 ,1
,1The Genetic Diversity, Pathogenicity, and Toxigenic Chemotypes of Fusarium graminearum Species Complex Causing Maize Ear Rot
WANG BaoBao1, GUO Cheng2, SUN SuLi1, XIA YuSheng1, ZHU ZhenDong1, DUAN CanXing,1通讯作者:
责任编辑: 岳梅
收稿日期:2020-03-26接受日期:2020-05-6网络出版日期:2020-12-01
| 基金资助: |
Received:2020-03-26Accepted:2020-05-6Online:2020-12-01
作者简介 About authors
王宝宝,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (1071KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
王宝宝, 郭成, 孙素丽, 夏玉生, 朱振东, 段灿星. 玉米穗腐病致病禾谷镰孢复合种的遗传多样性、致病力与毒素化学型分析[J]. 中国农业科学, 2020, 53(23): 4777-4790 doi:10.3864/j.issn.0578-1752.2020.23.005
WANG BaoBao, GUO Cheng, SUN SuLi, XIA YuSheng, ZHU ZhenDong, DUAN CanXing.
开放科学(资源服务)标识码(OSID):

0 引言
【研究意义】玉米是我国重要的粮食作物、饲料作物和能源植物,而穗腐病是玉米生产上最为重要的病害之一,严重影响玉米的产量和品质[1]。我国玉米穗腐病致病菌复杂多样,真菌和细菌均可引起穗腐病,在真菌中,尤以禾谷镰孢复合种(Fusarium graminearum species complex,FGSC)和拟轮枝镰孢(Fusarium verticillioides)引起的穗腐病分布最广,危害最重[2]。2018年我国玉米产量25 717.4万吨(1 材料与方法
1.1 材料
2014—2018年,在中国13个省(直辖市、自治区)(河北、河南、安徽、山东、陕西、重庆、四川、云南、北京、辽宁、广西、贵州、黑龙江)采集玉米病穗样本,并用组织分离法对采集到的玉米样品进行病原菌分离鉴定[2,5-6],确定菌种的构成。为分析分离致病菌中禾谷镰孢复合种的遗传多样性,从不同省(直辖市、自治区)选取具代表性的禾谷镰孢复合种45株,根据地理来源将其分为7大类群,分别为华北、华中、华东、东北、华南、西北和西南地区。马铃薯葡萄糖琼脂(potato dextrose agar,PDA)培养基:DifcoTM Potato Dextrose Agar 39 g、蒸馏水1.0 L;合成低营养琼脂(spezieller nahrstoffarmer agar,SNA)液体培养基:KH2PO4 1.0 g、KNO3 1.0 g、MgSO4·7H2O 0.5 g、KCl 0.5 g、蔗糖0.2 g、葡萄糖0.2 g、蒸馏水1.0 L。
真菌基因组DNA提取试剂盒(索莱宝科技有限公司,北京);超微量核酸蛋白测定仪(Q5000,美国QUAWELL公司);凝胶成像系统(Tanon 4100,南京);2×TaqPCR Master Mix、100 bp DNA Ladder(天根生化科技有限公司,北京);玉米穗腐病图像采集与数据处理系统(国家农业信息化工程技术研究中心定制);水平电泳槽(DYCP-36A,北京);立式压力蒸汽灭菌器(LS-75HD型,济南);锥形瓶(150 mL)、超净工作台、显微镜、血球计数板、无菌注射器。
1.2 DNA提取
将选取的单菌株转移到贴有玻璃纸的PDA培养基上,25℃培养5—7 d,刮取菌丝并自然晾干,取3—5 g干菌丝体装于2.0 mL离心管中。用真菌基因组DNA提取试剂盒(北京索莱宝科技有限公司)提取DNA。用Q5000超微量核酸蛋白测定仪测定提取DNA的浓度。1.3 分子标记和基因测序引物
使用22对SSR引物、10对VNTR引物(表1)对所选镰孢菌进行遗传多样性分析。25 μL反应体系:DNA 模板2 μL、上下游引物各1 μL,12.5 μL 2×TaqPCR Master Mix,ddH2O补足至25 μL。PCR反应程序:94℃预变性4 min;94℃变性45 s,退火温度(视不同引物而定)45 s,72℃延伸1 min,36个循环;最后72℃延伸7 min。Table 1
表1
表1本试验所用引物
Table 1
| 引物 Primer | 序列 Sequence (5′ to 3′) | 退火温度 Tm (℃) | 扩增片段大小 Product size (bp) | 参考文献 Reference |
|---|---|---|---|---|
| SSR-L1 | R: AGAAGAAGGGCAAATGG | 57 | 378 | [19] |
| F: TGTCCGAGTCGTCTGAAT | ||||
| SSR-L2 | R: GGTGGTGGTTTCCGTGAG | 60 | 214 | [19] |
| F: GCGTTGAGATACCCTTTCG | ||||
| SSR-L3 | R: CTGCTGCTATCACTTCCAC | 58 | 344 | [19] |
| F: TTCACAACGACCGTATTTC | ||||
| SSR-L4 | R: GTTGTTGGGTTGCTTGTG | 59 | 160 | [19] |
| F: GTCTCGTCCAGATTCCTATT | ||||
| SSR-L5 | R: GGTGGTGGTTTCCGTGAG | 60 | 214 | [19] |
| F: GCGTTGAGATACCCTTTCG | ||||
| SSR-L6 | R: AATAGTGGCGGTGCGTAG | 60 | 386 | [19] |
| F: GTCTCCCGAGTAGTTGGC | ||||
| SSR-L7 | R: TAGTGGCGGTGCGTAGTG | 62 | 379 | [19] |
| F: GTCTCCCGAGTAGTTGGC | ||||
| SSR-L8 | R: GGTCCGCAACATAGAGGG | 60 | 403 | [19] |
| F: CCGAAGAGGAGAACGAGC | ||||
| SSR-G11 | R: TTTTGGGTGTTGAAGAAGCC | 56 | 200-300 | [20] |
| F: TTTTTGTTTGCGTCGTTCTG | ||||
| SSR-G12 | R: AATGCAGTCGATGGGAAACT | 56 | 200-300 | [20] |
| F: GGGTTCCTTGTAAGTGGCTG | ||||
| SSR-G13 | R: AGTATCAGACAGGTTGGCCG | 56 | 200-300 | [20] |
| F: GTATGCAAAGCAGCGTCGTA | ||||
| SSR-G14 | R: GCCCATTACGTTGAGCAAAT | 56 | 200-300 | [20] |
| F: TTTTTCGGTCTGGCTATTGG | ||||
| SSR-G15 | R: CTCTAGTGGCAGACCCTTCG | 56 | 200-300 | [20] |
| F: CTTGACATGTCGCGCTTTTA | ||||
| SSR-G16 | R: CTGATCTGCGGACATCTTCA | 56 | 200-300 | [20] |
| F: GTTTTGGTTTGCCTGTCGTT | ||||
| SSR-G17 | R: TCTGTCGTTGACAAGCAAGC | 56 | 200-300 | [20] |
| F: ACGGACCGACGACATAACTC | ||||
| SSR-G18 | R: GCGTCGACTAAAGGAACCAG | 56 | 200-300 | [20] |
| F: TCCCGACGTTAGAGTGGAGT | ||||
| SSR-G19 | R: CCTTGTTCTTTCCACCCTGA | 56 | 200-300 | [20] |
| F: ACGAGGACGAACTTGTTGCT | ||||
| 引物 Primer | 序列 Sequence (5′ to 3′) | 退火温度 Tm (℃) | 扩增片段大小 Product size (bp) | 参考文献 Reference |
| SSR-G20 | R: AAGCTTGGGGGCTAACATCT | 56 | 200-300 | |
| F: CAGCTTTGGCGGACATTATT | [20] | |||
| SSR-G21 | R: TCACATATTCAACCGACCCA | 56 | 200-300 | [20] |
| F: GCTCCGTGTCCTTTCATTTC | ||||
| SSR-G22 | R: GACGGATCGTCGGATAGGTA | 56 | 200-300 | [20] |
| F: CACTAAGCTGCTCCTCCACC | ||||
| VNTR-F1 | F: ACAGGCATCCAAGGACATTT | 56 | 286 | [21] |
| R: GTTTGATGGCGCATTCAAAG | ||||
| VNTR-F2 | F: GCAGGACCTGGATGATGAA | 56 | 234 | [21] |
| R: ATGTGTGCAGCCATGAGATT | ||||
| VNTR-F3 | F: ATCTCCCAAGCTGGCTAATT | 56 | 234 | [21] |
| R: AGAACCGGCAAAGTTCGATT | ||||
| VNTR-F4 | F: TCCGAAGGTAGAAGCGTTGT | 56 | 298 | |
| R: TCAAGCCCATCTATGCTGTT | [21] | |||
| VNTR-F5 | F: GAGATGGCAACATTATTTGCA | 56 | 252 | |
| R: ATTGGCAGCAGGGCTTGATT | [21] | |||
| VNTR-F6 | F: AAGAGGGCGTGTCTCTGTTTT | 56 | 215 | |
| R: CGCTTCCTTCCTTTCAATTC | [21] | |||
| VNTR-F7 | F: AAGACTGGTCAGCAGTAGGGA | 56 | 248 | |
| R: TGAGAGCGAGACTGAGCATGA | [21] | |||
| VNTR-F8 | F: AAACGTAAACGGATCAACGG | 56 | 210 | |
| R: AGATTCGCAACTTTGTGCTG | [21] | |||
| VNTR-F9 | F: TGGATATGGTTCCCCAGCT | 56 | 239 | |
| R: TACTGACCTTGAGGAGCACCATAC | [21] | |||
| VNTR-F10 | F: TATGATGCAGCGAATGCAAC | 56 | 221 | [21] |
| R: TAGAGACCTGGCCCATACCA | ||||
| TEF-1α | R: ATGGGTAAGGAR-GACAAGAC | 58 | 680 | [22] |
| F: GGAR-GTACCAGTSATCATGTT | ||||
| β-tubulin | R: GGTAACCAAATCGGTGCTGCTTTC | 55 | 380 | [23] |
| F: ACCCTCAGTGTAGTGACCCTTGGC | ||||
| RPB2 | R: CCCATRGCTTGYTTRCCCAT | 55 | 900 | [24] |
| F: GGGGWGAYCAGAAGAAGGC | ||||
| Tri13a/b | R: CTCSACCGCATCGAAGASTCTC | 58 | 583, 644, 859 | [25] |
| F: GAASGTCGCARGACCTTGTTTC | ||||
| Tri13F/R | R: GGTGTCCCAGGATCTGCG | 57 | 415 | [26] |
| F: TACGTGAAACATTGTTGGC | ||||
| Tri303F/R | R: GCCGGACTGCCCTATTG | 52 | 586 | [27] |
| F: GATGGCCGCAAGTGGA | ||||
| Tri315F/R | R: GTCTATGCTCTCAACGGACAAC | 58 | 864 | [27] |
| F: CTCGCTGAAGTTGGACGTAA | ||||
新窗口打开|下载CSV
采用TEF-1α、β-tubulin以及RPB2基因序列(表1)对所选菌株进行分析。TEF-1α的50 μL PCR反应体系:DNA模板2 μL、上下游引物各2 μL、2×TaqPCR Master Mix 25 μL、ddH2O 19 μL。PCR反应程序:94℃预变性 5 min;95℃变性45 s,58℃退火45 s,72℃延伸1 min,36个循环:72℃延伸10 min,4℃保存产物。β-tubulin的反应体系同上,PCR反应程序:94℃预变性 4 min;95℃变性45 s,55℃退火45 s,72℃延伸 1 min,36个循环;72℃延伸7 min,4℃保存产物,DNA扩增片段大小约380 bp。RPB2反应体系同上,PCR反应程序:95℃预变性2 min;95℃变性30 s,55℃退火1 min,72℃延伸2 min,40个循环;72℃延伸7 min,4℃保存产物,DNA 扩增片段大小约800 bp。于1.2%琼脂糖凝胶电泳后检验有条带的扩增产物由生工生物工程(上海)股份有限公司进行测序,测序拼接结果在NCBI的BLAST模块进行比对分析。
1.4 致病力测定
