 ,1,2, 刘威生1, 王兴东1, 孙斌1, 刘修丽1, 杨艳敏1, 魏鑫1, 杨玉春1, 张舵1, 刘成,1, 李天忠,2
,1,2, 刘威生1, 王兴东1, 孙斌1, 刘修丽1, 杨艳敏1, 魏鑫1, 杨玉春1, 张舵1, 刘成,1, 李天忠,2Identification of F1 Hybrids in Blueberry (Vaccinium corymbosum L.) Based on Specific-Locus Amplified Fragment Sequencing (SLAF-seq)
LIU YouChun,1,2, LIU WeiSheng1, WANG XingDong1, SUN Bin1, LIU XiuLi1, YANG YanMin1, WEI Xin1, YANG YuChun1, ZHANG Duo1, LIU Cheng,1, LI TianZhong,2通讯作者:
责任编辑: 赵伶俐
收稿日期:2020-04-26接受日期:2020-07-30网络出版日期:2021-01-16
| 基金资助: |
Received:2020-04-26Accepted:2020-07-30Online:2021-01-16
作者简介 About authors
刘有春,Tel:18641713730,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (794KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
刘有春, 刘威生, 王兴东, 孙斌, 刘修丽, 杨艳敏, 魏鑫, 杨玉春, 张舵, 刘成, 李天忠. 基于简化基因组测序的越橘杂交后代鉴定[J]. 中国农业科学, 2021, 54(2): 370-378 doi:10.3864/j.issn.0578-1752.2021.02.012
LIU YouChun, LIU WeiSheng, WANG XingDong, SUN Bin, LIU XiuLi, YANG YanMin, WEI Xin, YANG YuChun, ZHANG Duo, LIU Cheng, LI TianZhong.
开放科学(资源服务)标识码(OSID):

0 引言
【研究意义】在遗传育种研究中,获得继承双亲基因的真杂种后代是进行品种改良、遗传分析及遗传图谱构建等研究的前提和基础[1],为了使杂交后代如实反映双亲和群体的遗传特征,初期对杂交后代的真实性鉴定十分必要,以避免或降低非杂交后代对群体的影响。在育种实践中,出现非杂交后代的可能性有如下几种:1)异花授粉中非选定父本花粉混入,导致此类后代缺少选定父本的遗传信息而混入其他材料的遗传信息;2)母本植株具有一定的自花受精习性,杂交过程中人工去雄不及时、不彻底可能会产生自交后代;3)杂交种子收集及幼苗管理过程中误引入非双亲杂交后代。由上述原因导致的非杂交后代混杂在群体中,在植物形态特征上不易辨别。而基于DNA的变异分析不受外界环境影响,能真实反映分离群体分子水平上的遗传信息,可靠性高。因此,群体在基因组水平上反映的遗传差异可借鉴于非杂交个体鉴别研究中,但筛选鉴别策略至关重要。【前人研究进展】对于植物,早期主要通过植株形态学、细胞学以及同工酶进行杂交后代的鉴定,但均存在一定不足之处,如形态学鉴定周期长、易受环境影响、准确率低,细胞学鉴定程序繁琐、分辨率低[2],同工酶则受酶种类限制不能反映全部结构基因的信息,存在基因位点少、多态性水平低等[3]问题。分子标记技术的发展使得杂种鉴定的准确性大幅提高,AFLP[4]、RAPD[5,6]、SRAP[7]、SSR[1,8-9]等分子标记已应用于果树非杂交后代鉴定工作中,但这一类分子标记已均存在自身通量小、耗时耗力、成本高等局限性。此类技术主要以亲本基因型作为判断依据,即通过亲本DNA的扩增产物多态性(片段长度或碱基差异)筛选出具有分辨能力的分子标记,并扫描杂交群体的基因型,经比对统计后代异于亲本的等位基因类型开展非杂交后代差异分析。以SNP为代表的第三代分子标记技术,相对于第一、二代分子标记,具有多态性高、能广泛分布于全基因组的特点,可鉴别更丰富的遗传信息,韩燕等[10]建立了利用亲本多态SNP位点设计引物,通过凝胶电泳鉴定花生F1代的方法。ZHANG等[11]、NIU等[12]通过检测样品中出现的非亲本类型异常SLAF标签鉴别无参考基因组物种的非杂交后代。然而,上述基于PCR扩增的DNA序列长度差异分析存在以下几个可能的误判情况:1)PCR扩增过程可能存在一定的非特异扩增,造成基因型的辨识困难(特别通过电泳凝胶成像);2)片段长短一致的扩增产物可能序列不同,所含遗传信息的差异难以检测,导致非杂交后代通过检测混入群体;3)由于序列的缺失、插入和重排等突变事件可以同时发生在亲本与其后代(包括非杂交后代),使鉴定试验存在误差。