 ,, 赵晋锋,, 成锴, 王振华, 张鹏, 刘鑫, 田岗, 赵太存, 王玉文,山西省农业科学院谷子研究所,山西长治 046011
,, 赵晋锋,, 成锴, 王振华, 张鹏, 刘鑫, 田岗, 赵太存, 王玉文,山西省农业科学院谷子研究所,山西长治 046011Screening and Analysis of Key Metabolic Pathways in Foxtail Millet During Different Water Uptake Phases of Germination
YU AiLi,, ZHAO JinFeng,, CHENG Kai, WANG ZhenHua, ZHANG Peng, LIU Xin, TIAN Gang, ZHAO TaiCun, WANG YuWen,Millet Research Institute, Shanxi Academy of Agricultural Sciences, Changzhi 046011, Shanxi通讯作者:
责任编辑: 李莉
收稿日期:2019-07-17接受日期:2020-02-2网络出版日期:2020-08-01
| 基金资助: |
Received:2019-07-17Accepted:2020-02-2Online:2020-08-01
作者简介 About authors
余爱丽,E-mail:
赵晋锋,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (3028KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
余爱丽, 赵晋锋, 成锴, 王振华, 张鹏, 刘鑫, 田岗, 赵太存, 王玉文. 谷子萌发吸水期关键代谢途径的筛选与分析[J]. 中国农业科学, 2020, 53(15): 3005-3019 doi:10.3864/j.issn.0578-1752.2020.15.002
YU AiLi, ZHAO JinFeng, CHENG Kai, WANG ZhenHua, ZHANG Peng, LIU Xin, TIAN Gang, ZHAO TaiCun, WANG YuWen.
0 引言
【研究意义】谷子为小粒作物,千粒重仅3 g左右,播种深度只有3—5㎝。在各种复杂多变气候土壤条件下,谷子种植常面临播种难、萌发出苗难、保苗难、苗不全等问题。种子萌发起始于干燥种子水分的吸收,涉及一系列形态结构、生理生化和分子水平的变化,并长成具有正常构造幼苗,其中,对水分的吸收与响应是植物生长周期起始的关键。因此,研究谷子萌发过程中不同吸水期的各种变化和调节机制,将有助于调控谷子萌发,提高出苗率和全苗率,并为谷子种子引发提供理论依据。【前人研究进展】种子萌发是植物生长周期的起点,也是农作物种植生产的开始。狭义的种子萌发,起始于水的吸收,终止于胚轴的延伸,以胚根突破种皮为萌发结束的标志[1]。广义的种子萌发,是指种胚恢复生长直至长成具有正常构造幼苗的过程,包括吸胀、萌动、发芽和幼苗形态建成[2]。种子萌发过程中水分吸收大致分为3个阶段:第一阶段开始快速吸水期;第二阶段滞缓吸水期;第三阶段重新大量吸水期。其中,涉及呼吸、可溶物的渗漏、以现存和新mRNA为模板的蛋白质合成起始、DNA修复、线粒体的修复和合成、细胞分化和DNA合成等过程[3]。目前,谷子萌发期的相关研究主要侧重于抗旱性鉴定、不同逆境的生理响应。如张锦鹏等[4]利用甘露醇为胁迫剂,以5个谷子品种为材料,开展了萌发期人工模拟胁迫谷子耐旱性的鉴定。朱学海[5]采用PEG-6000、甘露醇2种渗透剂模拟水分胁迫,确定了鉴定谷子芽期耐旱性的PEG-6000和甘露醇胁迫条件分别为-0.75和-1.00 MPa,种子萌发耐旱指数与相对根长宜作为谷子芽期耐旱性鉴定指标。秦岭等[6]鉴定了201份不同生态区主要育成谷子品种芽期耐旱性。2018年印度****BHEEMESH等[7]采用4种不同浓度的PEG对60个谷子品种进行萌发期干旱胁迫处理,筛选出4个高耐受性谷子品种。逆境生理响应方面,王春梅等[8]分析了不同浓度砷对谷子萌发的影响,发现As<4 mg·kg-1促进谷子萌发,高浓度(As≥4 mg·kg-1)抑制谷子萌发。裴帅帅等[9]研究了晋谷28、晋谷45、晋谷42和晋谷77-322萌发期对干旱胁迫的生理响应,结果表明,在18% PEG的胁迫下,谷子的可溶性蛋白、脯氨酸含量、超氧化物歧化酶(superoxide dismutase,SOD)和过氧化物酶(peroxidase,POD)活性、丙二醛(malonaldehyde,MDA)含量都有所升高,差异较显著。其中,晋谷45的脯氨酸含量、蛋白质含量、SOD和POD活性比对照分别提高462.34%、34.17%、113.84%和61.22%;晋谷28的MDA含量增幅最小,晋谷42增幅最大,比CK增加98.7%。杨净等[10]研究超重力对谷子种子萌发及生理生化特性的影响,结果表明,中低速离心处理促进种子萌发,增加了根长及鲜重,减小了芽长,降低了丙二醛含量,提高了SOD活性,除3 000 r/min离心60 min处理外,都提高了POD活性。谷子萌发期分子机理研究较少,2017年,TANG等[11]分析了干旱胁迫下抗、感谷子在生理和转录水平上的差异,鉴定出20个与苗期和萌发期抗旱相关的QTL。窦祎凝等[12]通过反向遗传学方法研究表明,谷子转录因子SiNAC18通过ABA信号途径正向调控干旱条件下的种子萌发。2018年,许冰霞等[13]对萌发10和18 h种子的对照组和PEG处理组构建cDNA文库并进行差异表达基因的表达谱分析,分别筛选出456和545个差异表达基因,其中87和267个上调表达基因,369和278个下调表达基因;KEGG富集分析表明,差异表达基因参与到苯丙烷生物合成途径和植物激素信号转导过程;苯丙烷生物合成途径的苯丙氨酸解氨酶(PAL)、肉桂酸-4-羟化酶(CA4H)基因在谷子萌发10和18 h均发生了下调表达。2019年,潘教文等[14]分析了盐条件下培养7 d,去掉胚根胚芽萌发种子的转录物组,发现NaCl处理下DEGs主要集中在胁迫响应、氧化还原、离子的跨膜转运、多胺、有机酸、苯丙烷的合成等过程。苯丙烷生物合成途径(phenylpropanoid biosynthesis)的上游为苯丙氨酸代谢途径(phenylalanine metabolism),其为苯丙烷类合成提供前体[15]。苯丙烷生物合成途径可形成一系列间产物,如反式肉桂酸、香豆酸等,也可进一步转化成香豆素、绿原酸、类黄酮、异黄酮、花青素、植物激素、木质素等[16,17]。苯丙烷类代谢物是植物特有的,在植物生长、发育、与环境互作过程中发挥重要作用[18,19];也可作为调节分子,在植物防御病原物、抵御捕食者、信号转导与其他生物互作过程中发挥重要作用[20,21]。【本研究切入点】目前,针对谷子萌发期主要侧重于干旱、盐等逆境胁迫条件下的萌发率、形态结构、生理生化和转录谱研究,但对谷子不同萌发吸水期的转录组和代谢物变化及其调控机理鲜见报道。【拟解决的关键问题】本研究采用转录组分析方法探寻谷子在快速吸水期、滞缓吸水期、和重新大量吸水期的差异表达基因和关键代谢途径,分析重要代谢物的含量变化,从转录水平和代谢水平探讨谷子萌发吸水的调控机理,为调节和引发谷子萌发,提高出苗率的技术创制提供可借鉴依据。1 材料与方法
1.1 试验材料
筛选成熟度好、大小均一的晋谷20为试验材料。用3% NaClO浸泡消毒20 min,其间不断混匀,开盖透气,然后用蒸馏水冲洗4—5次(合计20 min),均匀播种于已铺放3层发芽纸的9 cm培养皿中,每皿50粒,加入蒸馏水7 mL。放置于温度24℃—26℃,相对湿度40%—50%培养室中培养。每个取样时间点设3次重复,共重复2轮。依据已经绘制的相同试验条件和试验材料的种子萌发吸水规律图,选取吸水3和5 h收集开始快速吸水期样品,混合,两轮分别标记为CK2和CK2R;收集吸水9和11 h滞缓吸水期样品,混合,两轮分别标记为CK8和CK8R;采集吸水15和17 h重新大量吸水期样品,混合,两轮分别标记为CK14和CK14R;液氮速冻,-80℃保存,用于转录组分析。相应的开始快速吸水期、滞缓吸水期和重新大量吸水期,分别用PhaseⅠ、PhaseⅡ、PhaseⅢ表示。另外,于相同条件下,培养和收集吸水0、2、3、5、8、9、11、14、15和17 h的萌发种子,液氮速冻-80℃保存,用于基因表达定量分析和代谢物含量分析。1.2 总RNA的提取和检测
采用CTAB法提取总RNA,1%的琼脂糖凝胶电泳进行初步检测。由NanoPhotometer? spectrophotometer(IMPLEN, CA, USA)检测RNA的纯度,Agilent Bioanalyzer 2100 system(Agilent Technologies, CA, USA)分析RNA的完整度。合格的RNA进行cDNA文库构建。1.3 cDNA文库的构建及测序
cDNA文库的构建和测序由中国北京百迈客公司完成。每个样品取1 μg RNA,采用试剂盒NEBNext?UltraTM RNA Library Prep Kit for Illumina?(NEB, USA),依据其说明书,用AMPure XP system(Beckman Coulter, Beverly, USA)纯化的200—250 bp片段,构建cDNA文库。用Illumina Hiseq 2500开展高通量转录组测序。共进行6个样本的高通量测序建库。1.4 基因表达分析及功能注释
1.4.1 基因表达分析 采用Tophat2[22]将样品的Clean reads与参考基因组进行序列比对。利用FPKM[23]值来反应基因的表达丰度。将皮尔逊相关系数r(Pearson’s Correlation Coefficient)作为生物学重复相关性的评估指标[24],分析3个不同吸水阶段的相关性。通过K-Means方法进行基因表达聚类分析。1.4.2 差异表达基因分析 采用DESeq[25]进行差异表达分析,然后采用Benjamini-Hochberg方法对差异显著性P-values进行校正。用Fold Change≥2且FDR(false discovery rate)<0.01作为标准,筛选差异表达基因。开始快速吸水期和滞缓吸水期比较、滞缓吸水期与重新大量吸水期比较、开始快速吸水期与重新大量吸水期比较分别表示为PhaseⅠ-PhaseⅡ、PhaseⅡ-PhaseⅢ和PhaseⅠ-PhaseⅢ。