田间种植:于2019年6月在北京顺义田间种植150行玉米自交品系掖478,每行留10株苗,行距60 cm,株距25 cm,重复两次。孢子悬浮液的制备:配备合成低营养琼脂(SNA)液体培养基,并倒入60 mL于锥形瓶中,于立式压力蒸汽灭菌器中灭菌,在超净工作台上分别将培养好的禾谷镰孢刮取菌丝,转移至上述锥形瓶中,记下菌株信息,连续光照12 h,25℃下振动培养3—5 d。在显微镜下镜检,使用血球计数板将浓度调至1×106个分生孢子/mL,待用。待田间玉米花丝吐出7—10 d,用无菌注射器吸取孢子悬浮液,每株玉米主穗通过花丝通道接种法[28]注射菌液2 mL,每个菌株注射接种两行玉米,去除主穗外的其他穗并在每行玉米行头做好标记。在玉米蜡熟后期,收集所有接种的玉米果穗,去除苞叶,利用玉米穗腐病图像采集与数据处理系统(国家农业信息化工程技术研究中心定制),测定每个果穗的发病籽粒区域占整个果穗籽粒表面积的百分比,并计算每个菌株所接种果穗(共计20个)的平均发病面积占比。发病面积(m)百分比计算公式:m=(d/e)×100%,其中d为玉米穗部出现穗腐症状的籽粒表面积,e为玉米穗部全部籽粒的面积。平均发病面积(me)百分比计算公式:me=(∑mi)/n×100%,n为接种果穗数,mi中i选值从1到n,是每个玉米穗的发病面积百分比。1.5 毒素化学型测定
对禾谷镰孢复合种进行毒素化学型分子检测,选用引物Tri13a/b。PCR反应程序:94℃预变性4 min;95℃变性45 s,58℃退火45 s,72℃延伸1 min,36个循环;72℃延伸7 min,4℃保存产物,15ADON、3ADON和NIV毒素化学型的菌株扩增片段分别为583、644和859 bp[25],PCR反应程序同上。分别采用Tri13F/R,扩增片段415 bp,退火温度57℃进行NIV毒素化学型的重复性检测[26];采用Tri303F/R,退火温度为52℃,扩增片段586 bp,进行3ADON毒素化学型的重复性检验[27];采用Tri315F/R,退火温度为58℃,扩增片段864 bp,进行15ADON毒素化学型的重复性检验[27],以鉴定特异性引物的有效性。1.6 数据处理
穗腐病致病力数据先通过玉米穗腐病图像采集与数据处理系统进行预处理,得到个体的表型数据,利用Excel 2010进行统计分析与处理,计算各组的平均表型值,并进行方差分析。2 结果
2.1 微卫星引物的SSR扩增
SSR引物在45株禾谷镰孢复合种中共检测到等位位点数48个,多态性位点数39个,多态性条带比率为81.25%,每对引物扩增出的多态性条带为2—4条。对于不同菌株相同引物扩增出的条带,同一片段大小记为‘1’,在同一位置没有扩增条带的记为‘0’。统计条带信息,利用NTsys2.1采用非加权对数算术平均法(UPGMA)构建系统发育树。2.2 不同地理种群的遗传多样性
对中国7个地理种群的禾谷镰孢复合种菌群进行遗传多样性分析(表2)。东北地区菌群的遗传多样性最为丰富,多态性位点数为39,有效等位基因数1.79,Shannon’s信息指数为0.62,Nei’s遗传多样性指数为0.43。禾谷镰孢复合种7个地理种群的多态性位点数在10—39,平均Shannon’s信息指数为0.41,平均Nei’s遗传多样性指数为0.29,表明禾谷镰孢复合种具有较丰富的遗传多样性。7个地理种群禾谷镰孢复合种的Nei’s遗传相似度为0.6677—0.8797,其中东北和西北地区的最高,而华中与西南地区的最低,表明华中和西南地区的菌群表现出较高的遗传多样性;遗传距离为0.1282—0.4039,表明7个区域间的种群分布较为分散(表3)。Table 2
表2
表2禾谷镰孢复合种的遗传多样性
Table 2
| 种群 Population | 多态性位点数 Number of polymorphic loci | 多态性位点百分比 Percentage of polymorphic loci (%) | 等位基因数 Observed number of alleles | 有效等位基因数 Effective number of alleles | Shannon’s信息指数 Shannon’s information index | Nei’s遗传多样性指数 Nei’s genetic diversity index |
|---|---|---|---|---|---|---|
| 华北North China | 18 | 46.15 | 1.46 | 1.46 | 0.32 | 0.23 |
| 华中Central China | 10 | 25.64 | 1.26 | 1.26 | 0.18 | 0.13 |
| 华东East China | 14 | 35.90 | 1.36 | 1.36 | 0.25 | 0.18 |
| 东北Northeast China | 39 | 100.00 | 2.00 | 1.79 | 0.62 | 0.43 |
| 华南South China | 26 | 66.67 | 1.67 | 1.46 | 0.40 | 0.27 |
| 西北Northwest China | 37 | 94.87 | 1.95 | 1.65 | 0.55 | 0.37 |
| 西南Southwest China | 36 | 92.31 | 1.92 | 1.71 | 0.56 | 0.39 |
| 平均Average | 25.71 | 66.00 | 1.66 | 1.53 | 0.41 | 0.29 |
新窗口打开|下载CSV
Table 3
表3
表3禾谷镰孢复合种遗传相似度和遗传距离
Table 3
| 种群 Population | 华北 North China | 华中 Central China | 华东 East China | 东北 Northeast China | 华南 South China | 西北 Northwest China | 西南 Southwest China |
|---|---|---|---|---|---|---|---|
| 华北North China | — | 0.6888 | 0.7100 | 0.7979 | 0.7011 | 0.8120 | 0.7910 |
| 华中Central China | 0.3727 | — | 0.8489 | 0.8484 | 0.7067 | 0.6934 | 0.6677 |
| 华东East China | 0.3424 | 0.1638 | — | 0.8572 | 0.7782 | 0.7556 | 0.7452 |
| 东北Northeast China | 0.2257 | 0.1644 | 0.1540 | — | 0.8170 | 0.8797 | 0.8502 |
| 华南South China | 0.3551 | 0.3471 | 0.2508 | 0.2021 | — | 0.8196 | 0.8609 |
| 西北Northwest China | 0.2083 | 0.3662 | 0.2803 | 0.1282 | 0.1990 | — | 0.8775 |
| 西南Southwest China | 0.2345 | 0.4039 | 0.2942 | 0.1623 | 0.1498 | 0.1307 | — |
新窗口打开|下载CSV
2.3 禾谷镰孢复合种地理种群的聚类分析
依据统计结果用NTsys2.1对禾谷镰孢复合种7个地理种群遗传距离进行UPGMA聚类分析,菌群在相似系数为0.77时,被聚类为3个大的分支,华北地区的菌株单独聚为一类,东北、西北、西南、华南地区的菌株聚为一大类群,华中和华东地区的同在一分支(图1)。图1
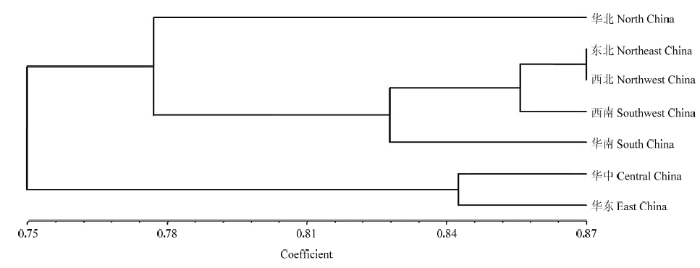 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1禾谷镰孢复合种7个地理种群的聚类分析图
Fig. 1Cluster analysis of 7 geographic-based FGSC populations
2.4 STRUCTURE群体结构分析
使用结构分析STRUCTURE2.3.4软件的贝叶斯聚类法对禾谷镰孢复合种地理种群的群体遗传结构进行分析,参数Length of Bunrin Period设为25 000,Numbers of MCMC Reps after Bunrin设为1 000 000,将K设置为1—10,K重复次数Number of Iterations设为10,将运行结果转换成压缩包文件在网站Structure Harvest(http://taylor0.biology.ucla.edu/structure Harvester/)中利用EVANNO等[29]ΔK方法计算LnP(K)值、ΔK值和K的最大可能值,结果图2所示,LnP(K)值随着K值增大而增大,表明镰孢菌群体间存在遗传分化,当K值为2时,ΔK取最大为150,表明7个地理群体的禾谷镰孢复合种分为两个类群最合适。图2
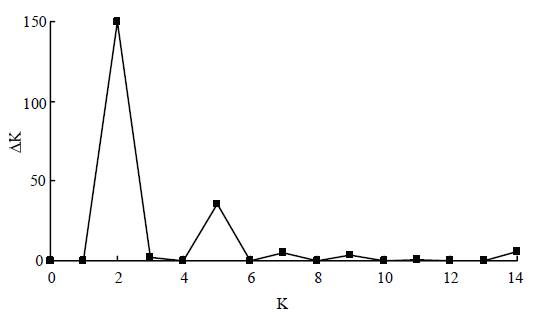 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2禾谷镰孢复合种取不同K值时的ΔK值曲线图
Fig. 2ΔK value curve of FGSC at different K values
进一步研究STRUCTURE软件分析得到的菌群和地理区域间的关系发现,禾谷镰孢复合种在两个类群中,西北地区绝大部分属于类群A,华中地区和华南地区菌株均属于类群B,东北地区50%以上菌株属于类群B(图3)。
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3禾谷镰孢复合种群体结构图(当K值为2时)
Fig. 3Population structure of FGSC (when K=2)
2.5 基于基因序列构建禾谷镰孢复合种树形图
通过TEF-1α、β-tubulin和RPB2基因测序对禾谷镰孢复合种的3个基因片段序列按相同顺序拼接起来,并用MEGA7.0软件构建树形图进行系统发育分析(图4)。最终,禾谷镰孢复合种被分为4大类群,分支标记为红色的以亚洲镰孢为主,包括广西、四川、陕西等相邻地区;分支标记为浅绿色的以布氏镰孢为主,分布在甘肃、黑龙江、北京等相邻地区;分支标记为蓝色的以南方镰孢为主,包括贵州、重庆、云南、广西和陕西等地区;分支标记为黑色(蓝色分支的下部)的以禾谷镰孢为代表,包括黑龙江、河南、辽宁、陕西、山东和北京地区。各地区之间菌种组成不尽相同,禾谷镰孢和布氏镰孢多分布在北方冷凉地区,亚洲镰孢和南方镰孢多分布于南方温湿地区,在北方地区也鲜有分布,从树形图来看,各镰孢菌种内部也存在小的分支,表明无论种间、种内遗传多样性均较为丰富。各地区间存在着丰富的遗传变异,尤其是相邻地区种间存在较频繁的基因交流与交换。使用MEGA7.0软件分析3个基因的序列,在全长1 800 bp的片段上,发现了142个单核苷酸多态性位点(SNP)。图4
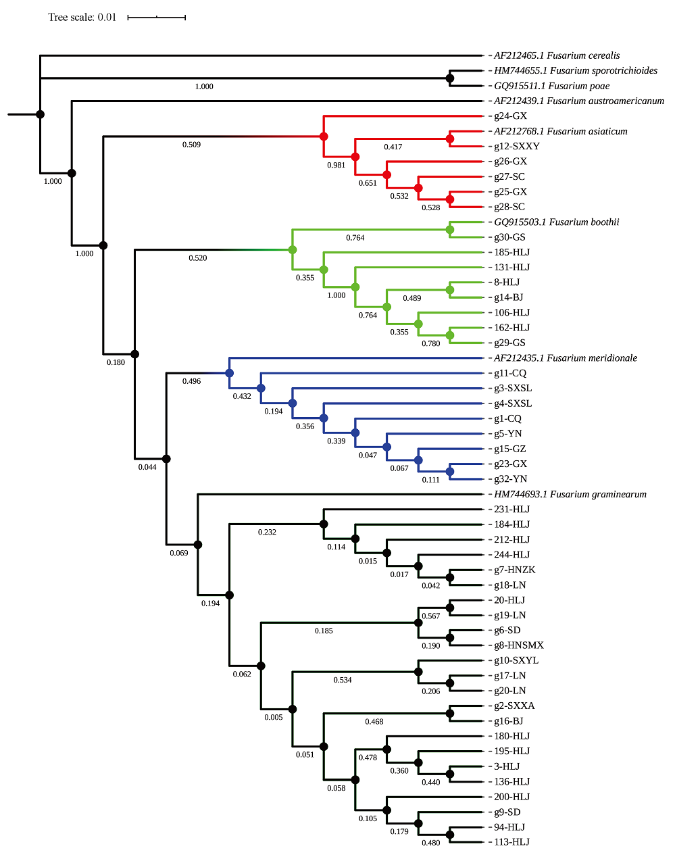 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4禾谷镰孢复合种RPB2+TEF-1α+β-tubulin基因聚类图
Fig. 4RPB2 + TEF-1α + β-tubulin gene clusters of FGSC
2.6 镰孢菌致病力测定与毒素化学型分析
禾谷镰孢复合种以15ADON毒素化学型为主,共32株,3ADON次之,共15株,NIV 5株;3ADON主要在西北、华南、西南地区分布,有两株菌能同时产生15ADON和3ADON,两株菌能同时产生3种毒素化学型,一株菌能同时产生15ADON和NIV;15ADON、NIV和3ADON平均发病面积百分比分别为17.87%、17.20%和12.37%,能同时产生3种毒素化学型的菌株平均发病面积百分比为16.36%,略高于3种类型总平均发病面积百分比。禾谷镰孢平均发病面积百分比最大,为20.79%;亚洲镰孢、布氏镰孢和南方镰孢的平均发病面积百分比分别为15.79%、11.77%和8.12%,禾谷镰孢的致病力在4种镰孢菌中最强。禾谷镰孢复合种在东北、华东、西南、华南、华北、华中和西北地区平均发病面积百分比分别为20.85%、18.00%、9.81%、15.22%、13.74%、13.65%和12.33%。禾谷镰孢毒素化学型为15ADON、3ADON、NIV和3ADON+ 15ADON+NIV 4种,以15ADON为主;布氏镰孢毒素化学型为15ADON、3ADON、NIV、3ADON +15ADON和3ADON+15ADON+NIV 5种,以15ADON为主;亚洲镰孢毒素化学型只有3ADON;南方镰孢毒素化学型为15ADON、3ADON、15ADON+NIV 3种,15ADON为优势类型(表4)。不同镰孢种引起的穗腐病平均发病面积占比存在显著差异(P=0.011<0.05),表明不同镰孢种之间的致病力存在较大差异(表5、表6)。禾谷镰孢复合种在7大地理区域的菌群构成、致病力和毒素化学型分布不均匀。Table 4
表4
表4禾谷镰孢复合种致病力与毒素化学型
Table 4
| 菌株 Strain | 菌种 Species | 发病面积 Diseased area (%) | 毒素类型 | 采集地点 Collection location | 地理区域 Geographical area | ||
|---|---|---|---|---|---|---|---|
| 3ADON | 15ADON | NIV | |||||
| 3HLJ | 禾谷镰孢F. graminearum | 10.70 | √ | 黑龙江肇源Zhaoyuan, Heilongjiang | 东北Northeast China | ||
| 8HLJ | 布氏镰孢F. boothii | 4.00 | √ | 黑龙江安达Anda, Heilongjiang | 东北Northeast China | ||
| 20HLJ | 禾谷镰孢F. graminearum | 10.09 | √ | 黑龙江尚志Shangzhi, Heilongjiang | 东北Northeast China | ||
| 94HLJ | 禾谷镰孢F. graminearum | 42.63 | √ | 黑龙江鹤岗Hegang, Heilongjiang | 东北Northeast China | ||
