 ,1, 盛子璐1, 解振强2, 黄雨晴1, 巩培杰1, 张川1, 郑婷1, 王晨,1, 房经贵1
,1, 盛子璐1, 解振强2, 黄雨晴1, 巩培杰1, 张川1, 郑婷1, 王晨,1, 房经贵1Function Analysis of vvi-miR172s and Their Target Genes Response to Gibberellin Regulation of Grape Berry Development
XUAN XuXian,1, SHENG ZiLu1, XIE ZhenQiang2, HUANG YuQing1, GONG PeiJie1, ZHANG Chuan1, ZHENG Ting1, WANG Chen,1, FANG JingGui1通讯作者:
责任编辑: 赵伶俐
收稿日期:2020-06-1接受日期:2020-11-19网络出版日期:2021-03-16
| 基金资助: |
Received:2020-06-1Accepted:2020-11-19Online:2021-03-16
作者简介 About authors
宣旭娴,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (4962KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
宣旭娴, 盛子璐, 解振强, 黄雨晴, 巩培杰, 张川, 郑婷, 王晨, 房经贵. vvi-miR172s及其靶基因响应赤霉素调控葡萄果实发育的作用分析[J]. 中国农业科学, 2021, 54(6): 1199-1217 doi:10.3864/j.issn.0578-1752.2021.06.011
XUAN XuXian, SHENG ZiLu, XIE ZhenQiang, HUANG YuQing, GONG PeiJie, ZHANG Chuan, ZHENG Ting, WANG Chen, FANG JingGui.
开放科学(资源服务)标识码(OSID):

0 引言
【研究意义】葡萄(Vitis vinifera L.)是世界四大水果之一,在我国广泛栽培。生产上,通常利用GA3调控葡萄花果发育,提高果实品质从而增加其经济效益。miR172是一个重要的保守miRNA家族,其家族成员的功能具有多样性,在植物成花作用[1]、种胚发育[2]以及非生物胁迫的响应[3]等方面发挥重要作用。近期研究发现,miR172可能响应GA参与葡萄花果发育的调控[4],但其对葡萄果实不同组织的调控作用尚不清楚。从miR172角度研究GA调控葡萄果实发育的作用机制,为深入认识vvi-miR172s的功能及其应答GA的分子调控机制提供重要信息。【前人研究进展】miR172广泛存在于植物中,不同物种中具有多个MIR172,可以在不同的组织和不同的发育阶段形成保守的成熟miR172序列。模式植物拟南芥中5个MIRNA172加工后形成3个miR172成熟体[5];杨树有9个MIRNA172和4个miR172成员[6];水稻含4个MIRNA172和2个miR172[7];油菜含有4个miR172成员[1];玉米中仅包括一个miR172成员[8]。拟南芥中miR172的过表达会导致非正常花的发育,还会使植物提早开花[9];miR172对拟南芥果实(长角果)的生长具有促进作用;miR172的过表达会使苹果果实显著变小[10];麻疯树种JcmiR172a的过表达增强木质部的发育,减少韧皮部的发育[7];据报道,miR172调控小麦穗型和脱粒率研究中发现miR172的过表达会诱导穗的加长和降低脱粒率[10]。另有研究发现,OsmiR172b是调控水稻种子发育表型最显著的成员[2],对种子胚发育具有负调节作用。这些结果表明,miR172家族在不同植物中的功能具有多样性。成熟miR172可以通过序列互补靶向AP2基因家族的mRNA,并且可以通过抑制翻译或引发靶mRNA的降解来抑制靶基因的表达。目前,在拟南芥[5]、水稻[3]、油菜[1]等植物中均发现AP2家族转录因子是miR172的靶基因。miR172及其靶基因在开花时间和花器官分化中起关键作用。在拟南芥中,除TOE3外的所有miR172靶基因过量表达均延迟开花[11,12,13]。相反,花椰菜中toe1toe2双重突变体,toe1toe2smzsnz四重突变体和toe1toe2toe3smzsnzapap六倍体突变体均导致提早开花,且开花时间随突变基因数量的增加而延长[14,15]。另有研究发现AP2类转录因子还参与番茄果实的转色[16]、成熟[17]及类胡萝卜素[18]积累过程,且拟南芥中AtERF055、AtAP2[19]可调控种子的发育。此外,AP2/ERF类转录因子还被视为植物激素信号连接的关键调节器,可参与赤霉素、生长素、细胞分裂素、茉莉酸、乙烯、脱落酸等多种激素信号转导途径[20]。【本研究切入点】赤霉素(GA)是调控葡萄果实发育如无核膨大等的关键生长调节剂,但其相关分子机制尚不甚清晰[21]。如上所述,miR172和AP2的研究主要集中在植物开花时间和花器官的发育方面。在拟南芥[22,23]和大豆[24]的研究中已证明AP2类转录因子与种子发育相关,但其参与果皮、果肉发育的研究甚少,更未见miR172通过应答GA靶向AP2类转录因子参与葡萄果皮、果肉等膨大发育关键组织调控的报道。【拟解决的关键问题】本试验以miRBase数据库为基础,鉴定葡萄中miR172c家族成员及其靶基因,系统分析miR172成员及其靶基因的染色体定位、进化关系、基因结构、保守基序和上游顺式作用元件;采用实时荧光定量PCR(qRT-PCR)技术,结合芯片数据,明确vvi-miR172成员及其靶基因应答GA3在葡萄果实不同组织中的表达特性,为深入研究其在果皮、果肉组织发育过程中的调控机制提供重要参考。1 材料与方法
试验于2017年在江苏省句容农博园葡萄基地进行。1.1 材料与处理
本试验以6年生优质‘白罗莎里奥’葡萄为试验材料,于盛花前10 d用50 mg·L-1的GA3浸蘸花穗30 s,以清水处理为对照。每组处理随机选定12株长势较为一致的植株,分别于幼果期(花后10 d,10 DAF)、硬核期(35 DAF)、第2次膨大期(60 DAF)、近成熟期(85 DAF)随机采集生长较为一致的葡萄果实,将果皮、果肉分离,用液氮速冻并保存于-80℃冰箱中。1.2 葡萄miR172s成熟体序列及前体序列的克隆鉴定
在miRBase数据库(http://www.mirbase.org/)搜索并下载葡萄miR172s(vvi-miR172a/b/c/d)前体及成熟体序列。基于成熟体序列,参考WANG等[25]的方法,设计特异性引物,利用miR-RACE技术在‘白罗莎里奥’果实组织中克隆vvi-miR172a/b/c/d的成熟体序列,测序后鉴定其精确序列。在miRBase数据库下载水稻(Oryza sativa Japonica)、玉米(Zea mays)、拟南芥(Arabidopsis thaliana)、大豆(Glycine max)、杨树(Populus trichocarpa)、烟草(Nicotiana tabacum)、马铃薯(Solanum tuberosum)和番茄(Solanum lycopersicum)miR172家族的成熟体序列,与葡萄miR172s(vvi-miR172a/b/c/d)进行序列比对。利用下载的vvi-miR172a/b/c/d前体序列,分别向上、向下扩展约200 bp,预测葡萄miR172a/b/c/d的前体基因序列,并设计特异性引物进行PCR扩增(表1)。反应体系为50 μL:上、下游引物各2 μL,cDNA 2 μL,10×PCR buffer(Mg2+ plus)5 μL,dNTP Mixture 4 μL,Ex-Taq酶0.50 μL,ddH2O 34.5 μL。反应程序:95℃ 5 min;95℃ 30 s,59℃ 30 s,72℃ 30 s,35个循环;72℃ 10 min。扩增产物用1.5%的琼脂糖凝胶电泳检测正确后回收目的片段,连接至pMD19T载体进行TA克隆,克隆后的纯化产物由上海生工生物工程股份有限公司进行测序。Table 1
