 ,1, 范元婵,1, 蒋海宾1, 王杰1, 范小雪1, 祝智威1, 隆琦1, 蔡宗兵1, 郑燕珍1, 付中民1,2, 徐国钧1, 陈大福1,2, 郭睿,1,2
,1, 范元婵,1, 蒋海宾1, 王杰1, 范小雪1, 祝智威1, 隆琦1, 蔡宗兵1, 郑燕珍1, 付中民1,2, 徐国钧1, 陈大福1,2, 郭睿,1,2Improvement of Nosema ceranae Genome Annotation Based on Nanopore Full-Length Transcriptome Data
CHEN HuaZhi,1, FAN YuanChan,1, JIANG HaiBin1, WANG Jie1, FAN XiaoXue1, ZHU ZhiWei1, LONG Qi1, CAI ZongBing1, ZHENG YanZhen1, FU ZhongMin1,2, XU GuoJun1, CHEN DaFu1,2, GUO Rui,1,2通讯作者:
责任编辑: 岳梅
收稿日期:2020-05-6接受日期:2020-05-28网络出版日期:2021-03-16
| 基金资助: |
Received:2020-05-6Accepted:2020-05-28Online:2021-03-16
作者简介 About authors
陈华枝,E-mail:
范元婵,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (7681KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
陈华枝, 范元婵, 蒋海宾, 王杰, 范小雪, 祝智威, 隆琦, 蔡宗兵, 郑燕珍, 付中民, 徐国钧, 陈大福, 郭睿. 基于纳米孔全长转录组数据完善东方蜜蜂微孢子虫的基因组注释[J]. 中国农业科学, 2021, 54(6): 1288-1300 doi:10.3864/j.issn.0578-1752.2021.06.018
CHEN HuaZhi, FAN YuanChan, JIANG HaiBin, WANG Jie, FAN XiaoXue, ZHU ZhiWei, LONG Qi, CAI ZongBing, ZHENG YanZhen, FU ZhongMin, XU GuoJun, CHEN DaFu, GUO Rui.
开放科学(资源服务)标识码(OSID):

0 引言
【研究意义】东方蜜蜂微孢子虫(Nosema ceranae)是细胞内寄生的单细胞真菌,特异性侵染成年蜜蜂的中肠上皮细胞,对蜜蜂幼虫也具有侵染性。目前,由于成熟的转基因操作技术平台缺失和公共数据库中微孢子虫基因注释信息的匮乏,东方蜜蜂微孢子虫的基因组注释很不完善。利用全长转录组数据对东方蜜蜂微孢子虫的完整开放阅读框(open reading frame,ORF)、简单重复序列(simple sequence repeat,SSR)及未注释基因和转录本进行鉴定,对已注释基因进行结构优化,可丰富和完善东方蜜蜂微孢子虫的参考基因组注释,为后续的生物信息学分析和分子生物学研究提供可靠的参考信息,也能为其他物种的基因组注释信息完善提供思路和方法借鉴。【前人研究进展】第一代测序技术即Sanger测序技术具有准确性高的优点,过去被成功用于人类[1]、家蚕(Bombyx mori)[2]和西方蜜蜂(Apis mellifera)[3]等物种的基因组测序和组装,但由于高成本和低通量的限制,逐渐被基于边合成边测序原理的第二代测序技术取代。近十几年来,以Illumina为代表的二代测序技术凭借高通量和成本持续下降的优势,在动物[4,5]、植物[6,7]和微生物[8,9]的基因组测序方面得到广泛应用,较大幅度地提升了物种的基因组组装质量。但二代测序具有GC偏好性且测序读段较短(不超过300 bp),需要通过生物信息学方法对短读段进行拼接,在测定重复序列方面劣势明显[10]。目前,除人类、小鼠和果蝇(Drosophila)等极少数模式物种的基因组组装到染色体水平外,绝大多数物种的基因组仅组装到contig或scafford水平,而且不同物种的基因组质量参差不齐[11,12,13]。近年来,随着以PacBio单分子实时(single- molecule real-time,SMRT)测序技术和Oxford纳米孔(nanopore)长读段测序技术为代表的三代测序技术的兴起与应用,人们通过纯三代测序或三代测序结合二代测序将越来越多物种基因组组装到染色体水平[14,15,16]。然而,目前三代测序的成本依然高昂,对于一些基因组较大的物种或经费有限的实验室,利用三代测序技术进行基因组测序还存在较大的困难。相对于基因组测序,利用三代测序技术进行转录组测序的成本较低且周期较短。利用PacBio SMRT测序数据完善小麦(Triticum aestivum)和锡兰勾虫(Ancylostoma ceylanicum)基因组注释的研究已见报道[11,17]。CORNMAN等[18]通过454焦磷酸测序技术对东方蜜蜂微孢子虫的纯净孢子进行测序,利用CABOG软件拼接出5 465条contig,组装的基因组(assembly ASM18298v1)大小为7.86 Mb,GC含量为25.3%,包含2 060个蛋白编码基因。2015年,PELIN等[8]利用Illumina HiSeq技术重新测序并组装了东方蜜蜂微孢子虫的基因组(assembly ASM98816v1),其大小为8.82 Mb,包含110条contig,目前为NCBI Genome数据库推荐的参考基因组版本。但上述两个东方蜜蜂微孢子虫的基因组版本都只组装到contig水平,远未达到染色体水平,因而会对基于这两个基因组版本的生物信息学分析产生影响。因此,通过多组学数据对东方蜜蜂微孢子虫的基因序列和功能注释进行补充和完善尤为必要。【本研究切入点】前期研究中,笔者所在团队已利用Oxford Nanopore测序技术对东方蜜蜂微孢子虫的纯净孢子进行测序,基于高质量的测序数据构建和注释了东方蜜蜂微孢子虫的首个全长转录组[19];并对东方蜜蜂微孢子虫基因的可变剪接和可变多聚腺苷酸化进行了系统鉴定和分析[20]。【拟解决的关键问题】利用已获得的全长转录组数据对东方蜜蜂微孢子虫参考基因组的完整ORF进行预测,对已注释基因进行结构优化,对未注释的SSR进行挖掘,并对未注释的新基因和新转录本进行鉴定和功能注释。1 材料与方法
试验于2019年在福建农林大学动物科学学院(蜂学学院)蜜蜂保护实验室完成。1.1 全长转录组数据来源
前期研究中,笔者所在团队利用Oxford Nanopore测序技术对东方蜜蜂微孢子虫的纯净孢子进行测序,获得了高质量的全长转录组数据,共测得6 988 795条原始读段(raw reads),居中长度(N50)、平均读长和最大读长分别为971、881和96 051 bp,共鉴定出10 243条非冗余全长转录本,N50、平均长度和最大长度分别为1 042、894和4 855 bp[19]。高质量的全长转录组数据可为本研究中的完整ORF预测、已注释基因的结构优化、SSR位点鉴定与分析、新基因鉴定与功能注释,以及新转录本的鉴定和功能注释提供可靠的数据支撑。1.2 基因结构优化
