0 引言
【研究意义】OFP(OVATE family protein)蛋白家族是一类植物特异转录因子家族,其C端包含保守的OVATE结构域,又称DUF623域,因其在调节植物生长和发育过程起到重要作用而被熟知,属植物生长抑制蛋白[1,2,3,4]。研究表明,OFP蛋白在拟南芥、水稻和番茄中已有较多研究,越来越多的OFP被克隆鉴定。研究苹果基因组中OFP家族基因成员的分布、系统进化关系、基因结构、组织表达模式及诱导表达差异,为进一步探索其在植物逆境生长发育过程中的功能提供理论依据。【前人研究进展】OVATE被首次鉴定是作为一个数量性状位点,主要控制番茄果实的形状,作为植物生长抑制调节蛋白还负调控番茄叶片和花的生长发育[1]。拟南芥、水稻和番茄基因组中分别含有18、31和31个OFP转录因子家族成员,它们控制着植物生长发育的多个方面[3,5-6]。其中,多数AtOFP在拟南芥瞬时原生质体表达系统中起到转录抑制的作用[3];水稻中的31个OsOFP具有不同的组织表达模式,且OsOFP多数在种子发育阶段表达量较高,暗示该家族成员可能参与调控水稻生长发育的多个过程,尤其是种子发育过程[6,7,8]。TALE(Three amino acid loop extension)蛋白家族是由3个氨基酸组成的典型环状结构,此环状结构可与其他蛋白分子发生特异性作用。酵母双杂交试验发现,拟南芥9个AtOFP蛋白可与TALE同源结构域蛋白相互作用[9],其中,AtOFP1和AtOFP5可共同调控TALE同源结构域蛋白BLH1(BEL1-Like Homeodomain)的亚细胞定位模式,当AtOFP1和AtOFP5在本生烟叶片中共表达时,BLH1由细胞核被重新定位到细胞质内[9]。赤霉素的积累能够在一定程度上恢复地上部组织长度变短的性状,AtOFP1的过表达会抑制赤霉素合成基因AtGA20ox1的表达,从而使该性状不能正常恢复[2,9]。另外,AtOFP1还能与DNA双链断裂修复相关基因AtKu相互作用,共同参与调控DNA双链断裂修复过程[10]。AtOFP5作为BLH1- KNAT3的负调节子在胚囊发育前期起作用,二者协同调控胚囊发育[11],而AtOFP4可通过与KNAT7蛋白间的相互作用,参与调节植物次生细胞壁形成过程[12]。AtOFP1和AtOFP4能够共同调节次生细胞壁形成过程所需的BLH6-KNAT7多蛋白复合体结构[13]。与番茄OVATE相反,缺失拟南芥OFP发现,单独敲除AtOFP1、AtOFP4、AtOFP8、AtOFP10、AtOFP15和AtOFP16,突变体并未出现生长缺陷,暗示以上基因具有功能冗余的特性[3]。上述研究表明,OFP蛋白可能通过直接或间接影响目的基因的转录调节过程,来调节植物的生长和发育。【本研究切入点】苹果是最重要的全球性经济作物之一,但苹果MdOFP基因家族未见相关报道,而苹果基因组的测序完成为分析苹果基因组序列中MdOFP基因家族成员的相关信息提供了基础[14]。【拟解决的关键问题】本研究通过生物信息学方法分析鉴定苹果全基因组序列中的所有MdOFP家族成员,从基因组水平上分析OFP在苹果中的基因数目、基因结构、染色体定位、系统进化关系和芯片表达谱模式;利用qRT-PCR技术进行组织表达模式及诱导表达分析,为进一步研究苹果MdOFPs的生物学功能提供参考。1 材料与方法
1.1 苹果OFP基因家族成员鉴定
苹果全基因组数据下载于植物基因组数据库(plantGDB: http://www.plantgdb.org/),拟南芥OFP基因序列和蛋白序列下载自TAIR(The Arabidopsis Information Resource,http://arabidopsis.org/)[15,16,17]。首先利用苹果全基因组序列,构建本地BLAST数据库,以拟南芥OFP转录因子家族基因序列执行本地BLAST(1e-003)搜索;同时使用Pfam数据库工具建立苹果全基因组蛋白结构域模型,利用perl程序筛选含有OFP典型结构域(OVATE结构域,DUF623域)的蛋白序列[18,19]。合并上述两部分结果,删除重复基因,所得结果利用PFAM及NCBI-CDD工具进行蛋白结构预测[18,19,20,21,22],删除不含OVATE结构域的蛋白,同时手工剔出无完整读码框的序列。利用ExPASy网站(http://expasy.org/)对所有苹果OFP蛋白氨基酸序列进行等电点、分子量预测等[22]。
1.2 苹果OFP基因家族系统进化树的构建
通过MUSCLE程序对拟南芥和苹果OFP蛋白进行多序列比对,选取OVATE结构域序列,再使用MEGA5.0(http://megasoftware.net)程序采用邻接法(Neighbor-Joining,NJ)生成MdOFP的系统进化树,校验参数Bootstrap重复1 000次[23,24]。1.3 苹果OFP基因结构及染色体定位分析
利用perl程序解析苹果基因组信息文件(assembly gff3 file),选取MdOFP的染色体位置信息及基因结构信息,利用MapDraw工具进行染色体定位作图,同时通过GSDS(http://gsds.cbi.pku.edu.cn/)工具进行基因外显子-内含子结构作图[25,26,27]。1.4 苹果OFP基因芯片表达谱分析
从EBI芯片数据库中下载苹果组织表达芯片数据,以苹果MdOFPs对苹果探针序列进行BLAST比对,选取完全匹配探针代表该MdOFP,搜索匹配探针代表MdOFP的表达量,采用Cluster3.0进行芯片聚类,Java Treeview查看芯片聚类结果并作图,进行基因组织表达分析。1.5 ‘嘎拉’苹果总RNA的提取和qRT-PCR分析
从2016年5月起,取栽种在山东农业大学园艺试验站的5年生‘嘎拉’组培生根苹果树,取当年新生根、幼茎、新生叶、花(初花期)和花后30 d的幼果为材料,取样后液氮冷冻,并置于-80℃超低温冰箱保存,用于RNA提取。将‘嘎拉’苹果组培苗进行生根处理,生长2周后将生根的组培苗分别用含有150 mmol?L-1 NaCl和10% PEG6000的B5营养液进行水培处理,6 h后分别取根部和地上部样品;高温39℃和低温4℃处理‘嘎拉’苹果组培苗,6 h后取材,用液氮冷冻保存,用于qRT-PCR分析。苹果总RNA提取采用RNA plant plus Reagent试剂盒提取,通过琼脂糖凝胶电泳检测RNA完整性,使用Thermo Nano Drop 2000检测RNA纯度和浓度。用DNase I(TaKaRa)除去基因组DNA后,取1 μg RNA用于反转录合成cDNA第一链,反转录过程按照PrimeScript? RT reagent Kit With gDNA Eraser(TaKaRa)的操作说明进行,获得的苹果各种cDNA样品在-20℃冰箱保存,备用。
qRT-PCR反应体系为:2×SYBR PCR mix 10 μL,上、下游引物(10 mmol?L-1)各1 μL,cDNA模板1 μL,ddH2O 7 μL,总体系为20 μL。qRT-PCR反应条件为:94℃ 1 min;94℃ 10 s,60℃ 10 s,72℃ 10 s,40个循环。每次循环第3步进行荧光采集。所有PCR反应都设3次重复。采用2-ΔΔCT法对数据进行定量分析,用Excel作图。qRT-PCR试验所需的引物通过Beacon Designer 8软件设计,采用DNAMAN和Primer BLAST软件验证引物特异性。Md18S作为内参,试验所用的引物见表1。所有引物由上海生工生物工程股份有限公司合成。
Table 1
表1
表1qRT-PCR试验的引物序列
Table 1The primers used in the qRT-PCR
