 ,1, 涂丽琴2, 沈建国3, 杜振国1, 蔡伟4, 季英华,2, 高芳銮,1
,1, 涂丽琴2, 沈建国3, 杜振国1, 蔡伟4, 季英华,2, 高芳銮,1The Evolutionary Dynamics and Adaptive Evolution of Tomato Chlorosis Virus
ZOU LinFeng,1, TU LiQin2, SHEN JianGuo3, DU ZhenGuo1, CAI Wei4, JI YingHua,2, GAO FangLuan,1通讯作者:
责任编辑: 岳梅
收稿日期:2020-07-2接受日期:2020-07-24网络出版日期:2020-12-01
| 基金资助: |
Received:2020-07-2Accepted:2020-07-24Online:2020-12-01
作者简介 About authors
邹林峰,E-mail:
涂丽琴,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (1756KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
邹林峰, 涂丽琴, 沈建国, 杜振国, 蔡伟, 季英华, 高芳銮. 番茄褪绿病毒的进化动态与适应性进化特征[J]. 中国农业科学, 2020, 53(23): 4791-4801 doi:10.3864/j.issn.0578-1752.2020.23.006
ZOU LinFeng, TU LiQin, SHEN JianGuo, DU ZhenGuo, CAI Wei, JI YingHua, GAO FangLuan.
开放科学(资源服务)标识码(OSID):

0 引言
【研究意义】番茄褪绿病毒(tomato chlorosis virus,ToCV)是番茄生产上危害极大的植物病毒之一。ToCV寄主广泛,可侵染茄科、番杏科、十字花科等13个科30多种植物,但其中以茄科的番茄和辣椒最为普遍[1]。该病毒在自然条件下,主要通过烟粉虱(Bemisia tabaci)进行半持久性传播[2,3,4]。在生产上,ToCV可与番茄黄化曲叶病毒(tomato yellow leaf curl virus,TYLCV)、番茄严重皱纹病毒(tomato severe rugose virus,ToSRV)和凤果花叶病毒(pepino mosaic virus,PepMV)等复合侵染植物寄主[5,6,7],从而导致严重的经济损失。在温室条件下,该病毒对番茄植株的感染率最高可达100%。ToCV侵染番茄后不仅导致其品质下降,产量也受很大影响,严重时可造成减产高达40%[1]。【前人研究进展】ToCV为长线形病毒科(Closteroviridae)毛形病毒属(Crinivirus)成员,其基因组由两条正单链的RNA(RNA1和RNA2)组成[8]。其中RNA1包含有4个开放读码框(open reading frame,ORF),主要编码与复制相关的蛋白,RNA2包含有9个ORF,主要编码与病毒外壳包被、胞间运动以及介体传播相关的蛋白[5,9-11]。ToCV的外壳蛋白有大小两个(即major and minor protein,CP and CPm)。其中,CP蛋白主要参与病毒粒体组装及病毒侵染[12]。ToCV自1998年在美国佛罗里达州[7]首次被报道发现后,已在全球30多个国家被相继报道发现。在我国,ToCV于2004年在台湾首次被报道,其后2012年相继在山东和北京[13,14]被报道,现已扩散至河北、广东和湖南等10多个省份[13,15-18],并有进一步蔓延的趋势。因此,生产上迫切需要开展ToCV的流行等研究。RNA病毒没有化石记录可用于分子钟校准,但其变异率高、世代时间短,容易形成可测量的进化群体。在一定时间跨度内,当病毒基因组累积足够的遗传变异,并且在数据集中能检测时间信号时,采样时间可以直接用于分子钟校准。通过对具有时间信号数据进行贝叶斯系统发育分析,不仅可以进行时间尺度上的推断,还可以结合离散或连续性状的信息(如采集点的地理信息)在空间尺度上进行量化分析。然而,截至目前相关研究主要集中于流感病毒(influenza virus,IFV)、登革热病毒(dengue virus,DENV)、寨卡病毒(Zika virus,ZKV)等重要人畜共患病毒[19,20,21],在植物RNA病毒中的报道相对较少,如日本****通过贝叶斯系统发育分析找到芜菁花叶病毒(turnip mosaic virus,TuMV)由欧洲传到大洋洲的证据,随后发现该病毒最早可能源于德国的野生兰花病毒,并在南欧传到小亚细亚半岛期间发生了蔓延和扩散,推断这一事件发生时间不超过700年前[22,23]。而在国内,DUAN等[24]分析了全球马铃薯S病毒(potato virus S,PVS)的进化动态,发现该病毒最先发生于南美洲(约在1325年左右),欧洲在该病毒的流行和扩散过程中起着极为重要的桥梁作用;GAO等[25]以烟草花叶病毒(tobacco mosaic virus,TMV)为对象,应用贝叶斯系统发育分析方法解析了TMV在我国主要烟草产区间的迁移特征,发现该病毒约在1935年前后传入我国西南烟草产区。这些研究为开展包括ToCV在内的其他植物病毒相关研究提供了重要的借鉴。【本研究切入点】ToCV最早报道于美国,但该病毒源于何地并于何时传入我国,目前尚未有相关报道。【拟解决的关键问题】对采自1997—2019年间的ToCV CP基因序列进行检测,确认是否存在足够的时间信号,以用于分子定年分析,并推断ToCV引入我国的时间以及替代速率。同时阐明地理因素对ToCV适应性进化的影响。1 材料与方法
试验于2017年11月至2019年5月在江苏省农业科学院植物保护研究所和福建农林大学植物病毒研究所完成。1.1 材料
供试病毒病样随机采自江苏的发病番茄叶片,样品采集后经血清学检测确定为ToCV,新鲜样本用液氮冷冻存放于-80℃冰箱。1.2 方法
1.2.1 病样总RNA的提取及cDNA合成 采用Trizol法提取样品总RNA:称取病叶0.1 g于已灭菌的2 mL离心管中,在液氮中冷冻处理后置于研磨机中将样品研磨成粉末,加入1 mL TRIzol Reagent混合,室温放置5 min。加200 μL氯仿、异戊醇混合液(氯仿:异戊醇=24:1,体积比),振荡15 s,室温放置2—3 min 后,4℃ 12 000 r/min离心15 min。取上清液至1.5 mL离心管中,加等体积的冰异丙醇,混匀,-20℃冰箱放置20 min以上。4℃ 12 000 r/min离心10 min后弃上清液,用l mL 75%乙醇清洗沉淀两次,洗涤后4℃ 5 000 r/min离心5 min,弃上清液,晾干,将RNA溶于35 μL DEPC水中,检测RNA浓度和纯度后,置于-20℃保存备用。以样品总RNA为模板进行反转录合成第一链cDNA,反转录体系(10 μL):模板RNA 0.5 μL,5×prime script master mix 2 μL,加RNase-Free H2O 7.5 μL补足10 μL。反应条件:37℃保温 15 min,85℃变性5 s,4℃保存。1.2.2 CP基因克隆 根据ToCV基因组序列设计CP扩增引物ToCVCP-F(5′-ATGGAG AACAGTGCTGTT GC-3′)和ToCVCP-R(5′-TTAGCA ACCAGT TATCG ATGC-3′),以上述cDNA为模板进行PCR扩增,反应体系:10×PCR Buffer(Mg2+ free)2.5 μL,1 mmol·L-1 MgCl2,0.05 mmol·L-1 dNTP,上下游引物各0.4 μmol·L-1,cDNA 1.0 μL,TaqDNA聚合酶 0.5 μL,ddH2O 17.5 μL,总体积25.0 μL。反应条件:94℃预变性 5 min;94℃变性 45 s,52℃退火45 s,72℃延伸50 s,34个循环;最后72℃充分延伸 10 min;PCR产物于1%琼脂糖凝胶电泳检测,按Axygen纯化回收试剂盒说明书纯化目的片段,16℃过夜连接pMD18-T载体,转化大肠杆菌DH5α感受态细胞,筛选出阳性克隆委托南京金斯瑞生物公司进行测序。
1.2.3 序列分析与重组信号检测 测序获得的序列采用DNAMAN(Lynnon BioSoft,Canada)、BLAST等软件进行序列分析。除了新测序获得的13条CP基因序列外,从GenBank下载带有采样时间和地理信息的90个分离物相应的核苷酸序列合并得到103条CP基因序列(表1)。使用PhyloSuite 1.16[26]中的MAFFT算法[27]基于密码子进行多重序列比对。由于重组可能导致过快或假阳性的进化速率[28],为排除重组对进化速率的影响,采用两种不同的方法检测ToCV CP中的重组事件。首先应用SplitsTree 4.13.1[29]基于邻接网络算法重建系统发育网络以检测数据集是否存在重组,随后通过RDP 4.97软件包的7种算法(RDP、GENECONV、BOOTSCAN、MAXCHI、CHIMAERA、SISCAN和3SEQ)[30]进行重组分析。
Table 1
表1
表1本研究用到的ToCV分离物信息
Table 1
| 分离物 Isolate | 国家 Country | 采样日期 Sampling date | 序列登录号 Accession number | 分离物 Isolate | 国家 Country | 采样日期 Sampling date | 序列登录号 Accession number | |
|---|---|---|---|---|---|---|---|---|
| BR: Taquara:15 | 巴西Brazil | 2015 | KY569401 | Gr-535 | 希腊Greece | 2008 | EU284744 | |
| ToC-Br2 | 巴西Brazil | 2006 | JQ952601 | To-Il1970 | 希腊Greece | 2011 | HG380085 | |
| BJ | 中国China | 2012-10-26 | KC311375 | To-Il1857 | 希腊Greece | 2011 | HG380088 | |
| SDSG | 中国China | 2012-10 | KC709510 | To-Il2002 | 希腊Greece | 2011 | HG380089 | |
| Nanjing | 中国China | 2014-05-12 | KJ815045 | To-Il2010 | 希腊Greece | 2011 | HG380090 | |
| 14HD-12 | 中国China | 2014-09-25 | KP335046 | To-Rh1933 | 希腊Greece | 2011 | HG380084 | |
| 15HD-8 | 中国China | 2014-09-25 | KR184675 | To-Rh1835 | 希腊Greece | 2011 | HG380087 | |
| 15-PG-9 | 中国China | 2015-03-25 | KT751008 | To-Se2042 | 希腊Greece | 2011 | HG380086 | |
| NGXJZ | 中国China | 2014-08-03 | KX272755 | 1 | 毛里求斯Mauritius | 2007-02 | FM206381 | |
| HBCZ | 中国China | 2014-08-03 | KP217195 | 2 | 毛里求斯Mauritius | 2007-02 | FM206382 | |
| HBHS | 中国China | 2014-08-03 | KP217196 | BK2-2 | 韩国South Korea | 2017-06-27 | MG001345 | |
| HBLF | 中国China | 2014-08-03 | KP217199 | BS-4 | 韩国South Korea | 2017-06-27 | MG001346 | |
| HBSJZ | 中国China | 2014-08-03 | KP217200 | N-2 | 韩国South Korea | 2017-06-17 | MG001347 | |
| HNAYHX | 中国China | 2014-08-04 | KP264983 | JN2 | 韩国South Korea | 2013 | MG813910 | |
| HNZZZM1 | 中国China | 2014-08-04 | KP264984 | NS | 韩国South Korea | 2013 | MG813911 | |
| HNZZZM2 | 中国China | 2014-08-04 | KP264985 | IS17 | 韩国South Korea | 2013 | KP114525 | |
| JLCC | 中国China | 2015-08-15 | KU306111 | YG | 韩国South Korea | 2013 | KP114528 | |
| LNDL | 中国China | 2015-08-15 | KU204707 | IS29 | 韩国South Korea | 2013 | KP114529 | |
| LNLZ | 中国China | 2016-10-18 | MF278016 | JJ3 | 韩国South Korea | 2013 | KP114533 | |
| NMHHHT | 中国China | 2015-07-15 | KU204709 | JJ5 | 韩国South Korea | 2013 | KP114534 | |
| ToCV-YL1 | 中国China | 2016-11-20 | MF346383 | JN1 | 韩国South Korea | 2013 | KP114536 | |
| SDTAFC-Bemisia tabaci | 中国China | 2012-10-20 | KC812621 | HP | 韩国South Korea | 2013 | KP114537 | |
| Shandong 12-1 | 中国China | 2015-11-01 | KY679886 | HS | 韩国South Korea | 2013 | KP137099 | |
| Shandong 28-2 | 中国China | 2015-11-01 | KY679887 | JJ | 韩国South Korea | 2013 | KP137101 | |
| Shandong 18-3 | 中国China | 2015-11-01 | KY679888 | 2.5 | 西班牙Spain | 2010 | KJ200305 | |
| SDTADP | 中国China | 2012-10-20 | KC812619 | MM8 | 西班牙Spain | 2005 | KJ200307 | |
| SDTAFC | 中国China | 2012-10-20 | KC812620 | Pl-1-2 | 西班牙Spain | 1997 | KJ200309 | |
| SDLC | 中国China | 2012-10-20 | KC812622 | AT80/99-IC | 西班牙Spain | 2014 | KJ740257 | |
| JD | 中国China | 2016-03-30 | KX900412 | AT80/99 | 西班牙Spain | 2006 | DQ136146 | |
| JD-H | 中国China | 2014-03-30 | KX987242 | XS | 中国China | 2011-11-19 | KY618797 | |
| FQ-J | 中国China | 2014-03-30 | KY471019 | TN11 | 中国China | 1998 | MF795557 | |