由于软件和数据本身的局限性,导致多数基因组的基因结构信息不够精确,需要进一步优化。利用gffcompare软件将本研究鉴定到的全长转录本与东方蜜蜂微孢子虫参考基因组注释的转录本进行比较,对基因组注释的基因结构信息进行补充。如果在注释基因边界之外的区域有比对上的读段(mapped reads)支持,则将基因的UTR向上游或下游延伸,修正基因的边界。1.3 SSR位点的鉴定及分析
MISA(MIcroSAtellite identification tool)软件[21]可以通过对转录本序列的分析,鉴定出7种类型的SSR,包括单核苷酸重复(p1)、双核苷酸重复(p2)、三核苷酸重复(p3)、四核苷酸重复(p4)、五核苷酸重复(p5)、六核苷酸重复(p6)、混合SSR(c,即两个SSR之间的距离<100 bp)。从去冗余的全长转录本中筛选长度在500 bp以上的全长转录本,利用MISA软件预测SSR位点,采用默认参数。1.4 新基因和新转录本的鉴定及数据库注释
以东方蜜蜂微孢子虫参考基因组(assembly ASM98816v1)[8](gff文件)和本研究中去冗余后的全长转录本文件为基础,获得一个数据格式与注释文件相同的gff文件,利用gffcompare软件将2个gff文件进行比较,对于在参考基因组上没有注释信息的基因和转录本,将其定义为新基因和新转录本。利用Blast工具将上述新基因和新转录本分别比对Nr、KOG、eggNOG、GO和KEGG数据库,从而获得相应的功能注释。1.5 ORF预测
TransDecoder(v3.0.0)软件可基于ORF长度、对数似然函数值、氨基酸序列与Pfam数据库蛋白质结构域序列的比对等信息,从转录本序列中识别可靠的潜在编码区序列(coding sequence,CDS)。采用TransDecoder(v3.0.0)软件对上述新转录本的CDS及其对应氨基酸序列进行识别,从而预测ORF。同时预测到起始密码子和终止密码子的ORF为完整ORF。2 结果
2.1 东方蜜蜂微孢子虫的基因结构优化
共对东方蜜蜂微孢子虫的2 340个基因的结构进行优化,其中5′端延长的基因有1 182个,3′端延长的基因有1 158个。部分基因的结构优化信息详见表1。Table 1
表1
表1东方蜜蜂微孢子虫参考基因组中10个基因的结构优化信息概要
Table 1
| 基因ID Gene ID | 基因位置 Gene locus | 正负链 Plus and minus strand | 末端 End | 原位置 Original site | 优化后位置 Optimized site |
|---|---|---|---|---|---|
| Gene1175 | NW_020169317.1:49652-50638 | - | 3′ | 50341 | 50638 |
| Gene326 | NW_020169413.1:1478-3041 | - | 3′ | 2683 | 3041 |
| Gene503 | NW_020169431.1:1208-2193 | - | 5′ | 1461 | 1208 |
| Gene503 | NW_020169431.1:1208-2193 | - | 3′ | 1634 | 2193 |
| Gene1591 | NW_020169298.1:96744-98975 | + | 5′ | 96746 | 96744 |
| Gene1591 | NW_020169298.1:96744-98975 | + | 3′ | 98671 | 98975 |
| Gene588 | NW_020169310.1:2436-3030 | + | 3′ | 2741 | 3030 |
| Gene350 | NW_020169307.1:13073-18338 | - | 5′ | 17277 | 13073 |
| Gene36 | NW_020169296.1: 54153-55245 | - | 5′ | 54289 | 54153 |
| Gene2363 | NW_020169300.1:40046-41314 | - | 5′ | 40510 | 40046 |
新窗口打开|下载CSV
2.2 东方蜜蜂微孢子虫的SSR鉴定及分析
如表2所示,在8 265 494 bp的序列中共鉴定到1 658个SSR,含有SSR超过1个的基因数为212个,以混合物形式存在的SSR有65个;此外,单核苷酸重复、双核苷酸重复、三核苷酸重复、四核苷酸重复的数量分别为1 622、23、7和6个。进一步分析SSR的类型分布,结果显示p1类型的SSR密度最大,达到182.32个/Mb,其次为c、p2和p3,分别达到6.90、2.78和0.73个/Mb。Table 2
表2
表2基于MISA的东方蜜蜂微孢子虫SSR的搜索结果
Table 2
| MISA搜索项目 MISA searching item | 数量 Number |
|---|---|
| 搜索基因 Searching gene | 8021 |
| 搜索基因的总序列长度 Total sequence length of searching gene (bp) | 8265494 |
| 鉴定到的SSR位点 Identified SSR loci | 1658 |
| 鉴定到的SSR总序列长度 Total sequence length of identified SSRs (bp) | 1405 |
| 含有1个以上SSR的基因 Genes containing more than one SSR | 212 |
| 混合SSR Mixed SSR | 65 |
| 单核苷酸重复Single nucleotide repeat | 1622 |
| 双核苷酸重复Dinucleotide repeat | 23 |
| 三核苷酸重复Trinucleotide repeat | 7 |
| 四核苷酸重复Tetranucleotide repeat | 6 |
新窗口打开|下载CSV
2.3 东方蜜蜂微孢子虫的新基因鉴定及数据库注释
共鉴定出954个新基因,其中分别有951、333、371、422和321个新基因可注释到Nr、KOG、eggNOG、GO和KEGG数据库。新基因注释数量最多的物种是东方蜜蜂微孢子虫(798),其次为蜜蜂微孢子虫(Nosema apis)(55)和按蚊微孢子虫(Anncaliia algerae)(43)(图1-A)。新基因可注释到KOG数据库的25个功能类别,包括翻译后修饰、蛋白折叠和分子伴侣(43),转录(41),一般功能预测(41),复制、重组和修复(39),以及翻译、核糖体结构和生物合成(36)等(图1-B)。此外,新基因可注释到eggNOG数据库的25个功能类别,包括未知功能(94),翻译、核糖体结构和生物合成(48),复制、重组和修复(47),翻译后修饰、蛋白折叠和分子伴侣(36),以及转录(35)等(图1-C)。括号内的数字代表注释上的新基因数量。图1
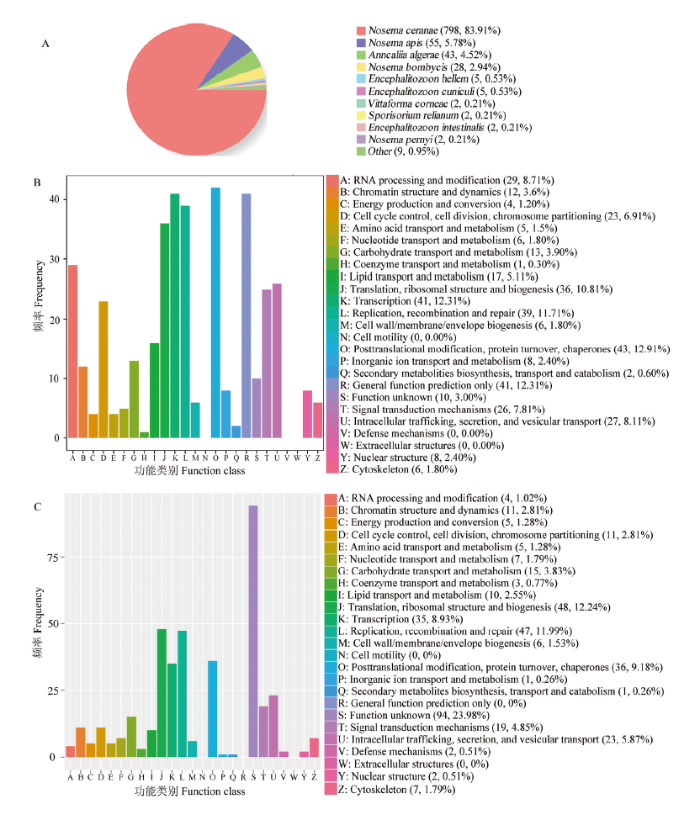 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1东方蜜蜂微孢子虫新基因的Nr(A)、KOG(B)和eggNOG(C)数据库注释