1.4.3 功能注释 依据NR(NCBI non-redundant protein sequences)[26]、Swiss-Prot(A manually annotated and reviewed protein sequence database)[27]、KEGG(Kyoto Encyclopedia of Genes and Genomes)[28]、COG(Clusters of Orthologous Groups of proteins)[29]和GO(Gene Ontology)[30]在线数据库,对获得的差异表达基因进行功能注释。并进行差异表达基因的GO富集和KEGG通路富集。利用topGO[31]软件展示富集的GO节点(Term)及其层级关系,然后用ReviGO[32]对富集GO term去冗余和进一步聚类。
1.5 实时荧光定量PCR分析
选取关键代谢途径的4个差异表达基因,用实时荧光定量技术验证转录组分析数据的可靠性。通过CTAB法提取样品总RNA,使用荧光定量SYBR Premix Ex TaqⅡ试剂盒(TaKaRa)和Thermal Cycler Dice Real Time System(TaKaRa Code.TP800)进行试验。扩增体系为12.5 μL SYBR Premix Ex TaqⅡ、1 μL引物(10 μmol·L-1)、8.5 μL无菌蒸馏水和2 μL的cDNA模板。扩增程序为95℃ 30 s;95℃ 5 s,60℃ 30 s,40个循环。各基因特异引物具体见表1。以谷子组成型表达基因β-Actin(Seita.7G294000)作为内参基因。采用双标准曲线法计算基因的相对表达量[33],每个样品设3个技术重复。Table 1
表1
表1苯丙烷类相关基因qRT-PCR表达分析引物
Table 1
| 基因名称 Gene name | 基因ID Gene ID | 正向引物 Forward primer (5'-3') | 反向引物 Reverse primer (5'-3') |
|---|---|---|---|
| β-Actin | Seita.7G294000 | TGTGCCGGCCATGTATGT | CACACCATCACCAGAGTCCAA |
| Cationic peroxidase SPC4 precursor | Seita.3G004800 | CGGCATCGTCAGGGATTT | AGGCGGGCTTTGTTGGT |
| 4-coumarate--CoA ligase 3 | Seita.1G065800 | TGGAGTTCGCCGAGGTGAT | CGAGTTGAGCGAGTAGATGTGG |
| Beta-glucosidase 6 precursor | Seita.9G492600 | CCGGCGTGTCACTAATGCT | GTCTTCCCTCTCCCATCTTCCT |
| Peroxidase 4 precursor | Seita.5G145500 | TCTCGACGCTCCTGTCCAT | TTGGTGGCGGTGTCGTT |
新窗口打开|下载CSV
1.6 苯丙烷类关键代谢物含量分析
采用HPLC方法[34],分析不同萌发时期谷子中的肉桂酸、对香豆酸、咖啡酸、阿魏酸和芥子酸的含量变化。2 结果
2.1 谷子萌发3个吸水阶段转录物组分析
采用高通量测序方法分析3个吸水阶段的6个混合样品的转录物组。去除低质量数据和接头序列,共获得34.51 Gb Clean bases,各样品Clean bases均达到4.89 Gb以上,碱基质量值Q30在85.13%及以上。与谷子参考基因组进行序列比对,比对效率为79.47%—81.57%(表2)。开始快速吸水期样本CK2和CK2R分别含有4.89和4.98 Gb的Clean bases,滞缓吸水期样本CK8和CK8R含有7.07和5.47 Gb的Clean bases,重新大量吸水期样本CK14和CK14R含有5.90和6.17 Gb的Clean bases。3个不同萌发吸水阶段的相关性分析结果表明,开始快速吸水期和滞缓吸水期的相关性为0.73—0.76,开始快速吸水期与重新大量吸水期的相关性为0.32—0.34;滞缓吸水期与重新大量吸水期的相关性为0.65—0.69,说明不同吸水阶段涉及大量不同基因和代谢途径。Table 2
表2
表2测序结果及其与参考基因组比对分析
Table 2
| 分类 Classification | 开始快速吸水期PhaseⅠ | 滞缓吸水期PhaseⅡ | 重新大量吸水期PhaseⅢ | |||
|---|---|---|---|---|---|---|
| CK2 | CK2R | CK8 | CK8R | CK14 | CK14R | |
| 总reads Clean reads | 38845760 | 39679812 | 56164700 | 43525742 | 46838670 | 49151796 |
| 总核苷酸 Clean bases | 4894565760 | 4986871932 | 7076752200 | 5472201146 | 5901672420 | 6178141042 |
| GC含量 GC content (%) | 58.72 | 57.83 | 57.53 | 56.81 | 57.21 | 56.36 |
| 碱基质量值 Q30 (%) | 89.74 | 85.29 | 90.33 | 85.17 | 91.62 | 85.13 |
| 匹配的reads Mapped reads | 31007621 (79.82%) | 31533133 (79.47%) | 45812237 (81.57%) | 35213766 (80.90%) | 38024006 (81.18%) | 39595878 (80.56%) |
| 唯一位点匹配reads Unique mapped reads | 26519605 (68.27%) | 27126368 (68.36%) | 42124995 (75.00%) | 31824703 (73.12%) | 35017461 (74.76%) | 35922812 (73.09%) |
| 多位点匹配reads Multiple mapped reads | 4488016 (11.55%) | 4406765 (11.11%) | 3687242 (6.57%) | 3389063 (7.79%) | 3006545 (6.42%) | 3673066 (7.47%) |
新窗口打开|下载CSV
依据RPKM(reads per kilobase per million mapped reads)来反映基因的表达丰度,共获得33 643个基因。以RPKM≥0.01为筛选标准,开始快速吸水期样本CK2和CK2R分别含29 181和27 026个基因,滞缓吸水期样本CK8和CK8R分别含27 655和26 247个基因,重新大量吸水期样本CK14和CK14R有28 666和26 864个基因。以K-Means方法分析基因表达规律,共识别9个不同表达模式的基因簇(图1-A)。其中基因簇3含227个基因,从开始快速吸水期,经滞缓吸水期,到重新大量吸水期,基因表达量显著下降;而基因簇4含281个基因,从开始快速吸水期至重新大量吸水期,基因表达量显著上调,表明这些基因在谷子种子吸水萌发过程中具有不同的作用。同时以各基因簇的各个时期的所有样品的RPKM平均值为依据进行聚类分析,结果表明,1、3、5、9基因簇聚为一大簇,基因表达呈下调趋势,2、4、6、7和8基因簇聚成另一簇,基因表达呈上调趋势(图1-B)。
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1基因表达谱K-Means聚类图
A:具有不同表达模式的9个基因簇,纵坐标表示中心化的基因表达量的对数值;B:9个基因簇基因表达热图
Fig. 1K-Means clustering map of gene expression profiles
A: Maps of nine clusters with different expression pattern. The Y-axis indicates the centered log2(FPKM+1) in the maps of A; B: A heat map plot of gene expression related to nine clusters
2.2 谷子萌发3个吸水阶段的差异表达基因分析
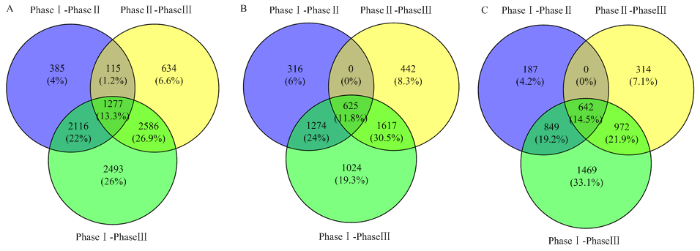
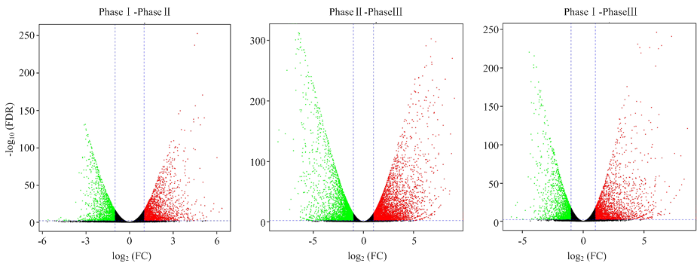
将Fold Change≥2和FDR<0.01作为筛选标准,对不同吸水阶段比较结果进行差异表达基因筛选,开始快速吸水期与滞缓吸水期比较(PhaseⅠ-PhaseⅡ)识别了3 893个差异表达基因,滞缓吸水期与重新大量吸水期之间(PhaseⅡ-PhaseⅢ)有4 612个差异表达基因,开始快速吸水期与重新大量吸水期比较(PhaseⅠ-PhaseⅢ)中识别8 472个差异表达基因(图2)。3个比较之间共有差异表达基因1 277个,占所有差异表达基因的13.3%,其中,共有上调基因625个,下调基因642个。PhaseⅠ-PhaseⅢ和PhaseⅡ-PhaseⅢ的差异表达基因的数量大于PhaseⅠ-PhaseⅡ,表明在重新大量吸水期中存在大量复杂的与谷子萌发发育相关的合成代谢途径(图2)。并且火山图(图3)也直观整体地展示基因在各比较的2个时期之间表达水平存在显著差异,在开始快速吸水期与重新大量吸水期比较中差异倍数大的基因最多。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2差异表达基因韦恩图
A:所有差异表达基因;B:上调差异表达基因;C:下调差异表达基因
Fig. 2A venn diagram of DEGs
A: All of DEGs; B: Up-regulated DEGs; C: Down-regulated DEGs
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3差异表达基因火山图