| SDTAMZ | 中国China | 2012-10-20 | KC812623 | Merkez | 土耳其Turkey | 2015-02-03 | KY419527 | |
| PD-KJ1 | 中国China | 2015-12-15 | KY206012 | Kas | 土耳其Turkey | 2015-02-03 | KY419528 | |
| PD-KJ2 | 中国China | 2015-12-15 | KY206014 | AKSU8 | 土耳其Turkey | 2012 | MF576337 | |
| SDQD | 中国China | 2015-08-16 | KT809400 | ALANYA42 | 土耳其Turkey | 2012 | MF576338 | |
| SDZB | 中国China | 2014-08-22 | KR072213 | ANTALYA6 | 土耳其Turkey | 2012 | MF576339 | |
| SXJZ | 中国China | 2016-08-20 | KX853540 | ANTALYA22 | 土耳其Turkey | 2012 | MF576340 | |
| ToCV-TJ6 | 中国China | 2014-08-07 | KP219722 | FETHIYE72 | 土耳其Turkey | 2012 | MF576341 | |
| ToCV-YN | 中国China | 2016-10-20 | KY471138 | FINIKE1 | 土耳其Turkey | 2012 | MF576342 | |
| BB6* | 中国China | 2019-03-21 | MN939023 | GAZIPASA4 | 土耳其Turkey | 2012 | MF576343 | |
| EB1* | 中国China | 2017-04-06 | MN939024 | KEMER20 | 土耳其Turkey | 2012 | MF576344 | |
| EB2* | 中国China | 2019-03-14 | MN939025 | KUMLUCA32 | 土耳其Turkey | 2012 | MF576345 | |
| EB6* | 中国China | 2019-03-14 | MN939026 | KUMLUCA39 | 土耳其Turkey | 2012 | MF576346 | |
| XMF3* | 中国China | 2017-04-12 | MN939027 | MANAVGAT6 | 土耳其Turkey | 2012 | MF576347 | |
| XMF4* | 中国China | 2017-03-30 | MN939028 | SERIK48 | 土耳其Turkey | 2012 | MF576348 | |
| XMF5* | 中国China | 2017-03-30 | MN939029 | TRAntToCV | 土耳其Turkey | 2018-03 | MK248741 | |
| XMF7* | 中国China | 2017-04-17 | MN939030 | Florida | 美国United States | 2005 | AY903448 | |
| XMF8* | 中国China | 2017-04-17 | MN939031 | Amelia | 美国United States | 2010-11-03 | HQ879840 | |
| XS3* | 中国China | 2019-03-14 | MN939032 | FL47 | 美国United States | 2010-11-03 | HQ879841 | |
| XS5* | 中国China | 2019-03-14 | MN939033 | Tygress | 美国United States | 2010-11-03 | HQ879842 | |
| YRDR03* | 中国China | 2017-04-27 | MN939034 | Shanty | 美国United States | 2010-11-03 | HQ879843 | |
| YRDR12* | 中国China | 2017-04-27 | MN939035 |
新窗口打开|下载CSV
1.2.4 时间信号检测 应用MURRAY等[31]的参考R脚本针对数据集中的遗传距离和采样时间差异进行Mantel 检验以确定数据集中时间信号与遗传结构是否存在混杂现象(即:相关序列的采样时间相同或相近)。如果数据集存在显著的系统发育-时间簇,则应用基于聚类排列的日期随机化检验(date- randomization test,DRT)检测数据集中的时间信号(将相同采样时间的序列先归簇后分析)。反之,则应用标准的DRT检测数据集是否存在时间信号(相同采样时间的序列直接分析不归簇)。DRT分析在BEAST 1.10.4[32]中完成,分析用实际采样时间和10个随机化日期的数据集进行比较分析。DRT有两个不同的标准:当实际数据集(根据实际采样时间)推断的替代速率平均值与日期随机化数据集推断的替代速率95%置信区间(95% CI)之间没有重叠,表明数据集通过DRT的标准1(criterion 1,CR1);而当实际数据集和日期随机化数据集两者替代速率的95% CI没有重叠,则通过DRT的标准2(criterion 2,CR2)[33]。1.2.5 替代速率和最近共祖时间推断 数据集最适的核苷酸替换模型应用PhyloSuite 1.16[26]中的ModelFinder[34]根据贝叶斯信息标准(Bayesian information criterion,BIC)进行选择,贝叶斯系统发育分析使用BEAST 1.10.4[32]软件。应用路径采样法(path sampling,PS)和垫脚石采样法(stepping-stone sampling,SS)[35]比较6个不同的模型组合,包括不同的分子钟模型(严格分子钟和对数正态分布的宽松分子钟)和溯祖树先验模型(恒定群体大小、指数增长和贝叶斯天际线)。为确保所有参数的收敛性(有效样品大小ESS>200),运行3次独立的马尔科夫链蒙特卡洛(Markov Chain Monte Carlo,MCMC)链,每次MCMC运行设置链长为 1亿代,每隔1万代抽取一次,获得的样本树弃去前10%老化样本进行分析。1.2.6 地理因素与ToCV适应性进化关联分析 将不同国家采集的ToCV分离物根据地区来源分组,对前一步贝叶斯系统发育分析生成的样本树经重采样后获得子样本。应用BaTS 2.0[36]通过计算性状随机化的1 000棵树获得关联系数(association index,AI)、简约分值(parsimony score,PS)和最大单系分支(maximum monophyletic clade,MC)3个关键参数的统计检验。地理因素与ToCV适应性进化之间是否存在关联以及关联强度根据计算获得的P 值进行判定:当所有P值<0.05,且AI较低时,表明地理因素与适应性进化存在很强的关联。
2 结果
2.1 CP的RT-PCR扩增与序列分析
应用特异性引物对(ToCVCP-F/ToCVCP-R)对20个随机抽取的ToCV分离物进行RT-PCR扩增,获得的PCR产物片段略>750 bp,与预计的目的片段大小一致(图1),阳性对照也可以扩增到目的片段,但阴性对照(健康番茄植株)和空白对照均未扩增到相应的片段。进一步抽取其中的13个分离物进行分子克隆、测序,获得的序列经DNAMAN、BLAST等分析确定目的片段大小均为774 bp。进一步序列比对分析显示,这些分离物的核苷酸序列与已报道的ToCV分离物的CP基因序列一致性均在98%以上,表明均为ToCV CP基因序列(GenBank登录号为MN939023—MN939035,表1)。两组重组检测方法均未检测显著的重组信号,因此完整的数据集全部应用于随后的分析。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1ToCV CP的RT-PCR扩增
Fig. 1RT-PCR amplification of CP from ToCV
M:DNA分子量标准 Marker DNA (2000 bp);1—20:随机抽取的ToCV分离物PCR products of CP of ToCV randomly sampled isolates;21—23:阳性对照、阴性对照和空白对照Positive control (with known ToCV infected plant), negative control 1 (healthy plant) and negative control 2 (water), respectively
2.2 时间信号检测
Mantel检验结果显示,数据集存在很强的系统发育-时间簇,共形成81个簇,遗传距离与时间距离的线性回归 P值<0.05(图2),表明数据集中时间信号与遗传结构存在混杂情形,因此,本研究数据集需要应用基于聚类排列的DRT进行时间信号的检测。基于聚类排列的DRT结果显示,通过实际采样时间和日期随机化的数据集分别推断获得的替代速率95%置信区间之间没有存在重叠,即DRT通过更为严格的CR2,表明数据集具有足够的时间信号(图3)。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2时间距离与遗传距离混杂的Mantel检验
左图为系统发育-时间簇的聚类结果,右图x和y轴分别为遗传距离和采样年份的差异
Fig. 2Mantel test of confounding of temporal and genetic distances
The left figure shows the results for phylo-temporal cluster and the x- and y-axes on the right figure indicate differences in sampling years and genetic distance, respectively
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3时间信号的日期随机化检测
Real为非日期随机化的原数据集,Rep1-10为10个不同的日期随机化数据集;实线段和虚线段显示根据实际采样日期和随机化日期计算获得的替代速率的95%置信区间,黑色实心点和圆圈显示实际采样日期和随机化日期计算获得的平均替代速率
Fig. 3Date-randomization test for temporal signal
Real indicates the non-randomized data set, Rep 1-10 are 10 different randomizations of the dates in the data set; The 95% credibility intervals of the rate estimates from the original and the randomization data set are shown by the solid and dashed lines, respectively. The mean rate estimates from the original and the randomization data set are shown by black solid dot and hollow circles, respectively
2.3 ToCV的最近共祖时间和替代速率推断
通过比较不同分子钟和不同溯祖树先验模型的对数边际似然值可知(表2),数据集最适合的模型组合为对数正态分布的宽松分子钟模型与贝叶斯天际线溯祖树先验模型。Table 2
表2
表2分子钟模型和树先验不同组合的对数边际似然值
Table 2
| 分子钟模型Molecular clock model | 树先验模型Tree prior | 对数边际似然值Log marginal likelihood |
|---|---|---|
| 不相关对数正态分布的宽松分子钟 Uncorrelated lognormal relaxed clock | 贝叶斯天际线 Bayesian skyline | -3347.81/-3347.15 |
| 不相关对数正态分布的宽松分子钟 Uncorrelated lognormal relaxed clock | 恒定大小 Constant size | -3348.44/-3349.36 |
| 不相关对数正态分布的宽松分子钟 Uncorrelated lognormal relaxed clock | 指数增长 Exponential growth | -3353.59/-3355.58 |
| 严格分子钟 Strict clock | 贝叶斯天际线Bayesian skyline | -3369.38/-3366.75 |
| 严格分子钟 Strict clock | 恒定大小Constant size | -3368.88/-3365.99 |
| 严格分子钟 Strict clock | 指数增长Exponential growth | -3372.35/-3371.24 |
新窗口打开|下载CSV
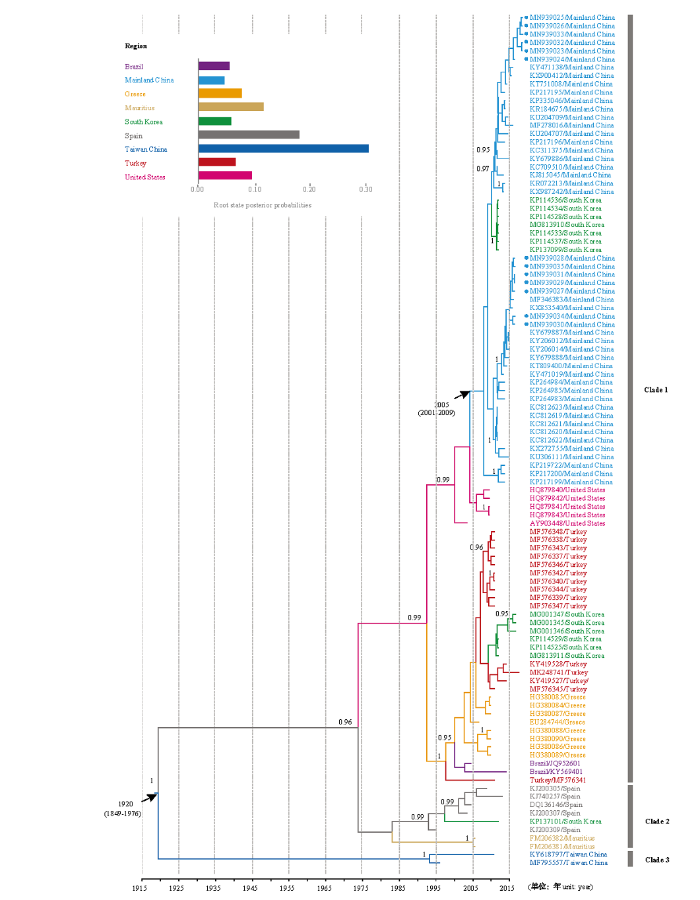
贝叶斯系统发育分析结果显示,103个ToCV分离物以非常高的后验概率(>0.95)可以聚为3个大簇(Clade 1、Clade 2和Clade 3,图4)。Clade 1多样性最高,我国大陆的所有ToCV分离物都聚在Clade 1中,Clade 2主要由西班牙分离物等组成,Clade 3仅由两个我国台湾分离物组成。ToCV最近共祖(the most recent common ancestor,MRCA)为1920年(95% CI:1849—1976),我国ToCV大陆分离物的分歧时间约为2005年(95% CI:2001—2009),并从美国传入我国大陆,而我国ToCV台湾分离物的分歧时间为1997年(95% CI:1995—1998),与我国大陆ToCV分离物的来源不同(图4)。贝叶斯系统发育分析显示(表3、图4),ToCV的CP基因替代速率约为1.12×10-3替代/位点/年(95% CI:6.08×10-4—1.73×10-3)。该替代速率与动物RNA病毒的相当,表明其正处于快速进化动态中。
图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4基于ToCV CP基因序列贝叶斯分析重建的系统发育树
分节节点上的为后验概率(仅显示>0.95)。时间轴表示枝长,为时间单位(年)。分支和端点的颜色为推断的地区状态,其根状态后验概率显示在左上角插入的面板中。蓝色加点显示的为本研究新测的13 个ToCV分离物
Fig. 4Phylogenetic tree inferred by Bayesian analysis of gene sequences of the CP of ToCV
Posterior probability values are given at each node (only >0.95 is shown). Branch lengths are scaled in units of time, as indicated by the time axis. Branch and tip colors denote inferred location states. The root state posterior probabilities estimated for each country are shown in the inset panel on the top-left corner. Blue solid dots indicate 13 ToCV isolates sequenced in this study
Table 3
表3
表3替代速率和最近共祖时间的估算
Table 3