Fig. 1Nr (A), KOG (B) and eggNOG (C) database annotations of novel genes of N. ceranae
GO数据库注释结果显示,东方蜜蜂微孢子虫的新基因还能注释到生物学进程大类的15个条目,包括细胞进程(237)、代谢进程(225)和单一组织进程(114)等;分子功能大类的10个条目,包括催化活性(219)、结合(207)和结构分子活性(10)等;细胞组分大类的10个条目,包括细胞(214)、细胞组件(212)和细胞器(158)等(图2)。此外,上述新基因还可注释到KEGG数据库的58条通路,注释基因数最多的前5位通路分别是真核生物核糖体的生物合成(24),嘧啶代谢(17),抗生素的生物合成(16),内质网的蛋白加工(16)和嘌呤代谢(15)(图3)。括号内的数字代表注释上的新基因数量。
图2
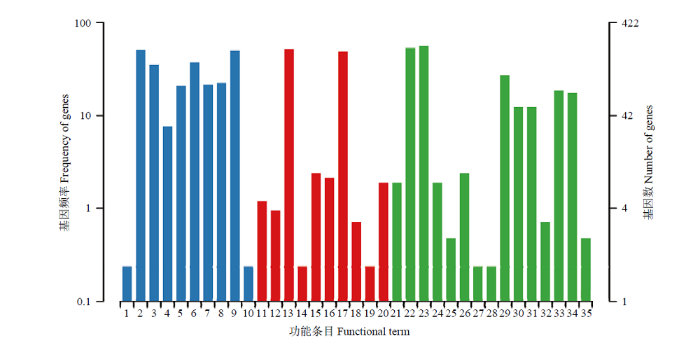 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2东方蜜蜂微孢子虫新基因的GO数据库注释
1:细胞外区域Extracellular region;2:细胞Cell;3:细胞膜Cell membrane;4:细胞膜内腔Membrane-enclosed lumen;5:高分子复合物Macromolecular complex;6:细胞器Organelle;7:细胞器组件Organelle part;8:细胞膜组件Cell membrane part;9:细胞组件Cell part;10:超分子复合物Supramolecular complex;11:转录因子活性,蛋白质结合Transcription factor activity, protein binding;12:核酸结合转录因子活性Nucleic acid binding transcription factor activity;13:催化活性Catalytic activity;14:信号转导因子活性Signal transducer activity;15:结构分子活性Structural molecule activity;16:转运活性Transporter activity;17:结合Binding;18:电子载体活性Electron carrier activity;19:抗氧化活性Antioxidant activity;20:分子功能调节因子Molecular function regulator;21:繁殖Reproduction;22:代谢进程Metabolic process;23:细胞进程Cellular process;24:生殖进程Reproductive process;25:生物黏附Biological adhesion;26:信号Signaling;27:发育进程Developmental process;28:生长Growth;29:单一组织进程Single-organism process;30:应激反应Response to stimulus;31:定位Localization;32:多组织进程Multi-organism process;33:生物调控Biological regulation;34:细胞成分组织或生物合成Cellular component organization or biogenesis;35:解毒Detoxification
Fig. 2GO database annotation of novel genes of N. ceranae
图3
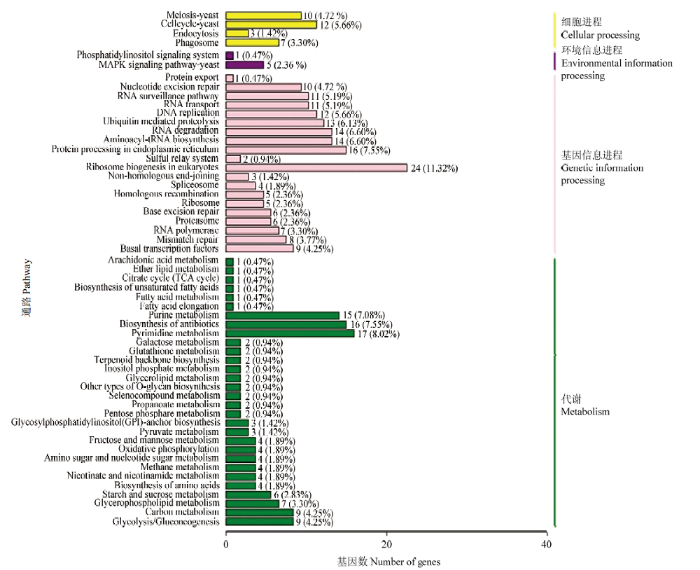 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3东方蜜蜂微孢子虫新基因的KEGG数据库注释
Fig. 3KEGG database annotation of novel genes of N. ceranae
2.4 东方蜜蜂微孢子虫的新转录本鉴定及数据库注释
共鉴定出6 164条新转录本,其中分别有6 141、2 808、2 932、3 196和2 585条新转录本可注释到Nr、KOG、eggNOG、GO和KEGG数据库。新转录本注释数量最多的物种是东方蜜蜂微孢子虫(5 512),其次为蜜蜂微孢子虫(263)和家蚕微孢子虫(Nosema bombycis)(156)(图4-A)。新转录本可注释到KOG数据库的25个功能类别,注释转录本数最多的是翻译、核糖体结构和生物合成(370),其次是转录(337),翻译后修饰、蛋白折叠和分子伴侣(327),复制、重组和修复(319)及RNA的加工与修饰(281)(图4-B)。此外,新转录本可注释到eggNOG数据库的25个功能类别,注释转录本数最多的是未知功能(557),其次是翻译、核糖体结构和生物合成(433),复制、重组和修复(391),转录(320)及翻译后修饰、蛋白折叠和分子伴侣(297)(图4-C)。括号内的数字代表注释上的新转录本数量。图4
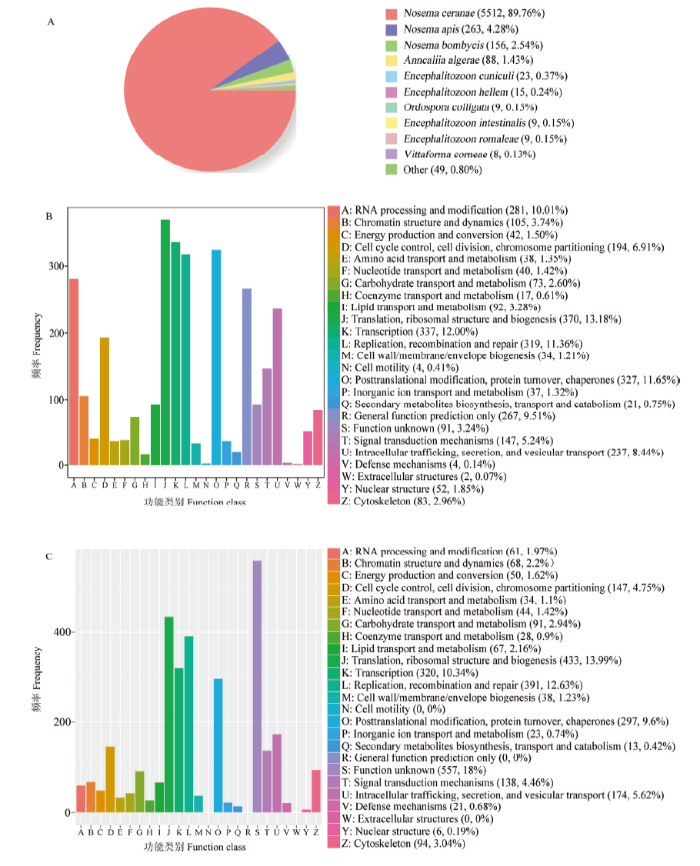 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4东方蜜蜂微孢子虫新转录本的Nr(A)、KOG(B)和eggNOG(C)数据库注释
Fig. 4Nr (A), KOG (B) and eggNOG (C) database annotations of novel transcripts of N. ceranae
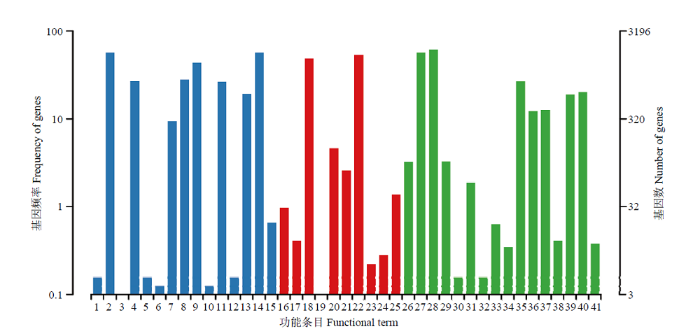
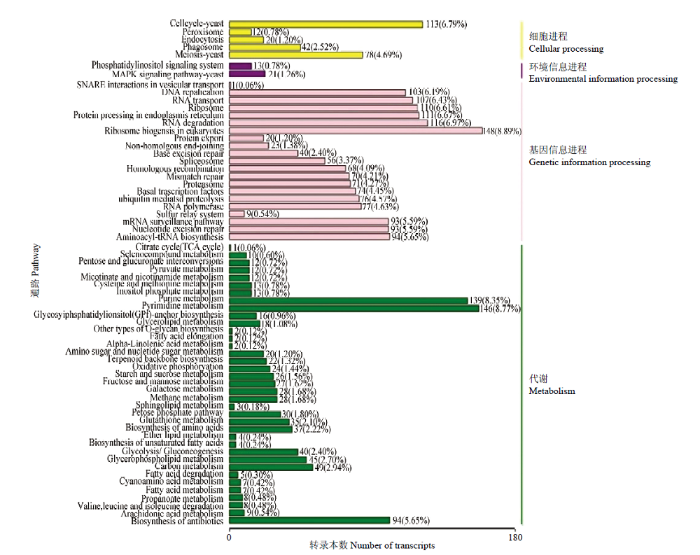
GO数据库注释结果显示,东方蜜蜂微孢子虫的新转录本还能注释到生物学进程大类的16个条目,包括细胞进程(1 973)、代谢进程(1 814)和单一组织进程(856)等;分子功能大类的10个条目,包括结合(1 711)、催化活性(1 561)和结构分子活性(147)等;细胞组分大类的15个条目,包括细胞组件(1 819)、细胞(1 816)和细胞器(1 392)等(图5)。此外,上述新基因还可注释到KEGG数据库的58条通路,包括真核生物核糖体的生物合成(148)、嘧啶代谢(146)、嘌呤代谢(139)、RNA降解(116)及细胞周期-酵母(113)等(图6)。括号内数字代表注释上的新转录本数量。
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5东方蜜蜂微孢子虫新转录本的GO数据库注释