对于SNP标记可存在的误判则多来自一种碱基或核苷酸被另一种替换,或者因碱基插入或缺失等点突变事件,造成基因型变化。上述情况在试验操作中存在偶然性、随机性,单一标记位点造成的误差权重可随分子标记数量的增加而相应降低。此类情况若发生在分子标记数量较少的试验中,会在一定程度上干扰最终判断。而基于异常SLAF标签进行非杂交后代筛选的方法[11,12],其SLAF标签的产生基于酶切产生SLAF片段的相似性,无基因组信息参照,且相似性聚类和异常SLAF标签的判定很大程度上受阈值影响。【本研究切入点】随着生物技术的快速发展,NGS(next generation sequencing)测序成本不断降低,三代测序和Hi-C技术也广泛应用于遗传群体测序,如何从测序产生的SNP大数据中准确鉴别非杂交后代十分重要,相关方法鲜见报道。由于非杂交后代所含遗传信息不源于或部分源于亲本(如自交后代),会呈现出遗传关系较远和等位基因分离异常等现象。因此,本研究以多年生果树越橘(Vaccinium corymbosum)的正、反交F1代群体为研究对象,通过高通量简化测序获取大量样本(亲本和子代)基因组序列和遗传变异信息,基于子代特有稀有等位变异为核心,重点揭示子代与群体间(不以亲本为标准)的遗传关系以鉴定非杂交后代。【拟解决的关键问题】探索适合于高通量测序数据的快速、准确的非杂交后代鉴别方法,排除假阳性样本干扰,为遗传图谱构建、性状定位、遗传育种及高通量分子标记开发等相关研究奠定基础。1 材料与方法
测序试验于2019年在北京百迈客生物科技有限公司进行,数据统计及验证分析于2020年在辽宁省果树科学研究所完成。1.1 试验材料与DNA提取
供试群体取自辽宁省果树科学研究所蓝莓杂交圃,为南高丛越橘品种‘N6’(Vaccinium. corymbosum SHB)和北高丛越橘品种‘Berkeley’(V. corymbosum NHB)的杂交F1后代,其中正交组合‘Berkeley’בN6’群体133株,群体代号FM_133;反交组合‘N6’× ‘Berkeley’群体185株,群体代号MF_185。试材采集群体及亲本幼嫩叶片液氮速冻后存于-80℃冰箱备用,CTAB法[13]提取基因组DNA,用Nanodrop 2000C(Thermo Fisher)和Qubit 2.0荧光计(Thermo Fisher)进行DNA的质量和浓度检测,以确保所提基因组DNA质量达到测序文库构建要求:OD260与OD280的比值分布在1.8—2.0,DNA浓度达到30 ng?μL-1。1.2 越橘基因组遗传变异数据收集
1.2.1 DNA酶切预测与测序文库构建 为了保证酶切片段在基因组上分布均匀,同时避开重复序列区域,利用越橘近缘物种蔓越莓参考基因组[14](https:// www.ncbi.nlm.nih.Gov /bioproject/PRJNA245813)随机选取2个亲本和10个子代,以Rsa I+Hae III、Hae III+Hpy 166II和Hpy 166II三种酶切方案进行电子酶切预测试验,根据开发的标签数等确定酶切方案。供试亲本及群体DNA经ddH2O稀释到100 ng?μL-1浓度后,利用确定的酶切组合(HaeIII+Hpy166II)双酶切并过夜,酶切产物在37℃下用Klenow 片段(3′→5′)(NEB)和dATP进行末端加A,之后T4连接酶连接区分样品的标签(barcode)和测序接头序列。常规PCR进行片段扩增,上游引物为5′-AATGATACGGC GACCACCGA-3′,下游引物为5′-CAAGCAGAAGA CGGCATACG-3′(Life Technologies,Gaithersburg,MD,United States),扩增循环数为8。最后利用试剂盒QIAquick gel extraction kit(Qiagen,Hilden,Germany)进行切胶纯化,切胶范围为314—444 bp。切胶后将文库混合,加入一条流动槽(flowcell)中,cBot进行cluster生成,进行Illumina Hiseq 2500(Illumina,Inc.,San Diego,CA,United States)高通量测序。为了监控建库有效性,本试验以模式物种水稻(Oryza sativa)(http://rice. plantbiology.msu.edu/)为对照,同步进行平行试验。