| 参数Parameter | 数值Value |
|---|---|
| 序列长度Sequence length (nt) | |
| 日期范围Date range (year) | 1997-2019 |
| 样本大小Sample size | 103 |
| 最近共祖时间 The most recent common ancestor (year) | 1920 (1849-1976) |
| 替代速率 Substitution rate (subs/site/year) | 1.12×10-3 (6.08×10-4-1.73×10-3) |
新窗口打开|下载CSV
2.4 ToCV的地区适应性进化
BaTS分析结果显示(表4),关联系数(AI)、简约分值(PS)和最大单系分支(MC)的统计检验均呈极显著水平(P<0.01),表明ToCV分离物与其来源地区存在非常强的关联性,该病毒的适应性进化主要受地理因素所驱动。Table 4
表4
表4ToCV地理结构的系统发育-性状关联检验
Table 4
| 统计 Statistic | 观察平均值(95%置信区间) Observed mean (95% CI) | 零假设平均值(95%置信区间) Null mean (95% CI) | P值 P-value |
|---|---|---|---|
| AI | 0.20 (0.02-0.44) | 8.03 (7.36-8.71) | <0.001 |
| PS | 12.24 (11.00-14.00) | 50.11 (48.24-51.68) | <0.001 |
| MC (巴西Brail) | 1.93 (1.00-2.00) | 1.00 (1.00-1.00) | <0.01 |
| MC (中国大陆Mainland China) | 32.77 (20.00-50.00) | 3.64 (2.80-5.60) | <0.01 |
| MC (希腊Greece) | 2.87 (2.00-4.00) | 1.18 (1.00-1.83) | <0.01 |
| MC (毛里求斯Mauritius) | 2.00 (2.00-2.00) | 1.00 (1.00-1.00) | <0.01 |
| MC (韩国South Korea) | 7.00 (7.00-7.00) | 1.47 (1.10-2.01) | <0.01 |
| MC (西班牙Spain) | 4.17 (4.00-5.00) | 1.06 (1.00-1.25) | <0.01 |
| MC (中国台湾Taiwan, China) | 2.00 (2.00-2.00) | 1.00 (1.00-1.00) | <0.01 |
| MC (土耳其Turkey) | 10.39 (6.00-14.00) | 1.49 (1.03-2.05) | <0.01 |
| MC (美国United States) | 3.25 (2.00-5.00) | 1.05 (1.00-1.17) | <0.01 |
新窗口打开|下载CSV
3 讨论
ToCV在我国台湾首次被报道以来,相继在山东、北京、河北、天津、陕西、甘肃、广东、山西、内蒙古、辽宁、吉林、云南、湖南等多地被报道,已在局部地区造成番茄绝产,给我国番茄生产造成严重的经济损失[37]。本研究进化分析表明,该病毒的平均替代速率为1.12×10-3替代/位点/年(表3),与已报道的动物RNA病毒速率非常接近[38],表明其正处于快速进化动态中,这与ToCV近年来在我国多省份的发生趋势情况相一致。通过贝叶斯系统发育分析推断的ToCV我国大陆分离物和台湾分离物的起源时间不同(图4),两者的最近共祖时间分别为2005年(95% CI:2001—2009)和1997年(95% CI:1995—1998),均早于其首次报道的时间,表明ToCV在被报道前已在我国发生,且ToCV在我国台湾地区的发生时间早于大陆,台湾地区和大陆ToCV来源也不同。数据集是否具有足够的时间信号或时间结构是贝叶斯分子定年(Bayesian molecular dating)分析是否准确的重要基础,其检测方法主要有两种:一种是基于遗传距离及采样时间的线性回归计算方法,即根到端点分析(root-to-tip,RTT);另一种即本文所采用的DRT[39]。相比于DRT方法,RTT只是一种粗略的时间信号评估方法,因为它主要基于严格的分子钟假设,更常用于检测类似分子钟进化。因此,在实际应用中,RTT通常需要结合其他算法检测数据集的时间信号。MURRAY等[31]研究发现,当数据集中存在采样时间相同或相近的序列时(即时间信号与遗传结构存在混杂现象),所有标准的时间信号检测方法都会产生误导性的结果,而通过聚类排列的DRT方法[33]则可以有效地克服该问题。本文通过Mantel检验发现数据集中时间信号与遗传结构存在混杂情形(图2),应用基于聚类排列的DRT数据集也通过了更为严格的CR2(图3),表明其具有非常强的时间信号。因此,本文基于具有强时间信号的ToCV数据集的贝叶斯推断获得可信度高的MRCA和替代速率(表3、图4)。
RNA病毒高度变异,通过不断的变异以完成适应性进化。除了地理因素,生物因素(如寄主)也可能影响RNA病毒的适应性进化。目前研究显示马铃薯Y病毒属(Potyvirus)病毒的进化多数具有很强的地区适应性,如马铃薯Y病毒(potato virus Y,PVY)、辣椒脉斑驳病毒(chilli veinal mottle virus,ChiVMV)和夜来香花叶病毒(telosma mosaic virus,TeMV)[40,41,42]。然而,由于本研究涉及的ToCV分离物寄主均为番茄,因此本研究仅评估地理因素对该病毒适应性进化的影响。系统发育分析与性状关联分析结果显示ToCV分离物与其来源地区存在非常强的关联性(表4),表明该病毒的适应性进化也主要受地理因素所驱动,这是毛形病毒属病毒地区适应性的首次报道。但进一步的选择压力分析却未找到ToCV显著发生适应性进化的氨基酸位点。究其原因,主要可能有以下两点:一是由于本研究的ToCV序列数量仍然有限;二是当前用于检测正选择压力的方法,其灵敏度仍然有限(如PAML等)。
明晰ToCV引入我国后,如何在不同地区间进行传播和扩散,以及其群体经历了什么样的历史动态,将有助于预测我国ToCV的发生流行趋势。然而,由于本研究不同地区的ToCV分离物样本量存在不均衡,后续需要在更大时间跨度上采集更多地区代表性的病毒样本以开展ToCV的空间迁移研究,才能更为全面地揭示我国乃至全球ToCV群体的“前世与今生”。
4 结论
对103条具有时间信号的CP基因序列进行贝叶斯谱系动力学分析,推断ToCV最近共同祖先在1920年前后,并于2005年左右从美国传入我国。该病毒正处于快速进化动态,其适应性进化主要受地理因素驱动。(责任编辑 岳梅)
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1111/mpp.12847URLPMID:31267719 [本文引用: 2]

Tomato chlorosis virus (ToCV) causes an important disease that primarily affects tomato, although it has been found infecting other economically important vegetable crops and a wide range of wild plants. First described in Florida (USA) and associated with a 'yellow leaf disorder' in the mid-1990s, ToCV has been found in 35 countries and territories to date, constituting a paradigmatic example of an emergent plant pathogen. ToCV is transmitted semipersistently by whiteflies (Hemiptera: Aleyrodidae) belonging to the genera Bemisia and Trialeurodes. Whitefly transmission is highly efficient and cases of 100% infection are frequently observed in the field. To date, no resistant or tolerant tomato plants are commercially available and the control of the disease relies primarily on the control of the insect vector. TAXONOMY: Tomato chlorosis virus is one of the 14 accepted species in the genus Crinivirus, one of the four genera in the family Closteroviridae of plant viruses. VIRION AND GENOME PROPERTIES: The genome of ToCV is composed of two molecules of single-stranded positive-sense RNA, named RNA1 and RNA2, separately encapsidated in long, flexuous, rod-like virions. As has been shown for other closterovirids, ToCV virions are believed to have a bipolar structure. RNA1 contains four open reading frames (ORFs) encoding proteins associated with virus replication and suppression of gene silencing, whereas RNA2 contains nine ORFs encoding proteins putatively involved in encapsidation, cell-to-cell movement, gene silencing suppression and whitefly transmission. HOST RANGE: In addition to tomato, ToCV has been found to infect 84 dicot plant species belonging to 25 botanical families, including economically important crops. TRANSMISSION: Like all species within the genus Crinivirus, ToCV is semipersistently transmitted by whiteflies, being one of only two criniviruses transmitted by members of the genera Bemisia and Trialeurodes. DISEASE SYMPTOMS: Tomato 'yellow leaf disorder' syndrome includes interveinal yellowing and thickening of leaves. Symptoms first develop on lower leaves and then advance towards the upper part of the plant. Bronzing and necrosis of the older leaves are accompanied by a decline in vigour and reduction in fruit yield. In other hosts the most common symptoms include interveinal chlorosis and mild yellowing on older leaves. CONTROL: Control of the disease caused by ToCV is based on the use of healthy seedlings for transplanting, limiting accessibility of alternate host plants that can serve as virus reservoirs and the spraying of insecticides for vector control. Although several wild tomato species have been shown to contain genotypes resistant to ToCV, there are no commercially available resistant or tolerant tomato varieties to date.
DOI:10.1094/PD-90-0814URLPMID:30781245 [本文引用: 1]
Tomato chlorosis virus (ToCV), family Closteroviridae, genus Crinivirus, causes interveinal chlorosis, leaf brittleness, and limited necrotic flecking or leaf bronzing on tomato leaves. ToCV can cause a decline in plant vigor and reduce fruit yield. It is emerging as a serious production problem for field and greenhouse tomato growers, and has been increasing in prevalence in many parts of the world. The virus is unique among known whitefly-transmitted viruses, due to its ability to be transmitted by four whitefly vectors from two genera. Studies demonstrated that transmission efficiency and virus persistence in the vector varies significantly among the different whitefly vectors. Trialeurodes abutilonea and Bemisia tabaci biotype B are highly efficient vectors of ToCV. B. tabaci biotype A and T. vaporariorum are less efficient vectors, but are fully capable of transmission. ToCV persists for up to 5 days in T. abutilonea, 2 days in B. tabaci biotype B, and only 1 day in B. tabaci biotype A and T. vaporariorum. ToCV has a moderately wide host range, infecting 24 host plant species in seven families. A portion of the coat protein coding region of five geographically diverse ToCV isolates was compared and found to be highly conserved. This information, coupled with existing information on conservation within the heat shock protein 70 homologue coding region, suggests that many ToCV isolates throughout the world are related very closely, and may have been distributed on plant material.