表1
表1引物序列及用途
Table 1
| 引物名称 Primer name | 引物序列 Primer sequence | 引物用途 Primer use |
|---|---|---|
| vvi-miR172a-F | AAGCTTTCGTAGTTGTCTACAAAATGATCTCT | vvi-miR172a前体序列的扩增 Amplification of vvi-miR172a precursor |
| vvi-miR172a-R | GGATCCCATCGCTTTACATTTTAGCCACT | |
| vvi-miR172b-F | AAGCTTCGGCATCTTTCTCCTCATACC | vvi-miR172b前体序列的扩增 Amplification of vvi-miR172b precursor |
| vvi-miR172b-R | GGATCCGGGTTGAAGACGTACATAAATCTCA | |
| vvi-miR172c-F | AAGCTTTTTGAAGCGTTCTGCTGTC | vvi-miR172c前体序列的扩增 Amplification of vvi-miR172c precursor |
| vvi-miR172c-R | GGATCCCAGATGAAACACTTCCAAGAGA | |
| vvi-miR172d-F | AAGCTTGAAGCGAGATCCGTATCCTC | vvi-miR172d前体序列的扩增 Amplification of vvi-miR172d precursor |
| vvi-miR172d-F | GGATCCATCTCAAATTACTGCATGATTTCAA | |
| Actin-F | TACAATTCCATCATGAAGTGTGATG | Actin内参引物 Actin internal reference primer |
| Actin-R | TTAGAAGCACTTCCTGTGAACAATG | |
| VvAP2 qRT-F | CTCGCGCATATGATAGGGCT | VvAP2定量RT-PCR引物 Real-time PCR of VvAP2 |
| VvAP2 qRT-R | ACTTTGTCGGCGAAGGACAT | |
| VvRAP2-1 qRT-F | CAACACTGGGGGTTCATCCA | VvRAP2-1定量RT-PCR引物 Real-time PCR of VvRAP2-1 |
| VvRAP2-1 qRT-R | TTTTGCCATGCCCAGTTTGG | |
| VvRAP2-2 qRT-F | CTGACTACCGTGCAAGGGTT | VvRAP2-2定量RT-PCR引物 Real-time PCR of VvRAP-22 |
| VvRAP2-2 qRT-R | TTTGATCACCCTGGCCTAGC | |
| VvRAP2-3 qRT-F | GGGCTAGCAATGACATCGGA | VvRAP2-3定量RT-PCR引物 Real-time PCR of VvRAP2-3 |
| VvRAP2-3 qRT-R | CATTTGCCACCCCCAGTTTG |
新窗口打开|下载CSV
1.3 葡萄miR172s靶基因预测及生物信息学分析
基于鉴定的vvi-miR172a/b/c/d成熟体序列,由在线软件psRNA Target(1.4 葡萄miR172s及其靶基因启动子作用元件分析
利用葡萄基因组数据库CRIBI(1.5 芯片数据分析
从NCBI Genesis Expression Omnibus(GEO)下载GSE36128系列(1.6 vvi-miR172家族成员及其靶基因的表达特性分析
采用CTAB法提取4个不同发育时期葡萄果皮、果肉中的RNA,利用Primer 3 Input(http://bioinfo.ut. ee/primer3-0.4.0/)设计引物(表1),以反转录后的cDNA为模板,以Actin为内参基因,对vvi-miR172s前体基因及4个靶基因进行qRT-PCR检测。扩增体系及程序参考TaKaRa公司的SYBR? Premix Ex TaqTM Ⅱ试剂盒说明书,设置3次生物学重复。采用2-ΔΔCt计算各基因的相对表达量,采用SPSS软件对数据进行统计及差异显著性分析(P<0.05)。2 结果
2.1 赤霉素处理对‘白罗莎里奥’葡萄果实生长发育的影响
GA3处理后,果实横径无明显变化,果实纵径在花后10、35及60 d显著增长,且各时期没有明显的果核出现,仅在种子区域留下一条木质化的细线;而未经GA3处理的对照组种子正常发育,在花后35 d可观察到果核,说明GA3处理可以高效诱导‘白罗莎里奥’葡萄产生无核果实。另外,也观察到外源GA3处理诱导葡萄无核果实的同时,促进了无籽果实的膨大,使其发育成正常商品果大小,甚至更大(图1),表明GA3在果实发育过程中起着重要调控作用。图1
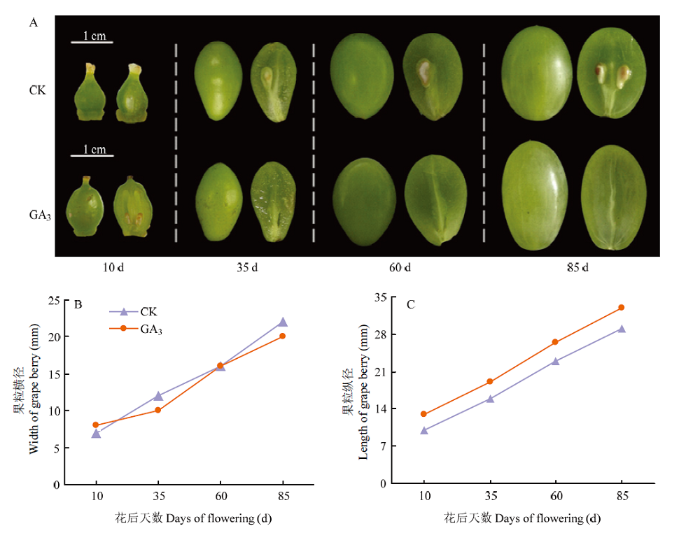 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1GA3和CK处理下‘白罗莎里奥’葡萄果实生长发育情况
Fig. 1Growth and development of White Rosario grape under GA3 and CK treatments
2.2 vvi-miR172家族成员成熟体及前体序列的克隆鉴定
为确定vvi-miR172家族成员在‘白罗莎里奥’葡萄中的真实序列,对其成熟体序列和前体序列进行克隆鉴定。通过miR-RACE获得vvi-miR172a/b/c/d的成熟体序列,克隆所得的序列与miRBase中的同源序列一致。克隆鉴定的vvi-miR172a/b/c/d前体基因分别为507、495、546和507 bp(图2-A)。染色体定位结果显示vvi-miR172家族成员分布在葡萄3条染色体上,vvi-miR172b和vvi-miR172c位于Chr13上,而vvi-miR172a和vvi-miR172d分别位于Chr6和Chr8上(图2-B)。4种miRNA前体结构显示出一定差异,但均可形成稳定的茎环结构,且成熟序列均位于茎环结构的3′臂上(图2-C),表明vvi-miR172a/b/c/d在前体基因上的位置高度保守。多序列比对结果显示,除vvi- miR172d含23个碱基外,vvi-miR172a、vvi-miR172b以及vvi-miR172c均为21个碱基,且vvi-miR172a和vvi-miR172b仅存在1个碱基的差异(图2-D)。vvi-miR172四个成熟体序列存在相对保守的区域,介于4—19 bp和21—22 bp,表明miR172成熟体高度保守。vvi-miR172c与ptr-miR172g和sly-miR172d的成熟体序列相同,表明其可能具有相似的生物学功能。图2
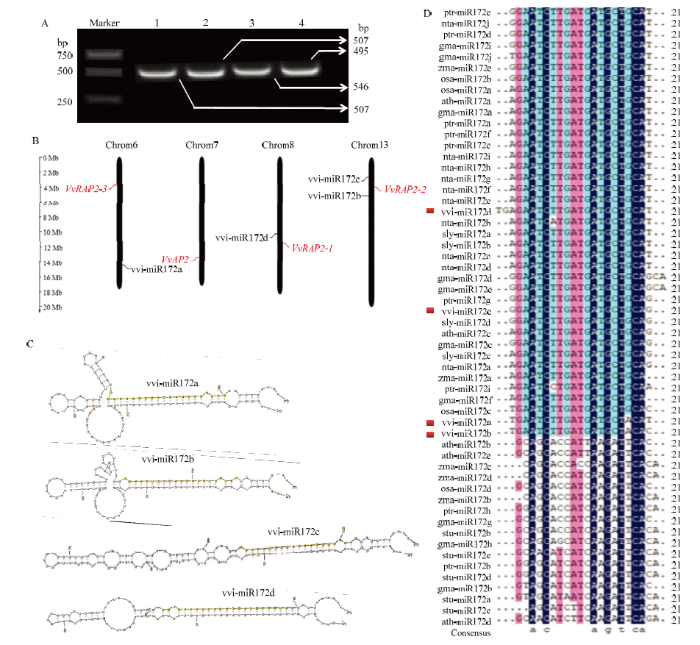 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2vvi-miR172a/b/c/d前体序列的克隆鉴定及不同物种miR172成熟体序列比对