1:细胞外区域Extracellular region;2:细胞Cell;3:拟核Nucleoid;4:细胞膜Cell membrane;5:病毒Virion;6:细胞连接Cell junction;7:细胞膜内腔Membrane-enclosed lumen;8:高分子复合物Macromolecular complex;9:细胞器Organelle;10:胞外区组件Extracellular region part;11:细胞器组件Organelle part;12:病毒组件Virion part;13:细胞膜组件Cell membrane part;14:细胞组件Cell part;15:超分子复合物Supramolecular complex;16:转录因子活性,蛋白质结合Transcription factor activity, protein binding;17:核酸结合转录因子活性Nucleic acid binding transcription factor activity;18:催化活性Catalytic activity;19:信号转导因子活性Signal transducer activity;20:结构分子活性Structural molecule activity;21:转运活性Transporter activity;22:结合Binding;23:电子载体活性Electron carrier activity;24:分子功能调控器Molecular function regulator;25:抗氧化活性Antioxidant activity;26:繁殖Reproduction;27:代谢进程Metabolic process;28:细胞进程Cellular process;29:生殖进程Reproductive process;30:生物黏附Biological adhesion;31:信号Signaling;32:多组织进程Multicellular organismal process;33:发育进程Developmental process;34:生长Growth;35:单一组织进程Single-organism process;36:应激反应Response to stimulus;37:定位Localization;38:多细胞组织进程Multi-organism process;39:生物调控Biological regulation;40:细胞成分组织或生物合成Cellular component organization or biogenesis;41:解毒Detoxification
Fig. 5GO database annotation of novel transcripts of N. ceranae
图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6东方蜜蜂微孢子虫新转录本的KEGG数据库注释
Fig. 6KEGG database annotations of novel transcripts of N. ceranae
2.5 东方蜜蜂微孢子虫的完整ORF预测
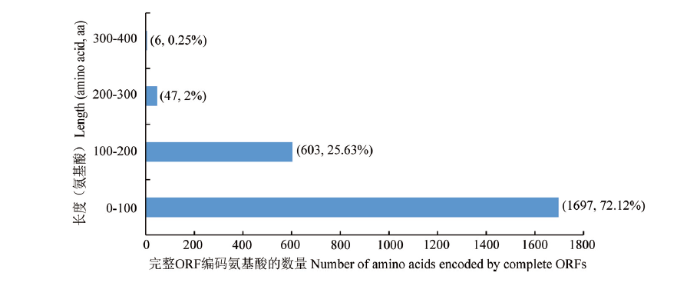
利用软件共预测出2 353个完整ORF,它们的长度分布介于0—400 aa,其中分布在0—100 aa的ORF数量最多,为1 697个;分布在100—200、200—300和300—400 aa的ORF分别有603、47和6个(图7)。图7
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7东方蜜蜂微孢子虫的完整ORF编码氨基酸的长度分布
Fig. 7Length distribution of amino acids encoded by complete ORFs of N. ceranae
3 讨论
近十几年来,二代测序技术的迅速发展和应用有力推动了动物、植物和微生物的基因组和转录组研究,存储于公共数据库(如NCBI SRA数据库)的海量二代转录组测序数据已成为完善物种基因组序列和功能注释的宝贵资源[22,23]。相对于一代和二代测序技术,Nanopore长读段测序技术具有超长读长(平均读长可达15 kb)的显著优势,不需要对测序读段进行拼接就能获得转录本的全长序列,所测即所得[24]。本研究利用前期已获得的全长转录组数据对东方蜜蜂微孢子虫的基因组注释进行完善,预测出2 353个完整ORF,分别延长了1 182和1 158个基因的5′ UTR和3′ UTR,发掘出1 658个SSR位点,此外鉴定到954个新基因和6 164条新转录本并对它们进行了功能注释。此为利用三代转录组测序数据完善蜜蜂病原基因组注释的首例报道。需要注意的是,本研究使用的全长转录组数据来源于东方蜜蜂微孢子虫的纯净孢子,而孢子是病原的休眠态,仅维持必要的低水平代谢[25],表达的转录本必然与病原在侵染过程表达的转录本存在差异。目前,笔者团队已获得东方蜜蜂微孢子虫感染7 d和10 d的意大利蜜蜂(Apis mellifera ligusctica,简称意蜂)和中华蜜蜂(Apis cerana cerana,简称中蜂)工蜂中肠的Nanopore长读段测序数据(未发表数据),下一步将从上述混合数据中筛滤出纯净的病原全长转录组数据,从而进一步对现有的参考基因组注释进行补充和完善。真核生物的基因表达调控与mRNA的UTR密切相关,例如mRNA的5′ UTR不仅能通过与反式作用因子结合调控翻译起始,还能通过控制mRNA的半衰期影响其稳定性;miRNA的种子序列能够与mRNA的3′ UTR靶向结合,从而抑制mRNA的翻译或使其降解[26]。前期研究中,笔者团队利用Illumina HiSeq技术对东方蜜蜂微孢子虫的纯净孢子进行测序,基于218 468 218条有效读段(clean reads)分别延长了6个已注释基因的5′ UTR和4个已注释基因的3′ UTR[27]。本研究基于东方蜜蜂微孢子虫的Nanopore全长转录组数据分别对1 182和1 158个基因的5′ UTR和3′ UTR进行了延长,说明三代测序数据较之二代测序数据可以大幅度提高已注释基因的结构优化质量,经优化的5′ UTR和3′ UTR对于深入研究东方蜜蜂微孢子虫的基因表达调控具有重要意义。
SSR是以1—6个核苷酸为重复单元组成的简单串联重复序列,作为第二代分子标记,SSR具有共显性遗传、重复性好、实验操作易及多态性高等优点[28]。SSR开发的传统方法以文库构建法为主,过程繁杂、费时费力且效率低下[29]。随着二代测序技术的不断进步和转录组数据的持续增多,人们开始利用测序得到的和公共数据库存储的二代转录组数据大规模开发SSR[30,31]。笔者团队前期也利用二代转录组数据大规模开发和验证了蜜蜂球囊菌(Ascosphaera apis)、中蜂和意蜂的SSR[32,33,34],证实了该方法的可行性。目前,东方蜜蜂微孢子虫的SSR严重缺乏。本研究基于东方蜜蜂微孢子虫的高质量全长转录组数据发掘出1 658个未注释的SSR位点,为现有的参考基因组的注释提供了有益补充。在前期研究中,利用蜜蜂球囊菌的二代转录组数据开发出7 968个SSR,其中最主要的重复类型为三核苷酸重复(53.15 %);此外,鉴定出13 448个中蜂SSR和6 312个意蜂SSR,其中最丰富的重复类型均为双核苷酸重复,占比分别达到58.03%和54.42%。本研究发现,东方蜜蜂微孢子虫的SSR中单核苷酸重复最为丰富,占比高达97.83%,与球囊菌和中蜂的研究结果存在差异,说明不同物种SSR的重复类型具有物种特异性。然而,对于沙葱萤叶甲(Galeruca daurica)[35]、扶桑绵粉蚧(Phenacoccus solenopsis)[31]和黄粉虫(Tenebrio molitor)[30]等昆虫,SSR的主要重复类型为单核苷酸重复,与本研究中东方蜜蜂微孢子虫SSR的主要重复类型一致,说明有些物种SSR的重复类型具有共性。此外,通过比较本研究鉴定到的SSR位点数与前期基于二代测序数据鉴定到的SSR位点数[32,33,34],发现前者的数量明显少于后者。对于哪种方法的准确性更高、假阳性更低,仍需要进一步深入研究。未来的工作重点是针对发掘出的SSR位点批量设计特异性引物,通过PCR扩增和毛细管电泳验证SSR的有效性及多态性,并将经验证的SSR应用于养蜂生产中东方蜜蜂微孢子虫的菌株鉴定、遗传分化及基因定位等研究。
