0 引言
【研究意义】柑橘叶斑驳病毒(Citrus leaf blotch virus,CLBV)为乙型线形病毒科(Betaflexiviridae)柑橘病毒属(Citrivirus)的代表成员,可以通过嫁接侵染大多数的柑橘品种,还可以通过种子传播[1],因而具有一定的传播流行风险。构建CLBV侵染性克隆将有助于了解其分子特性及致病机理,对于CLBV的流行防控也具有重要作用。另外,CLBV在多数柑橘品种上不会引起明显的病毒感染症状,有望开发成为具有广泛应用前景的病毒载体。【前人研究进展】1968年,在美国加利福尼亚发现橘橙(‘Dweet’tangor,Citrus tangerine × C. sinensis)感染某种病毒后,叶片出现斑驳状,将其病毒命名为橘橙斑驳病毒(Dweet mottle virus,DMV)[2],后来在西班牙采用嫁接鉴定技术研究发现金橘(Fortunella margarita)感染CLBV[3],进一步的研究确认DMV和CLBV为同种病毒的不同分离物[4,5]。随后从多个国家的柑橘及近缘种上发现此病毒。1997年,日本报道了猕猴桃嫁接传染性病害但未对其病原开展研究[6];新西兰于2013年首次在猕猴桃上发现[7];中国于2016年在猕猴桃上发现[8]且在甜樱桃首次检测到[9],后续在柠檬上也有报道[10]。CLBV基因组大小约为8.7 kb,包含3个开放阅读框(open reading frame,ORF),分别编码复制相关蛋白、运动蛋白及外壳蛋白,其中运动蛋白为CLBV的沉默抑制子[11],5′端有甲基化帽子结构,3′端有poly A尾巴[12]。感染CLBV的金橘和枳壳(Poncirus trifoliate)可导致芽接合部失调[13],橘橙叶片斑驳以及香橼(‘Etrog’citron,C. medica)茎痘[14]。CLBV的某些分离物可引起甜橙(‘Pineapple’)叶片脉明[3,5,13]。此外,来自中国不同地区CLBV的病株分离物间有较大的分子变异,外壳蛋白(coat protein,CP)基因核苷酸和氨基酸序列最大差异达到近18%和12%[15],这些分离物与中国报道的CLBV樱桃分离物核苷酸和氨基酸序列的相似性也很低。【本研究切入点】侵染性克隆是深入研究病毒分子特性及病毒与寄主互作的重要基础,但目前国内外构建侵染性克隆面临重要问题之一是较大病毒基因组cDNA的组装及其在大肠杆菌中的不稳定性[16],导致侵染性克隆构建费时费力,且成功率较低。【拟解决的关键问题】通过构建基于三元穿梭载体的酵母同源重组系统,提高病毒基因组全长cDNA的组装效率,进而通过直接转化农杆菌来克服病毒cDNA在大肠杆菌中可能的不稳定性,从而快速构建CLBV的侵染性克隆。1 材料与方法
试验于2017年4—11月在西南大学柑桔研究所国家柑桔苗木脱毒中心实验室进行。1.1 供试材料
自湖北省宜都市采集检测具有柑橘叶斑驳病毒的柑橘枝条,将其嫁接于其他常见柑橘病毒检测均呈阴性的代代酸橙(Citrus aurantium‘Daidai’)、西蒙斯甜橙(C. sinensis)的砧木上,获得柑橘叶斑驳病毒病株,作为毒源植株。本生烟(Nicotiana benthamiana)、锦橙(C. sinensis‘Jincheng’)幼苗由脱毒中心实验室提供。PVX全长cDNA侵染性克隆由笔者实验室提供。酵母菌株YPH501、农杆菌菌株C58C1由法国Thierry Candresse教授惠赠。1.2 酶与试剂
限制性内切酶Sac II、Stu I、Sma I 购自北京NEB公司,PrimerSTAR Max Premix、In-Fusion HD Cloning Kit、PrimeScriptTM II 1st Strand cDNA Synthesis Kit、BM JM109购自大连TaKaRa公司;Trizol试剂、GeneArt® pYES1L Vector购自美国Invitrogen公司。1.3 引物设计
根据NCBI中已报道CLBV全基因组序列(GenBank登录号为AJ318061),利用Primer Priemer 5.0软件设计扩增CLBV基因序列的特异引物。其中pCY-CLBV1F与CLBV1R、CLBV2F与pCY-CLBV2R两对特异引物分别用于扩增大片段CLBV1、CLBV2;PYES2117F和PYES2117R用于扩增构建穿梭载体上包含酵母能识别的复制起始位点的片段;pCY-PVX-F和pCY-PVX-R用于扩增PVX全长以验证穿梭载体的有效性。在引物设计时,CLBV1R与CLBV2F、pCY-CLBV1F与酶切后的pCY载体、pCY-CLBV2R与酶切后的pCY载体、pCY-PVX-R与酶切后的pCY载体、pCY-PVX-F与酶切后的pCY载体之间都分别有25—30 bp的重叠序列,以便于在酵母中完成同源重组。引物均由英骏(上海)生物技术有限公司合成(表1)。Table 1