DOI:10.1017/S0007485318000974URLPMID:30616696 [本文引用: 1]
Insect-borne plant viruses usually alter the interactions between host plant and insect vector in ways conducive to their transmission ('host manipulation hypothesis'). Most studies have tested this hypothesis with persistently and non-persistently transmitted viruses, while few have examined semi-persistently transmitted viruses. The crinivirus Tomato chlorosis virus (ToCV) is semi-persistently transmitted virus by whiteflies, and has been recently reported infecting potato plants in Brazil, where Bemisia tabaci Middle East Asia Minor 1 (MEAM1) is a competent vector. We investigated how ToCV infection modifies the interaction between potato plants and B. tabaci in ways that increase the likelihood of ToCV transmission, in two clones, one susceptible ('Agata') and the other moderately resistant (Bach-4) to B. tabaci. Whiteflies alighted and laid more eggs on ToCV-infected plants than mock-inoculated plants of Bach-4. When non-viruliferous whiteflies were released on ToCV-infected plants near mock-inoculated plants, adults moved more intensely towards non-infected plants than in the reverse condition for both clones. Feeding on ToCV-infected plants reduced egg-incubation period in both clones, but the egg-adult cycle was similar for whiteflies fed on ToCV-infected and mock-inoculated plants. Our results demonstrated that ToCV infection in potato plants alters B. tabaci behaviour and development in distinct ways depending on the host clone, with potential implications for ToCV spread.
DOI:10.1094/PHYTO.1998.88.5.402URLPMID:18944918 [本文引用: 1]
Tomato chlorosis virus (ToCV) is the second whitefly-transmitted, phloem-limited, bipartite closterovirus described infecting tomato. ToCV is distinct from tomato infectious chlorosis virus (TICV), based on lack of serological and nucleic acid cross-reactions and differences in vector specificity. TICV is transmitted only by the greenhouse whitefly (Trialeurodes vaporariorum), whereas ToCV is transmitted by the greenhouse whitefly, the banded-wing whitefly (T. abutilonea), and Bemisia tabaci biotypes A and B (B. argentifolii). Double-stranded (ds) RNA analyses of ToCV show two prominent dsRNAs of approximately 7,800 and 8,200 bp, with several small dsRNAs. Digoxigenin-11-UTP-labeled riboprobes derived from cDNA clones representing portions of RNAs 1 and 2 were used in Northern blot hybridizations to detect two large nonhomologous dsRNAs and a subset of smaller dsRNAs. These probes were used in dot blot hybridizations to detect ToCV in infected tomato. Inclusion bodies and cytoplasmic vesicles were consistently observed in phloem tissues of ToCV-infected Nicotiana clevelandii. Computer-assisted sequence analysis showed significant homology between ToCV clones that hybridize specifically with RNAs 1 and 2 and the lettuce infectious yellows virus methyltransferase of RNA 1 and the HSP70 heat shock protein homolog of RNA 2, respectively. Thus, ToCV is another member of the growing subgroup of bipartite closteroviruses transmitted by whiteflies.
DOI:10.1094/PDIS-92-5-0836BURLPMID:30769606 [本文引用: 2]
In May 2007, a survey was conducted to evaluate the phytosanitary status of grapevine propagating materials in a commercial nursery located in Valencia Province (eastern Spain). Fungal isolation was performed on 25 grafted plants (1-year-old grapevines cv. Tempranillo grafted onto 110 R rootstock) because they showed reduced root biomass and black discoloration of the xylem vessels. Sections (10 cm long) were cut from the basal end of the rootstocks, washed under running tap water, surface sterilized for 1 min in a 1.5% sodium hypochlorite solution, and washed twice with sterile distilled water. The sections were split longitudinally and small pieces of discolored tissues were placed onto malt extract agar (MEA) supplemented with streptomycin sulfate (0.5 g L(-1)). Plates were incubated at 25 degrees C in the dark for 14 to 21 days after which all colonies were transferred to potato dextrose agar (PDA). Togninia minima (Tul. & C. Tul.) Berl. (anamorph Phaeoacremonium aleophilum W. Gams, Crous, M.J. Wingf. & Mugnai) and another Phaeoacremonium sp. were consistently isolated from necrotic tissues. Single conidial isolates of this Phaeoacremonium sp. were grown on PDA and MEA in the dark at 25 degrees C for 2 to 3 weeks until colonies produced spores (3). Colonies were grayish brown on PDA and pinkish white on MEA. Conidiophores were mostly short and unbranched, 15 to 30 (mean 20.8) mum long, often consisting of an elongate-ampuliform phialide. Conidia were hyaline, oblong-ellipsoidal occasionally reniform or allantoid, 2.5 to 5.6 (mean 3.8) mum long, and 1 to 2.1 (mean 1.4) mum wide. On the basis of these characteristics, these isolates were identified as Phaeoacremonium scolyti L. Mostert, Summerb. & Crous (2,3). Identity of isolate Psc-1 was confirmed by PCR-restriction fragment length polymorphism of the internal transcribed spacer region using Phaeoacremonium-specific primers Pm1-Pm2 and restriction enzymes BssKI, EcoO109I, and HhaI (1). Additionally, the beta-tubulin gene fragment (primers T1 and Bt2b) of this isolate was sequenced (GenBank Accession No. EU260415). The sequence showed high similarity (98%) with the sequence of P. scolyti (GenBank Accession No. AY579292). Pathogenicity tests were conducted on 2-month-old grapevine seedlings (cv. Tempranillo) using the isolate Psc-1. Ten seedlings were inoculated when two to three leaves had emerged by watering the roots with 25 mL of a conidial suspension (10(6) conidia mL(-1)) harvested from 21-day-old cultures grown on PDA. Ten controls plants were inoculated with sterile distilled water. Seedlings were maintained in a greenhouse at 23 to 25 degrees C. Within 2 months of inoculation, symptoms developed on all of the inoculated plants as crown necrosis, chlorotic leaves, severe defoliation, and wilting. Control plants did not show any symptoms. The fungus was reisolated from internal tissues of the crown area and the stems of all inoculated seedlings, completing Koch's postulates. To our knowledge, this is the first report of P. scolyti causing Petri disease in Spain. References: (1) A. Aroca and R. Raposo. Appl. Environ. Microbiol. 73:2911, 2007. (2) L. Mostert et al. J. Clin. Microbiol. 43:1752, 2005. (3) L. Mostert et al. Stud. Mycol. 54:1, 2006.
