 ,, 石彩云,, 范小雪, 王杰, 祝智威, 蒋海宾, 范元婵, 陈华枝, 杜宇, 王心蕊, 熊翠玲, 郑燕珍, 付中民, 陈大福, 郭睿,福建农林大学动物科学学院(蜂学学院),福州 350002
,, 石彩云,, 范小雪, 王杰, 祝智威, 蒋海宾, 范元婵, 陈华枝, 杜宇, 王心蕊, 熊翠玲, 郑燕珍, 付中民, 陈大福, 郭睿,福建农林大学动物科学学院(蜂学学院),福州 350002The Mechanism Underlying MicroRNAs-Mediated Nosema ceranae Infection to Apis mellifera ligustica Worker
GENG SiHai,, SHI CaiYun,, FAN XiaoXue, WANG Jie, ZHU ZhiWei, JIANG HaiBin, FAN YuanChan, CHEN HuaZhi, DU Yu, WANG XinRui, XIONG CuiLing, ZHENG YanZhen, FU ZhongMin, CHEN DaFu, GUO Rui,College of Animal Sciences (College of Bee Science), Fujian Agriculture and Forestry University, Fuzhou 350002通讯作者:
责任编辑: 岳梅
收稿日期:2019-12-2接受日期:2019-12-28网络出版日期:2020-08-01
| 基金资助: |
Received:2019-12-2Accepted:2019-12-28Online:2020-08-01
作者简介 About authors
耿四海,E-mail:
石彩云,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (4596KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
耿四海, 石彩云, 范小雪, 王杰, 祝智威, 蒋海宾, 范元婵, 陈华枝, 杜宇, 王心蕊, 熊翠玲, 郑燕珍, 付中民, 陈大福, 郭睿. 微小RNA介导东方蜜蜂微孢子虫侵染意大利蜜蜂工蜂的分子机制[J]. 中国农业科学, 2020, 53(15): 3187-3204 doi:10.3864/j.issn.0578-1752.2020.15.018
GENG SiHai, SHI CaiYun, FAN XiaoXue, WANG Jie, ZHU ZhiWei, JIANG HaiBin, FAN YuanChan, CHEN HuaZhi, DU Yu, WANG XinRui, XIONG CuiLing, ZHENG YanZhen, FU ZhongMin, CHEN DaFu, GUO Rui.
0 引言
【研究意义】微孢子虫是一类细胞内寄生的真菌,其寄主范围非常广泛,包括哺乳动物、鱼类和昆虫等[1]。自从1857[2]首次在家蚕(Bombyx mori)中鉴定出家蚕微孢子虫(Nosema bombycis)以来,目前已在脊椎动物和无脊椎动物中鉴定出超过1 400种微孢子虫[3]。东方蜜蜂微孢子虫(Nosema ceranae)专性寄生成年蜜蜂中肠上皮细胞[4],可引起宿主肠道组织破坏、消化系统紊乱、免疫基因抑制、细胞凋亡抑制、寿命缩短以及工蜂进入采集工作的日龄提前等[5]。感染东方蜜蜂微孢子虫的蜂群罹患蜜蜂微孢子虫病,出现生产力下降和蜂群群势衰弱等现象[6],严重危害养蜂生产。1996年,FRIES等[7]在北京附近地区的东方蜜蜂(Apis cerana)样本中首次分离鉴定出东方蜜蜂微孢子虫,随后该微孢子虫迅速蔓延至欧洲地区和中国台湾等地的西方蜜蜂(Apis mellifera)蜂群[8,9],目前已广泛感染世界主要养蜂国家的蜂群。然而对于蜜蜂微孢子虫病,目前仍缺乏有效的治疗手段,东方蜜蜂微孢子虫侵染机制研究的滞后是最主要的限制因素。已有研究表明微小RNA(microRNA,miRNA)参与调控真菌的侵染、繁殖和毒力[10,11]。通过深度测序和组学分析解析东方蜜蜂微孢子虫在侵染意大利蜜蜂(Apis mellifera ligustica,简称意蜂)过程中的miRNA差异表达谱及调控网络,可为阐明东方蜜蜂微孢子虫的侵染机制提供数据基础和理论依据,并为蜜蜂微孢子虫病的治疗提供潜在的分子靶点。