traits is challenging as there are limited high-throughput phenotyping methods for roots in the field, and there is a risk that traits selected in the lab on young plants will not translate into mature root system traits in the field. We give an example of a breeding programme that combines laboratory and field phenotyping with proof of concept evaluation of the trait at the beginning of the selection programme. This would greatly enhance confidence in a high-throughput laboratory or field screen, and avoid investment in screens without yield value. This approach requires careful selection of field sites and years that allow expression of deep roots and increased yield. It also requires careful selection and crossing of germplasm to allow comparison of root expression among genotypes that are similar for other traits, especially flowering time and disease and toxicity resistances. Such a programme with field and laboratory evaluation at the outset will speed up delivery of varieties with improved root systems for higher yield. |
| [40] | . Better understanding of root system structure and function is critical to crop improvement in water-limited environments. The aims of this study were to examine root system characteristics of two wheat genotypes contrasting in tolerance to water limitation and to assess the functional implications on adaptation to water-limited environments of any differences found. The drought tolerant barley variety, Mackay, was also included to allow inter-species comparison. Single plants were grown in large, soil-filled root-observation chambers. Root growth was monitored by digital imaging and water extraction was measured. Root architecture differed markedly among the genotypes. The drought-tolerant wheat (cv. SeriM82) had a compact root system, while roots of barley cv. Mackay occupied the largest soil volume. Relative to the standard wheat variety (Hartog), SeriM82 had a more uniform rooting pattern and greater root length at depth. Despite the more compact root architecture of SeriM82, total water extracted did not differ between wheat genotypes. To quantify the value of these adaptive traits, a simulation analysis was conducted with the cropping system model APSIM, for a wide range of environments in southern Queensland, Australia. The analysis indicated a mean relative yield benefit of 14.5% in water-deficit seasons. Each additional millimetre of water extracted during grain filling generated an extra 55 kg ha-1 of grain yield. The functional implications of root traits on temporal patterns and total amount of water capture, and their importance in crop adaptation to specific water-limited environments, are discussed.. |
| [41] | . |
| [42] | . Objective : The main aim of the research was to evaluate genetic variability and interrelationships of mainly quantitative traits in 2-year population, and provide a basis for high-yield breeding of G. uralensis 2-year population, it could be used for breeding improvement of root yield. Objective : The main aim of the research was to evaluate genetic variability and interrelationships of mainly quantitative traits in 2-year population, and provide a basis for high-yield breeding of G. uralensis 2-year population, it could be used for breeding improvement of root yield. |
| [43] | [EB/OL]. . [EB/OL]. (in Chinese) |
| [44] | |
| [45] | Developing a deep root system is an important strategy for avoiding drought stress in . Using the 'basket' method, the ratio of deep rooting (; the proportion of total roots that elongated through the basket bottom) was calculated to evaluate deep rooting. A new major quantitative trait locus (QTL) controlling was detected on 9 by using 117 recombinant inbred lines () derived from a cross between the lowland cultivar IR64, with shallow rooting, and the upland cultivar Kinandang Patong (KP), with deep rooting. This QTL explained 66.6% of the total phenotypic variance in in the . A BC(2)F(3) line homozygous for the KP allele of the QTL had an of 40.4%, compared with 2.6% for the homozygous IR64 allele. Fine mapping of this QTL was undertaken using eight BC(2)F(3) recombinant lines. The QTL Dro1 (Deeper rooting 1) was mapped between the markers RM24393 and RM7424, which delimit a 608.4 kb interval in the reference cultivar Nipponbare. To clarify the influence of Dro1 in an upland field, the root distribution in different soil layers was quantified by means of core sampling. A line homozygous for the KP allele of Dro1 (Dro1-KP) and IR64 did not differ in root dry weight in the shallow soil layers (0-25 cm), but root dry weight of Dro1-KP in deep soil layers (25-50 cm) was significantly greater than that of IR64, suggesting that Dro1 plays a crucial role in increased deep rooting under upland field conditions. |