| 基因名称 Gene name | 上游引物(5′-3′) Forward primer (5′-3′) | 下游引物(5′-3′) Reverse primer (5′-3′) |
|---|---|---|
| MdOFP01 | TCCCGTTTAGTTCTTCATCTTTC | CAGTGTGTCAATCTCGTCATC |
| MdOFP03 | CGTGGTAGAAAGAGTGTGTC | GATAGCAAGCAAGCAGGTC |
| MdOFP04 | CAACAACAACTCGTCCTTACAG | TTCCTTCACCACCGCAATG |
| MdOFP06 | CGGAGGATGGGTTGGATTC | TTCTTGTTCTTCTTCAGTTCGC |
| MdOFP07 | CGGAGGATGGGTTGGATTC | TTCTTGTTCTTCTTCAGTTCGC |
| MdOFP08 | CAATGGAGGAAATGGTGGAATG | AGCAGAAAGAGCAAGAAGGAG |
| MdOFP11 | GGAGGAGAAGACGCAATAGAG | CCCATCACCACCATCATCAG |
| MdOFP12 | TTCAAGCCGTTCAGCCTCAG | GTTCATCACAAGACCGCCATC |
| MdOFP13 | AGAGAGAAACACCACCGAAG | TTGCCCTGAGCGAGAAAG |
| MdOFP16 | CTCCTTCTTGCTCTTTCTGC | CATCCTCCTCCTGACCTTG |
| MdOFP17 | AGGGTGAATGGTAAGAGCAATC | GACGACAGCGACGAAGAAG |
| MdOFP18 | TGCTGTGGTGAAGAAGTCG | CCCTGTGATGGTGTGTCG |
| MdOFP20 | TCCTCCTCGCAGTCACAC | TTCCTTCACCACCGCAATG |
| Md18S | ACACGGGGAGGTAGTGACAA | CCTCCAATGGATCCTCGTTA |
新窗口打开
2 结果
2.1 苹果OFP基因家族成员鉴定
利用生物信息学方法,从苹果全基因组中鉴定得到28个OFP转录因子家族成员,根据其染色体定位信息,对所有转录因子进行了系统编号,其中仅MdOFP28无匹配的染色体定位信息(表2)。通过PFAM及NCBI-CDD工具进行蛋白质结构分析,28个苹果OFP蛋白均含有OVATE结构域。为能利用模式植物拟南芥已有的研究成果进行苹果OFP蛋白的功能探索,随后通过序列比对寻找了28个苹果MdOFP在拟南芥中的最高同源基因;蛋白质生化属性分析发现,MdOFP蛋白长度在168 aa(MdOFP08和MdOFP13)—438 aa(MdOFP19)范围内,平均蛋白长度298 aa,等电点在5.28(MdOFP24)—9.51(MdOFP04)(表2)。Table 2
表2
表2苹果OFP基因家族信息
Table 2The Ovate proteins gene family in apple
| 基因名称Gene name | 基因ID号 Gene Identifier | 染色体定位 Chromosome location | 编码序列长度 CDS length (bp) | 外显子 数目 Exon no. | 大小 Size (aa) | 分子量Molecular weight (Da) | 等电点Isoelectric Point | 拟南芥同源基因 Best homologs in Arabidopsis |
|---|---|---|---|---|---|---|---|---|
| MdOFP01 | MDP0000243940 | chr2:4251394..4252275 | 882 | 1 | 293 | 33533.3 | 9.13 | AT5G66270.1 |
| MdOFP02 | MDP0000155311 | chr2:13187873..13188835 | 963 | 1 | 320 | 25246.9 | 9.10 | AT1G19860.1 |
| MdOFP03 | MDP0000684645 | chr3:4952419..4953570 | 1152 | 1 | 383 | 41697.8 | 8.99 | AT1G19860.1 |
| MdOFP04 | MDP0000141642 | chr3:16219260..16220244 | 675 | 2 | 224 | 25723.6 | 9.51 | AT1G19860.1 |
| MdOFP05 | MDP0000733399 | chr3:31034178..31034924 | 747 | 1 | 248 | 79142.9 | 5.58 | AT2G02160.1 |
| MdOFP06 | MDP0000134728 | chr4:17309604..17310695 | 1092 | 1 | 363 | 72112.4 | 6.00 | AT5G12440.3 |
| MdOFP07 | MDP0000147045 | chr4:17310063..17310721 | 627 | 2 | 208 | 26669.0 | 8.73 | AT3G12130.1 |
| MdOFP08 | MDP0000158931 | chr5:941980..942486 | 507 | 1 | 168 | 51797.0 | 8.27 | AT5G18550.1 |
| MdOFP09 | MDP0000241313 | chr5:5755844..5757320 | 1296 | 2 | 431 | 30624.6 | 9.44 | AT3G12130.1 |
| MdOFP10 | MDP0000402356 | chr7:5438947..5439702 | 756 | 1 | 251 | 40999.8 | 8.81 | AT3G08505 |
| MdOFP11 | MDP0000296199 | chr8:3579420..3580445 | 1026 | 1 | 341 | 47229.3 | 7.10 | AT2G41900.1 |
| MdOFP12 | MDP0000155705 | chr10:26964000..26965493 | 1296 | 2 | 431 | 79656.8 | 6.14 | AT3G51950 |
| MdOFP13 | MDP0000909475 | chr10:32149450..32149956 | 507 | 1 | 168 | 16845.6 | 8.27 | AT5G26749.1 |
| MdOFP14 | MDP0000156202 | chr11:4775252..4776394 | 1143 | 1 | 380 | 91640.3 | 7.22 | AT1G10320.1 |
| MdOFP15 | MDP0000311969 | chr11:22625025..22626007 | 549 | 2 | 182 | 75934.3 | 6.24 | AT1G30460.1 |
| MdOFP16 | MDP0000127146 | chr11:32348926..32350337 | 912 | 2 | 303 | 73772.2 | 6.10 | AT1G30460.1 |