横坐标表示某一个基因在两样品中表达量差异倍数的对数值;纵坐标表示基因表达量变化的统计学显著性的负对数值
Fig. 3A volcano plot of DEGs
The X-axis indicates the log2(FC) of DEGs (FC, fold change). The Y-axis indicates the -log10(FDR) of different expression genes (FDR, False Discovery Rate)
2.3 差异表达基因功能的注释和富集分析
依据数据库COG、GO和KEGG,对3个比较的差异表达基因进行功能注释、分类和富集。COG分类结果表明,开始快速吸水期和滞缓吸水期比较、滞缓吸水期与重新大量吸水期比较、开始快速吸水期与重新大量吸水期比较的差异表达基因归属最多的功能类别为碳水化合物转运和代谢(carbohydrate transport and metabolism)、转录(transcription)、复制重组和修复(replication,recombination and repair)、一般功能(general function prediction only)、信号转导机制(signal transduction mechanism)。PhaseⅠ-PhaseⅢ还包括翻译核糖体结构和生物合成(translation,ribosomal structure and biogenesis)、翻译后修饰蛋白质转换和伴侣(posttranslation modification,protein turnover,chaperones)。将3个不同吸水期间差异表达基因注释到GO数据库并进行功能分类,结果显示,PhaseⅠ-PhaseⅡ、PhaseⅡ-PhaseⅢ和PhaseⅠ-PhaseⅢ分别注释到生物过程(biological process)、分子功能(molecular function)和细胞组分(cellular component)的差异表达基因分别有2 028、1 182和330,2 082、1 219和335,2 498、1 572和434个。对比较间差异表达基因进行GO富集分析,以校正P-value≤0.01为标准筛选富集到的Term,结果显示,PhaseⅠ-PhaseⅡ、PhaseⅡ-PhaseⅢ和PhaseⅠ-PhaseⅢ分别注释到生物过程、细胞组分和分子功能的26、15和7,29、10和6,23、14和6个通路。对所有筛选到的富集Term进行分析,结果表明,3个比较共同富集至7个通路,反应于寒冷、缺水、缺氧、干枯、氧化压力、karrikin通路和油菜素类固醇生物合成过程;开始快速吸水期和滞缓吸水期比较特有10个通路;滞缓吸水期与重新大量吸水期比较特有12个通路;开始快速吸水期与重新大量吸水期比较特有3个通路。3个比较的GO富集结果经软件ReviGO去冗余和进一步聚类后,结果表明,PhaseⅠ-PhaseⅡ主要涉及对油菜素内脂的反应(response to brassinosteroid)、根毛伸长(root hair elongation)、甾醇生物合成(sterol biosynthesis)和水转运(water transport)4个通路;PhaseⅡ-PhaseⅢ主要集中氮的转运(nitrate transport)、油菜素类固醇生物合成(brassinosteroid biosynthesis)、真菌防御反应(defense response to fungus)、乙酰辅酶A代谢(acetyl-CoA metabolis)和细胞板形成引起的细胞分裂(cytokinesis by cell plate formation)通路;PhaseⅠ-PhaseⅢ主要与冷反应(response to cold)、植物型细胞壁松弛(plant-type cell wall loosening)、油菜素类固醇生物合成(brassinosteroid biosynthesis)、根毛伸长(root hair elongation)、DNA复制起始(DNA replication initiation)有关。可见在谷子萌发的不同吸水阶段启动了大量代谢通路,并且各有不同,过程非常复杂。
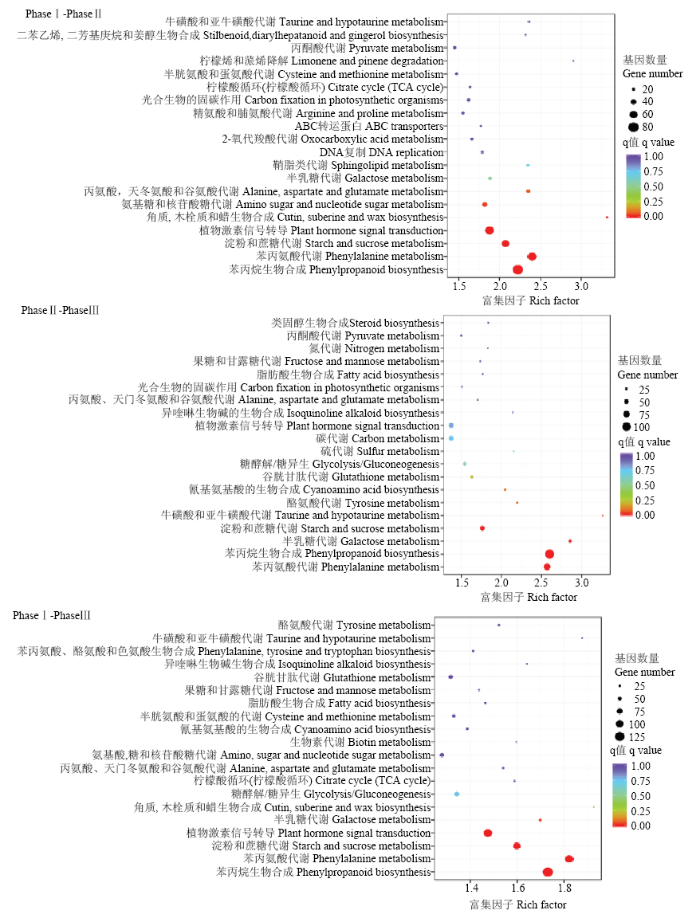
差异表达基因KEGG通路注释结果显示,开始快速吸水期和滞缓吸水期比较涉及111代谢通路,其中,苯丙氨酸代谢(phenylalanine metabolism)和苯丙烷生物合成(phenylpropanoid biosynthesis)分别有64个和80个差异表达基因,植物激素信号转导(plant hormone signal transduction)通路含65个差异表达基因,淀粉和蔗糖代谢(starch and sucrose metabolism)通路含56个。滞缓吸水期与重新大量吸水期比较的差异表达基因注释到117代谢通路,其中phenylalanine metabolism和phenylpropanoid biosynthesis分别有78个和106个差异表达基因,plant hormone signal transduction通路含54个差异表达基因,starch and sucrose metabolism通路含54个,碳代谢(Carbon metabolism)有55个差异表达基因。开始快速吸水期与重新大量吸水期比较涉及120代谢通路,其中phenylalanine metabolism和phenylpropanoid biosynthesis分别有105个和134个差异表达基因,plant hormone signal transduction通路含110个差异表达基因,starch and sucrose metabolism通路含93个,氨基酸生物合成(biosynthesis of amino acids)和carbon metabolism分别有86和83个,蛋白质在内质网中的加工(protein processing in endoplasmic reticulum)含72个。
KEGG富集结果进一步表明(图4),3个比较的差异表达基因全部显著富集于phenylalanine metabolism 和phenylpropanoid biosynthesis、starch and sucrose metabolism通路;Phase I-Phase II和Phase I-Phase III还显著富集于plant hormone signal transduction代谢通路。并且3个比较中富集于phenylalanine metabolism和phenylpropanoid biosynthesis代谢途径的基因数都最多。
图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4差异表达基因KEGG通路富集图
横坐标为富集因子(Rich Factor),表示差异表达基因中注释到某通路的基因比例与所有基因中注释到该通路的基因比例的比值。纵坐标表示通路名称;圆圈的大小表示通路中富集的基因数目,圆圈越大,表示基因越多;圆圈的颜色代表q value
Fig. 4KEGG enrichment map of DEGs
The x-axis indicated rich factor of DEGs,which represent percentages of DEGs belong to the corresponding pathway. The left y-axis represented the pathways. The sizes of bubble represent the number of DEGs in the corresponding pathway, and the colors of the bubble represent the enrichment q value of the corresponding pathway
2.4 苯丙烷生物合成和苯丙氨酸代谢途径相关基因分析
开始快速吸水期与滞缓吸水期比较中筛选到83个苯丙烷生物合成和苯丙氨酸代谢途径相关基因,滞缓吸水期与重新大量吸水期比较、开始快速吸水期与重新大量吸水期比较分别筛选到110、144个相关差异表达基因。其中3个比较中共有48个苯丙烷生物合成和苯丙氨酸代谢途径相关差异表达基因,PhaseⅠ-PhaseⅡ、PhaseⅡ-PhaseⅢ和PhaseⅠ-PhaseⅢ分别特有6、6和13个差异表达基因。如图5所示,各个苯丙烷生物合成和苯丙氨酸代谢相关差异表达基因在不同吸水时期样品中的表达量表明,大部分相关基因表达量上调,只有少量基因表达量下调,这说明此部分基因在谷子萌发吸胀启动中发挥重要作用,特别是下调基因可能在种子中有已形成mRNA储备。基因家族归类分析表明(图6),此部分基因中,过氧化物酶(peroxidase,EC:1.11.1.7)占59.2%,β-葡萄糖苷酶 (beta-glucosidase,EC:3.2.1.21)为14%,其次为4-香豆酰-辅酶A连接酶(4-coumarate--CoA ligase,EC:6.2.1.12)、肉桂醇脱氢酶(cinnamyl alcohol dehydrogenase,EC:1.1.1.195)和咖啡酸3-O-甲基转移酶(caffeic acid 3-O-methyltransferase,EC:2.1.1.68)。可见苯丙烷生物合成和苯丙氨酸代谢途径在谷子萌发过程中发挥重要作用,并通过关键基因表达调控种子萌发。图5
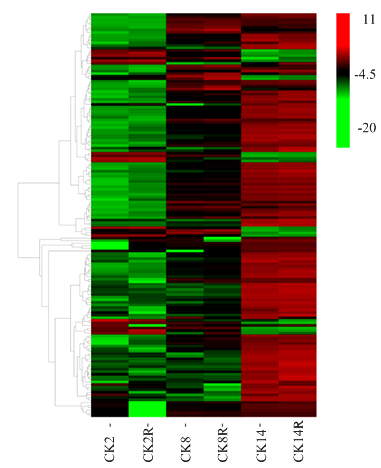 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5苯丙烷类相关的不同萌发吸水阶段间差异表达基因的表达分析
Fig. 5Expression analysis of phenylpropanoids-related DEGs between different water uptake stages of germination
图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6苯丙烷类相关的不同萌发吸水阶段间差异表达基因的功能分类