| [46] | . Abstract Drought is the most serious abiotic stress that hinders rice production under rainfed conditions. Breeding for deep rooting is a promising strategy to improve the root system architecture in shallow-rooting rice cultivars to avoid drought stress. We analysed the quantitative trait loci (QTLs) for the ratio of deep rooting (RDR) in three F090202 mapping populations derived from crosses between each of three shallow-rooting varieties ('ARC5955', 'Pinulupot1', and 'Tupa729') and a deep-rooting variety, 'Kinandang Patong'. In total, we detected five RDR QTLs on chromosomes 2, 4, and 6. In all three populations, QTLs on chromosome 4 were found to be located at similar positions; they explained from 32.0% to 56.6% of the total RDR phenotypic variance. This suggests that one or more key genetic factors controlling the root growth angle in rice is located in this region of chromosome 4. |
| [47] | Root growth angle (RGA) is an important trait that influences the ability of rice to avoid drought stress. DEEPER ROOTING 1 (DRO1), which is a major quantitative trait locus (QTL) for RGA, is responsible for the difference in RGA between the shallow-rooting cultivar IR64 and the deep-rooting cultivar Kinandang Patong. However, the RGA differences between these cultivars cannot be fully explained by DRO1. The objective of this study was to identify new QTLs for RGA explaining the difference in RGA between these cultivars. By crossing IR64 (which has a non-functional allele of DRO1) with Kinandang Patong (which has a functional allele of DRO1), we developed 26 chromosome segment substitution lines (CSSLs) that carried a particular chromosome segment from Kinandang Patong in the IR64 genetic background. Using these CSSLs, we found only one chromosomal region that was related to RGA: on chromosome 9, which includes DRO1. Using an F2 population derived from a cross between Kinandang Patong and the Dro1-NIL (near isogenic line), which had a functional DRO1 allele in the IR64 genetic background, we identified a new QTL for RGA (DRO3) on the long arm of chromosome 7. DRO3 may only affect RGA in plants with a functional DRO1 allele, suggesting that DRO3 is involved in the DRO1 genetic pathway. |
| [48] | . |
| [49] | . |
| [50] | . |
| [51] | |
| [52] | . Abstract Positive selection can be inferred from its effect on linked neutral variation. In the restrictive case when there is no recombination, all linked variation is removed. If recombination is present but rare, both deterministic and stochastic models of positive selection show that linked variation hitchhikes to either low or high frequencies. While the frequency distribution of variation can be influenced by a number of evolutionary processes, an excess of derived variants at high frequency is a unique pattern produced by hitchhiking (derived refers to the nonancestral state as determined from an outgroup). We adopt a statistic, H, to measure an excess of high compared to intermediate frequency variants. Only a few high-frequency variants are needed to detect hitchhiking since not many are expected under neutrality. This is of particular utility in regions of low recombination where there is not much variation and in regions of normal or high recombination, where the hitchhiking effect can be limited to a small (<1 kb) region. Application of the H test to published surveys of Drosophila variation reveals an excess of high frequency variants that are likely to have been influenced by positive selection. |
| [53] | . Abstract BACKGROUND AND AIMS: Most plant scientists, in contrast to animal scientists, study only half the organism, namely above-ground stems, leaves, flowers and fruits, and neglect below-ground roots. Yet all acknowledge roots are important for anchorage, water and nutrient uptake, and presumably components of yield. This paper investigates the relationship between domestication, and the root systems of landraces, and the parents of early, mid- and late green-revolution bread wheat cultivars. It compares the root system of bread wheat and 'Veery'-type wheat containing the 1RS translocation from rye. METHODS: Wheat germplasm was grown in large pots in sand culture in replicated experiments. This allowed roots to be washed free to study root characters. KEY RESULTS: The three bread wheat parents of early green-revolution wheats have root biomass less than two-thirds the mean of some landrace wheats. Crossing early green-revolution wheat to an F(2) of 'Norin 10' and 'Brevor', further reduced root biomass in mid-generation semi-dwarf and dwarf wheats. Later-generation semi-dwarf wheats show genetic variation for root biomass, but some exhibit further reduction in root size. This is so for some California and UK wheats. The wheat-rye translocation in 'Kavkaz' for the short arm of chromosome 1 (1RS) increased root biomass and branching in cultivars that contained it. CONCLUSIONS: Root size of modern cultivars is small compared with that of landraces. Their root system may be too small for optimum uptake of water and nutrients and maximum grain yield. Optimum root size for grain yield has not been investigated in wheat or most crop plants. Use of 1RS and similar alien translocations may increase root biomass and grain yield significantly in irrigated and rain-fed conditions. Root characters may be integrated into components of yield analysis in wheat. Plant breeders may need to select directly for root characters. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}