| 106HLJ | 布氏镰孢F. boothii | 6.88 | √ | 黑龙江穆棱Muling, Heilongjiang | 东北Northeast China | ||
| 113HLJ | 禾谷镰孢F. graminearum | 27.58 | √ | 黑龙江虎林Hulin, Heilongjiang | 东北Northeast China | ||
| 131HLJ | 布氏镰孢F. boothii | 11.63 | √ | √ | 黑龙江饶河Raohe, Heilongjiang | 东北Northeast China | |
| 136HLJ | 禾谷镰孢F. graminearum | 23.36 | √ | 黑龙江密山Mishan, Heilongjiang | 东北Northeast China | ||
| 162HLJ | 布氏镰孢F. boothii | 16.91 | √ | 黑龙江依安Yian, Heilongjiang | 东北Northeast China | ||
| 180HLJ | 禾谷镰孢F. graminearum | 26.54 | √ | 黑龙江嫩江Neijiang, Heilongjiang | 东北Northeast China | ||
| 184HLJ | 禾谷镰孢F. graminearum | 42.63 | √ | 黑龙江龙江Longjiang, Heilongjiang | 东北Northeast China | ||
| 185HLJ | 布氏镰孢F. boothii | 22.54 | √ | 黑龙江密山Mishan, Heilongjiang | 东北Northeast China | ||
| 195HLJ | 禾谷镰孢F. graminearum | 30.20 | √ | 黑龙江宁安Ningan, Heilongjiang | 东北Northeast China | ||
| 200HLJ | 禾谷镰孢F. graminearum | 39.27 | √ | 黑龙江穆棱Muling, Heilongjiang | 东北Northeast China | ||
| 212HLJ | 禾谷镰孢F. graminearum | 35.60 | √ | 黑龙江五常Wuchang, Heilongjiang | 东北Northeast China | ||
| 231HLJ | 禾谷镰孢F. graminearum | 4.29 | √ | 黑龙江哈尔滨Harbin, Heilongjiang | 东北Northeast China | ||
| 244HLJ | 禾谷镰孢F. graminearum | 13.70 | √ | 黑龙江绥化Suihua, Heilongjiang | 东北Northeast China | ||
| g1-CQ | 南方镰孢F. meridionale | 6.66 | √ | 重庆合川Hechuan, Chongqing | 西南Southwest China | ||
| g2-SXXA | 禾谷镰孢F. graminearum | 11.38 | √ | 陕西西安Xian, Shaanxi | 西北Northwest China | ||
| g3-SXSL | 南方镰孢F. meridionale | 9.67 | √ | 陕西商洛Shangluo, Shaanxi | 西北Northwest China | ||
| g4-SXSL | 禾谷镰孢F. graminearum | 25.33 | √ | √ | √ | 陕西商洛Shangluo, Shaanxi | 西北Northwest China |
| g5-YN | 南方镰孢F. meridionale | 6.56 | √ | 云南弥勒Mile, Yunnan | 西南Southwest China | ||
| g6-SD | 禾谷镰孢F. graminearum | 7.00 | √ | 山东兖州Yanzhou, Shandong | 华东East China | ||
| g7-HNZK | 禾谷镰孢F. graminearum | 12.44 | √ | 河南周口Zhoukou, Henan | 华中Central China | ||
| g8-HNSMX | 禾谷镰孢F. graminearum | 14.86 | √ | 河南三门峡Sanmenxia, Henan | 华中Central China | ||
| g9-SD | 禾谷镰孢F. graminearum | 29.00 | √ | 山东微山Weishan, Shandong | 华东East China | ||
| g10-SXYL | 禾谷镰孢F. graminearum | 2.75 | √ | 陕西榆林Yulin, Shaanxi | 西北Northwest China | ||
| g11-CQ | 南方镰孢F. meridionale | 4.71 | √ | 重庆秀山Xiushan, Chongqing | 西南Southwest China | ||
| g12-SXXY | 亚洲镰孢F. asiaticum | 12.36 | √ | 陕西乾县Qianxian, Shaanxi | 西北Northwest China | ||
| g14-BJ | 布氏镰孢F. boothii | 7.38 | √ | √ | √ | 北京昌平Changping, Beijing | 华北North China |
| g15-GZ | 南方镰孢F. meridionale | 6.00 | √ | 贵州遵义Zunyi, Guizhou | 西南Southwest China | ||
| g16-BJ | 禾谷镰孢F. graminearum | 20.10 | √ | 北京顺义Shunyi, Beijing | 华北North China | ||
| g17-LN | 禾谷镰孢F. graminearum | 17.60 | √ | 辽宁锦州Jinzhou, Liaoning | 东北Northeast China | ||
| g18-LN | 禾谷镰孢F. graminearum | 15.40 | √ | 辽宁沈阳Shenyang Liaoning | 东北Northeast China | ||
| g19-LN | 禾谷镰孢F. graminearum | 11.80 | √ | √ | 辽宁铁岭Tieling, Liaoning | 东北Northeast China | |
| g20-LN | 禾谷镰孢F. graminearum | 24.60 | √ | 辽宁沈阳Shenyang, Liaoning | 东北Northeast China | ||
| g23-GX | 南方镰孢F. meridionale | 13.11 | √ | 广西德保Debao, Guangxi | 华南South China | ||
| g24-GX | 亚洲镰孢F. asiaticum | 14.00 | √ | 广西河池Hechi, Guangxi | 华南South China | ||
| g25-GX | 亚洲镰孢F. asiaticum | 22.58 | √ | 广西大新Daxin, Guangxi | 华南South China | ||
| g26-GX | 亚洲镰孢F. asiaticum | 11.17 | √ | 广西田林Tianlin, Guangxi | 华南South China | ||
| g27-SC | 亚洲镰孢F. asiaticum | 16.64 | √ | 四川巴中Bazhong, Sichuan | 西南Southwest China | ||
| g28-SC | 亚洲镰孢F. asiaticum | 18.00 | √ | 四川浦江Pujiang, Sichuan | 西南Southwest China | ||
| g29-GS | 布氏镰孢F. boothii | 18.60 | √ | 甘肃张掖Zhangye, Gansu | 西北Northwest China | ||
| g30-GS | 布氏镰孢F. boothii | 6.20 | √ | 甘肃张掖Zhangye, Gansu | 西北Northwest China | ||
| g32-YN | 南方镰孢F. meridionale | 10.10 | √ | √ | 云南玉溪Yuxi, Yunnan | 西南Southwest China | |
新窗口打开|下载CSV
Table 5
表5
表5不同镰孢种引起的穗腐病发病面积占比统计分析
Table 5
| 菌种 Species | 菌株数 Number of strains | 发病面积和 Summary of diseased area (%) | 平均发病面积 Percentage of average diseased area (%) | 方差 Variance |
|---|---|---|---|---|
| 禾谷镰孢F. graminearum | 24 | 498.84 | 20.79±2.44A | 137.3899 |
| 亚洲镰孢F. asiaticum | 6 | 94.75 | 15.79±1.87B | 17.6138 |
| 布氏镰孢F. boothii | 8 | 94.12 | 11.77±2.56c | 46.2450 |
| 南方镰孢F. meridionale | 7 | 56.81 | 8.12±1.20d | 8.6493 |
新窗口打开|下载CSV
Table 6
表6
表6不同镰孢种致病力方差分析
Table 6
| 差异源 Source of difference | 平方和 SS | 自由度 df | 平均平方和 MS | 统计量 F | P值 P-value |
|---|---|---|---|---|---|
| 组间Intergroup | 1115.080 | 3 | 371.6934 | 4.206 | 0.011 |
| 组内Intragroup | 3623.646 | 41 | 88.3816 | ||
| 总计Total | 4738.727 | 44 |
新窗口打开|下载CSV
3 讨论
玉米穗腐病是一种在世界各玉米种植区频繁发生的真菌病害,主要病原真菌为镰孢菌。禾谷镰孢复合种可侵染玉米根、茎、穗、叶,引起根腐病、茎腐病、穗腐病、叶斑病等病害,其产生多种真菌毒素严重影响玉米籽粒的品质和安全,给生产者造成巨大损失[30,31]。目前,禾谷镰孢复合种至少包括16个种,我国已鉴定出10个种,本研究中的禾谷镰孢复合种由禾谷镰孢、布氏镰孢、南方镰孢和亚洲镰孢组成。禾谷镰孢在南北方均有分布;布氏镰孢主要分布在东北、西北和华北等较冷凉地区;亚洲镰孢多分布于华南地区,在东北、西南和西北地区也有少量分布;南方镰孢则主要分布在西南、华南等较温暖地区,与前人相关的研究结果基本一致[5]。本研究首次利用玉米穗腐病图像采集与数据处理系统(国家农业信息化工程技术研究中心定制)进行穗腐病发病面积调查与数据自动化处理,穗部发病面积占比由该系统通过AI识别测定后自动生成,结果比较精确;而以往玉米穗腐病发病情况调查均由人工观察估测,同一份材料由不同的人调查或同一个人调查不同材料的发病情况时,不可避免地产生不同程度的误差。通过该系统测定处理的数据,消除了人为造成的误差,能真实反映出不同镰孢菌株所致穗腐病的发病严重程度。马红霞等[15]报道在中国春玉米区和夏玉米区禾谷镰孢复合种的毒素化学型主要为DON和15ADON,温暖地区如长江流域则以3ADON和NIV为主;本研究组[2]于2018—2019年全面分析了黑龙江地区禾谷镰孢复合种的毒素化学型,发现禾谷镰孢与布氏镰孢均以产DON毒素衍生物为主,仅1株亚洲镰孢产NIV毒素;AAMOT等[32]报道了挪威小麦赤霉病病原菌禾谷镰孢以3ADON类型占主导;SNEIDERIS等[17]在57株禾谷镰孢中发现47株能产生15ADON。在本研究中,禾谷镰孢复合种的毒素化学型较为丰富,禾谷镰孢、布氏镰孢和南方镰孢均能产生15ADON、3ADON和NIV 3种毒素化学型,亚洲镰孢只发现3ADON型,总体以15ADON和3ADON型占主导,其中15ADON类型的最多,产生NIV的菌株较少,与前人相关研究对比不难发现,优势产毒类型的菌群已发生一定程度的变化。
有关禾谷镰孢复合种的致病力分析在小麦上报道较多,在玉米穗腐病上报道很少。WARD等[33]研究发现,小麦赤霉病病原菌禾谷镰孢产3ADON的菌株与产15ADON的菌株相比,单端孢霉烯含量更高,且繁殖力更强、生长速率更快;李伟等[34]利用30株小麦赤霉病的亚洲镰孢和1株禾谷镰孢进行致病力测定及毒素化学型分析,结果表明在长江流域,产3ADON的亚洲镰孢致病力最强,产15ADON的亚洲镰孢菌株在小麦品种上表现的致病力很弱;杜青等[35]对广西地区禾谷镰孢复合种进行毒素化学型分析未检测到产3ADON的菌株。本研究表明,对于玉米穗腐病而言,产15ADON毒素化学型菌株比产3ADON毒素化学型菌株的致病力强,结论有所不同,可能与李伟等[34]的研究中产15ADON的菌株样本量太少(仅1株)有关,也可能与寄主植物本身的特性有关。GAI等[14]对40株禾谷镰孢菌复合种致病力测定中,得到各菌株的病情指数在58.9—89.3,且致病力不存在地域性差异;但本试验发现北方区域菌株致病力较南方的强,总体而言,由北向南禾谷镰孢复合种致病力逐渐减弱。样本的数量选取对试验结果影响较大,尤其是遗传多样性分析,样本群体数量足够大,可能包含的基因越丰富,毒素化学型也更全面,本研究囿于材料等的限制(结合前人的研究,特定菌株在一些地区的玉米穗腐样本上分离频率比较低[2,5-6,15],本试验也如此,例如南方镰孢主要在南方地区分布,布氏镰孢主要在北方地区有分布,一些地区虽然分离了大量的禾谷镰孢,但为了保证试验均衡及代表性,去掉了同一地理来源的菌株),在样本选取上最大限度地保证禾谷镰孢复合种菌种的多样化以及每个菌株地理来源的差异性,因此,尽管菌株数量偏少,但所选菌株具有较好的代表性。下一步可以扩大样品的采集数目并对样本数量最优选取进行探讨,以选出最适合试验的菌群数。