1.2.2 基于越橘参考基因组的SNP标记获取 过滤后的样本序列(clean reads)经BWA-0.7.10软件比对到四倍体越橘参考基因组[15](http://gigadb.org/dataset/ 100537),用Picard 1.118软件http://picard.sourceforge. net)标记出来。用GATK 3.8软件[16]对碱基测序质量重新校正、序列重新比对,根据标准过滤参数分别对越橘亲本和正、反交群体(FM133和MF185)进行基因分型,获得SNP基因型数据。所有分析步骤按照GATK最优的执行方法进行操作(https://www. broadinstitute.org/partnerships/education/broade/best- practices-variant-calling-gatk-1)。设置SNP次等位基因频率(Minor allele frequency)在0.05处为阈值(低于5%则视为稀有位点),将SNP基因型数据划分到MAF>0.05 和MAF<0.05两个数据集,整理保留各自多态性位点进行后续相关分析。
1.3 越橘杂交群体中非杂交后代鉴别
1.3.1 供试群体遗传关系分析 利用GenoDive version 3.03[17]对SNP基因型数据(MAF>0.05)进行处理和分析。为避免缺失数据导致的偏差(bias)影响,数据经Filling-in Missing Data功能随机选取已有等位基因进行填充(Imputation)。采用Amova方法[18,19]对供试群体样品的SNP次等位基因进行K- Means聚类分析,设置模拟退火(Simulated Annealing,SA)算法[20]为50 000步,重复20次。主成分分析(principal component analysis,PCA)采用计算协方差方式对供试越橘样品进行统计,并整合K-Means聚类结果通过“scatterplot3d”[21]R分析绘制坐标图。1.3.2 供试群体稀有等位变异分析 统计符合MAF<0.05条件的SNP基因型数据中杂交后代个体稀有等位变异总数(Total rare-alleles)和个体特有的稀有等位变异数(Private rare-alleles)。利用“ggplot2”R分析包[22]的箱图(geom_boxplot)功能分析计数的分布与异常个体标注。
1.4 越橘非杂交后代验证
经上述分析获得的非杂交后代通过亲本特定的基因型进行验证分析。为避免受稀有等位变异干扰,笔者应用MAF>0.05的SNP数据筛选越橘正、反交群体中亲本为纯合显性的基因型数据,并统计后代群体中出现异于亲本基因型的SNP位点比率,利用箱图统计群体中后代异于母本(或父本)基因型的SNP位点比率,筛选各自的离群个体,与已获得的非杂交后代进行比较。2 结果
2.1 测序数据统计与评估
测序共获得 330.06 Mb reads(包含65.89 Gb)数据,平均Q30为95.04%,平均GC含量为39.72%,Q30和GC含量在供试材料间仅小幅波动(表1),说明GC分布正常,测序质量好,适合分析。Table 1
表1
表1越橘样品测序数据统计表
Table 1
| 样品名 Sample ID | 总测序条数 Total reads | 总碱基数量 Total bases | 高质量reads百分比 Q30 percentage (%) | GC含量百分比 GC percentage (%) |
|---|---|---|---|---|
| Berkeley | 10 350 711 | 2 067 990 444 | 95.12 | 39.14 |
| N6 | 10 914 992 | 2 180 687 340 | 94.88 | 39.45 |
| 杂交后代 Offspring | 971 057 | 193 850 201 | 95.04 | 39.72 |
| 水稻(对照样品) Rice (Control) | 1 047 399 | 209 302 386 | 94.59 | 40.46 |
| 总和 Total | 330 061 690 | 65 893 041 734 | 95.04 | 39.72 |
新窗口打开|下载CSV
2.2 基于参考基因组的SNP 标记开发
基于2019年发表的越橘参考基因组,对供试318个F1后代和2个亲本样本进行序列分值校正、局部重比对、SNP和INdel的发掘与基因分型。在MAF>0.05水平下,在MF_185群体和FM_133群体分别鉴定到70 243个和111 527个SNP,在MAF<0.05水平下分别鉴定到3 200个和3 324个SNP(表2)。Table 2
表2
表2不同越橘杂交群体中SNP标记数量统计
Table 2