| MdOFP17 | MDP0000669940 | chr12:3242911..3248219 | 957 | 3 | 318 | 20954.5 | 6.24 | AT3G48440.1 |
| MdOFP18 | MDP0000137053 | chr12:3317131..3317688 | 558 | 1 | 185 | 65503.5 | 9.21 | AT1G19860.1 |
| MdOFP19 | MDP0000456557 | chr12:14175298..14181532 | 1317 | 3 | 438 | 98062.9 | 5.78 | AT3G51120.1 |
| MdOFP20 | MDP0000692068 | chr12:21920389..21922396 | 1098 | 2 | 365 | 20469.1 | 9.34 | AT1G66810.1 |
| MdOFP21 | MDP0000238712 | chr12:21920806..21921922 | 1065 | 2 | 354 | 93378.1 | 8.34 | AT3G27700 |
| MdOFP22 | MDP0000332435 | chr12:25821429..25822584 | 1161 | 1 | 386 | 77290.9 | 6.31 | AT2G33835.1 |
| MdOFP23 | MDP0000410437 | chr13:18948503..18949252 | 750 | 1 | 249 | 76306.6 | 6.34 | AT2G41900.1 |
| MdOFP24 | MDP0000943578 | chr13:25670394..25671158 | 765 | 1 | 254 | 57322.6 | 5.28 | AT1G01350.1 |
| MdOFP25 | MDP0000243386 | chr14:4693458..4695714 | 1020 | 2 | 339 | 22465.2 | 6.99 | AT3G19360.1 |
| MdOFP26 | MDP0000140421 | chr15:11737514..11738332 | 822 | 1 | 273 | 44526 | 9.32 | AT3G47120.1 |
| MdOFP27 | MDP0000259366 | chr17:16535235..16536098 | 864 | 1 | 287 | 76587.0 | 5.93 | AT2G02160.1 |
| MdOFP28 | MDP0000921224 | chr0:61416319..61416894 | 576 | 1 | 191 | 172448.5 | 8.12 | AT1G21580.1 |
新窗口打开
2.2 苹果OFP基因家族系统进化及基因结构
为分析苹果OFP蛋白间的系统进化关系,采用MEGA5.0软件对28个MdOFP的全长氨基酸序列进行多序列比对,构建了系统进化树。如图1所示,根据亲缘关系远近可将MdOFP分为4组(A、B、C和D组),分别含有13、6、4和5个MdOFP;在系统进化树上发现了10对MdOFP的步长值(bootstrap values)高达99%,用红色阴影标记出来,也进一步说明该进化关系十分可靠(图1左)。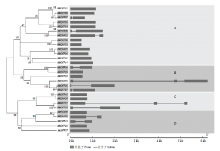 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图1苹果OFP基因家族进化树及基因结构
-->Fig. 1The unrooted neighbor joining phylogenetic tree and gene structure of OFP gene family in apple
-->
通过对苹果OFP家族成员的基因结构分析显示,该家族基因结构相对简单,外显子的数量从1(17个MdOFP,约占60.7%)到3(MdOFP17和MdOFP19),内含子子数目不高于2个,其中A组有10个成员不含有内含子(图1右)。结果表明,苹果OFP蛋白家族成员聚类关系较近的基因结构相对简单保守,且具有相似的外显子结构和长度。
2.3 苹果OFP基因家族染色体定位
根据苹果OFP基因位置信息得到28个OFP基因在苹果染色体上的定位图,它们分布在苹果17条染色体中的13条上,其中第12条染色体基因最多,含有6个MdOFP成员,其次是第3和第11条染色体各有3个MdOFP分布,而第7、8、14、15和17条染色体均仅有1个MdOFP,第1、6、9和16条染色体上没有MdOFP的分布;其中3对基因(MdOFP06/ MdOFP07、MdOFP17/MdOFP18及MdOFP20/MdOFP21)在染色体上紧密连锁在一起,MdOFP06/MdOFP07和MdOFP20/MdOFP21的步长值达到99,而MdOFP17与MdOFP18分别属于C组和B组。另外还存在6对串联重复基因,分别是MdOFP01/MdOFP26、MdOFP05/MdOFP16、MdOFP08/MdOFP13、MdOFP09/ MdOFP12、MdOFP10/MdOFP24和MdOFP17(MdOFP18)/MdOFP25(图2)。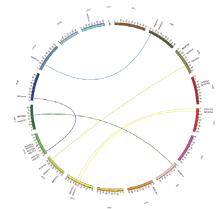 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图2苹果OFP基因家族成员在染色体上的位置
-->Fig. 2The chromosome location of the OFP gene family in apple
-->
基因比对是一种相对快速且有效的方法,能够帮助更好的理解基因组结构、功能和进化关系[28,29,30]。通过与已研究较清楚的模式植物拟南芥直接同源的基因进行比对就能推测出MdOFP在苹果中可能行使的功能。本研究对苹果和拟南芥OFP基因家族成员组间重复区的同线性分析,结果揭示苹果和拟南芥中最少10对MdOFP/AtOFP位于同线性基因组区域,分别是MdOFP02/ATOFP7、MdOFP07/ATOFP1、MdOFP14/ ATOFP2、MdOFP14/ATOFP3、MdOFP14/ATOFP4、MdOFP18/ATOFP10、MdOFP25/ATOFP15、MdOFP25/ ATOFP18、MdOFP26/ATOFP8、MdOFP27/ATOFP14(图3)。
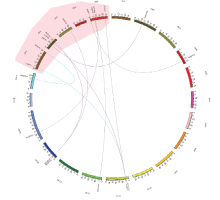 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图3苹果和拟南芥OFP基因的同线性分析
-->Fig. 3Synteny analysis of OFP genes between apple and Arabidopsis
-->
2.4 苹果MdOFP在不同组织中的芯片表达谱
基因的组织表达模式能够为研究基因功能提供重要的信息,为揭示苹果OFP转录因子家族基因在苹果组织中的表达情况,进而推测其可能在生长发育过程中所起的作用。根据EMBL-EBI(E-GEOD-42873和E-GEOD-51728)的基因芯片数据,能够在苹果不同组织(包括根、茎、叶、花、果实、种子及幼苗)中分析出苹果MdOFP的转录水平的表达量变化。在聚类图中用红色代表相对较高的表达水平,绿色表示基因表达相对较弱。结果表明,苹果MdOFP呈现出不同的组织表达模式,绝大多数苹果MdOFP在花、果实和叶子中表达量较高,而在根、茎、种子和幼苗期间表达量相对较低,说明这些基因可能在苹果的营养生长时期和生殖生长时期发挥不同作用(图4)。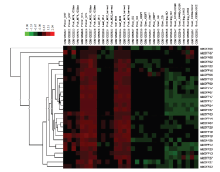 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图4苹果MdOFP在不同组织中的表达谱分析