禾谷镰孢复合种存在大量的单核苷酸多态性位点,具有高度适应环境的潜力,将遗传变异与表型变化联系起来仍然具有挑战性。LAURENT等[36]使用全基因组重测序在单核苷酸水平上研究了6个法国禾谷镰孢的基因组多样性,共检测到242 756个高度可信遗传变异位点,其中96%为单核苷酸多态性,约80%的禾谷镰孢编码蛋白的基因上产生了多态性。本研究通过SSR+VNTR分子标记结合TEF-1α、β-tubulin和RPB2基因测序对禾谷镰孢复合种进行遗传多样性分析,通过比较3个基因序列,共发现142个单核苷酸多态性位点,说明禾谷镰孢遗传变异具有高度的适应性。与传统的利用分子标记特异性扩增来分析镰孢菌的遗传多样性相比,利用基因测序,通过这些差异序列构建的聚类图能清楚显示出各种内与种间的遗传分化情况,构建树形图更精细,分支更多而精准,甚至还能比较任意两个菌株的单核苷酸多态性,但其不能覆盖菌株所有的染色体,将二者结合,进行局部和整体的全面分析,有利于深入研究。
马红霞等[15]研究发现,相同地理来源例如同属春玉米区或夏玉米区的菌株有较近的亲缘关系,且不同地理种群间存在基因交流,遗传多样性水平与地理来源有关。本研究发现在7大地理区域,菌群构成、致病力和毒素化学型分布都不均匀,华中和华东地区菌群亲缘关系较近,西南和东南、东北和西北地区菌群亲缘关系较近。AAMOT等[32]研究了挪威45株禾谷镰孢复合种的平均遗传多样性指数(H)在0.17—0.43,发现近些年采集的菌株要比前些年收集菌群的遗传多样性数值高。随着人们农业生产活动多元化,致病菌间的基因交流会愈加频繁,遗传多样性水平也会因此上升,是否会导致侵染性更强,需要多年持续跟踪的研究。
4 结论
禾谷镰孢复合种包括禾谷镰孢、布氏镰孢、亚洲镰孢和南方镰孢,其毒素化学型含15ADON、3ADON、NIV及其组合型,以15ADON和3ADON为主,禾谷镰孢的致病力在4种镰孢菌中最强,且致病力强弱与毒素化学型有关。基于SSR和VNTR分子标记结合TEF-1α、β-tubulin和RPB2基因测序分析,表明在7个地理区域内部或各区域之间,禾谷镰孢复合种群体各镰孢菌种间和种内均存在丰富的遗传变异,相邻区域间镰孢菌的基因交流与交换发生频繁;遗传多样性水平与地理来源、菌种构成、毒素化学型以及样本量均有关。(责任编辑 岳梅)
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.3864/j.issn.0578-1752.2020.02.006URL [本文引用: 1]

By reorganization of maize diseases and causal agents reported worldwide, a proposal for standardized translation of Chinese names of maize diseases and pathogens has been given in this article. A total of 185 fungal, oomycete, bacterial, viral and nematode diseases and 380 species/times causal agents in maize were gathered from books, including Compendium of Corn Diseases (4th edition, MUNKVOLD G P and WHITE D G, 2016), Maize Diseases (MCGEE D C, 1988), Field Crop Diseases (NYVALL R F, 1999), Illustrated Handbook of Maize Diseases, Insect Pests and Weeds in China (WANG X M and WANG Z Y, 2018), The American Phytopathological Society (APSnet, 2019), and some research articles. Some English names of maize diseases were originated from their pathogen species names. Accompanied by advances on scientific knowledge and identification techniques, 62 pathogen species were changed to new taxonomic category, which is different from the old taxonomic name, so the old disease name is not related with the new pathogen name that it will cause the confusion in practice. For international communication on maize diseases with universality, scientificity and normativity, 27 English names of maize diseases are proposed to modify with their new taxonomic names. 185 infectious diseases on maize were collected and reorganized including 123, 20, 26 and 16 diseases caused by fungi/oomycetes, bacteria, viruses and nematodes, respectively. The 185 maize diseases were caused by 286 pathogenic species/subspecies/varieries/pathovars, included 147 fungi, 34 oomycetes, 20 bacteria and 3 spitoplasm/phytoplasm, 38 viruses and 44 nematodes. The 185 Chinese names of maize diseases have been revised based on the principle of respecting the names published in historical and classic works of Chinese maize diseases or widely adopted in practice, including 96 new Chinese names based on the causal agent names and typical symptoms on maize. For the Chinese name of the maize pathogens, it should be given priority to the name of fungi, bacteria, viruses or nematodes in published plant and medical pathogen monographs, Flora Fungorum Sinicorum and research articles, and to give Chinese name first for the pathogen without Chinese name before based on the original meaning of pathogen’s Latin genus name and species epithet (English in virus). Standardized English names of maize diseases and determined taxonomic status of pathogens will be of benefit to scientific communication among maize disease researchers worldwide and to promote new researches. Under the determination of Chinese names of maize diseases and pathogens, the standardized names will be widely used for preventing translation between English-Chinese name of maize disease and pathogen optionally, and it will provide a correct list for research and teaching on maize diseases to avoid various ambiguities of the names and mistakes of disease-to-pathogen.
DOI:10.3864/j.issn.0578-1752.2020.02.006URL [本文引用: 1]
By reorganization of maize diseases and causal agents reported worldwide, a proposal for standardized translation of Chinese names of maize diseases and pathogens has been given in this article. A total of 185 fungal, oomycete, bacterial, viral and nematode diseases and 380 species/times causal agents in maize were gathered from books, including Compendium of Corn Diseases (4th edition, MUNKVOLD G P and WHITE D G, 2016), Maize Diseases (MCGEE D C, 1988), Field Crop Diseases (NYVALL R F, 1999), Illustrated Handbook of Maize Diseases, Insect Pests and Weeds in China (WANG X M and WANG Z Y, 2018), The American Phytopathological Society (APSnet, 2019), and some research articles. Some English names of maize diseases were originated from their pathogen species names. Accompanied by advances on scientific knowledge and identification techniques, 62 pathogen species were changed to new taxonomic category, which is different from the old taxonomic name, so the old disease name is not related with the new pathogen name that it will cause the confusion in practice. For international communication on maize diseases with universality, scientificity and normativity, 27 English names of maize diseases are proposed to modify with their new taxonomic names. 185 infectious diseases on maize were collected and reorganized including 123, 20, 26 and 16 diseases caused by fungi/oomycetes, bacteria, viruses and nematodes, respectively. The 185 maize diseases were caused by 286 pathogenic species/subspecies/varieries/pathovars, included 147 fungi, 34 oomycetes, 20 bacteria and 3 spitoplasm/phytoplasm, 38 viruses and 44 nematodes. The 185 Chinese names of maize diseases have been revised based on the principle of respecting the names published in historical and classic works of Chinese maize diseases or widely adopted in practice, including 96 new Chinese names based on the causal agent names and typical symptoms on maize. For the Chinese name of the maize pathogens, it should be given priority to the name of fungi, bacteria, viruses or nematodes in published plant and medical pathogen monographs, Flora Fungorum Sinicorum and research articles, and to give Chinese name first for the pathogen without Chinese name before based on the original meaning of pathogen’s Latin genus name and species epithet (English in virus). Standardized English names of maize diseases and determined taxonomic status of pathogens will be of benefit to scientific communication among maize disease researchers worldwide and to promote new researches. Under the determination of Chinese names of maize diseases and pathogens, the standardized names will be widely used for preventing translation between English-Chinese name of maize disease and pathogen optionally, and it will provide a correct list for research and teaching on maize diseases to avoid various ambiguities of the names and mistakes of disease-to-pathogen.