| 越橘杂交 群体 Population | SNP标记数量(MAF>0.05) Number of SNPs (MAF > 0.05) | SNP标记数量(MAF < 0.05) Number of SNPs (MAF < 0.05) |
|---|---|---|
| MF185 | 70 243 | 3 200 |
| FM133 | 111 527 | 3 324 |
新窗口打开|下载CSV
2.3 基于主成分分析和K-means聚类分析的非杂交后代筛选
利用MAF>0.05的SNP数据集,基于协方差矩阵的主成分分析表明,MF_185群体的亲本‘Berkeley’处于x轴右侧,距离亲本‘N6’与杂交群体均较远,多数杂交后代集中在x轴左侧(图1-A),其后代‘H194-180’偏离程度较远。FM_133群体中除H194-295和H194-297后代之外,均紧凑地聚在x轴左侧(图1-C)。利用K-means聚类对离群点敏感的特性,比较k=2和k=3时的聚类结果筛选杂交群体中的离群点。对于MF_185群体,k=2时,亲本分别处于不同聚类群(图1-A);k=3时,后代H194-169、H194-126和H194-180不同于亲本与其他后代,归为单独的聚类群(图1-B,cluster3,绿色),为离群点,视为非杂交后代。而对于FM_133群体,k=2时,亲本处在同一聚类群(图1-C);k=3时,后代H194-297不同于亲本与其他后代,归为单独的聚类群(图1-D,cluster3,绿色)为离群点,视为非杂交后代。图1
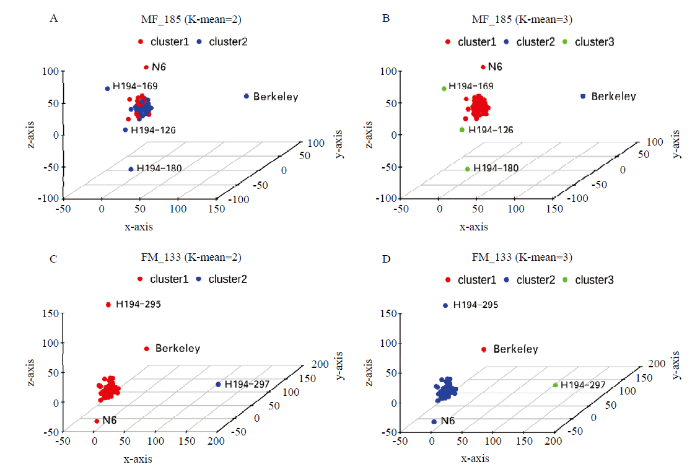 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1越橘不同杂交群体的主坐标分析(PCA)
A—B:MF185群体;C—D:FM133群体。聚群(cluster)1、2和3分别代表K-means在k=2和k=3的聚类结果
Fig. 1Principal component analysis in MF185 and FM133 populations in blueberry
A-B:MF185 population; C-D:FM133 population. Cluster1, 2 and 3 corresponded to K-means clustering result at k=2 and 3
2.4 基于稀有等位变异分析的非杂交后代筛选
双等位SNP数据(biallelic,MAF<0.05)可在正交FM_133群体和反交MF_185群体中分别产生6 648个和6 400个等位变异。试验分别统计个体稀有等位变异总数和个体特有的稀有等位变异数。个体在群体中产生的全部稀有等位变异数如图2-A所示,FM_133群体的个体稀有等位变异总数普遍高于FM_185,范围在2 594—4 802个,其中H194-300、H194-298和H194-231个体稀有等位变异数最多,分别为4 802个、4 578个和4 556个,且偏离群体,FM185群体中个体稀有等位变异数范围在2 098—3 606个,无离群个体。个体在群体中产生的稀有等位变异中异于其他群体成员及亲本的特有变异数如图2-B所示,FM_133群体中出现1个离群个体,即H194-297,特有的稀有等位变异数379个,与群体明显偏离;MF_185群体中离群个体共计9个,分别是H194-175、H194-169、H194-179、H194-126、H194-180,H194-107、H194-123、H194-170、H194-174,特有的稀有等位变异数为193—271个,视为非杂交后代。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2稀有等位变异在不同杂交群体中的分布与其异常值
●代表个体,*代表离群个体。下同
Fig. 2Boxplots analysis of total and private rare-allele in different populations
● represents each sample, * indicates outliers apart from population. The same as below
2.5 非杂交后代的基因型验证