【前人研究进展】东方蜜蜂微孢子虫在体外以休眠态的孢子形式存在,孢子壁由几丁质和孢壁蛋白组成,能够抵御外界的不良环境,当孢子被蜜蜂摄取之后,在蜜蜂中肠的特殊理化环境的刺激下萌发,内部高度压缩盘旋的极丝弹射而出,刺穿中肠上皮细胞并将有侵染性的孢原质注入,随后进入繁殖期,利用宿主的物质与能量合成系统进行增殖,孢子数量逐渐增多,最终导致宿主细胞裂解,释放出的成熟孢子继续感染临近的上皮细胞,或者随粪便排出体外,感染新的宿主[12]。MiRNA是一类长度在18—25 nt的单链小RNA,由细胞内源产生的发卡结构转录本加工而来。1993年,LEE等[13]首次在线虫中发现一种长度为22 nt的小RNA并命名为lin-4,lin-4可以调节线虫的发育。let-7为第二个被发现的miRNA,let-7同样可通过靶向结合靶mRNA调节线虫的发育[14]。随后在动植物中陆续鉴定出大量的miRNA[15,16]。越来越多的研究结果证实miRNA在生物体的新陈代谢、免疫应答、细胞分化、细胞周期等重要的生理生化过程中发挥着举足轻重的作用[15,16]。然而,真菌miRNA的研究相对滞后。LEE等[17]于2010年第一次在粗糙脉孢菌(Neurospora crassa)中发现并报道类微小RNA(microRNA-like),此后在绿僵菌(Metarhizium anisopliae)[18]、里氏木酶(Trichoderma reesei)[11]和蜜蜂球囊菌(Ascosphaera apis,简称球囊菌)[19]等中相继鉴定出miRNA。有研究报道miRNA参与调控真菌的菌丝生长和毒力,但关于miRNA如何参与调控尚未明确。目前,东方蜜蜂微孢子虫的miRNA研究极为有限,相关信息十分缺乏。近期,EVANS等[20]研究发现东方蜜蜂微孢子虫的5个miRNA不仅能靶向其本身的1 545个基因,还能靶向宿主的947个基因。MiRNA的形成过程需要Dicer酶进行加工,多数微孢子虫在进化过程中丢失了RNAi通路中Dicer、Argonaute和依赖RNA为模板的RNA聚合酶(RNA-dependent RNA polymerase,RdRP)基因等关键基因,但东方蜜蜂微孢子虫基因组中还保留有上述基因[21]。HUANG等[22]利用RNAi敲减东方蜜蜂微孢子虫的Dicer后,发现不但东方蜜蜂微孢子虫的增殖水平显著降低,而且侵染过程中病原基因的表达量也发生了变化,表明miRNA对东方蜜蜂微孢子虫的侵染和增殖起到重要的调节作用。【本研究切入点】意蜂作为西方蜜蜂的亚种之一,是我国养蜂生产使用的主要蜂种,具有重要的经济和生态价值。前期研究中,笔者团队利用RNA-seq技术和生物信息学分析方法在mRNA组学水平初步探究了东方蜜蜂微孢子虫对意蜂的侵染机制[23]。但东方蜜蜂微孢子虫的侵染机制尚未阐明,miRNA在东方蜜蜂微孢子虫侵染过程中的作用仍不清楚。【拟解决的关键问题】利用small RNA-seq(sRNA-seq)技术对东方蜜蜂微孢子虫纯化孢子及东方蜜蜂微孢子虫感染7 d和10 d的意蜂工蜂中肠进行深度测序,基于高质量的测序数据进行东方蜜蜂微孢子虫miRNA的生物信息学预测,结构特征和表达谱分析,以及差异表达miRNA(differentially expressed miRNA,DEmiRNA)的靶mRNA预测、功能注释、调控网络构建及分析,进一步筛选和探讨靶向病原毒力因子和侵染相关因子的DEmiRNA,以期在miRNA组学层面揭示DEmiRNA介导东方蜜蜂微孢子虫的侵染机制。1 材料与方法
试验于2017年9月至2019年8月在福建农林大学动物科学学院(蜂学学院)蜜蜂保护实验室完成。1.1 生物材料
意蜂工蜂取自福建农林大学动物科学学院(蜂学学院)教学蜂场;东方蜜蜂微孢子虫感染的意蜂外勤蜂取自福州市闽侯县荆溪源安养蜂场。1.2 东方蜜蜂微孢子虫孢子的纯化