现有的东方蜜蜂微孢子虫参考基因组(assembly ASM98816v1)共注释了3 264个基因,包括3 209个蛋白编码基因,35个tRNA基因,18个假基因和2个rRNA基因[8]。笔者团队前期基于东方蜜蜂微孢子虫的二代转录组数据仅鉴定出27个新基因[27]。本研究共鉴定到954个参考基因组未注释的新基因,占目前注释基因总数的约30%,说明基于Nanopore全长转录组数据能够高效挖掘新基因。本研究中,共有951个新基因可注释到Nr数据库,注释数量最多的物种是东方蜜蜂微孢子虫(798,83.91%),与实际情况相符,其次为蜜蜂微孢子虫(55),体现了二者同属Nosema属,亲缘关系近;分别有333、371、422和321个新基因可注释到KOG、eggNOG、GO和KEGG数据库,获得功能注释信息的新基因数量仍然偏少。一是由于目前还没有建立东方蜜蜂微孢子虫的转基因操作技术体系,导致绝大多数的基因功能尚未明确;二是上述4个数据库收录的东方蜜蜂微孢子虫及其近缘物种的功能注释信息还比较少,需要更多的研究数据对其进行持续补充。此外,本研究还鉴定出6 164条参考基因组未注释的新转录本,这些含有全长序列的新转录本为将来的基因克隆和功能研究提供了宝贵的数据资源。鉴定到的6 141条新转录本均能注释到Nr数据库,注释数量最多的物种仍为东方蜜蜂微孢子虫(5 512,89.76%),其次为蜜蜂微孢子虫(263,4.28%),与新基因的注释结果一致。分别有2 808、2 932、3 196和2 585条新转录本可注释到KOG、eggNOG、GO和KEGG数据库,这些功能注释信息可进一步完善现有的东方蜜蜂微孢子虫参考基因组的注释。
4 结论
利用高质量的Nanopore全长转录组数据对现有的东方蜜蜂微孢子虫参考基因组序列和功能注释进行了完善,为分子标记研究提供了大量SSR位点,补充了参考基因组的基因和转录本信息。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1038/nature06884URLPMID:18421352 [本文引用: 1]

The association of genetic variation with disease and drug response, and improvements in nucleic acid technologies, have given great optimism for the impact of 'genomic medicine'. However, the formidable size of the diploid human genome, approximately 6 gigabases, has prevented the routine application of sequencing methods to deciphering complete individual human genomes. To realize the full potential of genomics for human health, this limitation must be overcome. Here we report the DNA sequence of a diploid genome of a single individual, James D. Watson, sequenced to 7.4-fold redundancy in two months using massively parallel sequencing in picolitre-size reaction vessels. This sequence was completed in two months at approximately one-hundredth of the cost of traditional capillary electrophoresis methods. Comparison of the sequence to the reference genome led to the identification of 3.3 million single nucleotide polymorphisms, of which 10,654 cause amino-acid substitution within the coding sequence. In addition, we accurately identified small-scale (2-40,000 base pair (bp)) insertion and deletion polymorphism as well as copy number variation resulting in the large-scale gain and loss of chromosomal segments ranging from 26,000 to 1.5 million base pairs. Overall, these results agree well with recent results of sequencing of a single individual by traditional methods. However, in addition to being faster and significantly less expensive, this sequencing technology avoids the arbitrary loss of genomic sequences inherent in random shotgun sequencing by bacterial cloning because it amplifies DNA in a cell-free system. As a result, we further demonstrate the acquisition of novel human sequence, including novel genes not previously identified by traditional genomic sequencing. This is the first genome sequenced by next-generation technologies. Therefore it is a pilot for the future challenges of 'personalized genome sequencing'.