Fig. 6Function classification of phenylpropanoids-related DEGs between different water uptake stages of germination
2.5 差异表达基因的荧光定量PCR分析
挑选谷子苯丙烷生物合成途径关键基因进行qRT-PCR分析,以整体观测其在不同萌发时期的表达模式,验证转录物组分析结果,结果表明,在谷子萌发吸水的17 h过程中(图7),4-coumarate--CoA ligase 3(Seita.1G065800)呈先下调后上调再下调表达趋势;beta-glucosidase 6(Seita.9G492600)和cationic peroxidase SPC4(Seita.3G004800)呈上调趋势;peroxidase 4(Seita.5G145500)萌发后期呈上调趋势。这些基因在开始快速吸水期(3 h和5 h)、滞缓吸水期(9 h和11 h)、重新大量吸水期(15 h和17 h)表达趋势与转录物组分析结果表达趋势一致,除Seita.3G004800在15 h略有不同,分析其原因可能由于15 h样品全部为培养14 h后移苗1 h的样品,移苗可能对已经露白的萌发种子造成了伤害。再结合代谢图分析,Seita.1G065800为上游代谢基因,Seita.9G492600、Seita.3G004800和Seita.5G145500为下游代谢基因,说明在谷子萌发过程中Seita.1G065800发挥重要作用,并在种子中有所储备,以利于种子的萌发。图7
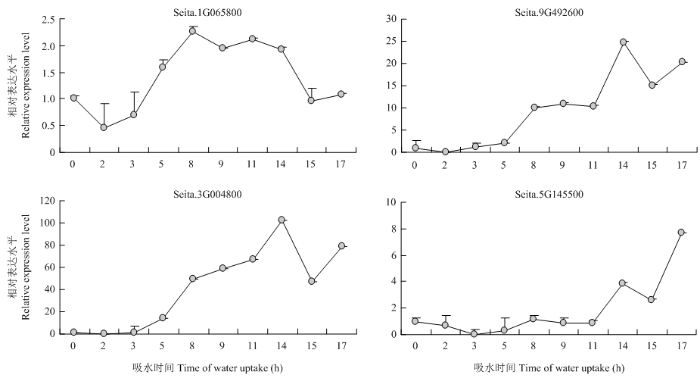 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7苯丙烷类相关基因在不同萌发吸水时期的qRT-PCR表达分析
Fig. 7Expression analysis of phenylpropanoids-related genes by qRT-PCR at different water uptake periods of germination
2.6 苯丙烷类关键代谢物含量分析
鉴于苯丙烷生物合成途径重要性,对其相关重要次生代谢物进行分析(图8),结果表明,芥子酸在种子中大量储备,在萌发0—17 h中呈下调趋势;肉桂酸在此阶段保持恒定;阿魏酸、对香豆酸和咖啡酸呈先上调后下调趋势。可见芥子酸和苯丙烷生物合成途径可能在种子休眠萌发过程中发挥重要作用。图8
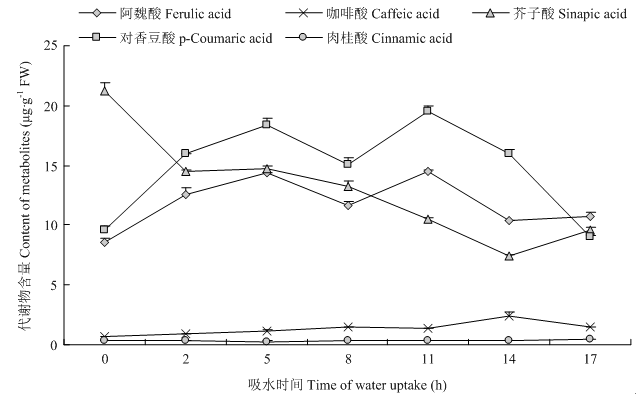 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8苯丙烷类相关代谢物不同萌发吸水时期含量变化
Fig. 8Changes in the content of phenylpropanoids-related metabolites during water uptake stage in germinating seeds
3 讨论
3.1 谷种萌发相关代谢途径
植物生长发育始于种子萌发。种子萌发是植物恢复新的生命周期的一个复杂过程,涉及一系列生长代谢和合成途径,以及分子、生理、生化和形态上的变化,如随着水吸入干种子细胞,呼吸强度明显增高、暂时的膜结构扰动、主要储存物质活化、多聚体的重聚、已形成mRNAs的翻译、新mRNA的合成、细胞膜和DNA的修复等[35,36]。本研究比较谷子种子不同吸水阶段的转录物组差异,发现大量表达差异表达基因COG富集于碳水化合物转运和代谢、转录、复制重组和修复、翻译后修饰、蛋白质转换和伴侣等途径。而GO富集更进一步将其聚焦于萌动种子对周围环境条件的感应,如对寒冷、缺水、缺氧、干枯、氧化压力、karrikin等的反应。并且各个阶段各有所侧重,如PhaseⅠ-PhaseⅡ主要涉及对油菜素内脂的反应、根毛伸长、甾醇生物合成和水转运4个通路;PhaseⅡ-PhaseⅢ主要集中氮的转运、油菜素类固醇生物合成、真菌防御反应、乙酰辅酶A代谢和细胞板形成引起的细胞分裂通路;PhaseⅠ-PhaseⅢ主要与冷反应、植物型细胞壁松弛、油菜素类固醇生物合成、根毛伸长、DNA复制起始。而KEGG富集将3个比较的差异表达基因显著富集于苯丙氨酸代谢、苯丙烷生物合成、淀粉和蔗糖代谢通路。这与许冰霞等[13]分析谷子萌发期响应干旱胁迫的基因表达谱结果一致,PEG胁迫处理10和18 h的谷子种子差异表达基因参与到苯丙烷生物合成过程。盐胁迫条件下,培养7 d去掉胚根胚芽的豫谷2和安04的萌发种子的KEGG富集分析也发现,上调DEGs主要富集于植物激素信号转导、精氨酸及脯氨酸代谢、淀粉及糖代谢、次生代谢物合成等途径;下调DEGs主要富集在氨基酸合成、甘油磷脂代谢、苯丙烷合成等代谢途径[14]。3.2 植物苯丙烷生物合成途径及其基因
苯丙氨酸代谢是苯丙烷生物合成的上游途径,为苯丙烷类合成提供前体。苯丙烷生物合成和苯丙氨酸代谢途径涉及一系列的基因,纵观其代谢图(K00940和K00360)主要基因有苯丙氨酸解氨酶(phenylalanine ammonia lyase,EC4.3.1.24和EC4.3.1.25)、4-香豆酸-CoA连接酶(4-coumarate-CoA ligase,EC6.2.1.12)、肉桂酰-CoA还原酶(Cinnamoyl-CoA reductase,EC1.2.1.44)、肉桂醇脱氢酶(cinnamyl alcohol dehydrogenase,EC1.1.1.195)、过氧化物酶(peroxidase,EC1.11.1.7)等。苯丙氨酸解氨酶(PAL)催化L-苯丙氨酸直接脱氨产生反式肉桂酸,是苯丙烷生物合成途径的关键酶和限速酶,在病原物侵害、伤害、营养缺乏、紫外线辐射、极端温度等条件下被诱导[16,21,39-45],并且涉及水杨酸的合成[46,47,48]。过氧化物酶是一个多基因家族,有非常多家族成员,催化H2O2和各种还原物的氧化还原反应,参与各种复杂生理过程[49,50],如激素代谢[51]、活性氧和活性氮代谢[52]、植物疾病防御等[51,53-54]。植物所特有的过氧化物酶为ClassⅢ过氧化物酶(POX,EC1.11.1.7),是苯丙烷生物合成途径的下游基因,涉及分泌途径、液泡[55,56],细胞壁化合物聚合、抵御病原体攻击、耐盐性、氧化应激、植物激素和生物碱代谢、棉纤维伸长和枝根发育等[50-51,57-60]。本研究谷子种子不同吸水阶段的差异表达基因功能富集分析结果表明,谷子萌发与苯丙氨酸代谢和苯丙烷生物合成途径密切相关,筛选到157个与2个途径相关的差异表达基因,如过氧化物酶(peroxidase,EC:1.11.1.7)、β-葡萄糖苷酶(beta-glucosidase,EC:3.2.1.21)、4-香豆酰-辅酶A连接酶(4-coumarate--CoA ligase,EC:6.2.1.12)、肉桂醇脱氢酶(cinnamyl alcohol dehydrogenase,EC:1.1.1.195)、咖啡酸3-O-甲基转移酶(caffeic acid 3-O-methyltransferase,EC:2.1.1.68)、苯丙氨酸解氨酶(phenylalanine ammonia lyase,PAL,EC4.3.1.24)和肉桂酰-CoA还原酶(cinnamoyl-CoA reductase,CCR,EC1.2.1.44)等。过氧化物酶(EC:1.11.1.7)基因共检测到157个,其中93个与苯丙烷生物合成和苯丙氨酸代谢相关,占全部苯丙烷生物合成和苯丙氨酸代谢途径相关差异表达基因的占59.2%。相似结果出现低温萌发蓖麻转录物组中,WANG等[15]发现苯丙烷生物合成相关基因phenylalanine ammonia-lyase、4-coumarate--CoA ligase 2、Peroxidase 52 precursor、Peroxidase 73 precursor、Cationic peroxidase 2 precursor出现显著下调。许冰霞等[13]分析PEG干旱胁迫处理10和18 h谷子萌发种子基因表达谱,获知PAL可能在PEG胁迫下调节种子的萌发。显然苯丙烷生物合成和苯丙氨酸代谢相关基因在谷子萌发吸水过程中被精密调控,特别是POX家族成员可能在谷子萌发木质素合成、细胞壁的形成、根系伸长过程中发挥重要作用。3.3 植物苯丙烷类相关代谢物
植物中苯丙烷生物合成途径的形成是植物防御非生物和生物胁迫的长期进化结果[37]。苯丙烷生物合成途径可催化苯丙氨酸形成反式肉桂酸、香豆酸、阿魏酸等多种酚类化合物。目前已知苯丙烷及其相关代谢物,如肉桂酸、阿魏酸、芥子酸等,是抑制种子萌发的重要的他感物质[38]。为此本研究分析了不同吸水阶段谷子萌发种子中几种他感物质的含量,发现在未萌发种子中含有大量芥子酸,在萌发过程中呈下调趋势,阿魏酸、对香豆酸和咖啡酸呈先上调后下调趋势,肉桂酸保持恒定,暗示芥子酸和苯丙烷生物合成途径可能在种子萌发启动过程中发挥重要作用。4 结论
谷子萌发开始快速吸水阶段、滞缓吸水阶段、重新大量吸水阶段的苯丙烷生物合成和苯丙氨酸代谢途径及其中间产物、基因在谷子萌发过程中发挥重要调控作用,特别是芥子酸可能参与调控种子的休眠与萌发,在未萌动种子中大量储备,萌发吸水过程中显著下调;上游基因4-香豆酰-辅酶A连接酶在未萌发种子中存在已形成mRNA,在萌发吸水阶段呈先下调后上调再下调表达趋势;下游基因过氧化物酶家族成员也参与谷子萌发过程。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1105/tpc.9.7.1055URLPMID:12237375 [本文引用: 1]
URL [本文引用: 1]

报道了在苗期快速鉴定谷子品种抗旱性的实验结果.通过比较种子在甘露醇渗透胁迫条件下的相对萌发率和芽生长抑制率、幼苗在适度控水条件下的相对含水量和水势、幼苗严重失水恢复供水后的存活率等几个指标,对5个谷子品种(鲁7060、羊角黄、豫谷5号、Jun 24和Mar 51)的苗期抗旱性进行鉴定.结果表明,Mar 51在所鉴定的品种中具有相对的抗旱优势,可以作为谷子苗期抗旱性研究的代表性材料.谷子种子在甘露醇渗透胁迫条件下的相对萌发率与幼苗在适度控水条件下相对含水量的变化趋势一致,初步认为这两个指标可以作为苗期快速鉴定谷子抗旱性的筛选指标.