表1
表1引物设计及其序列
Table 1Primers design and sequences
| 引物Primer | 序列Sequence (5′→3′) |
|---|---|
| PYES2117F | GCCGATTTTGAAACCGCGGAGTCAGTGAGCGAGGAAGCG |
| PYES2117R | CTGCCTGTGATCACCGCGGCATCTTTTACT TTCACCAGC |
| pCY-CLBV1F | ATATAAGGAAGTTCATTTCATTTGGAGAGGAGAAAAGCAACGAAAGCAACCTACACAAC |
| CLBV1R | AAAGGGTCACCCTCCAACCTTTCCTCCCTA |
| CLBV2F | TAGGGAGGAAAGGTTGGAGGGTGACCCTTT |
| pCY-CLBV2R | CGCGAGGAGGTGGAGATGCCATGCCGACCCTTTTTTTTTTTTTTTTTTTTGTCTAAA |
| pCY-PVX-F | ATATAAGGAAGTTCATTTCATTTGGAGAGGAGAAAACTAAACCATACACCACCAAC |
| pCY-PVX-R | CGCGAGGAGGTGGAGATGCCATGCCGACCCGGG(T)60ATTTAT |
| CLBV1F | AGCCATAGTTGAACCATTCCTC |
| CLBV5R | GCAGATCATTCACCACATGC |
| PVX-F | ATGTCAGCACCAGCTAGCAC |
| PVX-R | GGATCCTTATGGTGGTGGTAGAG |
| 01018-F | AGGTCTTCTTCGCCATTT |
| 01018-R | CCCTTCTTCCTCCAGTTT |
新窗口打开
1.4 穿梭载体的构建
采用限制性内切酶Sac II单酶切双元载体质粒DK1317-2,得到线性化的载体。酶切反应体系:质粒DK1317-2 12 μL,限制性内切酶Sac II 2.5 μL,10×NEB Buffer 5 μL,ddH2O 23 μL。酶切反应条件:37℃反应0.5 h。以GeneArt® pYES1L Vector(Invitrogen公司)为模板,PYES2117F、PYES2117R为引物扩增含有酵母相关复制起始位点的片段pYES1L-2117。反应体积为25 μL,包括ddH2O 8.5 μL,PrimerSTAR Max Premix(2×)12.5 μL,特异性上下游引物各1 μL,模板GeneArt® pYES1L Vector 1 μL。反应条件:98℃ 1 min,98℃ 10 s,58℃ 15 s,72℃ 2 min,30个循环;72℃ 5 min,4℃保存。然后利用DNA凝胶纯化试剂盒回收目的片段。采用In-Fusion HD Cloning Kit进行构建。重组反应体系:线性化DK1317-2载体加2 μL,插入片段pYES1L-2117加4 μL,In-Fusion Enzyme加2 μL,ddH2O补齐至10 μL。50℃,水浴30 min,4℃保存。转化大肠杆菌BM JM109,37℃过夜培养,挑选阳性克隆。得到可以在酵母-农杆菌-大肠杆菌中生长的穿梭载体pCY。1.5 穿梭载体的验证
1.5.1 PVX全长cDNA的扩增与酶切pCY载体 扩增PVX全长,反应体积为25 μL:ddH2O 8.5 μL,PrimerSTAR Max Premix(2×)12.5 μL,特异性上下游引物各1 μL,模板1 μL。反应条件:98℃ 1 min,98℃ 10 s,53.5℃ 15 s,72℃ 2 min,35个循环;72℃ 5 min,4℃保存。然后利用DNA凝胶纯化试剂盒回收目的片段。采用限制性内切酶Stu I、Sma I酶切质粒pCY,得到pCY线性化的载体。反应体系:质粒pCY 13 μL,限制性内切酶Sma I 1.0 μL,10×NEB Buffer 5 μL,ddH2O 31 μL。酶切反应条件:25℃反应0.5 h,然后再加入Stu I 1.0 μL,37℃孵育0.5 h。