-->Fig. 4Expression profile of MdOFP in different tissues by microarray analysis
-->
2.5 苹果MdOFP在不同组织和非生物胁迫下的表达
为分析苹果MdOFPs在不同组织器官中的表达情况,随机挑选了13个苹果MdOFP,其中A、B、C和D组分别选择了8、3、1和1个基因。采用qRT- PCR技术对其在苹果不同组织和非生物胁迫下的表达模式进行了检测。如图5所示,13个基因在所有检测组织中均有不同程度的表达,除MdOFP07在根和茎中表达量最高,MdOFP03、MdOFP04、MdOFP06、MdOFP08、MdOFP11、MdOFP13、MdOFP16、MdOFP17和MdOFP18在根中的表达量也较高外,其他基因均在花和果实中表达量最高,这与芯片表达谱分析的结果一致(图5)。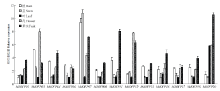 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图5MdOFP在苹果不同组织中的表达分析
-->Fig. 5Expression profile of MdOFP in different tissues in apple
-->
盐处理6 h后,根部检测到MdOFP上调2.0倍的较多,包括MdOFP01、MdOFP04、MdOFP12、MdOFP16、MdOFP18和MdOFP20,其中MdOFP20表达量最高达到对照的5倍,而仅有MdOFP04受到PEG的诱导,另外,MdOFP07和MdOFP16的表达量经PEG处理后明显下降(图6-A)。不同的是,地上部组织无论是NaCl处理还是PEG处理,除MdOFP04、MdOFP11和MdOFP20分别受NaCl、PEG和NaCl诱导外,其他基因的表达量变化均有明显的下调趋势,其中MdOFP01、MdOFP03、MdOFP06和MdOFP12在两种胁迫条件下都是明显下调,MdOFP13和MdOFP18在盐处理后表达量明显下降,而MdOFP04和MdOFP08在PEG处理后呈现下调(图6-B)。MdOFP04和MdOFP20在根部和地上部的表达变化均是NaCl处理明显上调,MdOFP01、MdOFP12和MdOFP18的表达情况则相反; PEG处理后,MdOFP04在根部和地上部表现出不同的变化趋势。
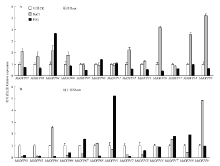 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图6NaCl和PEG处理后苹果MdOFP的表达分析
-->Fig. 6Expression analysis of the MdOFP under NaCl and PEG treatment in apple
-->
温度胁迫后MdOFPs表达量分析表明,39℃高温处理可以诱导MdOFP04、MdOFP06、MdOFP07、MdOFP12和MdOFP17的表达,其中MdOFP04表达量最高达到对照的4.53倍,MdOFP13和MdOFP16的表达量稍有下降。而经4℃低温处理后,检测到MdOFP03、MdOFP04、MdOFP13和MdOFP17表达量增加至对照的2.59—3.39倍,MdOFP06、MdOFP07和MdOFP20的表达下调明显,其中MdOFP04和MdOFP17在两种温度胁迫条件下都明显上调(图7)。
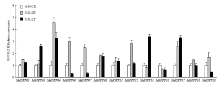 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图7高温和低温处理后苹果MdOFP的表达分析
-->Fig. 7Expression analysis of the MdOFP under high temperature (HT) and low temperature (LT) treatment in apple
-->
3 讨论
转录因子调节多种植物的生长和发育过程,包括分生组织功能和维持、侧生器官特异性以及叶子发育和延伸,并在非生物胁迫复杂的信号调控网络中发挥重要作用,因此,明确各转录因子基因在不同物种中的分布,确定各基因成员的组织表达和胁迫处理条件下的表达规律,将有助于探索基因的具体功能与胁迫响应调控的分子机理[31,32,33]。植物大部分生长和发育过程被认为是由于转录因子对细胞/组织/器官特异性的特殊基因的上调节或下调节作用调控。OFP基因家族是一种新型的植物特异性转录因子,在植物生长和发育过程中具有重要作用[2,10-13]。目前为止,苹果OFP转录因子家族基因的相关研究甚少,而模式植物中该基因家族的研究则很普遍,说明研究植物基因组中OFP家族基因十分必要[3,4,5,6]。本研究通过对苹果全基因组中含有OVATE保守结构域的基因进行筛选,首次鉴定得到28个MdOFP成员;依据MdOFP的蛋白质序列构建了系统进化树,可将MdOFP分为4组。根据MdOFP的系统进化关系和序列相似性,鉴定出了一系列MdOFP的串联重复序列。在苹果OFP转录因子家族成员间3对基因在染色体上紧密连锁、6对串联重复基因和10对MdOFP/AtOFP与拟南芥位于同线性基因组区域,表明MdOFP基因家族可能通过串联重复和片段重复来扩展,并且与拟南芥OFP转录因子家族成员间有较高的同源性。另外,MdOFP的内含子-外显子结构高度保守,但是通过SignalP、SMART和Pfam数据库对MdOFPs蛋白质的基序进行分析发现,除了C末端高度保守的OVATE结构域,C末端还存在其他的保守基序和不同的基序,进一步说明虽多数MdOFP具有相似的蛋白保守结构域和功能冗余特性,但仍存在基序的多样性,它们可能作为潜在的蛋白质-蛋白质间的相互作用结构域辅助OFP蛋白起转录因子的作用。
苹果MdOFP在不同组织中均有不同程度的表达,暗示着MdOFP在不同发育过程和不同组织中可能发挥不同作用,与特定的基因表达调控有关,具体功能还有待进一步验证。拟南芥中的AtOFP已经有较多的生长发育过程中的功能验证,根据进化关系,推测与拟南芥同缘关系较近的MdOFP会具有类似功能,但是前人研究结果多集中在AtOFP如何影响植物生长发育过程,对于参与胁迫响应的调控过程还报道较少。本研究表明苹果MdOFP受NaCl、PEG、高温和低温4种胁迫处理后,基因表达量有不同程度的变化差异,并且NaCl和PEG处理后根和地上部组织中的变化呈现胁迫响应的多样性,暗示苹果MdOFP可能参与非生物胁迫过程中的胁迫响应途径,具体的胁迫响应调节功能有待后续进行遗传转化等功能验证。苹果OFP转录因子家族如何调控苹果的生长发育过程,如何参与对逆境胁迫的响应等或将成为今后基因功能研究的重点与热点问题。
4 结论
本研究利用生物信息学方法对苹果OFP基因家族进行全基因组鉴定,共筛选获得28个MdOFP家族基因,可分为4组;染色体定位发现,第1、6、9和16号染色体没有OFP分布,其他的13条染色体上均有MdOFP,分布密度不同;不同MdOFP组织表达模式具有一定的时空特异性;另外,MdOFP不同程度地响应盐、干旱和温度胁迫,推测MdOFP可能参与调控苹果的盐、干旱与温度逆境响应过程。The authors have declared that no competing interests exist.