URL [本文引用: 1]
报道了在苗期快速鉴定谷子品种抗旱性的实验结果.通过比较种子在甘露醇渗透胁迫条件下的相对萌发率和芽生长抑制率、幼苗在适度控水条件下的相对含水量和水势、幼苗严重失水恢复供水后的存活率等几个指标,对5个谷子品种(鲁7060、羊角黄、豫谷5号、Jun 24和Mar 51)的苗期抗旱性进行鉴定.结果表明,Mar 51在所鉴定的品种中具有相对的抗旱优势,可以作为谷子苗期抗旱性研究的代表性材料.谷子种子在甘露醇渗透胁迫条件下的相对萌发率与幼苗在适度控水条件下相对含水量的变化趋势一致,初步认为这两个指标可以作为苗期快速鉴定谷子抗旱性的筛选指标.
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.11869/j.issn.100-8551.2014.10.1897URL [本文引用: 1]
对4个品种的谷子晋谷28、晋谷45、晋谷42和晋谷77-322进行种子萌发期抗旱性研究.试验首先筛选出合适的PEG浓度模拟干旱胁迫条件;在正常供水和模拟干旱条件下测定了种子发芽特性及生长、生理生化等指标.结果表明:18%PEG浓度为合适的干旱胁迫条件;随着干旱胁迫的加强,谷子的相对发芽势、相对发芽率、发芽指数和抗旱指数都呈下降趋势.在18%PEG的胁迫下,谷子的可溶性蛋白、脯氨酸含量、SOD和POD 活性、MDA含量都有所升高,差异较显著.其中,晋谷45的脯氨酸含量、蛋白质含量、SOD、POD活性比对照分别提高 462.34%、34.17%、113.84%、61,22%,晋谷28的SOD活性提高266.35%.晋谷42的POD活性比28、45、77-322三个品种分别降低了9.9%、7.5%、19.5%,MDA含量28增幅最小,42增幅最大比CK增加98.7%.经过隶属函数法分析谷子萌芽期抗旱性的强弱顺序为:晋谷45>晋谷28>晋谷77-322>晋谷42.研究为谷子抗旱种质资源鉴定评价及品种选育提供理论依据.
DOI:10.11869/j.issn.100-8551.2014.10.1897URL [本文引用: 1]
对4个品种的谷子晋谷28、晋谷45、晋谷42和晋谷77-322进行种子萌发期抗旱性研究.试验首先筛选出合适的PEG浓度模拟干旱胁迫条件;在正常供水和模拟干旱条件下测定了种子发芽特性及生长、生理生化等指标.结果表明:18%PEG浓度为合适的干旱胁迫条件;随着干旱胁迫的加强,谷子的相对发芽势、相对发芽率、发芽指数和抗旱指数都呈下降趋势.在18%PEG的胁迫下,谷子的可溶性蛋白、脯氨酸含量、SOD和POD 活性、MDA含量都有所升高,差异较显著.其中,晋谷45的脯氨酸含量、蛋白质含量、SOD、POD活性比对照分别提高 462.34%、34.17%、113.84%、61,22%,晋谷28的SOD活性提高266.35%.晋谷42的POD活性比28、45、77-322三个品种分别降低了9.9%、7.5%、19.5%,MDA含量28增幅最小,42增幅最大比CK增加98.7%.经过隶属函数法分析谷子萌芽期抗旱性的强弱顺序为:晋谷45>晋谷28>晋谷77-322>晋谷42.研究为谷子抗旱种质资源鉴定评价及品种选育提供理论依据.
[本文引用: 1]
[本文引用: 1]
URLPMID:28855520 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.3864/j.issn.0578-1752.2018.08.002URL [本文引用: 3]
【目的】谷子是一种耐旱作物,通过二代测序技术获得大量的谷子萌发期响应干旱胁迫的差异基因,进而挖掘谷子萌发期抵御干旱的关键基因及其相关的分子机制。【方法】以晋谷45为材料,谷子萌发期分别用18%PEG-6000干旱胁迫(处理组)和蒸馏水(对照组)处理种子并测定1、10和18 h种子的SOD、POD和CAT活性。SOD活性用氮蓝四唑(NBT)法测定,POD活性用愈创木酚法测定,CAT活性用比色法测定;对萌发10和18 h种子的对照组和处理组构建cDNA文库并进行差异基因表达谱分析;利用Bowtie将reads比对到参考基因组,采用RSEM对bowtie的比对结果进行表达量估计;使用DESeq进行差异表达基因分析;利用NR、Swiss-Prot、KEGG、COG和GO在线数据库对差异基因进行功能注释,挖掘调控谷子萌发的关键基因;利用qRT-PCR验证测序结果的可靠性。【结果】处理组的SOD活性整体比对照组高,而POD活性和CAT活性与之相反;随着萌发时间的变化,SOD活性在不断地增加,但CAT和POD活性逐渐减小。基因表达谱序列与所选参考基因组序列高度一致,基因表达呈现出高度不均一性。通过高通量测序最后获得35 470个基因,以RPKM≥0.01为筛选标准,对照样本中分别筛选出24 030和24 486个表达基因,PEG干旱胁迫处理10和18 h的样本分别筛选出24 019和23 877个表达基因;差异表达基因分析表明,谷子萌发10和18 h分别筛选出456和545个差异基因,其中87和267个上调表达基因,369和278个下调表达基因;GO功能显著性富集分析表明,差异基因主要涉及代谢过程,细胞进程和响应刺激;KEGG富集分析表明,差异基因参与到苯丙烷代谢和植物激素信号转导过程;通过qRT-PCR对5个差异基因在干旱胁迫下种子萌发时的表达分析表明,其表达趋势与表达谱分析结果基本一致。【结论】差异表达基因广泛涉及到糖、蛋白质、核酸等生物大分子代谢、次生代谢和能量代谢等过程;SnRK2和PAL可能在干旱胁迫下调节种子的萌发。
DOI:10.3864/j.issn.0578-1752.2018.08.002URL [本文引用: 3]
【目的】谷子是一种耐旱作物,通过二代测序技术获得大量的谷子萌发期响应干旱胁迫的差异基因,进而挖掘谷子萌发期抵御干旱的关键基因及其相关的分子机制。【方法】以晋谷45为材料,谷子萌发期分别用18%PEG-6000干旱胁迫(处理组)和蒸馏水(对照组)处理种子并测定1、10和18 h种子的SOD、POD和CAT活性。SOD活性用氮蓝四唑(NBT)法测定,POD活性用愈创木酚法测定,CAT活性用比色法测定;对萌发10和18 h种子的对照组和处理组构建cDNA文库并进行差异基因表达谱分析;利用Bowtie将reads比对到参考基因组,采用RSEM对bowtie的比对结果进行表达量估计;使用DESeq进行差异表达基因分析;利用NR、Swiss-Prot、KEGG、COG和GO在线数据库对差异基因进行功能注释,挖掘调控谷子萌发的关键基因;利用qRT-PCR验证测序结果的可靠性。【结果】处理组的SOD活性整体比对照组高,而POD活性和CAT活性与之相反;随着萌发时间的变化,SOD活性在不断地增加,但CAT和POD活性逐渐减小。基因表达谱序列与所选参考基因组序列高度一致,基因表达呈现出高度不均一性。通过高通量测序最后获得35 470个基因,以RPKM≥0.01为筛选标准,对照样本中分别筛选出24 030和24 486个表达基因,PEG干旱胁迫处理10和18 h的样本分别筛选出24 019和23 877个表达基因;差异表达基因分析表明,谷子萌发10和18 h分别筛选出456和545个差异基因,其中87和267个上调表达基因,369和278个下调表达基因;GO功能显著性富集分析表明,差异基因主要涉及代谢过程,细胞进程和响应刺激;KEGG富集分析表明,差异基因参与到苯丙烷代谢和植物激素信号转导过程;通过qRT-PCR对5个差异基因在干旱胁迫下种子萌发时的表达分析表明,其表达趋势与表达谱分析结果基本一致。【结论】差异表达基因广泛涉及到糖、蛋白质、核酸等生物大分子代谢、次生代谢和能量代谢等过程;SnRK2和PAL可能在干旱胁迫下调节种子的萌发。
DOI:10.3864/j.issn.0578-1752.2019.22.003URL [本文引用: 2]
【Objective】 Foxtail millet (Setaria italica L.) remarkably adapts to adverse ecologies, including drought, barren regions and high soil salinity. These characteristics promote it as a very promising model crop for exploring basic biology processes of plants. Unveil of mechanisms of foxtail millet under salinity is of immense significance. 【Method】 In this research, 14 millet varieties at germination stage were used for resistance screening under 150 mmol·L -1 NaCl. The germination rate, root and bud length of each variety were measured after been treated for 7 d, and the salt tolerance variety Yugu 2 and salt susceptible variety An 04 were obtained. These two varieties before and after salt treatment were used for transcriptomes analysis by RNA-seq sequencing. The functional and pathway enrichments of differentially expressed genes (DEGs) were also performed by GO and KEGG analysis. Fifteen DEGs were randomly selected for further qRT-PCR analysis to verify the RNA-seq data.【Result】 Totally 2 786 and 4 413 DEGs were identified in Yugu 2 and An 04, and there were 1 470 and 3 826 DEGs within each cultivar before and after salt treatment, respectively. GO and KEGG enrichment analysis suggested that DEGs were mainly clustered into signaling, antioxidant system, organic acid biosynthesis and transport, and secondary metabolism, and the DEGs participated in response to stress, ion transmembrane transport, redox homeostasis, secondary metabolism, organic acid, polyamine and phenylpropanoid biosynthetic process. The qRT-PCR results showed high correlation coefficient of 0.8817 with the data of RNA-seq. Especially the genes such as cation transporter (HKT8), peroxidase (POD), flavanone 3-dioxygenase (FL3H) and MYB transcription factors, which displayed higher vary degrees in Yugu2 under salinity may play key roles in salt response process of foxtail millet. 【Conclusion】There was difference in the response degree of salt tolerance variety and salt sensitive variety of millet under salinity. Moreover, the resistance trait formation and function to salt was mainly depends on the response degree instead of the number of DEGs under stress.