[本文引用: 5]
[本文引用: 5]
DOI:10.3390/toxins8060186URL [本文引用: 1]
DOI:10.3864/j.issn.0578-1752.2015.11.007URL [本文引用: 2]
Maize is one of the most important crops in the world. The percentage of maize yields is 38.12% in gross cereal yields of China. Ear rot is a serious disease of maize, which often leads to a decline in yield and quality, and that hazards animals and human health because of the toxins that are produced in the ear rot. The development and utilization of resistant cultivars is the most economical, safe, and effective method for controlling ear rot. So far, many studies on pathogens causing maize ear rot have been reported and more than 40 pathogenic fungi have been isolated and identified. The ear rots caused by Fusarium verticillioides, F. graminearum and Aspergillus flavus are very common and serious diseases. The four methods of resistance identification have been established, i.e. two toothpicks method, silk spraying, silk channel injection and kernel injection. Thousands of maize germplasm have been identified and evaluated for resistance to ear rot, and some accessions with resistance to F. verticillioides, F. graminearum or A. flavus also have been screened out. Resistance inheritance and gene mapping have been conducted for some materials, and more than 60 quantitative trait loci (QTL) associated with resistance to A. flavus infection and aflatoxin accumulation in maize have been mapped on chromosomes 1-8. Furthermore, 55, 29 and 16 QTLs resistance to F. verticillioides, F. graminearum and fumonisin and deoxynivalenol (DON) accumulation have been discovered, respectively. Based on QTL associated with ear rot resistance in maize, the meta-QTL analysis has been projected and some stable QTLs have been detected. Also, more than 10 ear rot resistant accessions have been developed and some new varieties with different levels of resistance have been bred. In addition, physical and biochemical resistance mechanisms were demonstrated, which mainly reflected in anti-infection and anti-diffusion. However, there are few successful cases that the resistance research and disease-resistant breeding results are applied to production practice, resulting in lack of maize cultivars with stable resistance as well as high yield and quality. In this paper, the advances including pathogens, identification of resistant maize germplasm, resistance inheritance, discovery and mapping of resistance genes, resistance mechanism, and breeding resistant varieties in research of maize resistance to ear rot are reviewed, and several important directions for the future researches are prospected. The main aim is to supply useful information so as to promote the development of the research on maize resistance to ear rot.
DOI:10.3864/j.issn.0578-1752.2015.11.007URL [本文引用: 2]
Maize is one of the most important crops in the world. The percentage of maize yields is 38.12% in gross cereal yields of China. Ear rot is a serious disease of maize, which often leads to a decline in yield and quality, and that hazards animals and human health because of the toxins that are produced in the ear rot. The development and utilization of resistant cultivars is the most economical, safe, and effective method for controlling ear rot. So far, many studies on pathogens causing maize ear rot have been reported and more than 40 pathogenic fungi have been isolated and identified. The ear rots caused by Fusarium verticillioides, F. graminearum and Aspergillus flavus are very common and serious diseases. The four methods of resistance identification have been established, i.e. two toothpicks method, silk spraying, silk channel injection and kernel injection. Thousands of maize germplasm have been identified and evaluated for resistance to ear rot, and some accessions with resistance to F. verticillioides, F. graminearum or A. flavus also have been screened out. Resistance inheritance and gene mapping have been conducted for some materials, and more than 60 quantitative trait loci (QTL) associated with resistance to A. flavus infection and aflatoxin accumulation in maize have been mapped on chromosomes 1-8. Furthermore, 55, 29 and 16 QTLs resistance to F. verticillioides, F. graminearum and fumonisin and deoxynivalenol (DON) accumulation have been discovered, respectively. Based on QTL associated with ear rot resistance in maize, the meta-QTL analysis has been projected and some stable QTLs have been detected. Also, more than 10 ear rot resistant accessions have been developed and some new varieties with different levels of resistance have been bred. In addition, physical and biochemical resistance mechanisms were demonstrated, which mainly reflected in anti-infection and anti-diffusion. However, there are few successful cases that the resistance research and disease-resistant breeding results are applied to production practice, resulting in lack of maize cultivars with stable resistance as well as high yield and quality. In this paper, the advances including pathogens, identification of resistant maize germplasm, resistance inheritance, discovery and mapping of resistance genes, resistance mechanism, and breeding resistant varieties in research of maize resistance to ear rot are reviewed, and several important directions for the future researches are prospected. The main aim is to supply useful information so as to promote the development of the research on maize resistance to ear rot.