URL [本文引用: 1]
Tomato chlorosis virus (ToCV), a species in the Crinivirus genus, was first reported in tomatoes in Brazil (state of Sao Paulo) in 2008. This was followed by reports in several other Brazilian states. Tomato plants with chlorotic spots and leaf roll symptoms are frequently observed in tomato fields with high whitefly populations in Central Brazil. These plants could be infected with a begomovirus, a crinivirus, or with both viruses. A survey of two selected tomato fields in the Federal District and Goias State was conducted in 2012 and 2013 specifically to determine the occurrence of begomoviruses and criniviruses. A total of 150 samples were collected and subjected to RT-PCR for ToCV detection and PCR for begomovirus detection. About 48% of the tested plants were infected with both viruses, 32% were infected with ToCV alone and 20% were only infected with the begomovirus ToSRV. The increasing incidence of ToCV associated with high whitefly populations in the field highlights the need for studying this virus disease to clarify its impact on tomato crops and minimize its potential damage.
[本文引用: 2]
DOI:10.1093/nar/gkx932URL [本文引用: 1]
DOI:10.1093/emboj/20.24.6997URLPMID:11742977 [本文引用: 1]
Diverse animal and plant viruses are able to translocate their virions between neighboring cells via intercellular connections. In this work, we analyze the virion assembly and cell-to-cell movement of a plant closterovirus and reveal a strong correlation between these two processes. The filamentous virions of a closterovirus possess a long body formed by the major capsid protein (CP) and a short tail formed by the minor capsid protein (CPm). Genetic and biochemical analyses show that the functions of these virion components are distinct. A virion body is required primarily for genome protection, whereas a tail represents a specialized device for cell-to-cell movement. Furthermore, tail assembly is mediated by the viral Hsp70 homolog (Hsp70h) that becomes an integral part of the virion. Inactivation of the ATPase domain of Hsp70h results in assembly of tailless virions that are incapable of translocation. A dual role for the viral molecular chaperone Hsp70h in virion assembly and transport, combined with the previous finding of this protein in intercellular channels, allowed us to propose a model of closteroviral movement from cell to cell.
DOI:10.1016/j.virusres.2006.02.002URLPMID:16529837
The largest extant RNA genomes are found in two diverse families of positive-strand RNA viruses, the animal Coronaviridae and the plant Closteroviridae. Comparative analysis of the viruses from the latter family reveals three levels of gene conservation. The most conserved gene module defines RNA replication and is shared with plant and animal viruses in the alphavirus-like superfamily. A module of five genes that function in particle assembly and transport is a hallmark of the family Closteroviridae and was likely present in the ancestor of all three closterovirus genera. This module includes a homologue of Hsp70 molecular chaperones and three diverged copies of the capsid protein gene. The remaining genes show dramatic variation in their numbers, functions, and origins among closteroviruses within and between the genera. Proteins encoded by these genes include suppressors of RNA silencing, RNAse III, papain-like proteases, the AlkB domain implicated in RNA repair, Zn-ribbon-containing protein, and a variety of proteins with no detectable homologues in the current databases. The evolutionary processes that have shaped the complex and fluid genomes of the large RNA viruses might be similar to those that have been involved in evolution of genomic complexity in other divisions of life.
DOI:10.1006/viro.2000.0638URLPMID:11112500 [本文引用: 1]
Assembly of the viral genome into virions is a critical process of the virus life cycle often defining the ability of the virus to move within the plant and to be transmitted horizontally to other plants. Closteroviridae virions are polar helical rods assembled primarily by a major coat protein, but with a related minor coat protein at one end. The Closteroviridae is the only virus family that encodes a protein with similarity to cellular chaperones, a 70-kDa heat-shock protein homolog (HSP70h). We examined the involvement of gene products of Citrus tristeza virus (CTV) in virion formation and found that the chaperone-like protein plus the p61 and both coat proteins were required for efficient virion assembly. Competency of virion assembly of different CTV mutants was assayed by their ability to be serially passaged in Nicotiana benthamiana protoplasts using crude sap as inoculum, and complete and partial virus particles were analyzed by serologically specific electron microscopy. Deletion mutagenesis revealed that p33, p6, p18, p13, p20, and p23 genes were not needed for virion formation. However, deletion of either minor- or major-coat protein resulted in formation of short particles which failed to be serially transferred in protoplasts, suggesting that both coat proteins are required for efficient virion assembly. Deletion or mutation of HSP70h and/or p61 dramatically reduced passage and formation of full-length virions. Frameshift mutations suggested that the HSP70h and p61 proteins, not the RNA sequences, were needed for virion assembly. Substitution of the key amino acid residues in the ATPase domain of HSP70h, Asp(7) to Lys or Glu(180) to Arg, reduced assembly, suggesting that the chaperone-like ATPase activity is involved in assembly. Both HSP70h and p61 proteins appeared to contribute equally to assembly, consistent with coordinate functions of these proteins in closterovirus virion formation. The requirement of two accessory proteins in addition to both coat proteins for efficient assembly is uniquely complex for helical virions.
DOI:10.1073/pnas.92.7.2470URLPMID:7708667 [本文引用: 1]
Elongated particles of simple RNA viruses of plants are composed of an RNA molecule coated with numerous identical capsid protein subunits to form a regular helical structure, of which tobacco mosaic virus is the archetype. Filamentous particles of the closterovirus beet yellow virus (BYV) reportedly contain approximately 4000 identical 22-kDa (p22) capsid protein subunits. The BYV genome encodes a 24-kDa protein (p24) that is structurally related to the p22. We searched for the p24 in BYV particles by using immunoelectron microscopy with specific antibodies against the recombinant p24 protein and its N-terminal peptide. A 75-nm segment at one end of the 1370-nm filamentous viral particle was found to be consistently labeled with both types of antibodies, thus indicating that p24 is indeed the second capsid protein and that the closterovirus particle, unlike those of other plant viruses with helical symmetry, has a
DOI:10.1111/jph.12236URL [本文引用: 2]
Tomato chlorosis virus (ToCV) is a whitefly-transmitted, phloem-limited, bipartite Crinivirus. In 2012, severe interveinal symptoms characteristic of ToCV infections were observed in greenhouse tomato plants in the Shandong province of China. High levels of infestation by whiteflies (Bemisia tabaci), which transmit ToCV, were also observed on tomato plants in all the greenhouses investigated. The presence of ToCV was confirmed by specific RT-PCR either in the sampled plants or in the whiteflies collected from the ventral surface of the leaves of diseased plants. The complete genomic nucleotide sequences (RNA1 and RNA2) of the Shandong isolate of ToCV (ToCV-SDSG) were determined and analysed. ToCV-SDSG RNA1 consisted of 8594 nucleotides encompassing four open reading frames (ORFs). ToCV-SDSG RNA2 consisted of 8242 nucleotides encompassing nine ORFs. Phylogenetic analysis suggests that the Chinese ToCV-SDSG isolate is most similar to the ToCV-Florida isolate.
DOI:10.1094/PDIS-12-12-1163-PDNURLPMID:30722472 [本文引用: 1]
In October 2012, a severe yellowing disease was found on greenhouse and plastic house tomato (Solanum lycopersicum) plants in Beijing, China. The disease incidence varied from 5 to 80% in each of six fields across Haidian and Daxing districts. The lower leaves showed symptoms of interveinal chlorosis, leaf brittleness, and limited brown necrotic flecks, similar to symptoms induced by Tomato chlorosis virus (ToCV) and Tomato infectious chlorosis virus (TICV) (two members of genus Crinivirus, family Closteroviridae) (4). A large number of whiteflies (Bemisia tabaci) were also observed. Leaf samples were taken from eight symptomatic and two asymptomatic tomato plants in two plastic houses in the Haidian district and total RNA was isolated from the 10 samples using TRIzol reagent (Tiangen, Beijing, China). Nested reverse transcription (RT)-PCR was performed to test the presence of ToCV and TICV with degenerate primers HS-11 and HS-12 and specific primers ToC-5/ToC-6 or TIC-3/TIC-4 for ToCV or TICV, respectively (1). With ToCV primers, a 463-bp specific fragment was amplified from eight symptomatic samples but not from two asymptomatic samples, and there was no amplification with TICV primers from any sample. Sequence analysis of the amplified fragment showed 99% nucleotide sequence identity with the heat shock protein 70 homolog (HSP70h) gene of ToCV isolates from Japan (GenBank Accession No. AB513442), Spain (DQ136146), Florida (AY903448), and Greece (EU284744). The presence of ToCV was confirmed by amplification of a 848-bp fragment covering the coat protein (CP) gene of ToCV with primers CP-F (5'-GAATCTTTTAGAAGCTTTGGTTTAAGG-3') and CP-R (5'-GATCCTCTTGATCCTCATAGATTTC-3') (3). The CP had 97 to 99% amino acid sequence identity to the above-mentioned four ToCV isolates. A sequence of the CP gene obtained from one isolate was deposited at GenBank (KC311375). Additionally, virions were isolated from 25 g of symptomatic samples followed Klaassen's method (2) and their lengths were estimated to be about 800 to 850 nm by transmission electronic microscopy To our knowledge, this is the first report of ToCV on tomato in mainland China. Tomato is one of the most widely cultivated crops in China and the spread of ToCV in China may cause significant economic losses. Further information on the prevalence and incidence of ToCV is required to assess the potential impact of this virus. References: (1) C. I. Dovas et al. Plant Dis. 86:1345, 2002. (2) V. A. Klaassen et al. J. Gen. Virol. 75:1525, 1994. (3) H. Tomoki et al. J. Gen. Plant Pathol. 76:168, 2010. (4) G. C. Wisler et al. Phytopathology 88:402, 1998.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/srep45505URLPMID:28378782 [本文引用: 1]
DENV-2 spread throughout the tropical and subtropical regions globally, which is implicated in deadly outbreaks of DHF and DSS. Since dengue cases have grown dramatically in recent years, about half of the world's population is now at risk. Our timescale analysis indicated that the most recent common ancestor existed about 100 years ago. The rate of nucleotide substitution was estimated to be 8.94 x 10(-4) subs/site/year. Selection pressure analysis showed that two sites 160 and 403 were under positive selection, while E gene is mainly shaped by stronger purifying selection. BSP analysis showed that estimating effective population size from samples of sequences has undergone three obvious increases, additionally, Caribbean and Puerto Rico maintained higher levels of genetic diversity relative to other 6 representative geographical populations using GMRF method. The phylogeographic analysis indicated that two major transmission routes are from South America to Caribbean and East&SouthAsia to Puerto Rico. The trunk reconstruction confirmed that the viral evolution spanned 50 years occurred primarily in Southeast Asia and East&South Asia. In addition, phylogeographic association-trait analysis indicated that the viral phenotypes are highly correlated with phylogeny in Nicaragua and Puerto Rico (P < 0.05).