A:vvi-miR172a/b/c/d 前体基因的PCR克隆产物;1:vvi-miR172a;2:vvi-miR172b;3:vvi-miR172c;4:vvi-miR172d;B:vvi-miR172s及其靶基因在染色体上的定位;C:vvi-miR172s前体的二级茎环结构;D:不同物种miR172成熟体序列比对
Fig. 2Identification of vvi-miR172a/b/c/d precursor sequences and mature sequences alignment of miR172 in different species
A: PCR products of vvi-miR172a/b/c/d primary sequences; 1: vvi-miR172a; 2: vvi-miR172b; 3: vvi-miR172c; 4: vvi-miR172d. B: Chromosomal location of vvi-miR172s and their targets genes; C: Hairpin structure of vvi-miR172s; D: Mature sequences alignment of miR172 in different species
2.3 vvi-miR172s靶基因的鉴定
2.3.1 vvi-miR172s靶基因的预测及匹配程度分析 根据所获得的vvi-miR172a/b/c/d成熟体序列,利用在线软件psRNA Target预测到4条靶基因VvAP2、VvRAP2-1、VvRAP2-2和VvRAP2-3,分别分布于葡萄基因组的4条染色体上(Chr7、Chr8、Chr13、Chr6)(图2-B),分析发现vvi-miR172a/b/c/d的靶区都在其靶基因的CDS区,且均通过裂解作用于靶基因(表2)。对vvi-miR172a/b/c/d与其靶基因的匹配程度进一步分析(图3),发现vvi-miR172a、vvi-miR172b与预测所得靶基因的匹配程度相同,错配率均为2.5;vvi-miR172c与VvRAP2-2和VvAP2的匹配程度最高,错配率为0.5,与VvRAP2-1和VvRAP2-3的错配率为1.0;vvi-miR172d与VvRAP2-2的错配率最高为2.5,与VvRAP2-3、VvRAP2-1、VvAP2的错配率分别为0.5、1.5和2.0(表2),说明vvi-miR172家族成员与靶基因间的作用强度存在差异。Table 2
表2
表2葡萄vvi-miR172s及靶基因信息
Table 2
| miRNAs | MiRNA成熟 体长度 Length of miRNA | 靶基因编号 Target_Acc. | 靶基因长度 Length of target | 靶区 Target regions | 靶区起始、 终止位点 Target start and stop sites | 错配率 Expectation | 允许不匹配 的最大能量 UPE | 切割方式 Interaction mode |
|---|---|---|---|---|---|---|---|---|
| vvi-miR172a/b | 21 | VIT_07s0031g00220(VvAP2) | 2565 | CDS:1870-1890 | C、C | 2.5 | 15.05 | 裂解Cleavage |
| 21 | VIT_08s0040g03180(VvRAP2-1) | 1730 | CDS:1331-1351 | C、T | 2.5 | 11.489 | 裂解Cleavage | |
| 21 | VIT_13s0019g03550(VvRAP2-2) | 2217 | CDS:1566-1586 | C、C | 2.5 | 17.172 | 裂解Cleavage | |
| 21 | VIT_06s0004g03590(VvRAP2-3) | 2347 | CDS:1806-1826 | C、T | 2.5 | 14.04 | 裂解Cleavage | |
| vvi-miR172c | 21 | VIT_07s0031g00220(VvAP2) | 2565 | CDS:1870-1890 | C、C | 0.5 | 15.05 | 裂解Cleavage |
| 21 | VIT_08s0040g03180(VvRAP2-1) | 1730 | CDS:1331-1351 | C、T | 1.0 | 11.489 | 裂解Cleavage | |
| 21 | VIT_13s0019g03550(VvRAP2-2) | 2217 | CDS:1566-1586 | C、C | 0.5 | 17.172 | 裂解Cleavage | |
| 21 | VIT_06s0004g03590(VvRAP2-3) | 2347 | CDS:1806-1826 | C、T | 1.0 | 14.04 | 裂解Cleavage | |
| vvi-miR172d | 23 | VIT_07s0031g00220(VvAP2) | 2565 | CDS:1870-1892 | C、A | 2.0 | 15.552 | 裂解Cleavage |
| 23 | VIT_08s0040g03180(VvRAP2-1) | 1730 | CDS:1331-1353 | C、G | 1.5 | 9.197 | 裂解Cleavage | |
| 23 | VIT_13s0019g03550(VvRAP2-2) | 2217 | CSS:1566-1588 | G、A | 2.5 | 22.9 | 裂解Cleavage | |
| 23 | VIT_06s0004g03590(VvRAP2-3) | 2347 | CDS:1806-1828 | C、A | 0.5 | 14.075 | 裂解Cleavage |
新窗口打开|下载CSV
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3vvi-miR172s与靶基因的匹配程度
Fig. 3Match degree of vvi-miR172s and their target genes
2.3.2 vvi-miR172s靶基因验证 利用RLM-RACE技术对vvi-miR172s剪切靶基因的作用位点进行验证,发现预测的4条靶基因(VvAP2、VvRAP2-1、VvRAP2-2和VvRAP2-3)均被验证到裂解产物,表明它们为vvi- miR172s的真实靶基因(图4);同时发现vvi-miR172s在VvAP2、VvRAP2-1、VvRAP2-2和VvRAP2-3上均只有一个酶切位点,4个靶基因的裂解位点位于vvi- miR172a/b 5′端的第9位碱基(U)和第10位碱基(A)之间,vvi-miR172c/d 5′端的第10位碱基(U)和第11位碱基(A)之间,表明miRNA切割位点的保守性。此外,VvRAP2-1切割频率最低,表明其与vvi-miR172a/ b/c/d间的作用强度较弱(图4)。
图4
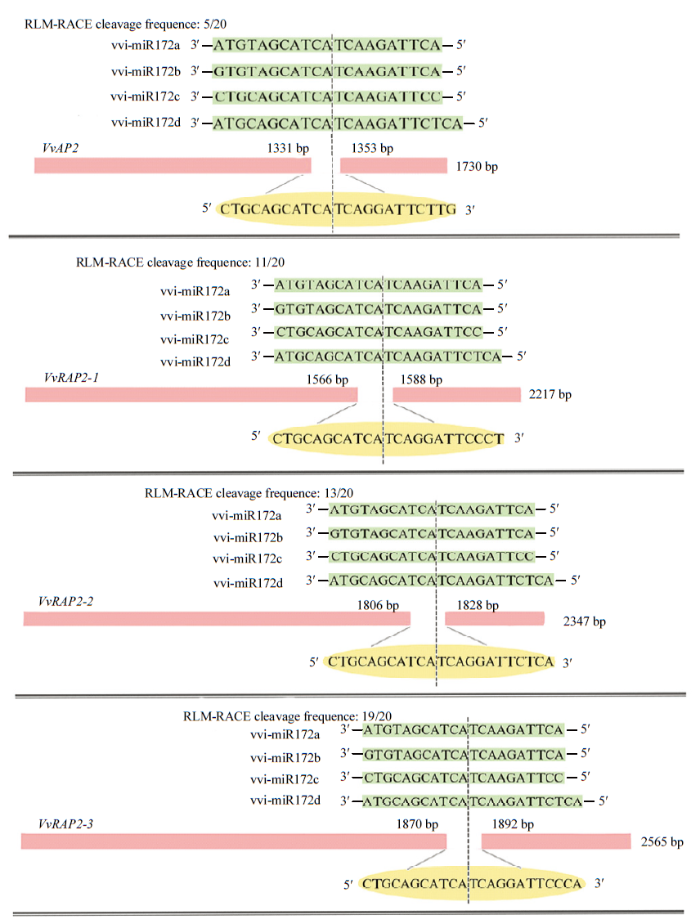 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4VvmiRNA172s剪切靶基因mRNA的切割位点
虚线代表裂解位点;数字代表mRNA片段5′末端克隆的频率
Fig. 4The cleavage site between vvi-miR172s and target genes
The dotted lines represent the cleavage sites; numbers indicate the frequency of clones at 5 termini of mRNA fragments