DOI:10.3864/j.issn.0578-1752.2019.22.003URL [本文引用: 2]
【Objective】 Foxtail millet (Setaria italica L.) remarkably adapts to adverse ecologies, including drought, barren regions and high soil salinity. These characteristics promote it as a very promising model crop for exploring basic biology processes of plants. Unveil of mechanisms of foxtail millet under salinity is of immense significance. 【Method】 In this research, 14 millet varieties at germination stage were used for resistance screening under 150 mmol·L -1 NaCl. The germination rate, root and bud length of each variety were measured after been treated for 7 d, and the salt tolerance variety Yugu 2 and salt susceptible variety An 04 were obtained. These two varieties before and after salt treatment were used for transcriptomes analysis by RNA-seq sequencing. The functional and pathway enrichments of differentially expressed genes (DEGs) were also performed by GO and KEGG analysis. Fifteen DEGs were randomly selected for further qRT-PCR analysis to verify the RNA-seq data.【Result】 Totally 2 786 and 4 413 DEGs were identified in Yugu 2 and An 04, and there were 1 470 and 3 826 DEGs within each cultivar before and after salt treatment, respectively. GO and KEGG enrichment analysis suggested that DEGs were mainly clustered into signaling, antioxidant system, organic acid biosynthesis and transport, and secondary metabolism, and the DEGs participated in response to stress, ion transmembrane transport, redox homeostasis, secondary metabolism, organic acid, polyamine and phenylpropanoid biosynthetic process. The qRT-PCR results showed high correlation coefficient of 0.8817 with the data of RNA-seq. Especially the genes such as cation transporter (HKT8), peroxidase (POD), flavanone 3-dioxygenase (FL3H) and MYB transcription factors, which displayed higher vary degrees in Yugu2 under salinity may play key roles in salt response process of foxtail millet. 【Conclusion】There was difference in the response degree of salt tolerance variety and salt sensitive variety of millet under salinity. Moreover, the resistance trait formation and function to salt was mainly depends on the response degree instead of the number of DEGs under stress.
[本文引用: 2]
[本文引用: 2]
URLPMID:15199968 [本文引用: 1]
URLPMID:20035037 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:24642849 [本文引用: 2]
DOI:10.1186/gb-2013-14-4-r36URLPMID:23618408 [本文引用: 1]
TopHat is a popular spliced aligner for RNA-sequence (RNA-seq) experiments. In this paper, we describe TopHat2, which incorporates many significant enhancements to TopHat. TopHat2 can align reads of various lengths produced by the latest sequencing technologies, while allowing for variable-length indels with respect to the reference genome. In addition to de novo spliced alignment, TopHat2 can align reads across fusion breaks, which can occur after genomic translocations. TopHat2 combines the ability to identify novel splice sites with direct mapping to known transcripts, producing sensitive and accurate alignments, even for highly repetitive genomes or in the presence of pseudogenes. TopHat2 is available at http://ccb.jhu.edu/software/tophat.
DOI:10.12688/f1000research.2-188.v2URLPMID:24555089 [本文引用: 1]
Alternative splicing is widely recognized for its roles in regulating genes and creating gene diversity. However, despite many efforts, the repertoire of gene splicing variation is still incompletely characterized, even in humans. Here we describe a new computational system, ASprofile, and its application to RNA-seq data from Illumina's Human Body Map project (>2.5 billion reads). Using the system, we identified putative alternative splicing events in 16 different human tissues, which provide a dynamic picture of splicing variation across the tissues. We detected 26,989 potential exon skipping events representing differences in splicing patterns among the tissues. A large proportion of the events (>60%) were novel, involving new exons (~3000), new introns (~16000), or both. When tracing these events across the sixteen tissues, only a small number (4-7%) appeared to be differentially expressed ('switched') between two tissues, while 30-45% showed little variation, and the remaining 50-65% were not present in one or both tissues compared. Novel exon skipping events appeared to be slightly less variable than known events, but were more tissue-specific. Our study represents the first effort to build a comprehensive catalog of alternative splicing in normal human tissues from RNA-seq data, while providing insights into the role of alternative splicing in shaping tissue transcriptome differences. The catalog of events and the ASprofile software are freely available from the Zenodo repository ( http://zenodo.org/record/7068; doi: 10.5281/zenodo.7068) and from our web site http://ccb.jhu.edu/software/ASprofile.
[本文引用: 1]
DOI:10.1186/gb-2010-11-10-r106URLPMID:20979621 [本文引用: 1]
High-throughput sequencing assays such as RNA-Seq, ChIP-Seq or barcode counting provide quantitative readouts in the form of count data. To infer differential signal in such data correctly and with good statistical power, estimation of data variability throughout the dynamic range and a suitable error model are required. We propose a method based on the negative binomial distribution, with variance and mean linked by local regression and present an implementation, DESeq, as an R/Bioconductor package.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
.
[本文引用: 1]
DOI:10.1371/journal.pone.0021800URLPMID:21789182 [本文引用: 1]
Outcomes of high-throughput biological experiments are typically interpreted by statistical testing for enriched gene functional categories defined by the Gene Ontology (GO). The resulting lists of GO terms may be large and highly redundant, and thus difficult to interpret.REVIGO is a Web server that summarizes long, unintelligible lists of GO terms by finding a representative subset of the terms using a simple clustering algorithm that relies on semantic similarity measures. Furthermore, REVIGO visualizes this non-redundant GO term set in multiple ways to assist in interpretation: multidimensional scaling and graph-based visualizations accurately render the subdivisions and the semantic relationships in the data, while treemaps and tag clouds are also offered as alternative views. REVIGO is freely available at http://revigo.irb.hr/.
[本文引用: 1]
DOI:10.1016/j.chroma.2004.10.068URL [本文引用: 1]
DOI:10.1016/j.crvi.2008.07.023URLPMID:18926496 [本文引用: 1]
Using post-genomic technologies, it is now possible to understand the molecular basis of complex developmental processes. In the case of seed germination, recent transcriptome- and proteome-wide studies led to new insights concerning the building up of the germination potential during seed maturation on the mother plant, the reversible character of the first phases of the germination process enabling the imbibed embryo to recapitulate the late maturation program for mounting defense response when confronted to environmental fluctuations, the timing of expression of genes playing a role in controlling radicle emergence, the role of plant hormones as abscisic acid and gibberellins in seed germination, and finally the global changes in proteome activity induced by redox regulation occurring in seed development and germination. In this way, post-genomic technologies help facilitating the advent of a systems approach to uncover novel features of seed quality, which can lead to potential applications, for example in selection programs.
DOI:10.1146/annurev-arplant-042811-105550URLPMID:22136565 [本文引用: 1]
Germination vigor is driven by the ability of the plant embryo, embedded within the seed, to resume its metabolic activity in a coordinated and sequential manner. Studies using
URLPMID:18272377 [本文引用: 1]
DOI:10.1007/s10886-007-9318-xURLPMID:17577597 [本文引用: 1]
This study investigated potential phytotoxic effects on germination and root growth of 21 plant secondary metabolites (sinapinic, syringic, vanillic, ferulic, p-coumaric, chlorogenic, gallic, gentisic, protocatechuic, p-hydroxybenzoic, and trans-cinnamic acids, and eucalyptol, quercetin, vanillin, syringaldehyde, rutin, 2-benzoxazolinone, protocatechualdehyde, tyrosol, juglone, and L-mimosine) in the plant model Arabidopsis thaliana. Eleven of the 21 molecules showed significant inhibitory effects on germination, and 17 inhibited root growth. Inhibitory effects on root growth were more evident when nutrients were not added. We present dose response curves for germination effects and IC50 values for each compound, along with possible explanations of the observed inhibitory actions in terms of molecular structure.
[本文引用: 1]
URLPMID:2760071
Phenylalanine ammonia-lyase (PAL) catalyzes the first reaction in the biosynthesis from phenylalanine of a wide variety of phenylpropanoid natural products including lignin, flavonoid pigments, and phytoalexins. In bean (Phaseolus vulgaris L.), PAL is encoded by a family of three genes. We show here by RNase protection with gene-specific probes that these genes are expressed differentially during development and in response to different environmental cues. While all three genes are expressed at high levels in roots, only PAL1 and PAL2 are expressed in shoots and only PAL1 is expressed in leaves. Strikingly, PAL2 is expressed at very high levels in petals, where PAL1 is only very weakly expressed and PAL3 is not expressed. All three genes are induced by mechanical wounding of hypocotyls, but fungal infection only activates PAL1 and PAL3. Illumination of etiolated hypocotyls activates PAL1 and PAL2 but not PAL3. Corresponding differential patterns of synthesis of specific PAL polypeptide isoforms were observed by two-dimensional gel electrophoretic analysis of in vitro translation products encoded by RNA isolated from hypocotyls stimulated by light, wounding, or infection. The specific isoforms encoded by transcripts of the three PAL genes were identified by inhibition of synthesis in vitro with gene-specific anti-sense transcripts followed by comparative two-dimensional gel electrophoretic analysis of the pattern of translation products. These data indicate that selective expression of PAL genes encoding functional variants is governed by a complex set of regulatory networks for developmental and environmental control of phenylpropanoid biosynthesis.
DOI:10.1073/pnas.86.23.9284URLPMID:2594769
A 1.1-kilobase promoter fragment of the bean (Phaseolus vulgaris L.) phenylalanine ammonia-lyase (EC 4.3.1.5) gene PAL2 was translationally fused to the beta-glucuronidase reporter gene and transferred to tobacco by Agrobacterium tumefaciens-mediated leaf disk transformation. The distribution of beta-glucuronidase activity in these transgenic plants is very similar to that of endogenous PAL2 transcripts in bean, with very high levels in petals; marked accumulation in anthers, stigmas, roots, and shoots; and low levels in sepals, ovaries, and leaves. Histochemical analysis of the spatial pattern of beta-glucuronidase activity showed that the PAL2 promoter is highly active in the shoot apical meristem, the zone of cell proliferation immediately adjacent to the root apical meristem, and in the early stages of vascular development at the inception of xylem differentiation. Wounding and light evoke specific changes in the spatial pattern of beta-glucuronidase activity in stems, including induction in the epidermis. These data indicate that the PAL2 promoter transduces a complex set of developmental and environmental cues into an integrated spatial and temporal program of gene expression to regulate the synthesis of a diverse array of phenylpropanoid natural products.
DOI:10.1104/pp.110.157370URLPMID:20566705
Phenylalanine ammonia-lyase (PAL) catalyzes the first step of the phenylpropanoid pathway, which produces precursors to a variety of important secondary metabolites. Arabidopsis (Arabidopsis thaliana) contains four PAL genes (PAL1-PAL4), but there has been no genetic analysis to assess the biological functions of the entire gene family. Here, we report the generation and analysis of combined mutations for the four Arabidopsis PAL genes. Contrary to a previous report, we found that three independent pal1 pal2 double mutants were fertile and generated yellow seeds due to the lack of condensed tannin pigments in the seed coat. The pal1 pal2 double mutants were also deficient in anthocyanin pigments in various plant tissues, which accumulate in wild-type plants under stress conditions. Thus, PAL1 and PAL2 have a redundant role in flavonoid biosynthesis. Furthermore, the pal1 pal2 double mutants were more sensitive to ultraviolet-B light but more tolerant to drought than wild-type plants. We have also generated two independent pal1 pal2 pal3 pal4 quadruple knockout mutants, which are stunted and sterile. The quadruple knockout mutants still contained about 10% of the wild-type PAL activity, which might result from one or more leaky pal mutant genes or from other unknown PAL genes. The quadruple mutants also accumulated substantially reduced levels of salicylic acid and displayed increased susceptibility to a virulent strain of the bacterial pathogen Pseudomonas syringae. These results provide further evidence for both distinct and overlapping roles of the Arabidopsis PAL genes in plant growth, development, and responses to environmental stresses.