参照GUO等[24,25]的方法进行东方蜜蜂微孢子虫孢子的粗提和纯化,流程简述如下:(1)抓取东方蜜蜂微孢子虫感染严重的意蜂外勤蜂若干只,拉取中肠放入研钵充分研磨,经滤网过滤后的滤液转移到干净的EP管,4℃,3 000 r/min离心15 min,弃上清;(2)加无菌水重悬底部白色沉淀,4℃,5 000 r/min离心15 min,弃上清,保留沉淀,如此重复两次;(3)将25%、50%、75%、100%的Percoll密度梯度溶液按密度从高到低的顺序依次缓缓加入干净的EP管,每种密度200 μL;用25%的Percoll溶液重悬孢子,并缓缓加在上述密度梯度溶液的上方,4℃,14 000 r/min离心30 min;(4)利用无菌的注射器针头小心吸取乳白色的孢子带,并转移到干净的EP管,即得到东方蜜蜂微孢子虫的纯净孢子。将纯化得到的纯净孢子分为两部分,一部分经液氮速冻后保存于-80℃冰箱,作为对照组(NcCK);另一部分保存于4℃冰箱,用于饲喂接种意蜂工蜂。1.3 东方蜜蜂微孢子虫孢子的饲喂接种及意蜂工蜂中肠样品的制备
按照本实验室已建立的方法[26,27,28]进行意蜂工蜂的饲喂接种及中肠样品的制备,流程简述如下:(1)选取群势较强且外观健康的蜂群,参照文献报道合成克什米尔蜜蜂病毒(KBV)[29]、黑蜂王台病毒(BQCV)[30]、囊状幼虫病病毒(SBV)[31]、蜜蜂急性麻痹病毒(ABPV)[29]、慢性蜜蜂麻痹病病毒(CBPV)[32]、以色列急性麻痹病毒(IAPV)[31]的特异性引物,同时设计合成蜜蜂微孢子虫(Nosema apis)[33]和东方蜜蜂微孢子虫[33]的特异性引物进行PCR检测,检测结果显示上述6种常见蜜蜂病毒、蜜蜂微孢子虫和东方蜜蜂微孢子虫均为阴性,将该蜂群作为本研究的实验蜂群;(2)将实验蜂群刚要出房的封盖子脾迅速提至实验室,放入(34±0.5)℃培养箱,将刚出房的工蜂(当天记为0 d)放入干净的塑料盒(35只/盒),共计6盒,每盒上方插入一支装有50%(w/v)无菌蔗糖溶液的饲喂器,(34±0.5)℃饲养过夜;(3)将上述工蜂饥饿处理2 h,然后其中3盒工蜂用于饲喂接种东方蜜蜂微孢子虫,每只工蜂饲喂接种5 μL蔗糖溶液(含106个东方蜜蜂微孢子虫孢子);(4)每日清理死去的蜜蜂,于接种后7 d和10 d分别快速拉取工蜂中肠,放入干净的1.5 mL RNA-Free离心管,每6只中肠置于1个离心管,经液氮速冻后保存于-80℃冰箱备用。将侵染意蜂后7 d和10 d工蜂中肠的东方蜜蜂微孢子虫分别命名为NcT1(NcT1-1、NcT1-2、NcT1-3)和NcT2(NcT2-1、NcT2-2、NcT2-3)。1.4 RNA提取、sRNA文库构建及深度测序
参照笔者团队已建立的流程[34],利用Trizol法提取上述3个孢子样品(NcCK1、NcCK2、NcCK3)和6个中肠样品的总RNA;通过琼脂糖凝胶电泳选择18—30 nt的片段进行切胶回收,并连接3′接头序列;连接产物通过15%的变性PAGE胶电泳进行分离,选择36—44 nt目的片段进行切胶回收,再连接5′接头序列,然后对已连接两侧接头序列的小RNA进行反转录,产物通过3.5%的琼脂糖凝胶电泳分离,进而切胶回收140—160 bp区域条带。委托广州基迪奥生物科技有限公司对建好的cDNA文库进行上机测序,测序平台为Illumina MiSeq。已将测序原始数据上传NCBI SRA数据库,BioProject号:PRJNA395264(NcCK)和PRJNA406998(NcT1、NcT2)。1.5 测序数据质控及处理组东方蜜蜂微孢子虫数据的筛滤
通过过滤原始数据中的低质量reads得到高质量reads,进而过滤掉不含3′接头序列的reads,3′和5′接头序列,短于18或长于30个核苷酸的reads及含ployA的reads,得到最终small RNA(sRNA)的tag序列。按照以下流程筛滤处理组东方蜜蜂微孢子虫(NcT1和NcT2)的数据:(1)利用Bowtie软件将中肠测序得到的混合数据映射西方蜜蜂参考基因组(Amel_4.5),去除比对上的数据;(2)继而将未比对上的clean tags和NcCK组的tags分别比对到GeneBank和Pfam数据库,过滤掉比对到核糖体RNA(rRNA)、细胞质小RNA(scRNA)、核仁小RNA(snoRNA)、核内小RNA(snRNA)和转运RNA(tRNA)的tags;(3)进而将未比对上的clean tags映射到东方蜜蜂微孢子虫参考基因组(Assembly ASM98816v1),比对上的数据即为NcT1和NcT2的数据。类似地,按照上述流程获得比对上东方蜜蜂微孢子虫参考基因组的NcCK数据。
1.6 MiRNA的预测与分析