URL [本文引用: 4]
Based on morphology, cultural characters and species-specific molecular detection, 130 kernel samples collected from 100 counties in 18 provinces of China were identified, and the species in Fusarium graminearum species complex were analyzed by comparing TEF-1α gene sequences. The results showed that the species of Fusarium were mainly pathogens (56.0%) causing maize ear rot in China, and other fungi were Penicilium spp., Aspergillus spp., Trichoderma spp. and Alternaria spp. In the 138 isolates of Fusarium, seven species were identified, including widely distributed F.verticillioides (56.5%) and F.graminearumspecies complex (37.7%), and others were F.culmorum (2.2%), F.proliferatum (1.5%), F.oxysporum species complex (0.7%), F.solani species complex (0.7%) and F.subglutinans (0.7%). In the isolates of F.graminearum species complex, three species were identified, including widely distributed F.graminearum sensu stricto with the frequency of 59.6%, F.meridionale (25.0%) only found in southern maize-growing zone and F.boothii (11.5%) in northern ecological zone.
URL [本文引用: 4]
Based on morphology, cultural characters and species-specific molecular detection, 130 kernel samples collected from 100 counties in 18 provinces of China were identified, and the species in Fusarium graminearum species complex were analyzed by comparing TEF-1α gene sequences. The results showed that the species of Fusarium were mainly pathogens (56.0%) causing maize ear rot in China, and other fungi were Penicilium spp., Aspergillus spp., Trichoderma spp. and Alternaria spp. In the 138 isolates of Fusarium, seven species were identified, including widely distributed F.verticillioides (56.5%) and F.graminearumspecies complex (37.7%), and others were F.culmorum (2.2%), F.proliferatum (1.5%), F.oxysporum species complex (0.7%), F.solani species complex (0.7%) and F.subglutinans (0.7%). In the isolates of F.graminearum species complex, three species were identified, including widely distributed F.graminearum sensu stricto with the frequency of 59.6%, F.meridionale (25.0%) only found in southern maize-growing zone and F.boothii (11.5%) in northern ecological zone.
[本文引用: 3]
[本文引用: 3]
DOI:10.3390/toxins11060320URL [本文引用: 1]
DOI:10.1016/j.cj.2019.06.003URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1094/PDIS-11-19-2473-SRURLPMID:32525450 [本文引用: 1]
Apple scab, caused by Venturia inaequalis, is a major fungal disease worldwide. Cultivation of scab-resistant cultivars would reduce the chemical footprint of apple production. However, new apple cultivars carrying durable resistances should be developed to prevent or at least slow the breakdown of resistance against races of V. inaequalis. One way to achieve durable resistance is to pyramid multiple scab resistance genes in a cultivar. The choice of the resistance genes to be combined in the pyramids should take into account the frequency of resistance breakdown and the geographical distribution of apple scab isolates able to cause such breakdowns. In order to acquire this information and to make it available to apple breeders, the VINQUEST project (www.vinquest.ch) was initiated in 2009. Ten years after launching this project, 24 partners from 14 countries regularly contribute data. From 2009 to 2018, nearly 9,000 data points have been collected. This information has been used to identify the most promising apple scab resistance genes for developing cultivars with durable resistance, which to date are: Rvi5, Rvi11, Rvi12, Rvi14, and Rvi15. As expected, Rvi1, together with Rvi3 and Rvi8, were often overcome, and have little value for scab resistance breeding. Rvi10 may also belong to this group. On the other hand, Rvi2, Rvi4, Rvi6, Rvi7, Rvi9, and Rvi13 are still useful for breeding, but their use is recommended only in extended pyramids of >/=3 resistance genes.
DOI:10.3390/toxins11040224URL [本文引用: 1]
DOI:10.3389/fmicb.2017.01543URLPMID:28861058 [本文引用: 1]
DOI:10.1007/s00122-019-03412-2URLPMID:31440772 [本文引用: 1]
KEY MESSAGE: Genetic mapping, genomic profiling and bioinformatic approaches were used to identify putative resistance genes for ear rots and low mycotoxin contamination in maize. Genomic selection seems to have good perspectives. Maize is globally an indispensable crop for humans and livestock. About 30% of yield is lost by fungal diseases with Gibberella, Fusarium and Aspergillus ear rots (ERs) having a high economic impact in most maize-growing regions of the world. They reduce not only yield, but also contaminate grains with mycotoxins like deoxynivalenol, zearalenone, fumonisins and aflatoxins, respectively. These mycotoxins pose serious health problems to humans and animals. A number of studies have been conducted to dissect the genetic architecture of resistance to these three major ear rots over the past decade. The review concentrates on studies carried out to locate quantitative trait loci (QTL) and candidate genes (CG) on the maize genome as well as the application of genomic selection in maize for resistance against Fusarium graminearum, Fusarium verticillioides and Aspergillus flavus. QTL studies by linkage or genome-wide association mapping, omic technologies (genomics, proteomics, transcriptomics and metabolomics) and bioinformatics are the methods used in the current studies to propose resistance genes against ear rot pathogens. Though a number of QTL and CG are reported, only a few specific genes were found to directly confer ER resistance in maize. A combination of two or more gene identification methods would provide a more powerful and reliable tool. Genomic selection seems to be promising for ER resistance breeding, but there are only a limited number of studies in this area. A strategy that can accurately validate and predict genotypes with major effect QTL and CG for selection will be worthwhile for practical breeding against ERs and mycotoxin contamination in maize.
DOI:10.1111/ppa.2017.66.issue-8URL [本文引用: 2]
DOI:10.3864/j.issn.0578-1752.2018.01.008URL [本文引用: 4]
【Objective】The objective of this study is to clarify the toxigenic chemotype and genetic diversity of Fusarium graminearum species complex (FGSC) causing maize ear rot from different provinces (autonomous region).【Method】The toxigenic chemotypes of 92 FGSC isolates collected from 11 provinces (autonomous region) were analyzed by using the specific primer designed based on Tri13 and Tri3 sequences. Thirteen primers with abundant bands, good repeatability, strong signal and clear background were screened from 100 universal primers developed by Columbia University (UBC) and used for ISSR-PCR amplification of these isolates. The Popgen32 software was used to calculate the percentage of polymorphic loci, Shannon’s diversity index, genetic distance and genetic similarity among populations. According to Nei’s genetic distance, UPGMA cluster analysis was carried out using NTsys2.10e software, and the dendrogram of different geographical populations was constructed. 【Result】 Four different toxigenic chemotypes including DON, 15-ADON, DON+15-ADON and NIV+15-ADON were detected among 92 FGSC isolates. One and 20 isolates represented the DON and 15-ADON chemotypes, respectively. One isolate produced both 15-ADON and NIV, and 55 isolates carried both 15-ADON and DON producing segments. The PCR amplification of all FGSC isolates was performed with 13 ISSR primers and a total of 102 fragments were obtained, of which 101 fragments displayed polymorphic and accounted for 99.02% in the total amplified fragments. The average number of fragments amplified per primer was 7.85. At population average level, the Nei’s gene diversity index (H) was 0.3129 and the Shannon’s information index (I) was 0.4774, which indicated that there was a high genetic diversity in FGSC isolates. The diversity existed among fungal populations from different geographic regions. Hebei, Shanxi, Heilongjiang and Jilin had the highest genetic diversity, while Anhui and Henan had the lowest genetic diversity. The genetic differentiation coefficient (Gst) of geographical populations was 0.2722, indicating that there were some genetic variations among different geographical populations, but most of the genetic variation (72.78%) occurred in the population. The Nm=1.3372 (>1) indicated that there was a certain gene flow among different geographical populations. The results of genetic relationship showed that the populations of Hebei, Shanxi, Heilongjiang, Jilin, Liaoning, Inner Mongolia and Gansu were relatively close, and the populations of Shandong, Jiangsu, Henan and Anhui also displayed a close relationship, whereas the population isolated from Anhui had the lowest similarity with population isolated from Inner Mongolia. Cluster analysis showed that the similarity coefficient of all isolates was 0.43-0.95. All the isolates were divided into two groups with the similarity coefficient of 0.43. Group 1 included 4 isolates of northern spring sowing region (Jilin, Shanxi and Zhangjiakou, Hebei), which did not produce NIV and DON. Group 2 consisted of the remaining 88 isolates which originated from northern spring sowing region (Shanxi, Heilongjiang, Jilin, Liaoning, Gansu and Hebei (Zhangjiakou and Tangshan)) and Huang-Huai-Hai summer sowing region (Henan, Shandong, Jiangsu, Anhui and Shijiazhuang, Hebei), and the toxins produced by these isolates were mainly DON and 15-ADON. When the similarity coefficient was 0.664, each group was divided into different subgroups, and the results of subgroups were related to the source of the isolates.【Conclusion】The main toxigenic chemotypes of FGSC from northern spring sowing region and Huang-Huai-Hai summer sowing region are DON and 15-ADON. There are abundant genetic variations in the populations of FGSC causing maize ear rot. There are certain gene exchanges among different geographical populations. The genetic diversity was related to the geographical origin.