DOI:10.1126/science.1102210URLPMID:15591204 [本文引用: 1]
We report a draft sequence for the genome of the domesticated silkworm (Bombyx mori), covering 90.9% of all known silkworm genes. Our estimated gene count is 18,510, which exceeds the 13,379 genes reported for Drosophila melanogaster. Comparative analyses to fruitfly, mosquito, spider, and butterfly reveal both similarities and differences in gene content.
DOI:10.1038/nature05260URLPMID:17073008 [本文引用: 1]
Here we report the genome sequence of the honeybee Apis mellifera, a key model for social behaviour and essential to global ecology through pollination. Compared with other sequenced insect genomes, the A. mellifera genome has high A+T and CpG contents, lacks major transposon families, evolves more slowly, and is more similar to vertebrates for circadian rhythm, RNA interference and DNA methylation genes, among others. Furthermore, A. mellifera has fewer genes for innate immunity, detoxification enzymes, cuticle-forming proteins and gustatory receptors, more genes for odorant receptors, and novel genes for nectar and pollen utilization, consistent with its ecology and social organization. Compared to Drosophila, genes in early developmental pathways differ in Apis, whereas similarities exist for functions that differ markedly, such as sex determination, brain function and behaviour. Population genetics suggests a novel African origin for the species A. mellifera and insights into whether Africanized bees spread throughout the New World via hybridization or displacement.
DOI:10.1186/gb-2013-14-12-r142URLPMID:24359881 [本文引用: 1]
BACKGROUND: Taxa that harbor natural phenotypic variation are ideal for ecological genomic approaches aimed at understanding how the interplay between genetic and environmental factors can lead to the evolution of complex traits. Lasioglossum albipes is a polymorphic halictid bee that expresses variation in social behavior among populations, and common-garden experiments have suggested that this variation is likely to have a genetic component. RESULTS: We present the L. albipes genome assembly to characterize the genetic and ecological factors associated with the evolution of social behavior. The de novo assembly is comparable to other published social insect genomes, with an N50 scaffold length of 602 kb. Gene families unique to L. albipes are associated with integrin-mediated signaling and DNA-binding domains, and several appear to be expanded in this species, including the glutathione-s-transferases and the inositol monophosphatases. L. albipes has an intact DNA methylation system, and in silico analyses suggest that methylation occurs primarily in exons. Comparisons to other insect genomes indicate that genes associated with metabolism and nucleotide binding undergo accelerated evolution in the halictid lineage. Whole-genome resequencing data from one solitary and one social L. albipes female identify six genes that appear to be rapidly diverging between social forms, including a putative odorant receptor and a cuticular protein. CONCLUSIONS: L. albipes represents a novel genetic model system for understanding the evolution of social behavior. It represents the first published genome sequence of a primitively social insect, thereby facilitating comparative genomic studies across the Hymenoptera as a whole.
DOI:10.1186/1471-2164-16-1URL [本文引用: 1]
DOI:10.1038/nature16548URLPMID:26814964 [本文引用: 1]
Seagrasses colonized the sea on at least three independent occasions to form the basis of one of the most productive and widespread coastal ecosystems on the planet. Here we report the genome of Zostera marina (L.), the first, to our knowledge, marine angiosperm to be fully sequenced. This reveals unique insights into the genomic losses and gains involved in achieving the structural and physiological adaptations required for its marine lifestyle, arguably the most severe habitat shift ever accomplished by flowering plants. Key angiosperm innovations that were lost include the entire repertoire of stomatal genes, genes involved in the synthesis of terpenoids and ethylene signalling, and genes for ultraviolet protection and phytochromes for far-red sensing. Seagrasses have also regained functions enabling them to adjust to full salinity. Their cell walls contain all of the polysaccharides typical of land plants, but also contain polyanionic, low-methylated pectins and sulfated galactans, a feature shared with the cell walls of all macroalgae and that is important for ion homoeostasis, nutrient uptake and O2/CO2 exchange through leaf epidermal cells. The Z. marina genome resource will markedly advance a wide range of functional ecological studies from adaptation of marine ecosystems under climate warming, to unravelling the mechanisms of osmoregulation under high salinities that may further inform our understanding of the evolution of salt tolerance in crop plants.
DOI:10.1038/srep19029URLPMID:26754549 [本文引用: 1]
Orchids make up about 10% of all seed plant species, have great economical value, and are of specific scientific interest because of their renowned flowers and ecological adaptations. Here, we report the first draft genome sequence of a lithophytic orchid, Dendrobium catenatum. We predict 28,910 protein-coding genes, and find evidence of a whole genome duplication shared with Phalaenopsis. We observed the expansion of many resistance-related genes, suggesting a powerful immune system responsible for adaptation to a wide range of ecological niches. We also discovered extensive duplication of genes involved in glucomannan synthase activities, likely related to the synthesis of medicinal polysaccharides. Expansion of MADS-box gene clades ANR1, StMADS11, and MIKC(*), involved in the regulation of development and growth, suggests that these expansions are associated with the astonishing diversity of plant architecture in the genus Dendrobium. On the contrary, members of the type I MADS box gene family are missing, which might explain the loss of the endospermous seed. The findings reported here will be important for future studies into polysaccharide synthesis, adaptations to diverse environments and flower architecture of Orchidaceae.
DOI:10.1111/1462-2920.12883URLPMID:25914091 [本文引用: 4]
Nosema ceranae is a microsporidian pathogen whose infections have been associated with recent global declines in the populations of western honeybees (Apis mellifera). Despite the outstanding economic and ecological threat that N. ceranae may represent for honeybees worldwide, many aspects of its biology, including its mode of reproduction, propagation and ploidy, are either very unclear or unknown. In the present study, we set to gain knowledge in these biological aspects by re-sequencing the genome of eight isolates (i.e. a population of spores isolated from one single beehive) of this species harvested from eight geographically distant beehives, and by investigating their level of polymorphism. Consistent with previous analyses performed using single gene sequences, our analyses uncovered the presence of very high genetic diversity within each isolate, but also very little hive-specific polymorphism. Surprisingly, the nature, location and distribution of this genetic variation suggest that beehives around the globe are infected by a population of N. ceranae cells that may be polyploid (4n or more), and possibly clonal. Lastly, phylogenetic analyses based on genome-wide single-nucleotide polymorphism data extracted from these parasites and mitochondrial sequences from their hosts all failed to support the current geographical structure of our isolates.