DOI:10.1016/S0140-6736(13)60938-1URL [本文引用: 1]
DOI:10.1371/journal.pntd.0002636URLPMID:24421913 [本文引用: 1]
Zika virus (ZIKV) is a mosquito-borne flavivirus first isolated in Uganda in 1947. Although entomological and virologic surveillance have reported ZIKV enzootic activity in diverse countries of Africa and Asia, few human cases were reported until 2007, when a Zika fever epidemic took place in Micronesia. In the context of West Africa, the WHO Collaborating Centre for Arboviruses and Hemorrhagic Fever at Institut Pasteur of Dakar (http://www.pasteur.fr/recherche/banques/CRORA/) reports the periodic circulation of ZIKV since 1968. Despite several reports on ZIKV, the genetic relationships among viral strains from West Africa remain poorly understood. To evaluate the viral spread and its molecular epidemiology, we investigated 37 ZIKV isolates collected from 1968 to 2002 in six localities in Senegal and Cote d'Ivoire. In addition, we included strains from six other countries. Our results suggested that these two countries in West Africa experienced at least two independent introductions of ZIKV during the 20(th) century, and that apparently these viral lineages were not restricted by mosquito vector species. Moreover, we present evidence that ZIKV has possibly undergone recombination in nature and that a loss of the N154 glycosylation site in the envelope protein was a possible adaptive response to the Aedes dalzieli vector.
DOI:10.1099/jgv.0.000007URL [本文引用: 1]
DOI:10.1038/s41598-017-01934-7URLPMID:28652582 [本文引用: 1]
Plant viruses have important global impacts on crops, and identifying their centre and date of emergence is important for planning control measures. Turnip mosaic virus (TuMV) is a member of the genus Potyvirus in the family Potyviridae and is a major worldwide pathogen of brassica crops. For two decades, we have collected TuMV isolates, mostly from brassicas, in Turkey and neighbouring countries. This region is thought to be the centre of emergence of this virus. We determined the genomic sequences of 179 of these isolates and used these to estimate the timescale of the spread of this virus. Our Bayesian coalescent analyses used synonymous sites from a total of 417 novel and published whole-genome sequences. We conclude that TuMV probably originated from a virus of wild orchids in Germany and, while adapting to wild and domestic brassicas, spread via Southern Europe to Asia Minor no more than 700 years ago. The population of basal-B group TuMVs in Asia Minor is older than all other populations of this virus, including a newly discovered population in Iran. The timescale of the spread of TuMV correlates well with the establishment of agriculture in these countries.
DOI:10.1016/j.virol.2018.09.022URLPMID:30296680 [本文引用: 1]
Potato virus S (PVS) is a major plant pathogen that causes considerable losses in global potato production. Knowledge of the evolutionary history and spatio-temporal dynamics of PVS is vital for developing sustainable management schemes. In this study, we investigated the phylodynamics of the virus by analysing 103 nucleotide sequences of the coat protein gene, sampled between 1985 and 2014. Our Bayesian phylogenetic analyses showed that PVS has been evolving at a rate of 3.32x10(-4) substitutions/site/year (95% credibility interval 1.33x10(-4)-5.58x10(-4)). We dated the crown group to the year 1325 CE (95% credibility interval 762-1743 CE). Our phylogeographic analyses pointed to viral origins in South America and identified multiple migration pathways between Europe and other regions, suggesting that Europe has been a major hub for PVS transmission. The results of our study have potential implications for developing effective strategies for the control of this pathogen.
DOI:10.1016/j.virol.2018.12.001URLPMID:30594790 [本文引用: 1]
Tobacco mosaic virus (TMV) is widespread in China and causes considerable economic losses to tobacco production. The molecular epidemiology of this virus is, however, poorly understood. In this study, we sequenced the genomes of 51 TMV isolates from five tobacco-producing regions in China and investigated the dispersal patterns of this virus. Our phylogenetic analysis showed that TMV might have been introduced to China in the early 1900s, probably first to southwest China. However, TMV then moved to the north of the country, where it expanded. The north became the main seeding region for the subsequent movements of the virus within China. The north-to-south movement of TMV coincides with a shift of major tobacco-producing areas from north to south in this century, suggesting a link between human activities and the dispersal of TMV in China.
DOI:10.1111/1755-0998.13096URLPMID:31599058 [本文引用: 2]
Multigene and genomic data sets have become commonplace in the field of phylogenetics, but many existing tools are not designed for such data sets, which often makes the analysis time-consuming and tedious. Here, we present PhyloSuite, a (cross-platform, open-source, stand-alone Python graphical user interface) user-friendly workflow desktop platform dedicated to streamlining molecular sequence data management and evolutionary phylogenetics studies. It uses a plugin-based system that integrates several phylogenetic and bioinformatic tools, thereby streamlining the entire procedure, from data acquisition to phylogenetic tree annotation (in combination with iTOL). It has the following features: (a) point-and-click and drag-and-drop graphical user interface; (b) a workplace to manage and organize molecular sequence data and results of analyses; (c) GenBank entry extraction and comparative statistics; and (d) a phylogenetic workflow with batch processing capability, comprising sequence alignment (mafft and macse), alignment optimization (trimAl, HmmCleaner and Gblocks), data set concatenation, best partitioning scheme and best evolutionary model selection (PartitionFinder and modelfinder), and phylogenetic inference (MrBayes and iq-tree). PhyloSuite is designed for both beginners and experienced researchers, allowing the former to quick-start their way into phylogenetic analysis, and the latter to conduct, store and manage their work in a streamlined way, and spend more time investigating scientific questions instead of wasting it on transferring files from one software program to another.
DOI:10.1093/molbev/mst010URL [本文引用: 1]
We report a major update of the MAFFT multiple sequence alignment program. This version has several new features, including options for adding unaligned sequences into an existing alignment, adjustment of direction in nucleotide alignment, constrained alignment and parallel processing, which were implemented after the previous major update. This report shows actual examples to explain how these features work, alone and in combination. Some examples incorrectly aligned by MAFFT are also shown to clarify its limitations. We discuss how to avoid misalignments, and our ongoing efforts to overcome such limitations.
DOI:10.1038/nrg3905URLPMID:25783448 [本文引用: 1]
Infections are one of the major selective pressures acting on humans, and host-pathogen interactions contribute to shaping the genetic diversity of both organisms. Evolutionary genomic studies take advantage of experiments that natural selection has been performing over millennia. In particular, inter-species comparative genomic analyses can highlight the genetic determinants of infection susceptibility or severity. Recent examples show how evolution-guided approaches can provide new insights into host-pathogen interactions, ultimately clarifying the basis of host range and explaining the emergence of different diseases. We describe the latest developments in comparative immunology and evolutionary genetics, showing their relevance for understanding the molecular determinants of infection susceptibility in mammals.
DOI:10.1093/bioinformatics/14.1.68URLPMID:9520503 [本文引用: 1]
MOTIVATION: Real evolutionary data often contain a number of different and sometimes conflicting phylogenetic signals, and thus do not always clearly support a unique tree. To address this problem, Bandelt and Dress (Adv. Math., 92, 47-05, 1992) developed the method of split decomposition. For ideal data, this method gives rise to a tree, whereas less ideal data are represented by a tree-like network that may indicate evidence for different and conflicting phylogenies. RESULTS: SplitsTree is an interactive program, for analyzing and visualizing evolutionary data, that implements this approach. It also supports a number of distances transformations, the computation of parsimony splits, spectral analysis and bootstrapping.
DOI:10.1093/ve/vev003URLPMID:27774277 [本文引用: 1]
RDP4 is the latest version of recombination detection program (RDP), a Windows computer program that implements an extensive array of methods for detecting and visualising recombination in, and stripping evidence of recombination from, virus genome sequence alignments. RDP4 is capable of analysing twice as many sequences (up to 2,500) that are up to three times longer (up to 10 Mb) than those that could be analysed by older versions of the program. RDP4 is therefore also applicable to the analysis of bacterial full-genome sequence datasets. Other novelties in RDP4 include (1) the capacity to differentiate between recombination and genome segment reassortment, (2) the estimation of recombination breakpoint confidence intervals, (3) a variety of 'recombination aware' phylogenetic tree construction and comparison tools, (4) new matrix-based visualisation tools for examining both individual recombination events and the overall phylogenetic impacts of multiple recombination events and (5) new tests to detect the influences of gene arrangements, encoded protein structure, nucleic acid secondary structure, nucleotide composition, and nucleotide diversity on recombination breakpoint patterns. The key feature of RDP4 that differentiates it from other recombination detection tools is its flexibility. It can be run either in fully automated mode from the command line interface or with a graphically rich user interface that enables detailed exploration of both individual recombination events and overall recombination patterns.
DOI:10.1111/2041-210X.12466URLPMID:27110344 [本文引用: 2]
'Dated-tip' methods of molecular dating use DNA sequences sampled at different times, to estimate the age of their most recent common ancestor. Several tests of 'temporal signal' are available to determine whether data sets are suitable for such analysis. However, it remains unclear whether these tests are reliable.We investigate the performance of several tests of temporal signal, including some recently suggested modifications. We use simulated data (where the true evolutionary history is known), and whole genomes of methicillin-resistant Staphylococcus aureus (to show how particular problems arise with real-world data sets).We show that all of the standard tests of temporal signal are seriously misleading for data where temporal and genetic structures are confounded (i.e. where closely related sequences are more likely to have been sampled at similar times). This is not an artefact of genetic structure or tree shape per se, and can arise even when sequences have measurably evolved during the sampling period. More positively, we show that a 'clustered permutation' approach introduced by Duchene et al. (Molecular Biology and Evolution, 32, 2015, 1895) can successfully correct for this artefact in all cases and introduce techniques for implementing this method with real data sets.The confounding of temporal and genetic structures may be difficult to avoid in practice, particularly for outbreaks of infectious disease, or when using ancient DNA. Therefore, we recommend the use of 'clustered permutation' for all analyses. The failure of the standard tests may explain why different methods of dating pathogen origins have reached such wildly different conclusions.