2.3.3 靶基因序列结构及其进化保守性分析 基于预测的4条靶基因cDNA编码的氨基酸序列,利用MEGA7.0软件对同源比对结果进行进化分析(图5-A),发现比对结果可分为3组,其中水稻OsRAP2成员和玉米ZmRAP2成员分别聚集为一组;VvRAP2-1、VvRAP2-2和VvRAP2-3属于同一组,而VvAP2在另一组。且VvAP2、VvRAP2-1和VvRAP2-2与杨树的亲缘关系较近,VvRAP2-3与大豆具有较高的同源性。利用在线软件GSDS对靶基因序列结构进行分析(图5-B),发现4个靶基因所含的内含子和外显子数量相同,均含有10个外显子和9个内含子。其中,VvAP2与其他3个靶基因的外显子长度相似,但内含子长度不同。通过MEME在线软件预测motif元件(图5-C),发现各物种内蛋白基序存在一定差异,但各类元件的种类和数量较为稳定,表明AP2及RAP2具有一定的功能保守性。
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5靶基因序列结构及其进化保守性分析
A:靶基因的系统进化;B:靶基因基因结构;C:靶基因保守基序
Fig. 5Phylogenetic relationship and gene structure and conserved motifs analysis of target genes
A: Phylogeny tree of target genes; B: Gene structure of target genes; C: The conserved motifs of target genes
2.3.4 靶蛋白的结构分析与亚细胞定位预测 利用在线软件PRABI对vvi-miR172s的靶蛋白进行二级结构预测,发现四者二级结构的表现形式均为α-螺旋(图6-A)。利用SWISS-MODEL进行三维结构同源建模,结果表明其蛋白三维空间排列所形成的蛋白质分子构象具有多样性[26],其中VvAP2、VvRAP2-1和VvRAP2-3的蛋白构象更相似(图6-B),表明其可能具有功能的相似性。利用在线软件CDD对靶基因蛋白及其同源序列进行蛋白结构域预测,发现各序列均含有两个排列顺序相近的AP2蛋白结构域(图6-C),由位置和数量相似的氨基酸编码。对靶蛋白及同源序列进行亚细胞定位预测(图6-D),发现4条靶蛋白均位于细胞核中,而GmRAP2-1和GmRAP2-2编码的蛋白还存在于线粒体中。
图6
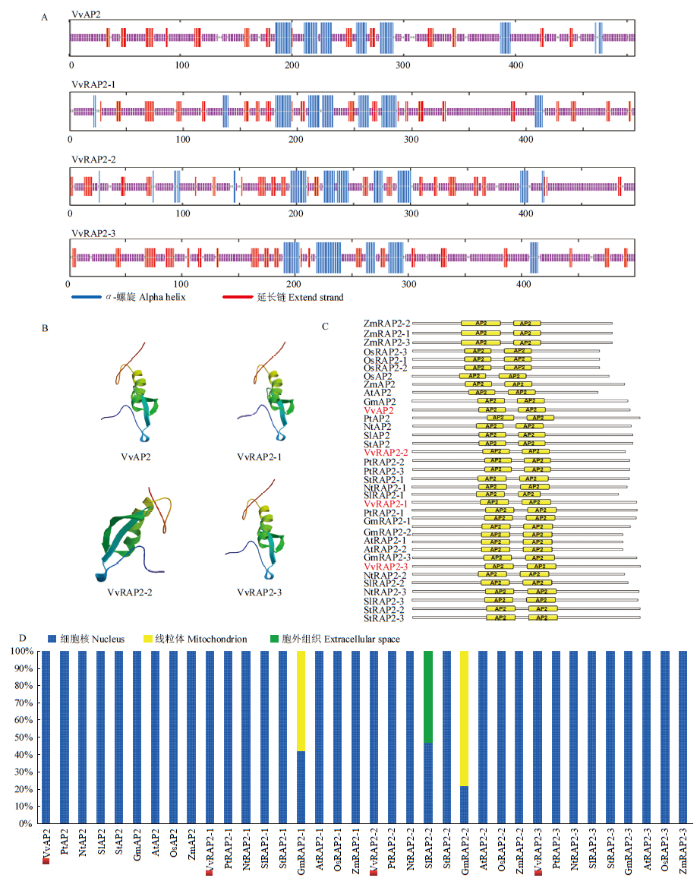 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6靶蛋白结构分析及亚细胞定位预测
A:靶蛋白的二级结构预测;B:靶蛋白的三级结构预测;C:靶蛋白保守结构域分析;D:靶蛋白的亚细胞定位预测
Fig. 6Structure analysis and subcellular localization prediction of target proteins
A: The secondary structure prediction of target proteins; B: The tertiary structure prediction of target proteins; C: The conserved domains prediction of target proteins; D: The subcellular localization prediction of target proteins
2.4 vvi-miR172s及其靶基因潜在功能分析
2.4.1 vvi-miR172s前体及靶基因的启动子作用元件分析 通过分析vvi-miR172s前体基因及其靶基因启动子作用元件,发现其作用元件大致可分为4类:光信号响应(Gap-box、Box4、GAT-motif等)、激素相关响应(ABRE、TGACG-motif、TATC-box等)、胁迫相关响应(MBS、ARE、GC-motif等)和昼夜周期节律(circadian等)相关元件。葡萄vvi-miR172家族成员及靶基因中光信号响应元件数量最多,作用位点最丰富;激素和胁迫相关响应作用元件数量次之;昼夜周期节律相关元件数量最少(图7-A)。此外,vvi-miR172s及其靶基因启动子作用元件的数量和类型也各不相同。在元件数量上,VvRAP2-2最多,VvAP2次之,而vvi-miR172a最少;在作用元件类型上,除VvRAP2-3含有昼夜周期节律相关元件外,其他基因只含有3种作用元件(图7-A)。为进一步认识vvi-miR172s及其靶基因应答激素反应的潜在机制,分析发现所有vvi-miR172家族成员及靶基因均含有激素响应作用元件,主要包括赤霉素(gibberellin,GA)、生长素(auxin,IAA)、水杨酸(salicylicacid,SA)、脱落酸(abscisic acid,ABA)及茉莉酸甲酯(methyljasmonate,MeJA)响应元件(图7-B)。但与激素相关的顺式作用元件的组成和数量在不同的vvi-miR172成员及其靶基因间均表现出一定的差异,其中,GA响应元件仅存在于VvRAP2-1和vvi-miR172c中;IAA响应元件仅存在于VvAP2和VvRAP2-3中;ABA响应元件存在于4个靶基因中;MeJA和SA响应元件分布较为广泛(图7-B)。上述作用元件分析表明vvi-miR172s及其靶基因不仅能响应光、胁迫和昼夜周期节律等外界环境信号,还可能通过参与不同激素信号途径调控果实的生长发育。图7
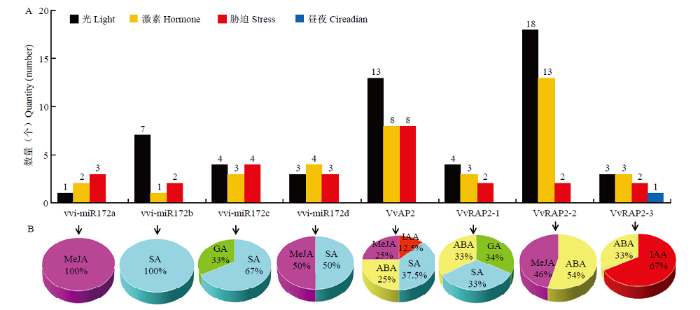 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7vvi-miR172s及其靶基因的顺式作用元件
A:vvi-miR172s及其靶基因启动子不同类型顺式作用元件汇总;B:vvi-miR172s及其靶基因激素相关顺式作用元件
Fig. 7Predicted cis-elements in the promoter of vvi-miR172s and their target genes
A: The total number of diverse types of cis-elements derived from vvi-miR172s and their target genes promoters; B: Hormone-related cis-elements of vvi-miR172s and their target genes
2.4.2 vvi-miR172s及靶基因在葡萄不同器官组织中的时空表达模式分析 为认识vvi-miR172家族及靶基因在葡萄不同器官不同组织的时空表达特征,本研究基于葡萄54个器官组织的转录组数据[27]和70个miRNA测序数据[28]对vvi-miR172s及靶基因的表达模式进行分析,发现vvi-miR172c及靶基因在13个不同组织或器官中均有表达。vvi-miR172c在花器官(心皮、花粉、雄蕊和花序)中的表达水平相对较高,在运输器官(穗轴和茎)和生殖器官(果实和种子)的表达相对较低;相比之下,靶基因VvAP2在所有组织中具有较高的表达水平,VvRAP2-1和VvRAP2-3在不同组织中的表达水平较低(图8-A)。进一步分析vvi-miR172c及其靶基因在花、果实、茎、叶基本组织中的表达,发现vvi-miR172c在茎、叶和果实中低表达,而靶基因VvAP2和VvRAP2-2呈现相反的表达模式(图8-B),表明vvi-miR172家族可能主要通过vvi-miR172c介导靶基因VvAP2和VvRAP2-2调控葡萄茎、叶、果实的发育过程。