筛选双亲基因型为纯合显性的SNP位点进行验证。统计结果显示,FM_133群体包含17 646个母本纯合SNP位点和20 906个父本纯合SNP位点,共占群体总SNP数据的34.56%;MF_185群体包含12 351个母本纯合SNP位点和15 012个父本纯合SNP位点,共占群体总SNP数据的38.95%。基于该SNP数据集,利用箱图统计群体后代拥有异于母本(或父本)基因型的SNP位点比率,筛选各自的离群个体(图3)。基于亲本纯合SNP位点中的异常SNP位点,正交FM_133群体中H194-297为离群个体,与图2-B中FM_133群体离群点一致;MF_185群体中,H194-169,H194-180、H194-175、H194-126、H194-107、H194-174、H194-173、H194-170、H194-160及H194-179为离群个体,与图2-B中MF_185鉴定结果相比,除H194-123外,其余非杂交后代样品与验证结果离群个体一致,即利用双亲基因型为纯合显性的SNP位点中异常SNP位点准确验证了基于等位基因频率鉴定的非杂交后代。图3
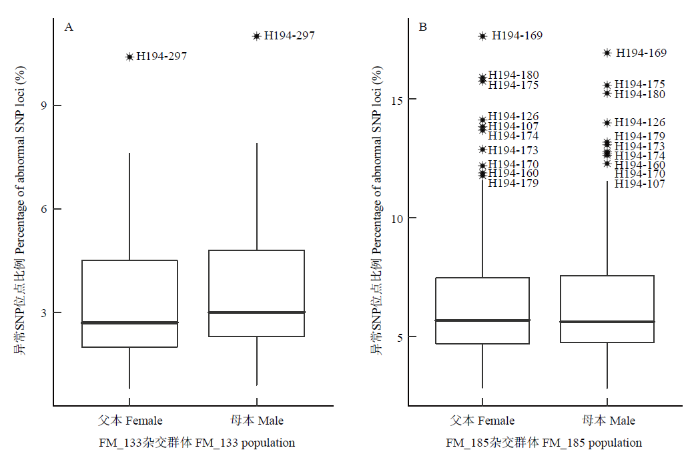 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3基于亲本基因型为纯合显性SNP的非杂交后代验证
Fig. 3The false-hybrids verification based on homozygous dominant SNP of parental genotype
3 讨论
3.1 利用群体的遗传关系鉴别非杂交后代
有关植物全基因组水平的遗传多样性、群体结构和亲缘关系等分析中[23,24,25],需要对次要等位基因频率进行降噪处理(MAF>0.05),减少低频率等位变异对数据整体造成的偏差影响。本研究中除噪后的SNP数据保留大量多态性位点,可以较为可靠地反映群体的遗传差异,并较为保守地用来筛选离群个体。采取PCA来推测离群点(outlier)是较为普遍和有效的办法,广泛地应用在各种数据集和样品集的过滤环节[26,27,28],是进一步数据分析的重要前提。本研究经PCA解析两个越橘正、反交群体的遗传差异结果表明,亲本‘N6’和‘Berkeley’无论在越橘的正交群体还是反交群体中的遗传差异均较明显,而它们的后代个体多集中分布于两个亲本之间,遗传差异有限。该结果有利于离群个体的筛选,可以较容易地通过观察PCA坐标中个体距离亲本和绝大多数后代的聚集位置远近来推测。然而,基于单一个体间协方差矩阵的PCA分析,不足以判断离群个体是否为非杂交后代。为此,笔者增加一种迭代求解的均值聚类办法,通过解析个体间在遗传水平上的相似性并归入差异群组,结合PCA结果进一步对离群个体加以讨论。K-means聚类对离群点较为敏感[29],对于MAF>0.05的SNP数据集,在PCA和K-means聚类分析中,当k=2转换为k=3时,群体MF185中的‘H194-169’‘H194-126’和‘H194-180’及群体FM133中的‘H194-297’单独成为聚类群(绿色)(图1),均表现出“与众不同”,因此视为非杂交后代。此外,自交个体因只携带母本遗传信息,不属于双亲的杂交后代,但在聚类时,遗传上更倾向母本,不易形成明显的离群点而被误为杂交后代,分析中须特别注意。本试验中越橘属于异花结实植物[30],也有报道发现越橘存在一定的自花结实现象[31],笔者对供试正、反交组合的亲本‘N6’和‘Berkeley’进行了自花结实性验证,发现均可自花结实并产生自交种子和自交后代。本研究正、反杂交群体的PCA和K-means聚类中并未发现与母本遗传十分相近的个体,说明供试群体中不存在由于自交产生的假杂种。
3.2 非杂交后代的特有稀有等位变异特征及鉴别
本研究将低频率的等位基因(MAF<0.05)视为稀有等位变异。稀有等位变异的形成存在几个可能:1)亲本的等位基因型在杂交后代的分离,出现严重偏分离情况(频率小于0.05);2)由于碱基点突变造成的低频率异常基因型出现;3)非杂交后代自身携带的未知(新)等位基因型混入。前两种可能中,遗传偏分离情况主要发生在整个群体,由双亲间遗传分化程度、基因相互作用和环境因素等影响[32];而植物组织细胞内的DNA碱基突变既可发生在世代繁衍和一般生长周期阶段,具有随机性和低频性;第三种情况的发生通常会包含第二种情况,因稀有等位变异的基因型来自外部,所以非杂交后代会携带大量区别于亲本和其他后代的特有稀有等位变异。本研究在群体FM_133和MF_185中分别检测到1个和9个个体,其特有的稀有等位变异多且偏离群体(箱体之外)(图2-B),视为非杂交后代。综上所述,PCA和K-means聚类分析对MAF>0.05数据集鉴定的4个非杂交后代全部重现在MAF<0.05数据集中个体特有稀有等位变异的鉴定结果中,说明以上两种鉴定方法均可有效用于越橘群体非杂交后代筛选,后者鉴定条件更严格。
3.3 应用群体稀有等位变异鉴定非杂交后代的可行性