DOI:10.3864/j.issn.0578-1752.2018.01.008URL [本文引用: 4]
【Objective】The objective of this study is to clarify the toxigenic chemotype and genetic diversity of Fusarium graminearum species complex (FGSC) causing maize ear rot from different provinces (autonomous region).【Method】The toxigenic chemotypes of 92 FGSC isolates collected from 11 provinces (autonomous region) were analyzed by using the specific primer designed based on Tri13 and Tri3 sequences. Thirteen primers with abundant bands, good repeatability, strong signal and clear background were screened from 100 universal primers developed by Columbia University (UBC) and used for ISSR-PCR amplification of these isolates. The Popgen32 software was used to calculate the percentage of polymorphic loci, Shannon’s diversity index, genetic distance and genetic similarity among populations. According to Nei’s genetic distance, UPGMA cluster analysis was carried out using NTsys2.10e software, and the dendrogram of different geographical populations was constructed. 【Result】 Four different toxigenic chemotypes including DON, 15-ADON, DON+15-ADON and NIV+15-ADON were detected among 92 FGSC isolates. One and 20 isolates represented the DON and 15-ADON chemotypes, respectively. One isolate produced both 15-ADON and NIV, and 55 isolates carried both 15-ADON and DON producing segments. The PCR amplification of all FGSC isolates was performed with 13 ISSR primers and a total of 102 fragments were obtained, of which 101 fragments displayed polymorphic and accounted for 99.02% in the total amplified fragments. The average number of fragments amplified per primer was 7.85. At population average level, the Nei’s gene diversity index (H) was 0.3129 and the Shannon’s information index (I) was 0.4774, which indicated that there was a high genetic diversity in FGSC isolates. The diversity existed among fungal populations from different geographic regions. Hebei, Shanxi, Heilongjiang and Jilin had the highest genetic diversity, while Anhui and Henan had the lowest genetic diversity. The genetic differentiation coefficient (Gst) of geographical populations was 0.2722, indicating that there were some genetic variations among different geographical populations, but most of the genetic variation (72.78%) occurred in the population. The Nm=1.3372 (>1) indicated that there was a certain gene flow among different geographical populations. The results of genetic relationship showed that the populations of Hebei, Shanxi, Heilongjiang, Jilin, Liaoning, Inner Mongolia and Gansu were relatively close, and the populations of Shandong, Jiangsu, Henan and Anhui also displayed a close relationship, whereas the population isolated from Anhui had the lowest similarity with population isolated from Inner Mongolia. Cluster analysis showed that the similarity coefficient of all isolates was 0.43-0.95. All the isolates were divided into two groups with the similarity coefficient of 0.43. Group 1 included 4 isolates of northern spring sowing region (Jilin, Shanxi and Zhangjiakou, Hebei), which did not produce NIV and DON. Group 2 consisted of the remaining 88 isolates which originated from northern spring sowing region (Shanxi, Heilongjiang, Jilin, Liaoning, Gansu and Hebei (Zhangjiakou and Tangshan)) and Huang-Huai-Hai summer sowing region (Henan, Shandong, Jiangsu, Anhui and Shijiazhuang, Hebei), and the toxins produced by these isolates were mainly DON and 15-ADON. When the similarity coefficient was 0.664, each group was divided into different subgroups, and the results of subgroups were related to the source of the isolates.【Conclusion】The main toxigenic chemotypes of FGSC from northern spring sowing region and Huang-Huai-Hai summer sowing region are DON and 15-ADON. There are abundant genetic variations in the populations of FGSC causing maize ear rot. There are certain gene exchanges among different geographical populations. The genetic diversity was related to the geographical origin.
DOI:10.1007/s10658-019-01913-wURL [本文引用: 1]
DOI:10.1007/s10658-018-1543-3URL [本文引用: 2]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1111/j.1471-8286.2007.02025.xURLPMID:21585853
Simple sequence repeat (SSR) markers for Fusarium pseudograminearum with 2 to 3 bp repeat motifs were identified by screening the genome database of the related species Fusarium graminearum. Twelve SSRs amplified single loci in both F. graminearum and F. pseudograminearum. Forty F. pseudograminearum and six F. graminearum individual isolates were screened to determine levels of polymorphism, with all SSRs displaying three to 14 alleles across all isolates. Eleven SSRs were polymorphic across F. pseudograminearum isolates tested proving the usefulness of genome databases of closely related species in identifying genetic markers.
DOI:10.1111/men.2004.4.issue-3URL
DOI:10.1016/j.fgb.2008.09.002URL
Abstract
A survey of Fusarium head blight (FHB)-contaminated wheat in Ethiopia recovered 31 isolates resembling members of the Fusarium graminearum species complex. Results of a multilocus genotyping (MLGT) assay for FHB species and trichothecene chemotype determination suggested that 22 of these isolates might represent a new species within the Fg complex. Phylogenetic analyses of multilocus DNA sequence data resolved the 22 Ethiopian isolates as a novel, phylogenetically distinct species. The new species also appears to be novel in that MLGT probe data and sequence analysis of both ends of the TRI-cluster identified 15ADON and NIV recombination blocks, documenting inter-chemotype recombination involving the chemotype-determining genes near the ends of the TRI-cluster. Results of pathogenicity experiments and analyses of trichothecene mycotoxins demonstrated that this novel Fg complex species could induce FHB on wheat and elaborate 15ADON in planta. Herein the FHB pathogen from Ethiopia is formally described as a novel species.DOI:10.3767/003158512X658123URL
DOI:10.1111/nph.14894URLPMID:29160900
Mycotoxin-producing Fusarium graminearum and related species cause Fusarium head blight on cultivated grasses, such as wheat and barley. However, these Fusarium species may have had a longer evolutionary history with North American grasses than with cultivated crops and may interact with the ancestral hosts in ways which are biochemically distinct. We assayed 25 species of asymptomatic native grasses for the presence of Fusarium species and confirmed infected grasses as hosts using re-inoculation tests. We examined seed from native grasses for the presence of mycotoxin-producing Fusarium species and evaluated the ability of these fungi to produce mycotoxins in both native grass and wheat hosts using biochemical analysis. Mycotoxin-producing Fusarium species were shown to be prevalent in phylogenetically diverse native grasses, colonizing multiple tissue types, including seeds, leaves and inflorescence structures. Artificially inoculated grasses accumulated trichothecenes to a much lesser extent than wheat, and naturally infected grasses showed little to no accumulation. Native North American grasses are commonly inhabited by Fusarium species, but appear to accommodate these toxigenic fungi differently from cultivated crops. This finding highlights how host identity and evolutionary history may influence the outcome of plant-fungal interactions and may inform future efforts in crop improvement.
DOI:10.3390/ijms9122495URLPMID:19330088 [本文引用: 1]
Fusarium graminearum clade pathogens cause Fusarium head blight (FHB) or scab of wheat and other small cereal grains, producing different kinds of trichothecene mycotoxins that are detrimental to human and domestic animals. Type B trichothecene mycotoxins such as deoxynivalenol, 3-acetyldeoxynivalenol (3-AcDON), 15-acetyldeoxynivalenol (15-AcDON) and nivalenol (NIV) are the principal Fusarium mycotoxins reported in China, as well as in other countries. A genomic polymerase chain reaction (PCR) to predict chemotypes was developed based on the structural gene sequences of Tri13 genes involved in trichothecene mycotoxin biosynthesis pathways. A single pair of primers derived from the Tri13 genes detected a 583 bp fragment from 15-AcDON-chemotypes, a 644 bp fragment from 3-AcDON-chemotypes and an 859 bp fragment from NIV-producing strains. Fusarium strains from China, Nepal, USA and Europe were identified by this method, revealing their mycotoxin chemotypes identical to that obtained by chemical analyses of HPLC or GC/MS and other PCR assays. The mycotoxin chemotype-specific fragments were amplified from a highly variable region located in Tri13 genes with three deletions for 15-AcDON-chemotypes, two deletions for 3-AcDON-chemotypes and no deletion for NIV-producers. This PCR assay generated a single amplicon and thus should be more reliable than other PCR-based assays that showed the absence or presence of a PCR fragment since these assays may generate false-negative results. The results with strains from several different countries as well as from different hosts further indicated that this method should be globally applicable. This is a rapid, reliable and cost-effective method for the identification of type B trichothecene mycotoxin chemotypes in Fusarium species and food safety controls.
DOI:10.1023/A:1026086510156URL [本文引用: 1]
The re-emergence of fusarium head blight throughout the world and especially in Western Europe prompted a survey of the situation in the Netherlands. To allow for a high throughput screening of large numbers of samples, a diagnostic PCR method was developed to detect the most common species of Fusarium occurring on wheat. Seven primer pairs were tested for their ability to identify isolates of Fusarium avenaceum, F. culmorum, F. graminearum, F. poae, F. proliferatum and Microdochium nivale var. majus and M. nivale var. nivale. Each primer pair only generated a PCR product with the corresponding Fusarium species and all PCR fragments had different molecular sizes. This allowed the generation of these amplicons using a mixture of all seven primer pairs. The robustness of this multiplex PCR encouraged us to screen a large series of isolates collected in 2000 and 2001. In both years 40 fields were sampled leading to a collection of 209 isolates from 2000 and 145 isolates from 2001. The results of the multiplex PCR demonstrated that F. graminearum was the most abundant species in the Fusarium complex on wheat in both years. This is in sharp contrast to reports from the 1980s and early 1990s, which found F. culmorum as the predominant species. Primers derived from the tri7 and tri13 genes, which are implicated in the acetylation and oxygenation of the C-4 atom of the backbone of the trichothecene molecule, were used to discriminate between deoxynivalenol and nivalenol (NIV) producers. The populations of F. culmorum and F. graminearum both showed a slight increase in NIV-producers in 2001.
DOI:10.1111/ppa.2004.53.issue-2URL [本文引用: 2]
[本文引用: 1]
DOI:10.1111/j.1365-294X.2005.02553.xURLPMID:15969739 [本文引用: 1]
The identification of genetically homogeneous groups of individuals is a long standing issue in population genetics. A recent Bayesian algorithm implemented in the software STRUCTURE allows the identification of such groups. However, the ability of this algorithm to detect the true number of clusters (K) in a sample of individuals when patterns of dispersal among populations are not homogeneous has not been tested. The goal of this study is to carry out such tests, using various dispersal scenarios from data generated with an individual-based model. We found that in most cases the estimated 'log probability of data' does not provide a correct estimation of the number of clusters, K. However, using an ad hoc statistic DeltaK based on the rate of change in the log probability of data between successive K values, we found that STRUCTURE accurately detects the uppermost hierarchical level of structure for the scenarios we tested. As might be expected, the results are sensitive to the type of genetic marker used (AFLP vs. microsatellite), the number of loci scored, the number of populations sampled, and the number of individuals typed in each sample.
DOI:10.1007/s10658-017-1331-5URL [本文引用: 1]
DOI:10.2135/cropsci2017.05.0322URL [本文引用: 1]
DOI:10.1007/s10658-015-0629-4URL [本文引用: 2]
DOI:10.1016/j.fgb.2007.10.003URL [本文引用: 1]
Abstract
Analysis of Fusarium head blight (FHB) pathogen diversity revealed that 3ADON producing Fusarium graminearum are prevalent in North America and identified significant population structure associated with trichothecene chemotype differences (FST > 0.285; P < 0.001). In addition, we identified a trichothecene chemotype cline in Canada and documented a recent and significant shift in FHB pathogen composition by demonstrating that the 3ADON chemotype frequency in western Canada increased more than 14-fold between 1998 and 2004. On average, isolates from 3ADON populations produced significantly (P < 0.05) more trichothecene and had significantly (P < 0.005) higher fecundity and growth rates than isolates from the 15ADON population. These results indicate that selection is driving the rapid spread of an introduced pathogen population that is more toxigenic and potentially more vigorous. The discovery of this previously unrecognized pathogen diversity has significant implications for food safety and cereal production in North America.URL [本文引用: 2]
EF-1α (translation elongation factor) gene and PHO (phosphate permease) gene of 31 Fusarium graminearum clade strains collected from the Yangtze basin were amplified and sequenced, and the phylogenetic analyses were made. The trichothecene mycotoxin chemotypes of these strains were assayed by PCR, and relative pathogenicity was determined on wheat cultivars Yangmai 158 and Annong 8455. Phylogenetic analysis based on sequences of EF-1α and PHO gene showed that most of strains were clustered with a distinct clade, i.e. Fusarium asiaticum, and only one strain 11027 clustered with F. graminearum. Twenty four of the 30 F. asiaticum strains were DON (deoxynivalenol) and 3-AcDON (3-Acetyldeoxynivalenol) type, and the other 6 strains were NIV (nivalenol) type, while the one F. graminearum strain 11027 was DON (deoxynivalenol) and 15-AcDON (15-Acetyldeoxynivalenol) type. On wheat cultivar Yangmai 158, there was differentiation in virulence between strains. Almost all of the high virulent strains were 3-AcDON producer, while virulence of NIV producers was generally weaker. In the Yangtze basin of China, population of F. asiaticum with a 3-AcDON chemotype is predominant. The wheat cultivar Yangmai 158 is a valuable material to assay the pathogenicity of the strains of F. graminearum clade.