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
| [1] | . A common, recurring theme in domesticated plants is the occurrence of pear-shaped fruit. A major quantitative trait locus (termed ovate) controlling the transition from round to pear-shaped fruit has been cloned from tomato. OVATE is expressed early in flower and fruit development and encodes a previously uncharacterized, hydrophilic protein with a putative bipartite nuclear localization signal, Von Willebrand factor type C domains, and an 鈮70-aa C-terminal domain conserved in tomato, Arabidopsis, and rice. A single mutation, leading to a premature stop codon, causes the transition of tomato fruit from round- to pear-shaped. Moreover, ectopic, transgenic expression of OVATE unevenly reduces the size of floral organs and leaflets, suggesting that OVATE represents a previously uncharacterized class of negative regulatory proteins important in plant development. |
| [2] | . Transcription factors regulate multiple aspects of plant growth and development. Here we report the identification and functional analysis of a plant-specific, novel transcription factor in Arabidopsis. We isolated a dominant, gain-of-function mutant that displays reduced lengths in all aerial organs including hypocotyl, rosette leaf, cauline leaf, inflorescence stem, floral organs and silique. Molecular cloning revealed that these phenotypes are caused by elevated expression of the Arabidopsis thaliana Ovate Family Protein 1 ( AtOFP1 ). This mutant was designated as Atofp1-1D . We show that the altered morphology of Atofp1-1D mutant is caused by reduced cell length resulting from reduced cell elongation, and demonstrate that a mutant harboring a transposon insertion that disrupts the OVATE domain of AtOFP1 is indistinguishable from wild-type plants. Plants overexpressing other closely related AtOFP genes phenocopy plants overexpressing AtOFP1 , implying a possible overlapping function among members of the AtOFP gene family. We found that AtOFP1 localizes in the nucleus, and that AtOFP1 functions as an active transcriptional repressor. Chromatin immunoprecipitation results indicated that AtGA20ox1 , a gene encoding the key enzyme in GA biosynthesis, is a target gene regulated by AtOFP1. Consistent with this, exogenous gibberellic acid can partially restore defects in cell elongation in plants overexpressing AtOFP1 , suggesting that such a reduced cell elongation is caused, in part, by the deficiency in gibberellin biosynthesis. Taken together, our results indicate that AtOFP1 is an active transcriptional repressor that has a role in regulating cell elongation in plants. |
| [3] | . The Arabidopsis genome contains 18 genes that are predicted to encode Ovate Family Proteins (AtOFPs), a protein family characterized by a conserved OVATE domain, an approximately 70-amino acid domain that was originally found in tomato OVATE protein. Among AtOFP family members, AtOFP1 has been shown to suppress cell elongation, in part, by suppressing the expression of AtGA20ox1, AtOFP4 has been shown to regulate secondary cell wall formation by interact with KNOTTED1-LIKE HOMEODOMAIN PROTEIN 7 (KNAT7), and AtOFP5 has been shown to regulate the activity of a BEL1-LIKEHOMEODOMAIN 1(BLH1)-KNAT3 complex during early embryo sac development, but little is known about the function of other AtOFPs.We demonstrated here that AtOFP proteins could function as effective transcriptional repressors in the Arabidopsis protoplast transient expression system. The analysis of loss-of-function alleles of AtOFPs suggested AtOFP genes may have overlapping function in regulating plant growth and development, because none of the single mutants identified, including T-DNA insertion mutants in AtOFP1, AtOFP4, AtOFP8, AtOFP10, AtOFP15 and AtOFP16, displayed any apparent morphological defects. Further, Atofp1 Atofp4 and Atofp15 Atofp16 double mutants still did not differ significantly from wild-type. On the other hand, plants overexpressing AtOFP genes displayed a number of abnormal phenotypes, which could be categorized into three distinct classes, suggesting that AtOFP genes may also have diverse functions in regulating plant growth and development. Further analysis suggested that AtOFP1 regulates cotyledon development in a postembryonic manner, and global transcript profiling revealed that it suppress the expression of many other genes.Our results showed that AtOFPs function as transcriptional repressors and they regulate multiple aspects of plant growth and development. These results provided the first overview of a previously unknown transcriptional repressor family, and revealed their possible roles in plant growth and development. |
| [4] | . . |
| [5] | . |
| [6] | . Abstract The Arabidopsis ovate family proteins (AtOFPs) have been shown to function as transcriptional repressors and regulate multiple aspects of plant growth and development. There are 31 genes that encode the full-length OVATE-domain containing proteins in the rice genome. In this study, the gene structure analysis revealed that OsOFPs are intron poor. Phylogenetic analysis suggested that OVATE proteins from rice, Arabidopsis and tomato can be divided into 4 groups (I-IV). Real-time quantitative polymerase chain reaction (RT-qPCR) analysis identified OsOFPs with different tissue-specific expression patterns at all stages of development in the rice plant. Interestingly, nearly half of the total number of OsOFP family was more highly expressed during the seed developmental stage. In addition, seed developmental cis-elements were found in the promoter region of the OsOFPs. Subcellular localization analysis revealed that YFP-OsOFP fusion proteins predominantly localized in the nucleus. Our results suggest that OsOFPs may act as regulatory proteins and play pivotal roles in the growth and development of rice. |
| [7] | . . |
| [8] | . . |
| [9] | . |
| [10] | . The Ku heterodimer, a DNA repair protein complex consisting of 70- and 80-kDa subunits, is involved in the non-homologous end-joining (NHEJ) pathway. Plants are thought to use the NHEJ pathway primarily for the repair of DNA double-strand breaks (DSBs). The Ku70/80 protein has been identified in many plants and been shown to possess several similar functions to its counter protein complex in mammals. In the present study, ovate family protein 1 ( At OFP1) was demonstrated to be a plant Ku-interacting protein by yeast two-hybrid screening and the GST pull-down assay. Truncation analysis revealed that the C-terminal domain of At Ku70 contains interacting sites for At OFP1. The electrophoretic mobility shift assay (EMSA) indicated that At OFP1 is also a DNA binding protein with its binding domain at the N-terminus. In 3-week-old seedlings, expression of the AtOFP1 gene increased after exposure to DNA-damaging agents (such as methyl methanesulfonate (MMS) and menadione) in a time dependent manner. Seedlings lacking the At OFP1 protein were more sensitive to MMS and menadione as compared with wild-type. Furthermore, similar to AtKu70 61 / 61 and AtKu80 61 / 61 , the AtOFP1 61 / 61 mutant showed relatively lower NHEJ activity in vivo. Taken together, these results suggest that At OFP1 may play a role in DNA repair through the NHEJ pathway accompanying with the At Ku protein. |