1.5.2 利用醋酸锂转化法转化酵母 参照YOUSSEF等采用的醋酸锂转化法[16,17],取制备好的酵母感受态细胞100 μL至2 mL的离心管中,按顺序加入以下试剂:PEG4000(50% w/v)240 μL,1 mol·L-1的LiAc 36 μL、10 mg·mL-1的鲑鱼精DNA 25 μL和pCY线性化的载体200 ng、PVX全长胶回收片段200 ng,振荡使转化体系中的组分充分混匀,在30℃摇床中250 r/min 摇菌30 min,42℃水浴热激15 min;6 000 r/min 离心3 min,弃去上清液,使用300 μL ddH2O重悬细胞,均匀涂布在Trp缺陷型筛选平板上,于30℃培养2—4 d。
1.5.3 转化农杆菌及接种本生烟 在PCR管中加入8 μL水,用牙签挑取酵母菌落的1/3到PCR管中,98℃ 10 min,然后取2 μL作为模板。用检测引物PVX-F、PVX-R检测。筛选到的阳性菌落用牙签蘸取后,加到含有5 mL YPD液体培养基10 mL离心管中。将离心管置于30℃的摇床中220 r/min摇培12—16 h,提取酵母质粒,质粒命名为pCY-PVX。将酵母质粒pCY-PVX电击转入农杆菌C58C1中。鉴定为阳性克隆的菌液加到含对应抗生素的LB液体培养基(20 mg·L-1 Rif,50 mg·L-1 Kan),200 r/min,28℃振荡培养12—16 h。离心收集菌体,用缓冲液悬浮(悬浮缓冲液:10 mmol·L-1 MgCl2,10 mmol·L-1 MES,200 μmol·L-1 As),使其OD600为0.8—1.0。静置2 h后使用注射器接种本生烟。以pCY空载质粒的农杆菌作为阴性对照;以实验室之前构建的PVX全长cDNA侵染性克隆为阳性对照。在接种7—10 d后用RT-PCR测定其侵染性。
1.6 CLBV侵染性克隆的构建
1.6.1 感病叶片总RNA的提取与CLBV序列的分段克隆 按照Trizol试剂说明书提取毒源植株的总RNA,琼脂糖凝胶电泳检测RNA完整性。以所提取感病叶片的总RNA为模版,使用PrimeScriptTM II 1st Strand cDNA Synthesis Kit(TaKaRa),合成cDNA的第一链。反转录后以cDNA作为模板,使用两对特异引物pCY-CLBV1F、CLBV1R与CLBV2F、pCY-CLBV2R对全长CLBV的cDNA分别进行PCR扩增,其序列分别命名为大片段CLBV1、CLBV2。PCR反应体系为25 μL:ddH2O 8.5 μL,PrimerSTAR Max Premix(2×)12.5 μL,特异性上下游引物各1 μL,模板1 μL。反应条件:98℃ 1 min,98℃ 10 s,55℃ 15 s,72℃ 2 min,35个循环;72℃ 5 min,4℃保存。然后利用DNA凝胶纯化试剂盒回收目的片段。1.6.2 酵母同源重组构建CLBV的侵染性克隆 利用1.5.2醋酸锂转化法转化酵母的方法,将CLBV1、CLBV2与PCY线性化载体在酵母体内进行重组。培养2—4 d后,利用引物CLBV1F、CLBV5R对酵母菌落进行鉴定。鉴定为阳性的菌落,提取酵母质粒,然后再利用pCY-CLBV1F、CLBV1R与CLBV2F、pCY-CLBV2R进行CLBV全长的鉴定,鉴定为全长的质粒,命名为pCY-CLBV。
1.6.3 全长序列测序 将所得的酵母质粒pCY-CLBV转化大肠杆菌感受态细胞BM JM 109后,送上海英骏生物技术有限公司测序。将测序结果利用软件DNAMAN(6.0)进行序列比对分析。利用DNAMAN 6.0(Lynnon Biosoft,Quebec,Canada)和MEGA6.0软件[18],对所获基因组序列及目前GenBank中已登录的CLBV序列进行比对分析。采用邻接法(neighbor-joining,NJ),重复次数1 000,构建系统进化树。
1.6.4 农杆菌转化及接种 将测序正确的克隆利用1.5.3中的方法转化农杆菌并接种本生烟,以pCY空载质粒的农杆菌作为阴性对照。每个克隆接种3株,接种植株置于光照培养箱中,于23℃、16 h光照下培养,接种20 d后统计发病情况。
1.6.5 RT-PCR法检测CLBV 以提取接种烟草的总RNA为模板,用引物CLBV1F、CLBV5R进行RT-PCR扩增检测。PCR反应体系:先后在PCR管中加入模板1 μL和ddH2O 1 μL,94℃解链3 min后置于冰上,加入ddH2O 2.3 μL,2×1 step buffer 5 μL,CLBV1F 0.2 μL,CLBV5R 0.2 μL,PrimeScript 1 step Enzyme Mix 0.3 μL。反应程序:50℃ 30 min,94℃ 2 min,94℃ 30 s,50℃ 30 s,72℃ 45 s,35个循环,72℃ 5 min,4℃停止。