图8
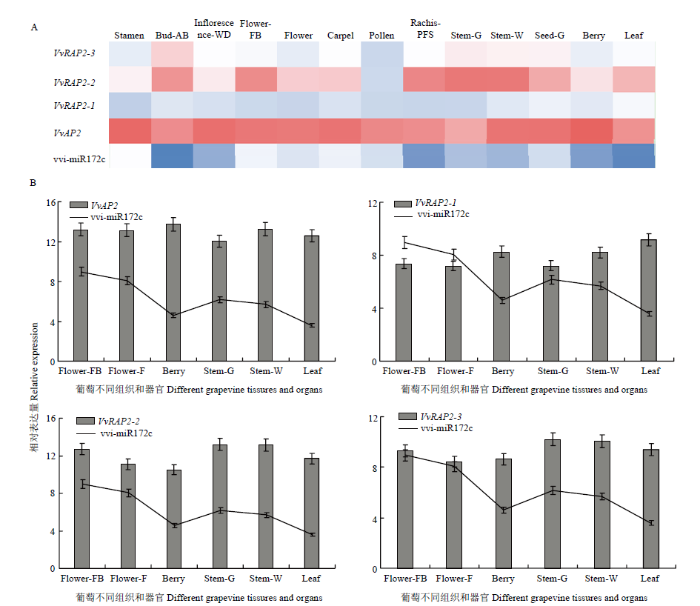 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8vvi-miR172s(GEO:GSE59802)及其靶基因(GEO:GSE36128)在葡萄不同器官、组织和发育阶段的表达水平
A:vvi-miR172c及其靶基因在葡萄不同器官、组织和发育阶段的表达谱;B:vvi-miR172c及其靶基因在葡萄不同组织的表达水平。Stamen:雄蕊;Bud-AB:萌发后的芽;Inflorescence-WD:发育良好的花序;Flower-FB:始花期;Flower-F:盛花期;Carpel:心皮;Pollen:花粉;Rachis-PFS:坐果后的穗轴;Stem-G:绿色的茎;Stem-W:木质化的茎;Seed-G:绿果的种子
Fig. 8Expression levels of vvi-miR172s (GEO:GSE59802) and target genes (GEO:GSE36128) in different orGA3ns, tissues and development stages
A: Expression profiles of vvi-miR172c and target genes in different orGA3ns, tissures and development satges; B: Expression of vvi-miR172c and target genes in different tissues. Bud-AB: Bud after brust; Inflorescence-WD: Well development inflorescence; Flower-FB: Flowering begins; Flower-F: Flowering; Rachis-PFS: Rachis post fruit set; Stem-G: Green stem; Stem-W: Woody stem; Seed-G: Seed collected from green berries
2.4.3 vvi-miR172s及靶基因在果实不同组织中的时空表达图谱分析 vvi-miR172a/b/d在其发育前期表达量高,在发育后期表达量低(图9-A),其中,vvi-miR172a和vvi-miR172d呈现出相似的表达模式,在幼果期(10 DAF)表达量最高,在近成熟期(85 DAF)表达量最低(图9-A),而vvi-miR172b在硬核期(35 DAF)果皮中有最高的表达,表明前两个成员可能与幼果期果皮发育相关,而后者可能与硬核期果皮发育相关性更强。而vvi-miR172c在果皮发育的各个阶段都表现出较低的表达水平(图9-A)。相比之下,靶基因VvRAP2-2在果皮发育中呈现幼果期较高表达,而近成熟期最高表达的趋势,与vvi-miR172a/b/d的表达趋势相反;VvAP2与vvi-miR172c呈相反的表达趋势,从幼果果皮到成熟期逐渐降低(图9-A)。这些结果表明,vvi-miR172s家族的不同成员可能分别参与葡萄果皮发育不同时期的调控。
图9
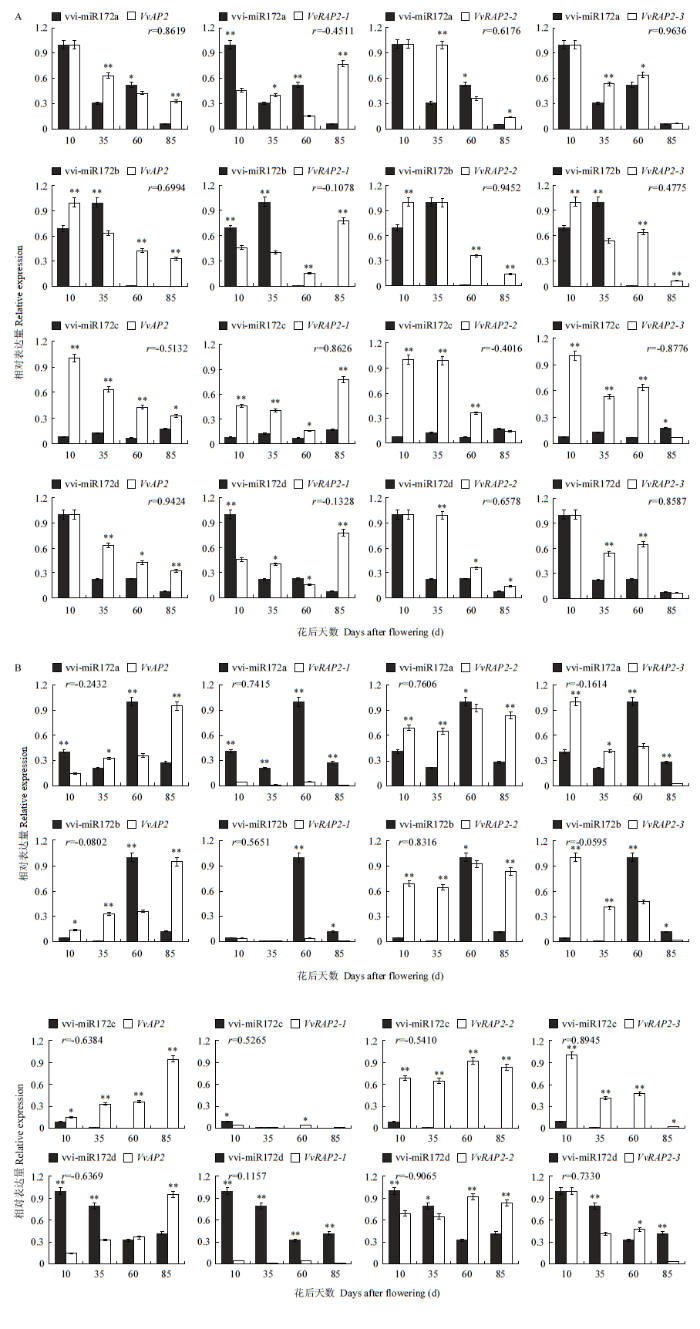 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图9vvi-miR172a/b/c/d及其靶基因在葡萄果皮(A)和果肉(B)中的表达水平
*、**分别代表差异显著(P<0.05)和极显著(P<0.01)。下同
Fig. 9Expression levels of vvi-miR172a/b/c/d and its target genes in berry pericarp (A) and flesh (B) of grape
*,** represented significant difference (P<0.05) and extremely significant difference (P<0.01), respectively. The same as below
如图9-B所示,与果皮中不同,果肉中vvi- miR172a/b具有相同的表达模式,均在果实第二次膨大期(60 DAF)表达水平最高,其他时期具有较低的表达;vvi-miR172d随着果肉发育逐渐降低,在幼果果肉中有最高表达,成熟期表达最低,而vvi- miR172c在整个果肉发育期的表达均较低。对比相关靶基因发现,Vv-RAP2-2在果肉不同发育时期均呈现较高表达,与vvi-miR172c呈相反表达趋势,而VvAP2在果肉发育过程中表达逐渐升高,相反于vvi-miR172d的表达趋势,表明vvi-miR172c/d可能负调控其靶基因介导葡萄果肉发育,而vvi-miR172a/b可能与葡萄果实膨大发育相关。
对比vvi-miR172s和靶基因的表达模式发现,葡萄果皮中,vvi-miR172a/b/d与靶基因VvRAP2-1呈较显著的负相关,与其他靶基因的相关性较差;而在葡萄果肉中,vvi-miR172d显著负调控VvAP2的表达(图9-B),表明vvi-miR172s及其靶基因的表达存在时空特异性。且在果实不同组织和不同发育时期,vvi-miR172家族成员对靶基因的调控作用存在差异,果皮组织中vvi-miR172家族可能主要通过vvi-miR172a/b/d介导VvRAP2-1起调控作用,而果肉中vvi-miR172d靶向VvAP2的调控作用更强。
2.4.4 vvi-miR172s及靶基因在果实不同组织发育过程中应答GA3的模式分析 赤霉素处理可诱导葡萄产生无核果实,为认识vvi-miR172s及其靶基因在葡萄果实发育过程中应答GA3的模式,分析其在GA3处理下的表达水平。果皮中外源GA3的施用使vvi-miR172c在果实第二次膨大期(60 DAF)的表达水平显著上调,vvi-miR172a/b/d在幼果期和硬核期的表达量显著下降(图10-A)。GA3处理显著下调了靶基因VvAP2/VvRAP2-2/VvRAP2-3的表达,但VvRAP2-1的表达在果实第二次膨大期显著上调。
图10
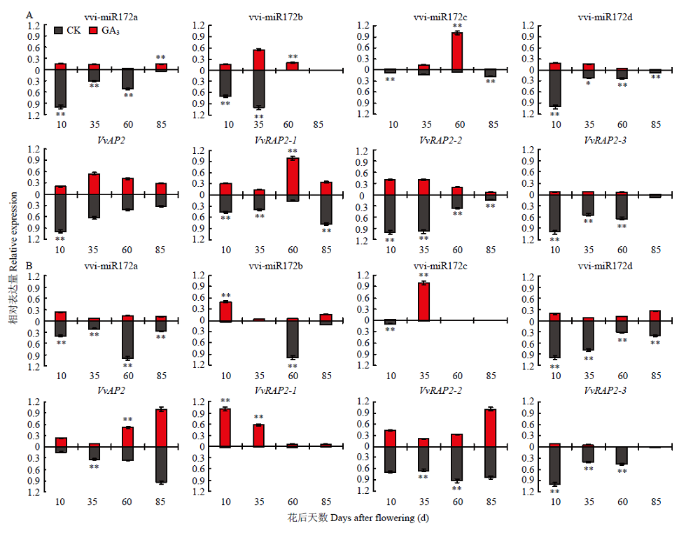 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图10GA3处理下vvi-miR172a/b/c/d及其靶基因在葡萄果皮(A)和果肉(B)中的表达水平
Fig. 10Expression levels of vvi-miR172a/b/c/d and target genes in berry pericarp (A) and flesh (B) of grape under GA3 treatment
相关性分析显示,GA3处理增强了果皮组织中vvi-miR172d对VvRAP2-1的负调控作用(图11-A)。GA3处理使果肉中vvi-miR172d在各个发育阶段呈下降趋势,但显著增加vvi-miR172c在果实第二次膨大期(60 DAF)的表达。靶基因VvAP2/VvRAP2-2/ VvVvRAP2-3在60 DAF显著下调,VvRAP2-1的表达在GA3处理下显著上调(图10-B)。对比赤霉素处理后vvi-miR172s和靶基因表达水平的相关性,发现在果肉中GA3同样增强了果实发育前期vvi-miR172d与VvRAP2-1的负相关性,同时诱导了vvi-miR172c对VvAP2/VvRAP2-2/VvVvRAP2-3的负调控作用(图11-B),表明vvi-miR172s及其靶基因可能存在不同的GA3应答模式参与葡萄果皮和果肉的发育过程,其中vvi-miR172c可能通过应答GA3介导相应靶基因参与葡萄果实第二次膨大发育的调控,而vvi-miR172d可能响应GA3介导其靶基因调控葡萄幼果期与硬核期果实的发育。