| [11] | In Arabidopsis thaliana, the female gametophyte is a highly polarized structure consisting of four cell types: one egg cell and two synergids, one central cell, and three antipodal cells. In this report, we describe the characterization of a novel female gametophyte mutant, eostre, which affects establishment of cell fates in the mature embryo sac. The eostre phenotype is caused by misexpression of the homeodomain gene BEL1-like homeodomain 1 (BLH1) in the embryo sac. It is known that BELL-KNAT proteins function as heterodimers whose activities are regulated by the Arabidopsis ovate family proteins (OFPs). We show that the phenotypic effect of BLH1 overexpression is dependent upon the class II knox gene KNAT3, suggesting that KNAT3 must be expressed and functional during megagametogenesis. Moreover, disruption of At OFP5, a known interactor of KNAT3 and BLH1, partially phenocopies the eostre mutation. Our study indicates that suppression of ectopic activity of BELL-KNOX TALE complexes, which might be mediated by At OFP5, is essential for normal development and cell specification in the Arabidopsis embryo sac. As eostre-1 embryo sacs also show nuclear migration abnormalities, this study suggests that a positional mechanism might be directing establishment of cell fates in early megagametophyte development. |
| [12] | The homeodomain transcription factor KNAT7 has been reported to be involved in the regulation of secondary cell wall biosynthesis. Previous work suggested that KNAT7 can interact with members of the Ovate Family Protein (OFP) transcription co-regulators. However, it remains unknown whether such an OFP–KNAT7 complex could be involved in the regulation of secondary cell wall biosynthesis in Arabidopsis. We re-tested OFP1 and OFP4 for their abilities to intact with KNAT7 using yeast two-hybrid assays, and verified KNAT7–OFP4 interaction but found only weak interaction between KNAT7 and OFP1. Further, the interaction of KNAT7 with OFP4 appears to be mediated by the KNAT7 homeodomain. We used bimolecular fluorescence complementation to confirm interactions and found that OFP1 and OFP4 both interact with KNAT7 in planta. Using a protoplast transient expression system we showed that KNAT7 as well as OFP1 and OFP4 act as transcriptional repressors. Furthermore, in planta interactions between KNAT7 and both OFP1 and OFP4 enhance KNAT7’s transcriptional repression activity. An ofp4 mutant exhibited similar irx and fiber cell wall phenotypes as knat7, and the phenotype of a double ofp4 knat7mutant was similar to those of the single mutants, consistent with the view that KNAT7 and OFP function in a common pathway or complex. Furthermore, the pleiotropic OFP1 and OFP4 overexpression phenotype was suppressed in a knat7 mutant background, suggesting that OFP1 and OFP4 functions depend at least partially on KNAT7 function. We propose that KNAT7 forms a functional complex with OFP proteins to regulate aspects of secondary cell wall formation. |
| [13] | Formation of secondary walls is a complex process that requires the coordinated and developmentally regulated expression of secondary wall biosynthetic genes. In Arabidopsis thaliana, a transcriptional network orchestrates the biosynthesis and deposition of the main SCW components in xylem and fiber cells. It was recently reported that interacting TALE homeodomain proteins BEL-LIKE HOMEODOMAIN6 (BLH6) and KNOTTED ARABIDOPSIS THALIANA7 (KNAT7) negatively regulate secondary cell wall formation in the interfascicular fibers of Arabidopsis inflorescence stems. Members of the Arabidopsis OVATE FAMILY PROTEIN (OFP) family of transcriptional regulators have been shown to physically interact in yeast with various KNAT and BLH proteins, forming a proposed TALE-OFP protein interaction network. This study presents molecular and genetic data indicating that OFP1 and OFP4, previously reported to interact with TALE homeodomain proteins, enhance the repression activity of BLH6, supporting a role for these OFPs as components of a putative multi-protein transcription regulatory complex containing BLH6 and KNAT7. |
| [14] | |
| [15] | . PlantGDB (http://www.plantgdb.org/) is a genomics database encompassing sequence data for green plants (Viridiplantae). PlantGDB provides annotated transcript assemblies for >100 plant species, with transcripts mapped to their cognate genomic context where available, integrated with a variety of sequence analysis tools and web services. For 14 plant species with emerging or complete genome sequence, PlantGDB's genome browsers (xGDB) serve as a graphical interface for viewing, evaluating and annotating transcript and protein alignments to chromosome or bacterial artificial chromosome (BAC)-based genome assemblies. Annotation is facilitated by the integrated yrGATE module for community curation of gene models. Novel web services at PlantGDB include Tracembler, an iterative alignment tool |
| [16] | |
| [17] | . The apple (Malus domestica) is one of the most economically important fruit crops in the world, due its importance to human nutrition and health. To analyze the function and evolution of different apple genes, we developed apple gene function and gene family database (AppleGFDB) for collecting, storing, arranging, and integrating functional genomics information of the apple. The AppleGFDB provides several layers of information about the apple genes, including nucleotide and protein sequences, chromosomal locations, gene structures, and any publications related to these annotations. To further analyze the functional genomics data of apple genes, the AppleGFDB was designed to enable users to easily retrieve information through a suite of interfaces, including gene ontology, protein domain and InterPro. In addition, the database provides tools for analyzing the expression profiles and microRNAs of the apple. Moreover, all of the analyzed and collected data can be downloaded from the database. The database can also be accessed using a convenient web server that supports a full-text search, a BLAST sequence search, and database browsing. Furthermore, to facilitate cooperation among apple researchers, AppleGFDB is presented in a user-interactive platform, which provides users with the opportunity to modify apple gene annotations and submit publication information for related genes. AppleGFDB is available at http://www.applegene.org or http://gfdb.sdau.edu.cn/. |
| [18] | . Abstract MOTIVATION: Automating the assignment of existing domain and protein family classifications to new sets of sequences is an important task. Current methods often miss assignments because remote relationships fail to achieve statistical significance. Some assignments are not as long as the actual domain definitions because local alignment methods often cut alignments short. Long insertions in query sequences often erroneously result in two copies of the domain assigned to the query. Divergent repeat sequences in proteins are often missed. RESULTS: We have developed a multilevel procedure to produce nearly complete assignments of protein families of an existing classification system to a large set of sequences. We apply this to the task of assigning Pfam domains to sequences and structures in the Protein Data Bank (PDB). We found that HHsearch alignments frequently scored more remotely related Pfams in Pfam clans higher than closely related Pfams, thus, leading to erroneous assignment at the Pfam family level. A greedy algorithm allowing for partial overlaps was, thus, applied first to sequence/HMM alignments, then HMM-HMM alignments and then structure alignments, taking care to join partial alignments split by large insertions into single-domain assignments. Additional assignment of repeat Pfams with weaker E-values was allowed after stronger assignments of the repeat HMM. Our database of assignments, presented in a database called PDBfam, contains Pfams for 99.4% of chains >50 residues. AVAILABILITY: The Pfam assignment data in PDBfam are available at http://dunbrack2.fccc.edu/ProtCid/PDBfam, which can be searched by PDB codes and Pfam identifiers. They will be updated regularly. |