DOI:10.1186/1471-2229-12-39URLPMID:22429339
BACKGROUND: Plant secondary metabolites, including phenylpropanoids and carotenoids, are stress inducible, have important roles in potato physiology and influence the nutritional value of potatoes. The type and magnitude of environmental effects on tuber phytonutrients is unclear, especially under modern agricultural management that minimizes stress. Understanding factors that influence tuber secondary metabolism could facilitate production of more nutritious crops. Metabolite pools of over forty tuber phenylpropanoids and carotenoids, along with the expression of twenty structural genes, were measured in high-phenylpropanoid purple potatoes grown in environmentally diverse locations in North America (Alaska, Texas and Florida). RESULTS: Phenylpropanoids, including chlorogenic acid (CGA), were higher in samples from the northern latitudes, as was the expression of phenylpropanoid genes including phenylalanine ammonia lyase (PAL), which had over a ten-fold difference in relative abundance. Phenylpropanoid gene expression appeared coordinately regulated and was well correlated with metabolite pools, except for hydroxycinnamoyl-CoA:quinatehydroxcinnamoyl transferase (HQT; r = -0.24). In silico promoter analysis identified two cis-acting elements in the HQT promoter not found in the other phenylpropanoid genes. Anthocyanins were more abundant in Alaskan samples and correlated with flavonoid genes including DFR (r = 0.91), UFGT (r = 0.94) and F3H (r = 0.77). The most abundant anthocyanin was petunidin-3-coum-rutinoside-5-glu, which ranged from 4.7 mg g-1 in Alaska to 2.3 mg g-1 in Texas. Positive correlations between tuber sucrose and anthocyanins (r = 0.85), suggested a stimulatory effect of sucrose. Smaller variation was observed in total carotenoids, but marked differences occurred in individual carotenoids, which had over a ten-fold range. Violaxanthin, lutein or zeaxanthin were the predominant carotenoids in tubers from Alaska, Texas and Florida respectively. Unlike in the phenylpropanoid pathway, poor correlations occurred between carotenoid transcripts and metabolites. CONCLUSION: Analysis of tuber secondary metabolism showed interesting relationships among different metabolites in response to collective environmental influences, even under conditions that minimize stress. The variation in metabolites shows the considerable phenotypical plasticity possible with tuber secondary metabolism and raises questions about to what extent these pathways can be stimulated by environmental cues in a manner that optimizes tuber phytonutrient content while protecting yields. The differences in secondary metabolites may be sufficient to affect nutritional quality.
DOI:10.1371/journal.pone.0062352URLPMID:23638048
In this study, a phenylalanine ammonia-lyase (PAL) gene was cloned from Dendrobium candidum using homology cloning and RACE. The full-length sequence and catalytic active sites that appear in PAL proteins of Arabidopsis thaliana and Nicotiana tabacum are also found: PAL cDNA of D. candidum (designated Dc-PAL1, GenBank No. JQ765748) has 2,458 bps and contains a complete open reading frame (ORF) of 2,142 bps, which encodes 713 amino acid residues. The amino acid sequence of DcPAL1 has more than 80% sequence identity with the PAL genes of other plants, as indicated by multiple alignments. The dominant sites and catalytic active sites, which are similar to that showing in PAL proteins of Arabidopsis thaliana and Nicotiana tabacum, are also found in DcPAL1. Phylogenetic tree analysis revealed that DcPAL is more closely related to PALs from orchidaceae plants than to those of other plants. The differential expression patterns of PAL in protocorm-like body, leaf, stem, and root, suggest that the PAL gene performs multiple physiological functions in Dendrobium candidum.
URLPMID:17612622 [本文引用: 1]
DOI:10.1105/tpc.8.2.203URLPMID:12239383 [本文引用: 1]
Arabidopsis ecotype Columbia (Col-0) seedlings, transformed with a phenylalanine ammonia-lyase 1 promoter (PAL1)-[beta]-glucuronidase (GUS) reporter construct, were inoculated with virulent and avirulent isolates of Peronospora parasitica. The PAL1 promoter was constitutively active in the light in vascular tissue but was induced only in the vicinity of fungal structures in the incompatible interaction. A double-staining procedure was developed to distinguish between GUS activity and fungal structures. The PAL1 promoter was activated in cells undergoing lignification in the incompatible interaction in response to the pathogen. Pretreatment of the seedlings with 2-aminoindan-2-phosphonic acid (AIP), a highly specific PAL inhibitor, made the plants completely susceptible. Lignification was suppressed after AIP treatment, and surprisingly, pathogen-induced PAL1 promoter activity could not be detected. Treatment of the seedlings with 2-hydroxyphenylaminosulphinyl acetic acid (1,1-dimethyl ester) (OH-PAS), a cinnamyl alcohol dehydrogenase inhibitor specific for the lignification pathway, also caused a shift toward susceptibility, but the effect was not as pronounced as it was with AIP. Significantly, although OH-PAS suppressed pathogen-induced lignification, it did not suppress pathogen-induced PAL1 promoter activation. Salicylic acid (SA), supplied to AIP-treated plants, restored resistance and both pathogen-induced lignification and activation of the PAL1 promoter. Endogenous SA levels increased significantly in the incompatible but not in the compatible combination, and this increase was suppressed by AIP but not by OH-PAS. These results provide evidence of the central role of SA in genetically determined plant disease resistance and show that lignification per se, although providing a component of the resistance mechanism, is not the deciding factor between resistance and susceptibility.
[本文引用: 1]
DOI:10.1021/jf020953bURLPMID:12670161 [本文引用: 1]
It has been suggested that salicylic acid (SA) is a signal in acquired resistance to pathogens in several plants. Also, it has been suggested that infestation of plants causes an increase in the activity of phenylalanine ammonia-lyase (PAL), a key phenolic biosynthesis enzyme. The purpose of this work was to investigate whether the induction of SA and PAL activity is related to the susceptibility of barley to aphid infestation. The induction of free and conjugated SA in two barley cultivars that differ in susceptibility to aphids was analyzed. Analyses of several physiological parameters showed that cv. UNA-80 was more susceptible to the aphid Schizaphis graminum than cv. LM-109. Salicylic acid was not detected in noninfested plants. Levels of free and conjugated SA in cv. LM-109 and of conjugated SA in cv. UNA-80 increased with aphid infestation, whereas the levels of free SA in cv. UNA-80 remained high under all infestation degrees. Maximum values reached in both cultivars were not significantly different. With respect to PAL activity, cv. LM-109 showed a significantly higher specific activity than cv. UNA-80, the more susceptible cultivar. The relationship between the susceptibility of a plant to aphid and SA induction and PAL activity is discussed.
DOI:10.1016/j.phytochem.2004.06.023URLPMID:15279994 [本文引用: 1]
Plant peroxidases (class III peroxidases, E.C. 1.11.1.7) are secreted glycoproteins known to be involved in the mechanism of cell elongation, in cell wall construction and differentiation, and in the defense against pathogens. They usually form large multigenic families in angiosperms. The recent completion of rice (Oryza sativa japonica c.v. Nipponbare) genome sequencing allowed drawing up the full inventory of the genes encoding class III peroxidases in this plant. We found 138 peroxidase genes distributed among the 12 rice chromosomes. In contrast to several other gene families studied so far, peroxidase genes are twice as numerous in rice as in Arabidopsis. This large number of genes results from various duplication events that were tentatively traced back using a phylogenetic tree based on the alignment of conserved amino acid sequences. We also searched for peroxidase encoding genes in the major phyla of plant kingdom. In addition to gymnosperms and angiosperms, sequences were found in liverworts, mosses and ferns, but not in unicellular green algae. Two rice and one Arabidopsis peroxidase genes appeared to be rather close to the only known sequence from the liverwort Marchantia polymorpha. The possible relationship of these peroxidases with the putative ancestor of peroxidase genes is discussed, as well as the connection between the development of the class III peroxidase multigenic family and the emergence of the first land plants.
DOI:10.1007/s00299-005-0972-6URL [本文引用: 2]
Plant peroxidases (class III peroxidases) are present in all land plants. They are members of a large multigenic family. Probably due to this high number of isoforms, and to a very heterogeneous regulation of their expression, plant peroxidases are involved in a broad range of physiological processes all along the plant life cycle. Due to two possible catalytic cycles, peroxidative and hydroxylic, peroxidases can generate reactive oxygen species (ROS) (
 OH, HOO), polymerise cell wall compounds, and regulate H2O2 levels. By modulating their activity and expression following internal and external stimuli, peroxidases are prevalent at every stage of plant growth, including the demands that the plant meets in stressful conditions. These multifunctional enzymes can build a rigid wall or produce ROS to make it more flexible; they can prevent biological and chemical attacks by raising physical barriers or by counterattacking with a large production of ROS; they can be involved in a more peaceful symbiosis. They are finally present from the first hours of a plant
OH, HOO), polymerise cell wall compounds, and regulate H2O2 levels. By modulating their activity and expression following internal and external stimuli, peroxidases are prevalent at every stage of plant growth, including the demands that the plant meets in stressful conditions. These multifunctional enzymes can build a rigid wall or produce ROS to make it more flexible; they can prevent biological and chemical attacks by raising physical barriers or by counterattacking with a large production of ROS; they can be involved in a more peaceful symbiosis. They are finally present from the first hours of a plant s life until its last moments. Although some functions look paradoxical, the whole process is probably regulated by a fine-tuning that has yet to be elucidated. This review will discuss the factors that can influence this delicate balance.
s life until its last moments. Although some functions look paradoxical, the whole process is probably regulated by a fine-tuning that has yet to be elucidated. This review will discuss the factors that can influence this delicate balance.DOI:10.1093/pcp/pce061URLPMID:11382811 [本文引用: 3]
Class III plant peroxidase (POX), a plant-specific oxidoreductase, is one of the many types of peroxidases that are widely distributed in animals, plants and microorganisms. POXs exist as isoenzymes in individual plant species, and each isoenzyme has variable amino acid sequences and shows diverse expression profiles, suggesting their involvement in various physiological processes. Indeed, studies have provided evidence that POXs participate in lignification, suberization, auxin catabolism, wound healing and defense against pathogen infection. Little, however, is known about the signal transduction for inducing expression of the pox genes. Recent studies have provided information on the regulatory mechanisms of wound- and pathogen-induced expression of some pox genes. These studies suggest that pox genes are induced via different signal transduction pathways from those of other known defense-related genes.