1.6.6 Northern blot法检测CLBV 探针的制备与标记:采用PCR制备探针,体系为PCR buffer with MgCl2 5 μL,PCR DIG Probe Synthesis Mix 5 μL,01018-F 0.5 μL,01018-R 0.5 μL,Enzyme mix 0.75 μL,模板为样品CLBV反转录cDNA 1 μL,ddH2O补充至50 μL。反应程序:预变性94℃ 5 min,变性94℃ 30 s,退火53℃ 30 s,延伸72℃ 30 s,35个循环;再延伸72℃ 5 min,琼脂糖凝胶电泳检测结果,胶回收之后保存备用。
膜制备、杂交及信号检测:制备1%甲醛变性凝胶电泳,25 V恒压、4℃、电泳过夜。再将凝胶中的DNA转至尼龙膜。预杂交:取10.0 mL DIG Easy Hyb,加入杂交管中,50℃杂交炉中预杂交2 h;排尽预杂交液,在10.0 mL DIG Easy Hyb加入新变性好的探针,混匀,50℃杂交仪中杂交过夜。然后洗膜,最后用凝胶成像系统扫描尼龙膜检测Northern blot杂交信号。
2 结果
2.1 穿梭载体pCY的构建及验证
以双元载体DK1317-2为骨架进行了三元穿梭载体的构建。首先通过内切酶Sac II对质粒DK1317-2酶切后进行胶回收。以GeneArt® pYES1L Vector为模板,PYES2117F、PYES2117R为引物扩增包括酵母能识别的复制起始位点的片段pYES1L-2117。采用In-Fusion HD Cloning Kit重组连接DK1317-2酶切后的骨架和片段pYES1L-2117(图1)。转化大肠杆菌,然后挑选单克隆进行测序验证,测序成功的穿梭载体命名为pCY。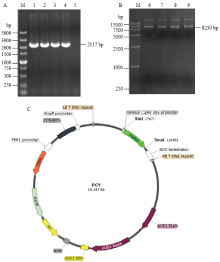 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图1穿梭载体pCY的构建
A:PCR扩增2117 bp 片段(1—4)PCR amplification of 2117 bp fragment (1-4);B:DK1317-2质粒的Sac II 酶切(6—9)DK1317-2 plasmid digested with Sac II (6-9);C:穿梭载体pCY质粒图谱Map of shuttle vector pCY。5:空白对照 Blank control ddH2O;M:DNA分子标准 DNA marker
-->Fig. 1Construction of shuttle vector pCY
-->
为测定穿梭载体的有效性,设计特异引物pCY- PVX-F、pCY-PVX-R扩增PVX全长。采用限制性内切酶StuⅠ、SmaⅠ酶切质粒pCY后胶回收。利用醋酸锂转化法将上述两个大片段进行重组(图2)。
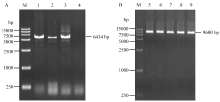 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图2PVX全长cDNA克隆及穿梭载体pCY StuⅠ、SmaⅠ酶切电泳
-->Fig. 2Electrophoresis of PVX full-length cDNA clone and shuttle vector pCY digested with Stu I and Sma I
-->
将验证为阳性的pCY-PVX质粒转化农杆菌C58C1并通过农杆菌注射浸润本生烟,接种10 d后的观察结果显示,注射pCY-PVX的烟草症状和阳性对照的完全一样,与阴性对照形成明显的对比(图3)。这说明PVX得到了高效表达,基于三元穿梭载体pCY的酵母重组克隆体系构建成功,可用于其他病毒的侵染性克隆构建。
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图3农杆菌介导pCY-PVX接种本生烟后的症状
-->Fig. 3Symptoms on N. benthamiana leaves by agro-mediated recombined plasmids of pCY-PVX
-->
2.2 柑橘叶片总RNA的提取及CLBV序列的分段扩增