图11
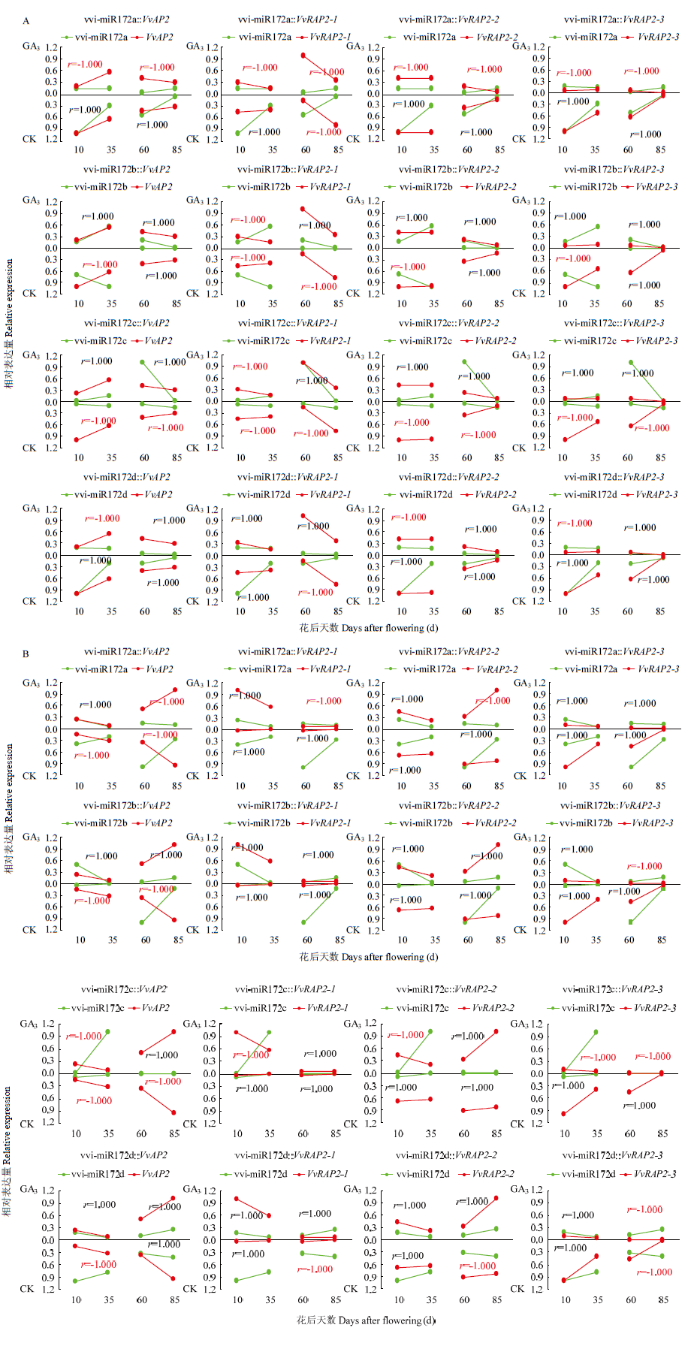 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图11葡萄果皮(A)和果肉(B)中vvi-miR172s及靶基因在GA3处理下的差异表达和相关性分析
Fig. 11Differential expression and correlation analysis of each member of vvi-miR172 family and target genes in response to GA3 treatment in berry pericarp (A) and flesh (B) of grape
3 讨论
MicroRNAs是一类内源性的非编码小RNA,在基因表达的转录后水平起重要的调控作用。其中,高度保守的miR156和miR172被认为是植物营养生长阶段变化的主要调控因子[8]。而miR172最早是在拟南芥中通过小RNA测序获得,由于其具有高度保守性,目前已在许多植物中鉴定到miR172的存在。本研究发现葡萄中miR172有4个成员(miR172a/b/c/d),和水稻、油菜中的成员数量相同,拟南芥miR172有5个成员,麻疯树有2个成员,表明miR172家族成员在不同物种中存在很大的差异。miR172是第一个主要通过抑制翻译来调节其靶标的植物miRNA,但在葡萄中vvi-miR172s通过裂解作用介导其靶基因的表达。miR172s的靶基因主要为AP2转录因子基因家族成员,AP2/ERF蛋白是一个转录因子大家族,在生物和非生物胁迫以及发育阶段起关键的作用,目前已在拟南芥[29]、水稻[30]、大麦、小麦[31]、油菜[1]、杨树[32]和花生[33]中鉴定到其存在。MiR156-miR172-AP2被认为是一种调控营养生长向生殖生长转变的途径,进而影响植物开花时间和花的发育[34]。AP2蛋白含有一到两个AP2结构域,AP2域包括60—70个高度保守的氨基酸(aa)和两个保守的YRG和RAYD motif元件,分别在AP2域的N和C末端[1]。本研究中葡萄miR172s的4条靶基因(VvAP2、VvRAP2-1、VvRAP2-2和VvRAP2-3),均含有两个AP2蛋白结构域,但不同物种中靶基因存在差异,油菜和拟南芥中分别含有19和6个AP2类靶基因。靶基因的进化分析显示,相比于拟南芥和水稻,葡萄中miR172靶基因的亲缘关系较远,推测葡萄中miR172靶基因VvAP2、VvRAP2-1、VvRAP2-2、VvRAP2-3的进化程度较大,其功能可能更加多样化。MicroRNA及其靶基因在不同组织中的表达具有时空特异性。JOFUKU等[35]指出,与其他花同源基因不同,AP2在拟南芥的非花组织(茎和叶)和花组织(萼片、花瓣、雄蕊、心皮、发育中的胚珠和花序分生组织)中均有表达。GIL-HUMANES等[31]研究了AP2类基因在小麦和大麦不同发育阶段根、茎、幼叶和穗中的表达,发现AP2类基因在所有组织中都有表达,在穗发育的早期达到高峰,并随着穗的成熟而逐渐减少。油菜中的研究表明大部分EuAP2在花器官中高表达,推测其在花发育过程中具有特殊的功能;且BnaAP2-1、BnaAP2-5和BnaTOE1-2在晚花材料中的表达水平均高于早花材料,表明它们可能具有抑花作用[1]。本试验的荧光定量分析显示,vvi-miR172s及其靶基因在果实发育过程中的表达存在显著差异,vvi-miR172c在果皮和果肉中的表达水平较低,在拟南芥中,miR172a/b/c的表达在生殖生长阶段升高,而miR172d/e基因的表达很低,并且不会随着植物的发育而改变[34];在水稻中,miR172c在幼苗中表达,但在发育中的谷物中不表达[36],表明在不同物种中miR172c的表达存在显著差异。本研究发现,外源GA3增强了果皮和果肉组织中vvi-miR172d对VvRAP2-1的负调控作用,同时诱导了果肉组织中vvi-miR172c对VvAP2/VvRAP2-2/VvVvRAP2-3的负调控作用。该研究结果初步表明vvi-miR172c/d可能介导VvAP2及VvRAP2-1调控葡萄果实的发育,为深入研究vvi-miR172s及其靶基因响应GA信号调控葡萄果实发育的分子机制奠定了重要的基础。
4 结论
葡萄miR172家族含有vvi-miR172a/b/c/d四个成员,均可通过裂解VvAP2、VvRAP2-1、VvRAP2-2、VvRAP2-3四条靶基因参与调控葡萄果皮、果肉发育。其中,vvi-miR172a/b/d和vvi-miR172c可能分别介导靶基因VvRAP2-1和VvAP2在果皮和果肉组织发育中起着更为重要的作用,而vvi-miR172c可能主要响应GA3信号介导VvAP2调控葡萄果实膨大期果皮、果肉的发育。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 6]
DOI:10.1111/j.1744-7909.2011.01062.xURLPMID:21676172 [本文引用: 2]

Plants have acquired sophisticated stress response systems to adapt to changing environments. It is important to understand plants' stress response mechanisms in the effort to improve crop productivity under stressful conditions. The AP2/ERF transcription factors are known to regulate diverse processes of plant development and stress responses. In this study, the molecular characteristics and biological functions of AP2/ERFs in a variety of plant species were analyzed. AP2/ERFs, especially those in DREB and ERF subfamilies, are ideal candidates for crop improvement because their overexpression enhances tolerances to drought, salt, freezing, as well as resistances to multiple diseases in the transgenic plants. The comprehensive analysis of physiological functions is useful in elucidating the biological roles of AP2/ERF family genes in gene interaction, pathway regulation, and defense response under stress environments, which should provide new opportunities for the crop tolerance engineering.