DOI:10.1093/molbev/mss075URL [本文引用: 2]
Computational evolutionary biology, statistical phylogenetics and coalescent-based population genetics are becoming increasingly central to the analysis and understanding of molecular sequence data. We present the Bayesian Evolutionary Analysis by Sampling Trees (BEAST) software package version 1.7, which implements a family of Markov chain Monte Carlo (MCMC) algorithms for Bayesian phylogenetic inference, divergence time dating, coalescent analysis, phylogeography and related molecular evolutionary analyses. This package includes an enhanced graphical user interface program called Bayesian Evolutionary Analysis Utility (BEAUti) that enables access to advanced models for molecular sequence and phenotypic trait evolution that were previously available to developers only. The package also provides new tools for visualizing and summarizing multispecies coalescent and phylogeographic analyses. BEAUti and BEAST 1.7 are open source under the GNU lesser general public license and available at http://beast-mcmc.googlecode.comandhttp://beast.bio.ed.ac.uk.
DOI:10.1093/molbev/msv056URLPMID:25771196 [本文引用: 2]
Rates and timescales of viral evolution can be estimated using phylogenetic analyses of time-structured molecular sequences. This involves the use of molecular-clock methods, calibrated by the sampling times of the viral sequences. However, the spread of these sampling times is not always sufficient to allow the substitution rate to be estimated accurately. We conducted Bayesian phylogenetic analyses of simulated virus data to evaluate the performance of the date-randomization test, which is sometimes used to investigate whether time-structured data sets have temporal signal. An estimate of the substitution rate passes this test if its mean does not fall within the 95% credible intervals of rate estimates obtained using replicate data sets in which the sampling times have been randomized. We find that the test sometimes fails to detect rate estimates from data with no temporal signal. This error can be minimized by using a more conservative criterion, whereby the 95% credible interval of the estimate with correct sampling times should not overlap with those obtained with randomized sampling times. We also investigated the behavior of the test when the sampling times are not uniformly distributed throughout the tree, which sometimes occurs in empirical data sets. The test performs poorly in these circumstances, such that a modification to the randomization scheme is needed. Finally, we illustrate the behavior of the test in analyses of nucleotide sequences of cereal yellow dwarf virus. Our results validate the use of the date-randomization test and allow us to propose guidelines for interpretation of its results.
DOI:10.1038/nmeth.4285URLPMID:28481363 [本文引用: 1]
Model-based molecular phylogenetics plays an important role in comparisons of genomic data, and model selection is a key step in all such analyses. We present ModelFinder, a fast model-selection method that greatly improves the accuracy of phylogenetic estimates by incorporating a model of rate heterogeneity across sites not previously considered in this context and by allowing concurrent searches of model space and tree space.
DOI:10.1093/molbev/mss084URL [本文引用: 1]
Recent developments in marginal likelihood estimation for model selection in the field of Bayesian phylogenetics and molecular evolution have emphasized the poor performance of the harmonic mean estimator (HME). Although these studies have shown the merits of new approaches applied to standard normally distributed examples and small real-world data sets, not much is currently known concerning the performance and computational issues of these methods when fitting complex evolutionary and population genetic models to empirical real-world data sets. Further, these approaches have not yet seen widespread application in the field due to the lack of implementations of these computationally demanding techniques in commonly used phylogenetic packages. We here investigate the performance of some of these new marginal likelihood estimators, specifically, path sampling (PS) and stepping-stone (SS) sampling for comparing models of demographic change and relaxed molecular clocks, using synthetic data and real-world examples for which unexpected inferences were made using the HME. Given the drastically increased computational demands of PS and SS sampling, we also investigate a posterior simulation-based analogue of Akaike's information criterion (AIC) through Markov chain Monte Carlo (MCMC), a model comparison approach that shares with the HME the appealing feature of having a low computational overhead over the original MCMC analysis. We confirm that the HME systematically overestimates the marginal likelihood and fails to yield reliable model classification and show that the AICM performs better and may be a useful initial evaluation of model choice but that it is also, to a lesser degree, unreliable. We show that PS and SS sampling substantially outperform these estimators and adjust the conclusions made concerning previous analyses for the three real-world data sets that we reanalyzed. The methods used in this article are now available in BEAST, a powerful user-friendly software package to perform Bayesian evolutionary analyses.
DOI:10.1016/j.meegid.2007.08.001URLPMID:17921073 [本文引用: 1]
Many recent studies have sought to quantify the degree to which viral phenotypic characters (such as epidemiological risk group, geographic location, cell tropism, drug resistance state, etc.) are correlated with shared ancestry, as represented by a viral phylogenetic tree. Here, we present a new Bayesian Markov-Chain Monte Carlo approach to the investigation of such phylogeny-trait correlations. This method accounts for uncertainty arising from phylogenetic error and provides a statistical significance test of the null hypothesis that traits are associated randomly with phylogeny tips. We perform extensive simulations to explore and compare the behaviour of three statistics of phylogeny-trait correlation. Finally, we re-analyse two existing published data sets as case studies. Our framework aims to provide an improvement over existing methods for this problem.
DOI:10.3864/j.issn.0578-1752.2019.02.005URL [本文引用: 1]
【Objective】 The objective of this study is to determine the pathogens, dominant viruses, distributions of Solanaceae, Cucurbitaceae, Leguminosae, and Cruciferae vegetables viral diseases, and to analyze the expansion trend of some important viruses in China.【Method】 Viral disease survey and virus detection using a combination of serological and molecular biology methods on Solanaceae, Cucurbitaceae, Leguminosae, and Cruciferae vegetables were carried out in 31 provinces (municipalities, autonomous regions) of mainland China from 2013 to 2017.【Result】 A total of 41 653 suspected virus disease samples of Solanaceae, Cucurbitaceae, Leguminosae and Cruciferae vegetables were collected from the 31 provinces (municipalities, autonomous regions) across the country. A total of 63 viruses were detected, of which 40 viruses were detected in solanaceous vegetable crops, as well as 26 viruses in cucurbitaceous vegetable crops, 19 viruses in leguminous vegetable crops, and 14 viruses in cruciferous vegetable crops. Among them, 33 and 25 viruses were detected in pepper and tomato of solanaceous samples, respectively; 22 and 19 viruses were detected in pumpkin and cucumber of cucurbitaceous samples, respectively; 14, 12, 12 and 7 viruses were detected in cowpea, kidney beans, radish and Chinese cabbage, respectively. Mixed infections were common in the vegetable crops with two to six viruses respectively, most of the co-infected crops were infected with two viruses and some of the pepper, tomato and eggplant plants were infected with six viruses. Tomato mottle mosaic virus (ToMMV), Zucchini tigre mosaic virus (ZTMV), Pepper vein yellows virus (PeVYV), Pepper cryptic virus 1 (PCV-1) and Pepper cryptic virus 2 (PCV-2) were firstly recorded in China. ToMMV, Melon aphid-borne yellows virus (MABYV), Cucurbit aphid-borne yellows virus (CABYV) were firstly detected on pepper plants, ZTMV and Tobacco mild green mosaic virus (TMGMV) were firstly recorded on cucumber and pumpkin plants, respectively. While Tomato spotted wilt tospovirus (TSWV) was firstly detected on celery, Datura stramonium, cowpea, pea, Codonopsis pilosula, dahlia, Tropaeolum majus, Solanum indicum, and so on. Chilli veinal mottle virus (ChiVMV) was firstly found infecting Solanum yingjiangense. Meanwhile, pepper and tomato plants were found as the new natural hosts of Tobacco bushy top virus (TBTV). 【Conclusion】 Cucumber mosaic virus (CMV), Tobacco mosaic virus (TMV), Turnip mosaic virus (TuMV) and Broad bean wilt virus 2 (BBWV2) are the dominant viruses currently in China due to their widely distribution and serious damage on vegetable crops nationwide. Among them, CMV occurs in all of the 31 provinces (municipalities, autonomous regions), and is also the dominant virus in the all regions. CMV and TMV are the dominant viruses on solanaceous vegetable crops, while CMV, Zucchini yellow mosaic virus (ZYMV), Cucumber green mottle mosaic virus (CGMMV) and TMV are the dominant viruses on cucurbitaceous vegetable crops. CMV and BBWV2 are the dominant viruses of leguminous vegetable crops, and TuMV, CMV, TMV are the dominant viruses of cruciferous vegetable crops. Tomato chlorosis virus (ToCV), TSWV, CGMMV and ToMMV, which occurring seriously in some provinces and spreading rapidly, may have the risk of national epidemic.
DOI:10.3864/j.issn.0578-1752.2019.02.005URL [本文引用: 1]
【Objective】 The objective of this study is to determine the pathogens, dominant viruses, distributions of Solanaceae, Cucurbitaceae, Leguminosae, and Cruciferae vegetables viral diseases, and to analyze the expansion trend of some important viruses in China.【Method】 Viral disease survey and virus detection using a combination of serological and molecular biology methods on Solanaceae, Cucurbitaceae, Leguminosae, and Cruciferae vegetables were carried out in 31 provinces (municipalities, autonomous regions) of mainland China from 2013 to 2017.【Result】 A total of 41 653 suspected virus disease samples of Solanaceae, Cucurbitaceae, Leguminosae and Cruciferae vegetables were collected from the 31 provinces (municipalities, autonomous regions) across the country. A total of 63 viruses were detected, of which 40 viruses were detected in solanaceous vegetable crops, as well as 26 viruses in cucurbitaceous vegetable crops, 19 viruses in leguminous vegetable crops, and 14 viruses in cruciferous vegetable crops. Among them, 33 and 25 viruses were detected in pepper and tomato of solanaceous samples, respectively; 22 and 19 viruses were detected in pumpkin and cucumber of cucurbitaceous samples, respectively; 14, 12, 12 and 7 viruses were detected in cowpea, kidney beans, radish and Chinese cabbage, respectively. Mixed infections were common in the vegetable crops with two to six viruses respectively, most of the co-infected crops were infected with two viruses and some of the pepper, tomato and eggplant plants were infected with six viruses. Tomato mottle mosaic virus (ToMMV), Zucchini tigre mosaic virus (ZTMV), Pepper vein yellows virus (PeVYV), Pepper cryptic virus 1 (PCV-1) and Pepper cryptic virus 2 (PCV-2) were firstly recorded in China. ToMMV, Melon aphid-borne yellows virus (MABYV), Cucurbit aphid-borne yellows virus (CABYV) were firstly detected on pepper plants, ZTMV and Tobacco mild green mosaic virus (TMGMV) were firstly recorded on cucumber and pumpkin plants, respectively. While Tomato spotted wilt tospovirus (TSWV) was firstly detected on celery, Datura stramonium, cowpea, pea, Codonopsis pilosula, dahlia, Tropaeolum majus, Solanum indicum, and so on. Chilli veinal mottle virus (ChiVMV) was firstly found infecting Solanum yingjiangense. Meanwhile, pepper and tomato plants were found as the new natural hosts of Tobacco bushy top virus (TBTV). 【Conclusion】 Cucumber mosaic virus (CMV), Tobacco mosaic virus (TMV), Turnip mosaic virus (TuMV) and Broad bean wilt virus 2 (BBWV2) are the dominant viruses currently in China due to their widely distribution and serious damage on vegetable crops nationwide. Among them, CMV occurs in all of the 31 provinces (municipalities, autonomous regions), and is also the dominant virus in the all regions. CMV and TMV are the dominant viruses on solanaceous vegetable crops, while CMV, Zucchini yellow mosaic virus (ZYMV), Cucumber green mottle mosaic virus (CGMMV) and TMV are the dominant viruses on cucurbitaceous vegetable crops. CMV and BBWV2 are the dominant viruses of leguminous vegetable crops, and TuMV, CMV, TMV are the dominant viruses of cruciferous vegetable crops. Tomato chlorosis virus (ToCV), TSWV, CGMMV and ToMMV, which occurring seriously in some provinces and spreading rapidly, may have the risk of national epidemic.