从感病柑橘叶片中用Trizol 法提取出总RNA后,用1.5%普通琼脂糖凝胶上进行电泳检测。结果显示,所提RNA效果较好,28S、18S的rRNA带形完整,条带明显,表明所提取植物总RNA可用于后续试验。以cDNA第一链为模版,利用两对特异引物分段扩增CLBV序列,获得的大片段CLBV1、CLBV2,大小分别为4 500和4 247 bp,符合预期大小并覆盖CLBV全长。2.3 CLBV全长cDNA的重组克隆及序列分析
切胶回收的CLBV1、CLBV2、酶切后的pCY载体共转化酵母菌YPH501(图4),并挑取单克隆菌落,利用特异引物CLBV1F、CLBV5R进行菌落PCR鉴定。结果表明,挑取的24个菌落中22个为PCR阳性。然后分片段PCR鉴定是否为全长cDNA,结果16个重组克隆包含CLBV全长cDNA,阳性率为71.4%。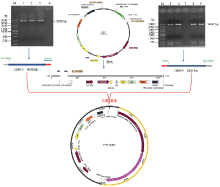 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图4利用酵母同源重组系统构建CLBV全长序列策略
-->Fig. 4Construction strategy of the full-length sequence of CLBV by yeast homologous recombination system
-->
随机选1个全长cDNA测序,测序结果表明CLBV全长为8 747 nt,包含3个开放阅读框,ORF1为5 889 nt、 ORF2为1 089 nt、ORF3为1 092 nt,5′末端有甲基化帽子结构,3′末端具有poly(A)。提交GenBank后获得登录号MG572236。序列比对结果显示,该序列与GenBank已登录的其他CLBV全长序列的核苷酸一致性为79%—98%,其中与柑橘来源的EU857540一致性最高,与猕猴桃来源的JN983454、JN983455、JN983456和JN900477的一致性均为79%。利用MEGA6软件构建系统发育树(图5),可见MG572236与柑橘来源的CLBV分离株聚为一簇。
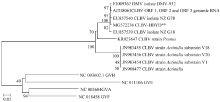 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图5CLBV全基因组核苷酸的系统进化树
-->Fig. 5Phylogenetic tree of whole genome sequences of CLBV isolates
-->
2.4 CLBV克隆的侵染性分析
将获得的CLBV全长cDNA克隆转化农杆菌,以pCY空载体为阴性对照注射接种本生烟。接种20 d后,抽提RNA进行RT-PCR检测,结果表明pCY-CLBV 1、2、3、5、8、9、12、13、14、15、16接种植株检测出CLBV特异性条带(图6)。说明CLBV侵染性克隆构建成功。 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图6CLBV侵染本生烟的RT-PCR检测
M:DNA分子标准DNA marker;1:ddH2O;2:阴性对照 Negative control;3—18:pCY-CLBV 1-16接种本生烟 N. benthamiana inoculated by pCY-CLBV 1-16;19:阳性对照 Positive control
-->Fig. 6Detection of CLBV in new leaves of N. benthamiana by RT-PCR
-->
有报道表明,本生烟接种CLBV并不表现明显症状。为进一步确定所构建克隆的侵染性,将RT-PCR检测阳性的本生烟样本随机选取5个进行Northern blot杂交验证,结果显示pCY-CLBV 1、2、3、14、15接本生烟可以检测到CLBV特异性条带,而对照样本未检测到任何条带(图7),表明pCY-CLBV 1、2、3、14、15为侵染性克隆。
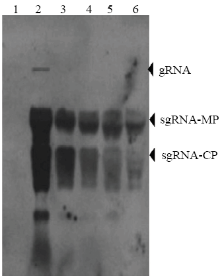 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图7Northern blot分析CLBV基因组RNA
1:健康对照Healthy control;2—6:农杆菌介导的pCY-CLBV 1、2、3、14、15侵染本生烟样品 N. benthamiana agroinfiltrated by pCY-CLBV 1, 2, 3, 14, 15