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
DOI:10.1371/journal.pbio.1000148URLPMID:19582143 [本文引用: 2]
A small mobile protein, encoded by the FLOWERING LOCUS T (FT) locus, plays a central role in the control of flowering. FT is regulated positively by CONSTANS (CO), the output of the photoperiod pathway, and negatively by FLC, which integrates the effects of prolonged cold exposure. Here, we reveal the mechanisms of regulation by the microRNA miR172 target SCHLAFMUTZE (SMZ), a potent repressor of flowering. Whole-genome mapping of SMZ binding sites demonstrates not only direct regulation of FT, but also of many other flowering time regulators acting both upstream and downstream of FT, indicating an important role of miR172 and its targets in fine tuning the flowering response. A role for the miR172/SMZ module as a rheostat in flowering time is further supported by SMZ binding to several other genes encoding miR172 targets. Finally, we show that the action of SMZ is completely dependent on another floral repressor, FLM, providing the first direct connection between two important classes of flowering time regulators, AP2- and MADS-domain proteins.
DOI:10.1093/nar/gkp1035URLPMID:19942686 [本文引用: 1]
MicroRNAs (miRNAs) are approximately 21 nt non-coding RNAs which regulate post-transcriptional gene expression. miRNAs are key regulators of nearly all essential biological processes. Aiming at understanding miRNA's functions in Euphorbiaceae, a large flowering plant family, we performed a genome-scale systematic study of miRNAs in Euphorbiaceae, by combining computational prediction and experimental analysis to overcome the difficulty of lack of genomes for most Euphorbiaceous species. Specifically, we predicted 85 conserved miRNAs in 23 families in the Castor bean (Ricinus communis), and experimentally verified and characterized 58 (68.2%) of the 85 miRNAs in at least one of four Euphorbiaceous species, the Castor bean, the Cassava (Manihot esculenta), the Rubber tree (Hevea brasiliensis) and the Jatropha (Jatropha curcas) during normal seedling development. To elucidate their function in stress response, we verified and profiled 48 (56.5%) of the 85 miRNAs under cold and drought stresses as well as during the processes of stress recovery. The results revealed some species- and condition-specific miRNA expression patterns. Finally, we predicted 258 miRNA:target partners, and identified the cleavage sites of six out of ten miRNA targets by a modified 5' RACE. This study produced the first collection of miRNAs and their targets in Euphorbiaceae. Our results revealed wide conservation of many miRNAs and diverse functions in Euphorbiaceous plants during seedling growth and in response to abiotic stresses.
DOI:10.1093/pcp/pcy175URLPMID:30541045 [本文引用: 2]
Jatropha curcas is a promising feedstock for biofuel production because its oil is highly suitable for processing bio-jet fuels and biodiesel. However, Jatropha exhibits a long juvenile stage in subtropical areas. miR172, a conserved small non-protein-coding RNA molecule with 21 nucleotides, regulates a wide range of developmental processes. To date, however, no studies have examined the function of miR172 in Jatropha. There are five miR172 precursors encoding two mature miR172s in Jatropha, which are expressed in all tissues, with the highest expression level in leaves, and the levels are up-regulated with age. Overexpression of JcmiR172a resulted in early flowering, abnormal flowers, and altered leaf morphology in transgenic Arabidopsis and Jatropha. The expression levels of miR172 target genes were down-regulated, and the flower identity genes were up-regulated in the JcmiR172a-overexpressing transgenic plants. Interestingly, we showed that JcmiR172 might be involved in regulation of stem vascular development through manipulating the expression of cellulose and lignin biosynthesis genes. Overexpression of JcmiR172a enhanced xylem development and reduced phloem and pith development. This study helped elucidate the functions of miR172 in perennial plants, a known age-related miRNA involved in the regulation of perennial plant phase change and organ development.
URL [本文引用: 2]
URL [本文引用: 1]
URL [本文引用: 1]
DOI:10.1080/15592324.2016.1156833URLPMID:26926448 [本文引用: 2]
microRNA172 (miR172) expression has been shown to have a positive effect on Arabidopsis fruit (siliques) growth. In contrast, over-expression of miR172 has a negative influence on fruit growth in apple, resulting in a dramatic reduction in fruit size. This negative influence is supported by the results of analyzing a transposable element (TE) insertional allele of a MIR172 gene that has reduced expression of the miRNA and is associated with an increase in fruit size. Arabidopsis siliques are a dry fruit derived from ovary tissues, whereas apple is a fleshy pome fruit derived mostly from hypanthium tissues. A model has been developed to explain the contrasting impact of miR172 expression in these two plant species based on the differences in their fruit structure. Transgenic apple plants with extremely high levels of miR172 overexpression produced flowers consisting of carpel tissues only, which failed to produce fruit. By comparison, in tomato, a fleshy berry fruit derived from the ovary, high level over-expression of the same miR172 resulted in carpel-only flowers which developed into parthenocarpic fruit. These results further indicate that the influence of miR172 on fruit growth in different plant species depends on its fruit type.
DOI:10.1038/nplants.2015.36URLPMID:27247036 [本文引用: 1]
Growth is a major factor in plant organ morphogenesis and is influenced by exogenous and endogenous signals including hormones. Although recent studies have identified regulatory pathways for the control of growth during vegetative development, there is little mechanistic understanding of how growth is controlled during the reproductive phase. Using Arabidopsis fruit morphogenesis as a platform for our studies, we show that the microRNA miR172 is critical for fruit growth, as the growth of fruit is blocked when miR172 activity is compromised. Furthermore, our data are consistent with the FRUITFULL (FUL) MADS-domain protein and Auxin Response Factors (ARFs) directly activating the expression of a miR172-encoding gene to promote fruit valve growth. We have also revealed that MADS-domain (such as FUL) and ARF proteins directly associate in planta. This study defines a novel and conserved microRNA-dependent regulatory module integrating developmental and hormone signalling pathways in the control of plant growth.
DOI:10.1126/science.1088060URLPMID:12893888 [本文引用: 1]
Plant microRNAs (miRNAs) show a high degree of sequence complementarity to, and are believed to guide the cleavage of, their target messenger RNAs. Here, I show that miRNA172, which can base-pair with the messenger RNA of a floral homeotic gene, APETALA2, regulates APETALA2 expression primarily through translational inhibition. Elevated miRNA172 accumulation results in floral organ identity defects similar to those in loss-of-function apetala2 mutants. Elevated levels of mutant APETALA2 RNA with disrupted miRNA172 base pairing, but not wild-type APETALA2 RNA, result in elevated levels of APETALA2 protein and severe floral patterning defects. Therefore, miRNA172 likely acts in cell-fate specification as a translational repressor of APETALA2 in Arabidopsis flower development.
DOI:10.1105/tpc.107.054528URLPMID:17890372 [本文引用: 1]
Regulated RNA metabolism appears to be a critical component of molecular mechanisms directing flowering initiation in plants. A group of RNA binding proteins exerts their roles through the autonomous flowering pathway. Posttranscriptional mechanisms regulated by microRNAs (miRNAs) also play a key role in flowering-time control. Here, we demonstrate that the GIGANTEA (GI)-regulated miR172 defines a unique genetic pathway that regulates photoperiodic flowering by inducing FLOWERING LOCUS T (FT) independent of CONSTANS (CO). A late-flowering mutant in which a miR172 target gene, TARGET OF EAT1, is constitutively activated by the nearby insertion of the cauliflower mosaic virus 35S enhancer normally responded to vernalization and gibberellic acid treatments. By contrast, its response to daylength changes was severely disrupted. In the mutant, FT was significantly repressed, but other flowering genes were unaffected. Notably, miR172 abundance is regulated by photoperiod via GI-mediated miRNA processing. Accordingly, miR172-overproducing plants exhibit early flowering under both long days and short days, even in the absence of functional CO, indicating that miR172 promotes photoperiodic flowering through a CO-independent genetic pathway. Therefore, it appears that GI-mediated photoperiodic flowering is governed by the coordinated interaction of two distinct genetic pathways: one mediated via CO and the other mediated via miR172 and its targets.