本研究将简化测序基于四倍体越橘参考基因组比对,获得基因组水平的SNP基因型数据,引入稀有等位变异作为判断标准,利用PCA和K-means聚类等不同方法着重解析群体子代遗传差异和亲缘关系,通过箱图反映离散个体从而鉴定非杂交后代。鉴定结果在利用亲本纯合显性SNP标记(基于群体与亲本遗传差异)进行验证时,绝大多数基于稀有等位变异的非杂交后代也被鉴定为离群个体,即准确通过验证,充分证明了本试验采用的基于基因组SNP基因型的个体稀有等位变异分析策略适用于越橘杂交群体的非杂交后代筛选和鉴定,这与前人[4,5,6,7,8,9,10]完全依赖于亲本基因型的鉴定策略不同。此外,由于稀有等位变异的非杂交后代鉴定策略是基于群体间的遗传差异进行分析,所以该策略可直接应用于亲本未知的群体进行非杂交后代鉴定,排除非该群体的后代。借助于参考基因组可靠、准确的基因分型,根据本研究所采用的策略和方法可简单、有效地对杂交群体的大规模测序数据进行质控处理,排除假阳性干扰。4 结论
本研究基于四倍体越橘参考基因组,利用个体特有的稀有等位变异分析和遗传关系分析的策略从不同角度反映数据特征,共鉴定出10个离群个体,确定为非杂交后代,在未来越橘植物的遗传图谱构建、性状定位和遗传育种等研究中应给予剔除或谨慎使用。通过该策略鉴定的非杂交后代绝大多数与基于双亲纯合显性SNP位点的验证结果一致,因此,对于有参考基因组物种的杂交群体,利用基于测序的SNP次等位基因频率(MAF)数据集,采用遗传关系和个体特有的稀有等位变异分析方法,从不同角度反映群体子代间的遗传关系以鉴别离群个体,是鉴定群体真假杂种的一种有效策略。致谢
感谢北京科技大学刘冬成研究员和中国农业大学许语辉博士对英文摘要的校正,感谢辽宁省果树科学研究所刘硕博士在数据分析中的指导和建议。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 2]
URL [本文引用: 2]
URL [本文引用: 2]
URL [本文引用: 2]
[本文引用: 2]
[本文引用: 2]
URL [本文引用: 2]
URL [本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
URLPMID:28725940 [本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:1644282 [本文引用: 1]
[本文引用: 1]
DOI:10.1126/science.220.4598.671URLPMID:17813860 [本文引用: 1]

There is a deep and useful connection between statistical mechanics (the behavior of systems with many degrees of freedom in thermal equilibrium at a finite temperature) and multivariate or combinatorial optimization (finding the minimum of a given function depending on many parameters). A detailed analogy with annealing in solids provides a framework for optimization of the properties of very large and complex systems. This connection to statistical mechanics exposes new information and provides an unfamiliar perspective on traditional optimization problems and methods.
URLPMID:24545891 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
[本文引用: 1]
URLPMID:27093601 [本文引用: 1]
URLPMID:28442746 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.16288/j.yczz.16-084URLPMID:27644741 [本文引用: 1]