-->Fig. 7Northern blot analysis of the CLBV genome RNA
-->
此外,随机选取5个侵染本生烟呈阳性的单克隆采用真空浸润法接种锦橙幼苗。接种40 d后,利用RT-PCR均可以在新发叶片中检测到CLBV特异性条带,这进一步确认了所构建CLBV克隆的侵染性。
3 讨论
病毒与寄主的相互关系通常是病毒学领域研究的重要课题,而该领域最有利的研究工具是病毒侵染性克隆的构建。在以前的研究报道中,利用大肠杆菌构建较大基因组RNA病毒全长cDNA侵染性克隆时,由于其自身编码的病毒蛋白会对宿主菌产生毒性作用,从而导致非特异性重组,出现不稳定现象[19,20]。这成为病毒侵染性克隆构建面临的最大困难和挑战,也成为开展病毒深入研究工作的限制因素。病毒全长基因组克隆在大肠杆菌中存在的不稳定现象的机制仍不清楚。通常利用以下几个方法解决:(1)将病毒序列分段克隆,侵染前再短暂的连接[21];(2)利用拷贝数目较低的载体[22];(3)调控细菌生长的环境(如降低培养温度等)来减少毒性[23];(4)利用插入内含子到病毒全基因组序列中来避免产生具有毒性的蛋白[24]。但这些方法均耗时耗力且成功率低。酵母体内同源重组是一种利用酵母细胞内高效的同源重组系统来实现多个相互存在同源序列DNA片段的组装方法。本研究为了克服病毒基因组序列克隆在E. coli中的不稳定性,首先通过In-Fusion HD Cloning Kit构建三元穿梭载体pCY,然后利用PVX侵染性克隆建立酵母重组克隆体系;省去了转化大肠杆菌的步骤,具有快速、便捷、成本低廉的优点。本研究在利用酵母同源重组系统构建CLBV的侵染性克隆时发现,通过酵母重组获得的CLBV全长cDNA克隆的效率可以达到70%以上。YOUSSEF等[16]在构建苹果褪绿叶斑病毒(Apple chlorotic leaf spot virus,ACLSV)时从36个经限制性内切酶酶切正确的大肠杆菌克隆中仅鉴定出1个有侵染性的克隆,表明ACLSV克隆在大肠杆菌细胞同样存在着不稳定性;庹德财等[25]在构建番木瓜畸形花叶病毒(Papaya leaf distortion mosaic virus,PLDMV)时,在酵母中完成拼接后转化大肠杆菌仍然存在不稳定现象,而当在PLDMV的P3基因中插入内含子时,经酵母重组后转化大肠杆菌能获得测序正确的克隆。产生此现象的原因可能是不同的病毒在大肠杆菌中的稳定性存在一定差异。因此,本研究为了克服病毒基因组序列克隆在大肠杆菌中的不稳定性,采用避免转化大肠杆菌细胞的策略,将同源重组获得的酵母质粒直接转化农杆菌,快速、便捷地构建稳定的适用于农杆菌注射接种的CLBV全长cDNA侵染性克隆。
利用酵母同源重组系统SHANG等[26]首次完成了苜蓿银纹夜蛾核型多角体杆状病毒(Autographa californica nucleopolyhedrovirus,AcMNPV)全长基因组145 kb的侵染性克隆;NIKIFORUK等[27]通过同源重组快速一步建立了中东呼吸系统综合症冠状病毒(Middle East Respiratory Syndrome-coronavirus,MERS-CoV)约32 kb全长基因组的侵染性克隆系统;赵光远等[28]也完成了番木瓜环斑病毒(Papaya ringspot virus,PRSV)约10 kb的侵染性克隆。对于本研究CLBV侵染性克隆构建成功更证实了酵母同源重组对于在侵染性克隆上应用的可行性。目前,酵母同源重组已在多个领域应用,如构建酿酒酵母新型表达质粒[29]、制备DNA大片段[30]、基因敲除等,相信酵母同源重组系统在将来会有更广阔的应用前景,在分子生物学研究中发挥越来越重要的作用。
4 结论
成功构建了一个三元穿梭载体pCY,该载体可在酵母细胞中通过同源重组快速克隆病毒全长cDNA,经农杆菌介导可直接接种寄主植物,还可在大肠杆菌中复制扩增;利用基于该载体的重组克隆体系,获得了中国CLBV分离株的适于农杆菌介导接种的基因组全长cDNA侵染性克隆。致谢:法国波尔多大学Thierry Candresse教授惠赠酵母菌株YPH501、农杆菌菌株C58C1;海南大学庹德财博士对试验进行了技术指导。在此一并表示感谢!
The authors have declared that no competing interests exist.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}