URL [本文引用: 2]
EF-1α (translation elongation factor) gene and PHO (phosphate permease) gene of 31 Fusarium graminearum clade strains collected from the Yangtze basin were amplified and sequenced, and the phylogenetic analyses were made. The trichothecene mycotoxin chemotypes of these strains were assayed by PCR, and relative pathogenicity was determined on wheat cultivars Yangmai 158 and Annong 8455. Phylogenetic analysis based on sequences of EF-1α and PHO gene showed that most of strains were clustered with a distinct clade, i.e. Fusarium asiaticum, and only one strain 11027 clustered with F. graminearum. Twenty four of the 30 F. asiaticum strains were DON (deoxynivalenol) and 3-AcDON (3-Acetyldeoxynivalenol) type, and the other 6 strains were NIV (nivalenol) type, while the one F. graminearum strain 11027 was DON (deoxynivalenol) and 15-AcDON (15-Acetyldeoxynivalenol) type. On wheat cultivar Yangmai 158, there was differentiation in virulence between strains. Almost all of the high virulent strains were 3-AcDON producer, while virulence of NIV producers was generally weaker. In the Yangtze basin of China, population of F. asiaticum with a 3-AcDON chemotype is predominant. The wheat cultivar Yangmai 158 is a valuable material to assay the pathogenicity of the strains of F. graminearum clade.
DOI:10.3864/j.issn.0578-1752.2019.11.005URL [本文引用: 1]
【Objective】 The objective of this study is to clarify the composition and toxigenic chemotype of Fusarium species causing maize ear rot in Guangxi, and to provide important guidance and theoretical basis for comprehensive control of maize ear rot and reasonable distribution of varieties and resistance breeding.【Method】 The diseased ear samples were collected from main maize producing areas in Guangxi between 2016 and 2018, a total of 138 isolates from 21 counties (districts) were obtained by tissue separation and single-spore purification. Fusarium species were identified and determined according to morphological characteristics and molecular methods. The phylogenetic tree was constructed based on TEF-1α gene sequences, and specific primers were used to detect toxigenic chemotype.【Result】 A total of 10 Fusarium species were identified and confirmed among 138 isolates, including F. verticillioides, F. proliferatum, F. kyushuense, F. meridionale, F. sacchari, F. fujikuroi, F. asiaticum, F. concentricum, F. incarnatum and F. graminearum, with the isolation frequencies of 50.72%, 12.32%, 10.87%, 8.70%, 6.52%, 3.62%, 3.62%, 1.45%, 1.45% and 0.72%, respectively. F. graminearum species complex (FGSC) contained three independent species, i.e. F. meridionale, F. asiaticum and F. graminearum. F. verticillioides was the predominant pathogen, FGSC, F. proliferatum and F. kyushuense were the secondary predominant pathogens. F. sacchari and F. concentricum were the first reported to be the pathogen of maize ear rot in China. The key gene FUM1 responsible for the biosynthesis of fumonisins was detected among 67 F. verticillioides, 13 F. proliferatum, 5 F. sacchari and 3 F. fujikuroi strains, respectively, which indicated the potential ability of producing fumonisins. FUM1 was not detected in the F. concentricum strains. Four toxigenic chemotypes including NIV, 15-ADON, NIV+15-ADON and DON+15-ADON were detected among FGSC, F. kyushuense and F. incarnatum strains. Strains containing NIV chemotype included 8 F. kyushuense, 2 F. asiaticum, 2 F. meridionale, and 1 F. incarnatum. Strains containing 15-ADON chemotype included 2 F. meridionale. The NIV+15-ADON chemotype was detected among 8 F. meridionale, 2 F. kyushuense, 1 F. asiaticum and 1 F. incarnatum strains. Only 1 F. graminearum strain carried DON+15-ADON chemotype. The 3-ADON chemotype was not detected among these strains.【Conclusion】 F. verticillioides is the predominant pathogen of maize ear rot in Guangxi. FGSC, F. proliferatum and F. kyushuense are the secondary predominant species. FUM1 was detected among F. verticillioides, F. proliferatum, F. sacchari and F. fujikuroi. The main toxigenic chemtype of FGSC in Guangxi is NIV+15-ADON, while NIV is the main toxigenic chemotype of F. incarnatum and some F. kyushuense. The population composition of Fusarium species causing maize ear rot in Guangxi is different from equivalent study of temperate zone in China, which may be due to the fact that Fusarium species adapt to the high temperature and humidity growth environment in tropical and subtropical Guangxi and thus leads to the differences of toxigenic chemotypes.
DOI:10.3864/j.issn.0578-1752.2019.11.005URL [本文引用: 1]
【Objective】 The objective of this study is to clarify the composition and toxigenic chemotype of Fusarium species causing maize ear rot in Guangxi, and to provide important guidance and theoretical basis for comprehensive control of maize ear rot and reasonable distribution of varieties and resistance breeding.【Method】 The diseased ear samples were collected from main maize producing areas in Guangxi between 2016 and 2018, a total of 138 isolates from 21 counties (districts) were obtained by tissue separation and single-spore purification. Fusarium species were identified and determined according to morphological characteristics and molecular methods. The phylogenetic tree was constructed based on TEF-1α gene sequences, and specific primers were used to detect toxigenic chemotype.【Result】 A total of 10 Fusarium species were identified and confirmed among 138 isolates, including F. verticillioides, F. proliferatum, F. kyushuense, F. meridionale, F. sacchari, F. fujikuroi, F. asiaticum, F. concentricum, F. incarnatum and F. graminearum, with the isolation frequencies of 50.72%, 12.32%, 10.87%, 8.70%, 6.52%, 3.62%, 3.62%, 1.45%, 1.45% and 0.72%, respectively. F. graminearum species complex (FGSC) contained three independent species, i.e. F. meridionale, F. asiaticum and F. graminearum. F. verticillioides was the predominant pathogen, FGSC, F. proliferatum and F. kyushuense were the secondary predominant pathogens. F. sacchari and F. concentricum were the first reported to be the pathogen of maize ear rot in China. The key gene FUM1 responsible for the biosynthesis of fumonisins was detected among 67 F. verticillioides, 13 F. proliferatum, 5 F. sacchari and 3 F. fujikuroi strains, respectively, which indicated the potential ability of producing fumonisins. FUM1 was not detected in the F. concentricum strains. Four toxigenic chemotypes including NIV, 15-ADON, NIV+15-ADON and DON+15-ADON were detected among FGSC, F. kyushuense and F. incarnatum strains. Strains containing NIV chemotype included 8 F. kyushuense, 2 F. asiaticum, 2 F. meridionale, and 1 F. incarnatum. Strains containing 15-ADON chemotype included 2 F. meridionale. The NIV+15-ADON chemotype was detected among 8 F. meridionale, 2 F. kyushuense, 1 F. asiaticum and 1 F. incarnatum strains. Only 1 F. graminearum strain carried DON+15-ADON chemotype. The 3-ADON chemotype was not detected among these strains.【Conclusion】 F. verticillioides is the predominant pathogen of maize ear rot in Guangxi. FGSC, F. proliferatum and F. kyushuense are the secondary predominant species. FUM1 was detected among F. verticillioides, F. proliferatum, F. sacchari and F. fujikuroi. The main toxigenic chemtype of FGSC in Guangxi is NIV+15-ADON, while NIV is the main toxigenic chemotype of F. incarnatum and some F. kyushuense. The population composition of Fusarium species causing maize ear rot in Guangxi is different from equivalent study of temperate zone in China, which may be due to the fact that Fusarium species adapt to the high temperature and humidity growth environment in tropical and subtropical Guangxi and thus leads to the differences of toxigenic chemotypes.
DOI:10.1186/s12864-017-3524-xURLPMID:28231761 [本文引用: 1]
BACKGROUND: Fusarium graminearum is one of the main causal agents of the Fusarium Head Blight, a worldwide disease affecting cereal cultures, whose presence can lead to contaminated grains with chemically stable and harmful mycotoxins. Resistant cultivars and fungicides are frequently used to control this pathogen, and several observations suggest an adaptation of F. graminearum that raises concerns regarding the future of current plant disease management strategies. To understand the genetic basis as well as the extent of its adaptive potential, we investigated the landscape of genomic diversity among six French isolates of F. graminearum, at single-nucleotide resolution using whole-genome re-sequencing. RESULTS: A total of 242,756 high-confidence genetic variants were detected when compared to the reference genome, among which 96% are single nucleotides polymorphisms. One third of these variants were observed in all isolates. Seventy-seven percent of the total polymorphism is located in 32% of the total length of the genome, comprising telomeric/subtelomeric regions as well as discrete interstitial sections, delineating clear variant enriched genomic regions- 7.5 times in average. About 80% of all the F. graminearum protein-coding genes were found polymorphic. Biological functions are not equally affected: genes potentially involved in host adaptation are preferentially located within polymorphic islands and show greater diversification rate than genes fulfilling basal functions. We further identified 29 putative effector genes enriched with non-synonymous effect mutation. CONCLUSIONS: Our results highlight a remarkable level of polymorphism in the genome of F. graminearum distributed in a specific pattern. Indeed, the landscape of genomic diversity follows a bi-partite organization of the genome according to polymorphism and biological functions. We measured, for the first time, the level of sequence diversity for the entire gene repertoire of F. graminearum and revealed that the majority are polymorphic. Those assumed to play a role in host-pathogen interaction are discussed, in the light of the subsequent consequences for host adaptation. The annotated genetic variants discovered for this major pathogen are valuable resources for further genetic and genomic studies.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}