| [19] | . 【Objective】Identification of LBD genes from tomato genome, and analysis of phylogeny,gene structure, chromosome location, phylogenetic and tissue expression pattern analysis of LBD family genes in tomato will be useful to the functions identification of plant LBD genes.【Method】Based on tomato genome database and bioinformatic method, tomato LBD family genes were identified and the genes were sequenced. A phylogenetic tree was created using the MEGA5 program. Gene structure and chromosomes location were done by Perl-based program, MapDraw and GSDS. Expression pattern of LBD genes at different development stages was analyzed based on the existing microarray database.【Result】A total of 46 LBD genes were systematically identified from tomato and classified into 2 classes (class I and class II), then was classified into 5 subfamilies (Ia, Ib, Ic, Id and II) according to the gene structure and conserved domain phylogeny relationship. They were distributed on 10 of tomato chromosomes, suggesting that they have an extensive distribution on the tomato chromosomes. Most of the LBD genes had differential expression pattern and response to external stimulation. 【Conclusion】Forty-six LBD family genes in tomato were identified by genome-wide screening. They were classified into 2 classes, 5 subfamilies and distributed on 10 chromosomes with different expression patterns in different tissues and developmental stages. They had special response to external stimulation. These results are helpful for the functional analysis of LBD genes in plants. . 【Objective】Identification of LBD genes from tomato genome, and analysis of phylogeny,gene structure, chromosome location, phylogenetic and tissue expression pattern analysis of LBD family genes in tomato will be useful to the functions identification of plant LBD genes.【Method】Based on tomato genome database and bioinformatic method, tomato LBD family genes were identified and the genes were sequenced. A phylogenetic tree was created using the MEGA5 program. Gene structure and chromosomes location were done by Perl-based program, MapDraw and GSDS. Expression pattern of LBD genes at different development stages was analyzed based on the existing microarray database.【Result】A total of 46 LBD genes were systematically identified from tomato and classified into 2 classes (class I and class II), then was classified into 5 subfamilies (Ia, Ib, Ic, Id and II) according to the gene structure and conserved domain phylogeny relationship. They were distributed on 10 of tomato chromosomes, suggesting that they have an extensive distribution on the tomato chromosomes. Most of the LBD genes had differential expression pattern and response to external stimulation. 【Conclusion】Forty-six LBD family genes in tomato were identified by genome-wide screening. They were classified into 2 classes, 5 subfamilies and distributed on 10 chromosomes with different expression patterns in different tissues and developmental stages. They had special response to external stimulation. These results are helpful for the functional analysis of LBD genes in plants. |
| [20] | . CDD, the Conserved Domain Database, is part of NCBI's Entrez query and retrieval system and is also accessible via http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml. CDD provides annotation of protein sequences with the location of conserved domain footprints and functional sites inferred from these footprints. Pre-computed annotation is available via Entrez, and interactive search services accept single protein or nucleotide queries, as well as batch submissions of protein query sequences, utilizing RPS-BLAST to rapidly identify putative matches. CDD incorporates several protein domain and full-length protein model collections, and maintains an active curation effort that aims at providing fine grained classifications for major and well-characterized protein domain families, as supported by available protein three-dimensional (3D) structure and the published literature. To this date, the majority of protein 3D structures are represented by models tracked by CDD, and CDD curators are characterizing novel families that emerge from protein structure determination efforts. |
| [21] | . |
| [22] | ExPASy (http://www.expasy.org) has worldwide reputation as one of the main bioinformatics resources for proteomics. It has now evolved, becoming an extensible and integrative portal accessing many scientific resources, databases and software tools in different areas of life sciences. Scientists can henceforth access seamlessly a wide range of resources in many different domains, such as proteomics, genomics, phylogeny/evolution, systems biology, population genetics, transcriptomics, etc. The individual resources (databases, web-based and downloadable software tools) are hosted in a 'decentralized' way by different groups of the SIB Swiss Institute of Bioinformatics and partner institutions. Specifically, a single web portal provides a common entry point to a wide range of resources developed and operated by different SIB groups and external institutions. The portal features a search function across 'selected' resources. Additionally, the availability and usage of resources are monitored. The portal is aimed for both expert users and people who are not familiar with a specific domain in life sciences. The new web interface provides, in particular, visual guidance for newcomers to ExPASy. |
| [23] | . BACKGROUND: In a previous paper, we introduced MUSCLE, a new program for creating multiple alignments of protein sequences, giving a brief summary of the algorithm and showing MUSCLE to achieve the highest scores reported to date on four alignment accuracy benchmarks. Here we present a more complete discussion of the algorithm, describing several previously unpublished techniques that improve biological accuracy and / or computational complexity. We introduce a new option, MUSCLE-fast, designed for high-throughput applications. We also describe a new protocol for evaluating objective functions that align two profiles. RESULTS: We compare the speed and accuracy of MUSCLE with CLUSTALW, Progressive POA and the MAFFT script FFTNS1, the fastest previously published program known to the author. Accuracy is measured using four benchmarks: BAliBASE, PREFAB, SABmark and SMART. We test three variants that offer highest accuracy (MUSCLE with default settings), highest speed (MUSCLE-fast), and a carefully chosen compromise between the two (MUSCLE-prog). We find MUSCLE-fast to be the fastest algorithm on all test sets, achieving average alignment accuracy similar to CLUSTALW in times that are typically two to three orders of magnitude less. MUSCLE-fast is able to align 1,000 sequences of average length 282 in 21 seconds on a current desktop computer. CONCLUSIONS: MUSCLE offers a range of options that provide improved speed and / or alignment accuracy compared with currently available programs. MUSCLE is freely available at http://www.drive5.com/muscle. |
| [24] | |