[本文引用: 1]
DOI:10.1105/tpc.110.075606URLPMID:20675573 [本文引用: 1]
The Arabidopsis thaliana transcription factor APETALA2 (AP2) has numerous functions, including roles in seed development, stem cell maintenance, and specification of floral organ identity. To understand the relationship between these different roles, we mapped direct targets of AP2 on a genome-wide scale in two tissue types. We find that AP2 binds to thousands of loci in the developing flower, many of which exhibit AP2-dependent transcription. Opposing, logical effects are evident in AP2 binding to two microRNA genes that influence AP2 expression, with AP2 positively regulating miR156 and negatively regulating miR172, forming a complex direct feedback loop, which also included all but one of the AP2-like miR172 target clade members. We compare the genome-wide direct target repertoire of AP2 with that of SCHLAFMUTZE, a closely related transcription factor that also represses the transition to flowering. We detect clear similarities and important differences in the direct target repertoires that are also tissue specific. Finally, using an inducible expression system, we demonstrate that AP2 has dual molecular roles. It functions as both a transcriptional activator and repressor, directly inducing the expression of the floral repressor AGAMOUS-LIKE15 and directly repressing the transcription of floral activators like SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1.
URL [本文引用: 1]
DOI:10.1105/tpc.105.036053URLPMID:16243903 [本文引用: 1]
Transcriptome profiling via cDNA microarray analysis identified 869 genes that are differentially expressed in developing tomato (Solanum lycopersicum) pericarp. Parallel phenotypic and targeted metabolite comparisons were employed to inform the expression analysis. Transcript accumulation in tomato fruit was observed to be extensively coordinated and often completely dependent on ethylene. Mutation of an ethylene receptor (Never-ripe [Nr]), which reduces ethylene sensitivity and inhibits ripening, alters the expression of 37% of these 869 genes. Nr also influences fruit morphology, seed number, ascorbate accumulation, carotenoid biosynthesis, ethylene evolution, and the expression of many genes during fruit maturation, indicating that ethylene governs multiple aspects of development both prior to and during fruit ripening in tomato. Of the 869 genes identified, 628 share homology (E-value < or = 1 x 10(-10)) with known gene products or known protein domains. Of these 628 loci, 72 share homology with previously described signal transduction or transcription factors, suggesting complex regulatory control. These results demonstrate multiple points of ethylene regulatory control during tomato fruit development and provide new insights into the molecular basis of ethylene-mediated ripening.
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1105/tpc.1.12.1195URLPMID:12359889 [本文引用: 1]
We have examined the floral morphology and ontogeny of three mutants of Arabidopsis thaliana, Ap2-5, Ap2-6, and Ap2-7, that exhibit homeotic changes of the perianth organs because of single recessive mutations in the AP2 gene. Homeotic conversions observed are: sepals to carpels in all three mutants, petals to stamens in Ap2-5, and petals to carpels in Ap2-6. Our analysis of these mutants suggests that the AP2 gene is required early in floral development to direct primordia of the first and second whorls to develop as perianth rather than as reproductive organs. In addition, our results support one of the two conflicting hypotheses concerning the structures of the calyx and the gynoecium in the Brassicaceae.
URLPMID:8281037 [本文引用: 1]
URLPMID:12368505 [本文引用: 1]
DOI:10.1371/journal.pone.0021259URLPMID:21829435 [本文引用: 1]
BACKGROUND: Alignment analysis of the Vv-miRNAs identified from various grapevine cultivars indicates that over 30% orthologous Vv-miRNAs exhibit a 1-3 nucleotide discrepancy only at their ends, suggesting that this sequence discrepancy is not a random event, but might mainly derive from divergence of cultivars. With advantages of miR-RACE technology in determining precise sequences of potential miRNAs from bioinformatics prediction, the precise sequences of vv-miRNAs predicted computationally can be verified with miR-RACE in a different grapevine cultivar. This presents itself as a new approach for large scale discovery of precise miRNAs in different grapevine varieties. METHODOLOGY/PRINCIPAL FINDINGS: Among 88 unique sequences of Vv-miRNAs from bioinformatics prediction, 83 (96.3%) were successfully validated with MiR-RACE in grapevine cv. 'Summer Black'. All the validated sequences were identical to their corresponding ones obtained from deep sequencing of the small RNA library of 'Summer Black'. Quantitative RT-PCR analysis of the expressions levels of 10 Vv-miRNA/target gene pairs in grapevine tissues showed some negative correlation trends. Finally, comparison of Vv-miRNA sequences with their orthologs in Arabidopsis and study on the influence of divergent bases of the orthologous miRNAs on their targeting patterns in grapevine were also done. CONCLUSION: The validation of precise sequences of potential Vv-miRNAs from computational prediction in a different grapevine cultivar can be a new way to identify the orthologous Vv-miRNAs. Nucleotide discrepancy of orthologous Vv-miRNAs from different grapevine cultivars normally does not change their target genes. However, sequence variations of some orthologous miRNAs in grapevine and Arabidopsis can change their targeting patterns. These precise Vv-miRNAs sequences validated in our study could benefit some further study on grapevine functional genomics.
URL [本文引用: 2]
URL [本文引用: 2]
[D].
[本文引用: 2]
[D].
[本文引用: 2]
URLPMID:25981679 [本文引用: 2]
[本文引用: 1]
URLPMID:17945171 [本文引用: 1]
[本文引用: 2]
DOI:10.1016/j.bbrc.2008.04.087URLPMID:18442469 [本文引用: 1]
Populus is a model system for investigating the wood development, crown formation, and disease resistance in perennial plants. AR2/ERF is a large family of transcription factors in plant, encoding transcriptional regulators with a variety of functions involved in the developmental and physiological processes. Here, starting from database of Populus genome, we identified 200 AP2/ERF genes by in silico cloning method using the AP2/ERF conserved domain amino acid sequence of Arabidopsis thaliana as probe. Based on the number of AP2/ERF domains and the function of the genes, those AP2/ERF genes from Populus were classified into four subfamilies named the AP2, DREB, ERF, RAV, and a soloist. Among these genes, the number genes of total AP2/ERF family genes, DREB subfamily, and ERF subfamily from Populus trichocarpa were about 1.4-1.6-fold than those from A. thaliana. The rates were very similar for the putative homologs between Populus and Arabidopsis.
URLPMID:18832187 [本文引用: 1]
DOI:10.3389/fpls.2019.00820URLPMID:31333689 [本文引用: 2]
In maize, shoot-borne roots dominate the whole root system and play essential roles in water and nutrient acquisition and lodging tolerance. Shoot-borne roots initiate at shoot nodes, including crown roots from the belowground nodes and brace roots from aboveground nodes. In contrast to crown roots, few genes for brace roots development have been identified. Here, we characterized a maize AP2/ERF transcription factor, ZmRAP2.7, to be involved in brace roots development. ZmRAP2.7 expressed in all types of roots, and the encoded protein localized in the nucleus with transcriptional activation activity. A maize transposon insert mutant RAP2.7-Mu defective in ZmRAP2.7 expression revealed a decreased number of brace roots but not crown roots. Maize Corngrass1 mutant, which showed an elevated expression of ZmRAP2.7, however, revealed an increased number of brace roots. The ZmRAP2.7-based association analysis in a maize panel further identified a SNP marker at the fifth exon of gene to be associated with number of brace roots. These results uncovered a function of ZmRAP2.7 in brace roots development and provided the valuable gene and allele for genetic improvement of maize root systems.
DOI:10.1105/tpc.6.9.1211URLPMID:7919989 [本文引用: 1]
APETALA2 (AP2) plays a central role in the establishment of the floral meristem, the specification of floral organ identity, and the regulation of floral homeotic gene expression in Arabidopsis. We show here that in addition to its functions during flower development, AP2 activity is also required during seed development. We isolated the AP2 gene and found that it encodes a putative nuclear protein that is distinguished by an essential 68-amino acid repeated motif, the AP2 domain. Consistent with its genetic functions, we determined that AP2 is expressed at the RNA level in all four types of floral organs--sepals, petals, stamens, and carpels--and in developing ovules. Thus, AP2 gene transcription does not appear to be spatially restricted by the floral homeotic gene AGAMOUS as predicted by previous studies. We also found that AP2 is expressed at the RNA level in the inflorescence meristem and in nonfloral organs, including leaf and stem. Taken together, our results suggest that AP2 represents a new class of plant regulatory proteins that may play a general role in the control of Arabidopsis development.
DOI:10.1093/jxb/erq295URLPMID:20952628 [本文引用: 1]
Since the discovery of miRNAs in plants it has become clear that they are central to the regulation of many aspects of plant development and responses to the environment. miR172 regulates expression of a small group of AP2-like transcription factors in an evolutionarily ancient interaction. miR172 functions in regulating the transitions between developmental stages and in specifying floral organ identity. These two roles are conserved across monocotyledons and dicotyledons. Investigations into the roles of miR172 and its targets in phase changes in the model plant Arabidopsis have illustrated that this process is governed by complex regulatory systems. In addition to its conserved roles, miR172 has also acquired specialized species-specific functions in other aspects of plant development such as cleistogamy and tuberization.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}