DOI:10.1093/jxb/ern277URLPMID:19073963 [本文引用: 1]
When plants are attacked by pathogens, they defend themselves with an arsenal of defence mechanisms, both passive and active. The active defence responses, which require de novo protein synthesis, are regulated through a complex and interconnected network of signalling pathways that mainly involve three molecules, salicylic acid (SA), jasmonic acid (JA), and ethylene (ET), and which results in the synthesis of pathogenesis-related (PR) proteins. Microbe or elicitor-induced signal transduction pathways lead to (i) the reinforcement of cell walls and lignification, (ii) the production of antimicrobial metabolites (phytoalexins), and (iii) the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS). Among the proteins induced during the host plant defence, class III plant peroxidases (EC 1.11.1.7; hydrogen donor: H(2)O(2) oxidoreductase, Prxs) are well known. They belong to a large multigene family, and participate in a broad range of physiological processes, such as lignin and suberin formation, cross-linking of cell wall components, and synthesis of phytoalexins, or participate in the metabolism of ROS and RNS, both switching on the hypersensitive response (HR), a form of programmed host cell death at the infection site associated with limited pathogen development. The present review focuses on these plant defence reactions in which Prxs are directly or indirectly involved, and ends with the signalling pathways, which regulate Prx gene expression during plant defence. How they are integrated within the complex network of defence responses of any host plant cell will be the cornerstone of future research.
DOI:10.1094/MPMI.2001.14.12.1411URLPMID:11768536 [本文引用: 1]
The rice bacterial blight pathogen Xanthomonas oryzae pv. oryzae is a vascular pathogen that elicits a defensive response through interaction with metabolically active rice cells. In leaves of 12-day-old rice seedlings, the exposed pit membrane separating the xylem lumen from the associated parenchyma cells allows contact with bacterial cells. During resistant responses, the xylem secondary walls thicken within 48 h and the pit diameter decreases, effectively reducing the area of pit membrane exposed for access by bacteria. In susceptible interactions and mock-inoculated controls, the xylem walls do not thicken within 48 h. Xylem secondary wall thickening is developmental and, in untreated 65-day-old rice plants, the size of the pit also is reduced. Activity and accumulation of a secreted cationic peroxidase, PO-C1, were previously shown to increase in xylem vessel walls and lumen. Peptide-specific antibodies and immunogold-labeling were used to demonstrate that PO-C1 is produced in the xylem parenchyma and secreted to the xylem lumen and walls. The timing of the accumulation is consistent with vessel secondary wall thickening. The PO-C1 gene is distinct but shares a high level of similarity with previously cloned pathogen-induced peroxidases in rice. PO-C1 gene expression was induced as early as 12 h during resistant interactions and peaked between 18 and 24 h after inoculation. Expression during susceptible interactions was lower than that observed in resistant interactions and was undetectable after infiltration with water, after mechanical wounding, or in mature leaves. These data are consistent with a role for vessel secondary wall thickening and peroxidase PO-C1 accumulation in the defense response in rice to X. oryzae pv. oryzae.
DOI:10.1111/j.1365-313X.2005.02372.xURLPMID:15807789 [本文引用: 1]
Bacterial speck caused by the pathogen Pseudomonas syringae pv. tomato (P. s. tomato) is a devastating disease of tomato plants. Here we show that inhibition of Ep5C gene expression, which encodes a secreted cationic peroxidase, is sufficient to confer resistance against P. s. tomato. The inhibition of Ep5C protein accumulation in antisense tomato plants established resistance that was not accompanied by the pre-activation of known defense pathways. Therefore, Ep5C inhibition represents a novel form of disease resistance based on a loss-of-gene function in the plant required for successful infection by a compatible bacterial pathogen. Ep5C expression is rapidly induced by H2O2, a reactive oxygen intermediate normally generated during the course of a plant-pathogen interaction. This was corroborated by monitoring the expression of an Ep5C-GUS gene in transgenic Arabidopsis plants. Collectively, these results identify a signaling pathway that uses early signals generated during the oxidative burst, such as H2O2, for the selective activation of host factors required for mounting a compatible interaction. Thus, Ep5C provides a new resource for developing bacterial speck disease-resistant varieties.
DOI:10.1016/0959-440X(92)90230-5URL [本文引用: 1]
DOI:10.1046/j.1432-1033.2002.03311.xURL [本文引用: 1]
DOI:10.1104/pp.105.069674URLPMID:16258008 [本文引用: 1]
The major basic peroxidase from Zinnia elegans (ZePrx) suspension cell cultures was purified and cloned, and its properties and organ expression were characterized. The ZePrx was composed of two isoforms with a M(r) (determined by matrix-assisted laser-desorption ionization time of flight) of 34,700 (ZePrx34.70) and a M(r) of 33,440 (ZePrx33.44). Both isoforms showed absorption maxima at 403 (Soret band), 500, and 640 nm, suggesting that both are high-spin ferric secretory class III peroxidases. M(r) differences between them were due to the glycan moieties, and were confirmed from the total similarity of the N-terminal sequences (LSTTFYDTT) and by the 99.9% similarity of the tryptic fragment fingerprints obtained by reverse-phase nano-liquid chromatography. Four full-length cDNAs coding for these peroxidases were cloned. They only differ in the 5'-untranslated region. These differences probably indicate different ways in mRNA transport, stability, and regulation. According to the k(cat) and apparent K(m)(RH) values shown by both peroxidases for the three monolignols, sinapyl alcohol was the best substrate, the endwise polymerization of sinapyl alcohol by both ZePrxs yielding highly polymerized lignins with polymerization degrees > or =87. Western blots using anti-ZePrx34.70 IgGs showed that ZePrx33.44 was expressed in tracheary elements, roots, and hypocotyls, while ZePrx34.70 was only expressed in roots and young hypocotyls. None of the ZePrx isoforms was significantly expressed in either leaves or cotyledons. A neighbor-joining tree constructed for the four full-length cDNAs suggests that the four putative paralogous genes encoding the four cDNAs result from duplication of a previously duplicated ancestral gene, as may be deduced from the conserved nature and conserved position of the introns.
DOI:10.1007/s00425-005-0153-4URLPMID:16284776
Two class III peroxidases from Arabidopsis, AtPrx33 and Atprx34, have been studied in this paper. Their encoding genes are mainly expressed in roots; AtPrx33 transcripts were also found in leaves and stems. Light activates the expression of both genes in seedlings. Transformed seedlings producing AtPrx33-GFP or AtPrx34-GFP fusion proteins under the control of the CaMV 35S promoter exhibit fluorescence in the cell walls of roots, showing that the two peroxidases are localized in the apoplast, which is in line with their affinity for the Ca(2+)-pectate structure. The role they can play in cell wall was investigated using (1) insertion mutants that have suppressed or reduced expression of AtPrx33 or AtPrx34 genes, respectively, (2) a double mutant with no AtPrx33 and a reduced level of Atprx34 transcripts, (3) a mutant overexpressing AtPrx34 under the control of the CaMV 35S promoter. The major phenotypic consequences of these genetic manipulations were observed on the variation of the length of seedling roots. Seedlings lacking AtPrx33 transcripts have shorter roots than the wild-type controls and roots are still shorter in the double mutant. Seedlings overexpressing AtPrx34 exhibit significantly longer roots. These modifications of root length are accompanied by corresponding changes of cell length. The results suggest that AtPrx33 and Atprx34, two highly homologous Arabidopsis peroxidases, are involved in the reactions that promote cell elongation and that this occurs most likely within cell walls.
DOI:10.1104/pp.107.107060URLPMID:18065566
Catharanthus roseus produces low levels of two dimeric terpenoid indole alkaloids, vinblastine and vincristine, which are widely used in cancer chemotherapy. The dimerization reaction leading to alpha-3',4'-anhydrovinblastine is a key regulatory step for the production of the anticancer alkaloids in planta and has potential application in the industrial production of two semisynthetic derivatives also used as anticancer drugs. In this work, we report the cloning, characterization, and subcellular localization of an enzyme with anhydrovinblastine synthase activity identified as the major class III peroxidase present in C. roseus leaves and named CrPrx1. The deduced amino acid sequence corresponds to a polypeptide of 363 amino acids including an N-terminal signal peptide showing the secretory nature of CrPrx1. CrPrx1 has a two-intron structure and is present as a single gene copy. Phylogenetic analysis indicates that CrPrx1 belongs to an evolutionary branch of vacuolar class III peroxidases whose members seem to have been recruited for different functions during evolution. Expression of a green fluorescent protein-CrPrx1 fusion confirmed the vacuolar localization of this peroxidase, the exact subcellular localization of the alkaloid monomeric precursors and dimeric products. Expression data further supports the role of CrPrx1 in alpha-3',4'-anhydrovinblastine biosynthesis, indicating the potential of CrPrx1 as a target to increase alkaloid levels in the plant.
DOI:10.1016/S1673-8527(08)60101-0URLPMID:19302970 [本文引用: 1]
The accumulation of reactive oxygen species (ROS) is involved in plant cell development. In plant, class III peroxidases are heme-containing enzymes encoded by a large multi-gene family participated in the release or consumption of ROS. The specific function of each member of the family is still elusive. Here, we showed that ROS was significantly generated during cotton fiber initiation and elongation, whereas, application of NADPH oxidase inhibitor diphenyleneiodonium (DPI) and peroxidase inhibitor salicylhydroxamic acid (SHAM) to the wild-type cotton ovule culture significantly suppressed fiber growth, respectively. Their inhibitory effects were caused by the reduction of superoxide radical (O(2)(-)). Ten GhPOX genes (cDNAs) encoding cotton class III peroxidases were isolated, among them eight GhPOX genes were reported for the first time. Microarray analyses indicated that GhPOX1 was the mostly predominantly expressed in fast-elongating cotton fiber cells. Real-time quantitative PCR analysis revealed the transcript level of GhPOX1 was over 400-fold higher in growing fiber cells than in ovules, flowers, roots, stems and leaves. To reveal the role of GhPOX1 in plant development, its Arabidopsis orthologue atpox13 mutant was demonstrated to be defective in branch root development. Taken together, the data suggest that GhPOX1 plays an important role during fiber cell elongation possibly by mediating production of reactive oxygen species.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}