| [25] | . MAPMAKER is one of the most widely used computer software package for constructing genetic linkage maps.However,the PC version,MAPMAKER 3.0 for PC,could not draw the genetic linkage maps that its Macintosh version,MAPMAKER 3.0 for Macintosh,was able to do.Especially in recent years,Macintosh computer is much less popular than PC.Most of the geneticists use PC to analyze their genetic linkage data.So a new computer software to draw the same genetic linkage maps on PC as the MAPMAKER for Macintosh to do on Macintosh has been crying for.Microsoft Excel,one component of Microsoft Office package,is one of the most popular software in laboratory data processing.Microsoft Visual Basic for Applications (VBA) is one of the most powerful functions of Microsoft Excel.Using this program language,we can take creative control of Excel,including genetic linkage map construction,automatic data processing and more.In this paper,a Microsoft Excel macro called MapDraw is constructed to draw genetic linkage maps on PC computer based on given genetic linkage data.Use this software,you can freely construct beautiful genetic linkage map in Excel and freely edit and copy it to Word or other application.This software is just an Excel format file.You can freely copy it from ftp://211.69.140.177 or ftp://brassica.hzau.edu.cn and the source code can be found in Excel鈥瞫 Visual Basic Editor.顎 |
| [26] | . We developed a web server GSDS (Gene Structure Display Server) for drawing gene structure schematic diagrams. Users can submit three types of data:CDS and genomic sequences,NCBI GenBank accession numbers or GIs,exon positions on a gene. GSDS uses this information to obtain the gene structure and draw diagram for it. Users can also designate some special regions to mark on the gene structure diagram. The output result will be PNG or SVG format picture. The corresponding sequence will be shown in a new window by clicking the picture in PNG format. A Chinese version for the main page is also built. The GSDS is available on http://gsds.cbi.pku.edu.cn/. |
| [27] | . We created a visualization tool called Circos to facilitate the identification and analysis of similarities and differences arising from comparisons of genomes. Our tool is effective in displaying variation in genome structure and, generally, any other kind of positional relationships between genomic intervals. Such data are routinely produced by sequence alignments, hybridization arrays, genome mapping, and genotyping studies. Circos uses a circular ideogram layout to facilitate the display of relationships between pairs of positions by the use of ribbons, which encode the position, size, and orientation of related genomic elements. Circos is capable of displaying data as scatter, line, and histogram plots, heat maps, tiles, connectors, and text. Bitmap or vector images can be created from GFF-style data inputs and hierarchical configuration files, which can be easily generated by automated tools, making Circos suitable for rapid deployment in data analysis and reporting pipelines. |
| [28] | . In addition to the genomes of Arabidopsis (Arabidopsis thaliana) and poplar (Populus trichocarpa), two near-complete rosid genome sequences, grape (Vitis vinifera) and papaya (Carica papaya), have been recently released. The phylogenetic relationship among these four genomes and the placement of their three independent, fractionated tetraploidies sum to a powerful comparative genomic system. CoGe, a platform of multiple whole or near-complete genome sequences, provides an integrative Web-based system to find and align syntenic chromosomal regions and visualize the output in an intuitive and interactive manner. CoGe has been customized to specifically support comparisons among the rosids. Crucial facts and definitions are presented to clearly describe the sorts of biological questions that might be answered in part using CoGe, including patterns of DNA conservation, accuracy of annotation, transposability of individual genes, subfunctionalization and /or fractionation of syntenic gene sets, and conserved noncoding sequence content. This pr茅cis of an online tutorial, CoGe with Rosids (http:// tinyurl.com/4a23pk), presents sample results graphically. |
| [29] | . Abstract MOTIVATION: DnaSP is a software package for a comprehensive analysis of DNA polymorphism data. Version 5 implements a number of new features and analytical methods allowing extensive DNA polymorphism analyses on large datasets. Among other features, the newly implemented methods allow for: (i) analyses on multiple data files; (ii) haplotype phasing; (iii) analyses on insertion/deletion polymorphism data; (iv) visualizing sliding window results integrated with available genome annotations in the UCSC browser. AVAILABILITY: Freely available to academic users from: (http://www.ub.edu/dnasp). |
| [30] | . The analysis of DNA sequence polymorphisms and SNPs (single nucleotide polymorphisms) can provide insights into the evolutionary forces acting on populations and species. Available population-genetic methods, and particularly those based on the coalescent theory, have become the primary framework to analyze such DNA polymorphism data. Here, I explain some essential analytical methods for interpreting DNA polymorphism data and also describe the basic functionalities of the DnaSP software. DnaSP is a multi-propose program that allows conducting exhaustive DNA polymorphism analysis using a graphical user-friendly interface. |
| [31] | . Drought stress is one of the major limitations to crop productivity. To develop crop plants with enhanced tolerance of drought stress, a basic understanding of physiological, biochemical and gene regulatory networks is essential. Various functional genomics tools have helped to advance our understanding of stress signal perception and transduction, and of the associated molecular regulatory network. These tools have revealed several stress-inducible genes and various transcription factors that regulate the drought-stress-inducible systems. Translational genomics of these candidate genes using model plants provided encouraging results, but the field testing of transgenic crop plants for better performance and yield is still minimal. Better understanding of the specific roles of various metabolites in crop stress tolerance will give rise to a strategy for the metabolic engineering of crop tolerance of drought. |
| [32] | . |
| [33] | . Plants would be more vulnerable to water stress and thereafter rewatering or a cycled water environmental change, which occur more frequently under climatic change conditions in terms of the prediction scenarios. Effects of water stress on plants alone have been well-documented in many reports. However, the combined responses to drought and rewatering and its mechanism are relatively scant. As we known, plant growth, photosynthesis and stomatal aperture may be limited under water deficit, which would be regulated by physical and chemical signals. Under severe drought, while peroxidation may be provoked, the relevant antioxidant metabolism would be involved to annihilate the damage of reactive oxygen species. As rewatering, the recoveries of plant growth and photosynthesis would appear immediately through growing new plant parts, re-opening the stomata, and decreasing peroxidation; the recovery extents (reversely: pre-drought limitation) due to rewatering strongly depend on pre-drought intensity, duration and species. Understanding how plants response to episodic drought and watering pulse and the underlying mechanism is remarkably helpful to implement vegetation management practices in climatic changing. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}