During natural hybridization and introgression, offspring may obtain alleles from both parents unevenly, resulting in allelic segregation that significantly deviated from the Mendelian ratio, referred to as segregation distortion (SD). Segregation distortion is a common phenomenon and a number of factors can influence the ratio and pattern of segregation distortion in hybrid descendants. However, knowledge on the evolutionary implication of SD is still very limited. In this review, we summarize the current knowledge regarding mechanisms of SD such as genetic interaction, genetic divergence, cytoplasmic background, and environmental effect, and analyze its implication for evolution. In short, SD affects evolutionary potential, sex ratio, genetic diversity and maintenance of stable genetic divergence. Therefore, the effect of transgene on the evolutionary potential of wild relative populations through SD and the changes of the pattern of SD in continuous generations should be paid attention to in future studies.
DOI:10.16288/j.yczz.16-084URLPMID:27644741 [本文引用: 1]
During natural hybridization and introgression, offspring may obtain alleles from both parents unevenly, resulting in allelic segregation that significantly deviated from the Mendelian ratio, referred to as segregation distortion (SD). Segregation distortion is a common phenomenon and a number of factors can influence the ratio and pattern of segregation distortion in hybrid descendants. However, knowledge on the evolutionary implication of SD is still very limited. In this review, we summarize the current knowledge regarding mechanisms of SD such as genetic interaction, genetic divergence, cytoplasmic background, and environmental effect, and analyze its implication for evolution. In short, SD affects evolutionary potential, sex ratio, genetic diversity and maintenance of stable genetic divergence. Therefore, the effect of transgene on the evolutionary potential of wild relative populations through SD and the changes of the pattern of SD in continuous generations should be paid attention to in future studies.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}