DOI:10.1016/j.virusres.2018.07.016URLPMID:30053416 [本文引用: 1]
The haemorrhagic disease of the gill is caused by Cyprinid herpesvirus 2 (CyHV2) infection, which often results in the severe economic losses in the farm of Allogynogenetic crucian carp (ACC). In this study, the genome of CyHV2 strain CyHV2-SY (SY) collected from Sheyang County, Jiangsu Province, China was sequenced. The complete genome of SY is at length of 290,455 bp with 154 potential open reading frames (ORFs) and a terminal direct repeat (TR) of 15,353 bp. Many variations were found by comparison the sequenced CyHV2 genomes. The ORF10, ORF107, ORF156 of SY genome have insertions or deletions of repeat sequences compared with ST-J1 and SY-C1, which may be used as the marks of SY strains. Besides, the promoter sequence analysis showed that 13 of the 150 ORFs have TATA-box elements and 119 of the 150 ORFs have SP1 cis-acting elements in the promoter region (350 bp upstream sequence of the initiation codon ATG). Twenty-eight viral miRNAs candidates were predicted in the CyHV2 genome and expression of 24 virogenes may be regulated by viral miRNAs. Horizontal transfer analysis indicated that 16 and 2 genes in the CyHV2 genome may transfer from the host and bacteria, respectively. By the genome sequencing and sequence mining, our results provided more new clues to further understand CyHV2 genome.
DOI:10.1016/j.gpb.2015.08.002URLPMID:26542840 [本文引用: 1]
Single-molecule, real-time sequencing developed by Pacific BioSciences offers longer read lengths than the second-generation sequencing (SGS) technologies, making it well-suited for unsolved problems in genome, transcriptome, and epigenetics research. The highly-contiguous de novo assemblies using PacBio sequencing can close gaps in current reference assemblies and characterize structural variation (SV) in personal genomes. With longer reads, we can sequence through extended repetitive regions and detect mutations, many of which are associated with diseases. Moreover, PacBio transcriptome sequencing is advantageous for the identification of gene isoforms and facilitates reliable discoveries of novel genes and novel isoforms of annotated genes, due to its ability to sequence full-length transcripts or fragments with significant lengths. Additionally, PacBio's sequencing technique provides information that is useful for the direct detection of base modifications, such as methylation. In addition to using PacBio sequencing alone, many hybrid sequencing strategies have been developed to make use of more accurate short reads in conjunction with PacBio long reads. In general, hybrid sequencing strategies are more affordable and scalable especially for small-size laboratories than using PacBio Sequencing alone. The advent of PacBio sequencing has made available much information that could not be obtained via SGS alone.
URLPMID:29495964 [本文引用: 2]
DOI:10.1038/ng.2875URL [本文引用: 1]
The hookworm Necator americanus is the predominant soil-transmitted human parasite. Adult worms feed on blood in the small intestine, causing iron-deficiency anemia, malnutrition, growth and development stunting in children, and severe morbidity and mortality during pregnancy in women. We report sequencing and assembly of the N. americanus genome (244 Mb, 19,151 genes). Characterization of this first hookworm genome sequence identified genes orchestrating the hookworm's invasion of the human host, genes involved in blood feeding and development, and genes encoding proteins that represent new potential drug targets against hookworms. N. americanus has undergone a considerable and unique expansion of immunomodulator proteins, some of which we highlight as potential treatments against inflammatory diseases. We also used a protein microarray to demonstrate a postgenomic application of the hookworm genome sequence. This genome provides an invaluable resource to boost ongoing efforts toward fundamental and applied postgenomic research, including the development of new methods to control hookworm and human immunological diseases.
DOI:10.1038/ng.769URLPMID:21336279 [本文引用: 1]
Genome evolution studies for the phylum Nematoda have been limited by focusing on comparisons involving Caenorhabditis elegans. We report a draft genome sequence of Trichinella spiralis, a food-borne zoonotic parasite, which is the most common cause of human trichinellosis. This parasitic nematode is an extant member of a clade that diverged early in the evolution of the phylum, enabling identification of archetypical genes and molecular signatures exclusive to nematodes. We sequenced the 64-Mb nuclear genome, which is estimated to contain 15,808 protein-coding genes, at approximately 35-fold coverage using whole-genome shotgun and hierarchal map-assisted sequencing. Comparative genome analyses support intrachromosomal rearrangements across the phylum, disproportionate numbers of protein family deaths over births in parasitic compared to a non-parasitic nematode and a preponderance of gene-loss and -gain events in nematodes relative to Drosophila melanogaster. This genome sequence and the identified pan-phylum characteristics will contribute to genome evolution studies of Nematoda as well as strategies to combat global parasites of humans, food animals and crops.
DOI:10.1038/nmeth.3454URLPMID:26121404 [本文引用: 1]
We present the first comprehensive analysis of a diploid human genome that combines single-molecule sequencing with single-molecule genome maps. Our hybrid assembly markedly improves upon the contiguity observed from traditional shotgun sequencing approaches, with scaffold N50 values approaching 30 Mb, and we identified complex structural variants (SVs) missed by other high-throughput approaches. Furthermore, by combining Illumina short-read data with long reads, we phased both single-nucleotide variants and SVs, generating haplotypes with over 99% consistency with previous trio-based studies. Our work shows that it is now possible to integrate single-molecule and high-throughput sequence data to generate de novo assembled genomes that approach reference quality.
DOI:10.1016/j.celrep.2018.05.014URLPMID:29874592 [本文引用: 1]
Ants are an emerging model system for neuroepigenetics, as embryos with virtually identical genomes develop into different adult castes that display diverse physiology, morphology, and behavior. Although a number of ant genomes have been sequenced to date, their draft quality is an obstacle to sophisticated analyses of epigenetic gene regulation. We reassembled de novo high-quality genomes for two ant species, Camponotus floridanus and Harpegnathos saltator. Using long reads enabled us to span large repetitive regions and improve genome contiguity, leading to comprehensive and accurate protein-coding annotations that facilitated the identification of a Gp-9-like gene as differentially expressed in Harpegnathos castes. The new assemblies also enabled us to annotate long non-coding RNAs in ants, revealing caste-, brain-, and developmental-stage-specific long non-coding RNAs (lncRNAs) in Harpegnathos. These upgraded genomes, along with the new gene annotations, will aid future efforts to identify epigenetic mechanisms of phenotypic and behavioral plasticity in ants.