DOI:10.1038/nrg2323URLPMID:18319742 [本文引用: 1]
Understanding the factors that determine the rate at which genomes generate and fix mutations provides important insights into key evolutionary mechanisms. We review our current knowledge of the rates of mutation and substitution, as well as their determinants, in RNA viruses, DNA viruses and retroviruses. We show that the high rate of nucleotide substitution in RNA viruses is matched by some DNA viruses, suggesting that evolutionary rates in viruses are explained by diverse aspects of viral biology, such as genomic architecture and replication speed, and not simply by polymerase fidelity.
DOI:10.1111/mec.13586URLPMID:26880113 [本文引用: 1]
Molecular dating of phylogenetic trees is a growing discipline using sequence data to co-estimate the timing of evolutionary events and rates of molecular evolution. All molecular-dating methods require converting genetic divergence between sequences into absolute time. Historically, this could only be achieved by associating externally derived dates obtained from fossil or biogeographical evidence to internal nodes of the tree. In some cases, notably for fast-evolving genomes such as viruses and some bacteria, the time span over which samples were collected may cover a significant proportion of the time since they last shared a common ancestor. This situation allows phylogenetic trees to be calibrated by associating sampling dates directly to the sequences representing the tips (terminal nodes) of the tree. The increasing availability of genomic data from ancient DNA extends the applicability of such tip-based calibration to a variety of taxa including humans, extinct megafauna and various microorganisms which typically have a scarce fossil record. The development of statistical models accounting for heterogeneity in different aspects of the evolutionary process while accommodating very large data sets (e.g. whole genomes) has allowed using tip-dating methods to reach inferences on divergence times, substitution rates, past demography or the age of specific mutations on a variety of spatiotemporal scales. In this review, we summarize the current state of the art of tip dating, discuss some recent applications, highlight common pitfalls and provide a 'how to' guide to thoroughly perform such analyses.
DOI:10.1007/s00705-016-2761-7URLPMID:26831930 [本文引用: 1]
Chilli veinal mottle virus (ChiVMV) is an important plant pathogen with a wide host range. The genetic structure of ChiVMV was investigated by analyzing the coat protein (CP) genes of 87 ChiVMV isolates from seven Asian regions. Pairwise F ST values between ChiVMV populations ranged from 0.108 to 0.681, indicating a significant spatial structure for this pathogen. In phylogeny-trait association analysis, the viral isolates from the same region tended to group together, showing a distinct geographic feature. These results suggest that geographic driven adaptation may be an important determinant of the genetic diversity of ChiVMV.
DOI:10.1111/eva.12459URLPMID:28352297 [本文引用: 1]
Potato virus Y (PVY) is an important plant pathogen causing considerable economic loss to potato production. Knowledge of the population genetic structure and evolutionary biology of the pathogen, particularly at a transnational scale, is limited but vital in developing sustainable management schemes. In this study, the population genetic structure and molecular evolution of PVY were studied using 127 first protein (P1) and 137 coat protein (CP) sequences generated from isolates collected from potato in China and Japan. High genetic differentiation was found between the populations from the two countries, with higher nucleotide diversity in Japan than China in both genes and a KST value of .216 in the concatenated sequences of the two genes. Sequences from the two countries clustered together according to their geographic origin. Further analyses showed that spatial genetic structure in the PVY populations was likely caused by demographic dynamics of the pathogen and natural selection generated by habitat heterogeneity. Purifying selection was detected at the majority of polymorphic sites although some clade-specific codons were under positive selection. In past decades, PVY has undergone a population expansion in China, whereas in Japan, the population size of the pathogen has remained relatively constant.
DOI:10.3864/j.issn.0578-1752.2017.24.006URL [本文引用: 1]
【Objective】 The objective of this study is to identify Telosma mosaic virus (TeMV) on passion fruit in Fujian orchard, develop a molecular method for specific and rapid identification of TeMV, and to provide a reference for prevention and control of the virus.【Method】The Fujian passion fruit samples were detected by using the serology method, electron microscopic observation, universal degenerate primers RT-PCR and specific primers RT-PCR. The PCR products of the positive sample were cloned and sequenced. A set of specific primes amplified the total length of coat protein (CP) gene was designed according to the sequences of reported TeMV and sequence determination of this study, and then specific primers RT-PCR detection method was established after optimizing reaction conditions. The sequence determination results were analyzed with BLAST program and DNAMAN software, and the phylogenetic tree was constructed based on the CP gene sequences obtained in this study by using Bayesian inference (BI) method implemented in MrBayes. 【Result】The serology results showed that one passion fruit sample exhibiting mosaic and crinkle symptom reacted with Potyvirus antiserum. The positive sample was found to have about 750 nm×12 nm linear virions by electron microscopic observation. The expected fragment was amplified from the positive sample by universal degenerate primers RT-PCR, and then cloned and sequenced. Sequence analysis showed that the obtained sequence from the positive sample was identical with the expected size (680 bp), and shared the highest nucleotide sequence identity (98.2%) with the reported TeMV gene sequence. The full-length sequence of CP gene obtained by specific primers RT-PCR was 816 bp (named BXGFJ-13 isolate), and the sequence of nucleotide and amino acid of BXGFJ-13 was 86.2%-98.4% and 88.2%-97.8% identity, respectively, with the reported TeMV isolates. The results of phylogenetic analysis showed that the 13 TeMV isolates could be divided into 3 groups, and the same area or host derived isolates preferentially clustered together, suggesting these isolates had a strong geographical and host specificity. BXGFJ-13 isolate obtained in this paper and Guangxi isolate of China (KJ789129) clustered into a branch with high posterior probability, and then clustered together with two Thailand isolates (AM409188, AM409187) into the 2nd group (Group II), showing that BXGFJ-13 and Guangxi isolate had the closest phylogenetic relationship. The specific primers RT-PCR showed good specificity, which only amplify the expected fragment from TeMV-infected passion fruit sample, and no expected fragment was obtained from Cucumber mosaic virus (CMV), Beet mosaic virus (BtMV), Soybean mosaic virus (SMV), Ornithogalum mosaic virus (OrMV), Onion yellow dwarf virus (OYDV), East Asian Passiflora virus (EAPV) and healthy control. The sensitivity results showed that the target fragment could be amplified from diluted 102 fold RNA.【Conclusion】According to the species demarcation criteria for the Potyvirus given by International Committee on Taxonomy (ICTV) and the results of serological detection and electron microscopic observation, TeMV on the passion fruit sample exhibiting mosaic and crinkle symptom in Fujian orchard was confirmed. The establishedassay of specific RT-PCR could be used for rapid detection of TeMV.
DOI:10.3864/j.issn.0578-1752.2017.24.006URL [本文引用: 1]
【Objective】 The objective of this study is to identify Telosma mosaic virus (TeMV) on passion fruit in Fujian orchard, develop a molecular method for specific and rapid identification of TeMV, and to provide a reference for prevention and control of the virus.【Method】The Fujian passion fruit samples were detected by using the serology method, electron microscopic observation, universal degenerate primers RT-PCR and specific primers RT-PCR. The PCR products of the positive sample were cloned and sequenced. A set of specific primes amplified the total length of coat protein (CP) gene was designed according to the sequences of reported TeMV and sequence determination of this study, and then specific primers RT-PCR detection method was established after optimizing reaction conditions. The sequence determination results were analyzed with BLAST program and DNAMAN software, and the phylogenetic tree was constructed based on the CP gene sequences obtained in this study by using Bayesian inference (BI) method implemented in MrBayes. 【Result】The serology results showed that one passion fruit sample exhibiting mosaic and crinkle symptom reacted with Potyvirus antiserum. The positive sample was found to have about 750 nm×12 nm linear virions by electron microscopic observation. The expected fragment was amplified from the positive sample by universal degenerate primers RT-PCR, and then cloned and sequenced. Sequence analysis showed that the obtained sequence from the positive sample was identical with the expected size (680 bp), and shared the highest nucleotide sequence identity (98.2%) with the reported TeMV gene sequence. The full-length sequence of CP gene obtained by specific primers RT-PCR was 816 bp (named BXGFJ-13 isolate), and the sequence of nucleotide and amino acid of BXGFJ-13 was 86.2%-98.4% and 88.2%-97.8% identity, respectively, with the reported TeMV isolates. The results of phylogenetic analysis showed that the 13 TeMV isolates could be divided into 3 groups, and the same area or host derived isolates preferentially clustered together, suggesting these isolates had a strong geographical and host specificity. BXGFJ-13 isolate obtained in this paper and Guangxi isolate of China (KJ789129) clustered into a branch with high posterior probability, and then clustered together with two Thailand isolates (AM409188, AM409187) into the 2nd group (Group II), showing that BXGFJ-13 and Guangxi isolate had the closest phylogenetic relationship. The specific primers RT-PCR showed good specificity, which only amplify the expected fragment from TeMV-infected passion fruit sample, and no expected fragment was obtained from Cucumber mosaic virus (CMV), Beet mosaic virus (BtMV), Soybean mosaic virus (SMV), Ornithogalum mosaic virus (OrMV), Onion yellow dwarf virus (OYDV), East Asian Passiflora virus (EAPV) and healthy control. The sensitivity results showed that the target fragment could be amplified from diluted 102 fold RNA.【Conclusion】According to the species demarcation criteria for the Potyvirus given by International Committee on Taxonomy (ICTV) and the results of serological detection and electron microscopic observation, TeMV on the passion fruit sample exhibiting mosaic and crinkle symptom in Fujian orchard was confirmed. The establishedassay of specific RT-PCR could be used for rapid detection of TeMV.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}