URLPMID:28581499 [本文引用: 1]
DOI:10.1186/s12864-015-2257-yURL [本文引用: 1]
DOI:10.1371/journal.ppat.1000466URLPMID:19503607 [本文引用: 1]
Recent steep declines in honey bee health have severely impacted the beekeeping industry, presenting new risks for agricultural commodities that depend on insect pollination. Honey bee declines could reflect increased pressures from parasites and pathogens. The incidence of the microsporidian pathogen Nosema ceranae has increased significantly in the past decade. Here we present a draft assembly (7.86 MB) of the N. ceranae genome derived from pyrosequence data, including initial gene models and genomic comparisons with other members of this highly derived fungal lineage. N. ceranae has a strongly AT-biased genome (74% A+T) and a diversity of repetitive elements, complicating the assembly. Of 2,614 predicted protein-coding sequences, we conservatively estimate that 1,366 have homologs in the microsporidian Encephalitozoon cuniculi, the most closely related published genome sequence. We identify genes conserved among microsporidia that lack clear homology outside this group, which are of special interest as potential virulence factors in this group of obligate parasites. A substantial fraction of the diminutive N. ceranae proteome consists of novel and transposable-element proteins. For a majority of well-supported gene models, a conserved sense-strand motif can be found within 15 bases upstream of the start codon; a previously uncharacterized version of this motif is also present in E. cuniculi. These comparisons provide insight into the architecture, regulation, and evolution of microsporidian genomes, and will drive investigations into honey bee-Nosema interactions.
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
URLPMID:12589540 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1093/gigascience/gix093URLPMID:29048480 [本文引用: 1]
Ginseng, which contains ginsenosides as bioactive compounds, has been regarded as an important traditional medicine for several millennia. However, the genetic background of ginseng remains poorly understood, partly because of the plant's large and complex genome composition. We report the entire genome sequence of Panax ginseng using next-generation sequencing. The 3.5-Gb nucleotide sequence contains more than 60% repeats and encodes 42 006 predicted genes. Twenty-two transcriptome datasets and mass spectrometry images of ginseng roots were adopted to precisely quantify the functional genes. Thirty-one genes were identified to be involved in the mevalonic acid pathway. Eight of these genes were annotated as 3-hydroxy-3-methylglutaryl-CoA reductases, which displayed diverse structures and expression characteristics. A total of 225 UDP-glycosyltransferases (UGTs) were identified, and these UGTs accounted for one of the largest gene families of ginseng. Tandem repeats contributed to the duplication and divergence of UGTs. Molecular modeling of UGTs in the 71st, 74th, and 94th families revealed a regiospecific conserved motif located at the N-terminus. Molecular docking predicted that this motif captures ginsenoside precursors. The ginseng genome represents a valuable resource for understanding and improving the breeding, cultivation, and synthesis biology of this key herb.
DOI:10.1007/s13592-018-0593-zURL [本文引用: 1]
URLPMID:22538991 [本文引用: 1]
[本文引用: 2]
[本文引用: 2]
URLPMID:21237902 [本文引用: 1]
DOI:10.1046/j.0962-1083.2001.01418.xURLPMID:11903900 [本文引用: 1]
In the last few years microsatellites have become one of the most popular molecular markers used with applications in many different fields. High polymorphism and the relative ease of scoring represent the two major features that make microsatellites of large interest for many genetic studies. The major drawback of microsatellites is that they need to be isolated de novo from species that are being examined for the first time. The aim of the present paper is to review the various methods of microsatellite isolation described in the literature with the purpose of providing useful guidelines in making appropriate choices among the large number of currently available options. In addition, we propose a fast and easy protocol which is a combination of different published methods.
URL [本文引用: 2]
黄粉虫Tenebrio molitor作为理想的模式研究生物, 虽然已围绕该昆虫在多个研究领域开展了诸多研究, 但是有关其分子和遗传方面的研究仍知之甚少。为此, 本研究基于前期构建的黄粉甲转录组数据库, 成功发掘获得1 249个微卫星序列。其中, 单碱基或三碱基序重复列最多, 分别占44.44%和41.15%; A/T型重复序列出现频率最高, 占42.70%。除单核苷酸重复序列外, 重复单元的重复次数以5次最多, 占30.90%。基于鉴定获得的微卫星序列, 共设计获得1 004对微卫星引物, 而且每对引物还设计了5对替代引物。研究获得的微卫星引物将有助于今后开展黄粉甲功能和比较基因组学方面的研究。
URL [本文引用: 2]
黄粉虫Tenebrio molitor作为理想的模式研究生物, 虽然已围绕该昆虫在多个研究领域开展了诸多研究, 但是有关其分子和遗传方面的研究仍知之甚少。为此, 本研究基于前期构建的黄粉甲转录组数据库, 成功发掘获得1 249个微卫星序列。其中, 单碱基或三碱基序重复列最多, 分别占44.44%和41.15%; A/T型重复序列出现频率最高, 占42.70%。除单核苷酸重复序列外, 重复单元的重复次数以5次最多, 占30.90%。基于鉴定获得的微卫星序列, 共设计获得1 004对微卫星引物, 而且每对引物还设计了5对替代引物。研究获得的微卫星引物将有助于今后开展黄粉甲功能和比较基因组学方面的研究。
URL [本文引用: 2]
【目的】扶桑绵粉蚧Phenacoccus solenopsis Tinsley是我国重要的检疫性害虫。简单重复序列(simple sequence repeat,SSR)研究在遗传图谱和物理图谱的构建、分子标记辅助育种、品种鉴定、基因定位、遗传多样性、动植物分类和进化等方面具有重要意义。筛选的SSR引物将为扶桑绵粉蚧遗传多样性分析、进化分析及入侵生物学等奠定基础。【方法】利用高通量搜索的方法对扶桑绵粉蚧转录组中28 120条unigenes的数据进行搜索。【结果】共找到1 781个SSR位点。扶桑绵粉蚧转录组中SSRs的主要重复类型是单核苷酸重复,占SSR总数的89.44%;其次是三核苷酸重复,占SSR总数的7.52%。单核苷酸重复里主要是A/T基序,占了总量的87.42%。基于筛选的SSRs,运用Primer 3软件进行引物的批量设计,共有481个unigenes成功设计引物,共设计出1 228对引物。【结论】研究表明利用扶桑绵粉蚧转录组数据开发SSR标记是可行的,本研究开发的引物将为扶桑绵粉蚧遗传多样性分析、进化分析及入侵生物学等奠定基础。
URL [本文引用: 2]
【目的】扶桑绵粉蚧Phenacoccus solenopsis Tinsley是我国重要的检疫性害虫。简单重复序列(simple sequence repeat,SSR)研究在遗传图谱和物理图谱的构建、分子标记辅助育种、品种鉴定、基因定位、遗传多样性、动植物分类和进化等方面具有重要意义。筛选的SSR引物将为扶桑绵粉蚧遗传多样性分析、进化分析及入侵生物学等奠定基础。【方法】利用高通量搜索的方法对扶桑绵粉蚧转录组中28 120条unigenes的数据进行搜索。【结果】共找到1 781个SSR位点。扶桑绵粉蚧转录组中SSRs的主要重复类型是单核苷酸重复,占SSR总数的89.44%;其次是三核苷酸重复,占SSR总数的7.52%。单核苷酸重复里主要是A/T基序,占了总量的87.42%。基于筛选的SSRs,运用Primer 3软件进行引物的批量设计,共有481个unigenes成功设计引物,共设计出1 228对引物。【结论】研究表明利用扶桑绵粉蚧转录组数据开发SSR标记是可行的,本研究开发的引物将为扶桑绵粉蚧遗传多样性分析、进化分析及入侵生物学等奠定基础。
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
URL [本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}