首先将东方蜜蜂微孢子虫的sRNA tags比对到miRBase中已知的miRNA,鉴定到保守miRNA。然后利用Mireap_v0.2软件预测上述未比对上的sRNA tags的发夹结构,并结合其在参考基因组的位置鉴定新 miRNA。统计miRNA的长度和首位碱基偏好性。已知miRNA和新miRNA在各样品中的表达量根据TPM(transcripts per million)算法计算。1.7 DEmiRNA及其靶mRNA分析
根据各组miRNA的表达量统计NcCK vs NcT1、NcCK vs NcT2和NcT1 vs NcT2比较组中的miRNA差异表达变化倍数(fold change,FC),按照 |log2 fold change|≥1且P≤0.05的标准筛选出显著DEmiRNA。利用Graph Prism 7软件统计DEmiRNA的个数并绘制柱状图。通过OmicShare在线平台(www.omicshare. com)的相关工具进行Venn分析,采用默认参数。联用RNAhybrid(v2.1.2)+ svm_light(v6.01)、Miranda(v3.3a)和TargetScan(Version: 7.0)软件对东方蜜蜂微孢子虫mRNA的3′-UTR进行靶向预测,取预测结果的交集作为可靠的预测结果。RNAhybrid软件设置的参数为-f 2,8 -b 1 -v 3 -u 3 -e -10 -n 24,之后选取比对区域5′端 1—9 nt为seed region,用程序筛选过滤掉两个连续GU matches的情况,再用svm_light和已经建立好的预测模型进行分类;Miranda(v3.3a)软件设置的参数为-sc 140 -en -10 -strict -go -4.0 -ge -9.0;TargetScan软件设置的参数:选取small RNA 5′端2—8 nt作为种子序列与转录本的3′-UTR区做预测。通过OmicShare在线平台相关工具对DEmiRNA的靶mRNA进行GO(Gene Ontology)和KEGG(Kyoto Encyclopedia of Genes and Genomes)数据库注释,采用默认参数。利用Cytoscape软件对DEmiRNA及其靶向结合的MAPK信号通路和糖酵解/糖异生通路相关mRNA的调控网络进行可视化。1.8 毒力因子和侵染相关因子相关DEmiRNA及靶mRNA调控网络的构建及分析
东方蜜蜂微孢子虫是专性寄生成年蜜蜂中肠上皮细胞的真菌病原,其增殖所需的物质和能量高度依赖宿主细胞提供。参考前人关于东方蜜蜂微孢子虫的基因组学、转录组学和分子生物学的研究报道[22,35-38],以及兔脑炎微孢子虫(Encephalitozoon cuniculi)[39]、斑马鱼微孢子虫(Pseudoloma neurophilia)[40]和家蚕微孢子虫[41]等其他微孢子虫的相关研究报道,筛选出细胞凋亡抑制因子、孢壁蛋白、蓖麻毒素和极管蛋白等毒力因子,以及己糖激酶、ATP/ADP移位酶、ABC转运蛋白和转录因子ste12等侵染因子。根据上述毒力因子和侵染因子相关mRNA及靶向结合关系预测出相应的DEmiRNA,利用Cytoscape软件对DEmiRNA及靶mRNA的调控网络进行可视化。1.9 DEmiRNA的Stem-loop RT-qPCR验证
随机在NcCK vs NcT1、NcCK vs NcT2和NcT1 vs NcT2中分别选取5、3和5个miRNA,根据成熟miRNA的序列,利用Premier 6软件设计Stem-loop引物、特异性上游引物和通用下游引物。委托上海生工生物工程股份有限公司合成引物(表1)。利用RNA提取试剂盒(TaKaRa公司,日本)抽提各样品的总RNA,利用Stem-loop引物和cDNA第一链合成试剂盒合成miRNA的cDNA,作为模板进行qPCR验证。qPCR体系(20 μL)包括:SYBR Green Dye 10 μL,上下游引物(4 μmol·L-1)各1 μL,cDNA模板1 μL,Rox 0.44 μL,DEPC水补至20 μL。选择东方蜜蜂微孢子虫的5S rRNA作为内参。反应体系在ABI QuantStudio 3荧光定量PCR系统(ABI公司,美国)中运行。反应程序:95℃ 1 min;95℃ 15 s,49℃ 30 s,共40个循环。每个样品进行3次重复。MiRNA的相对表达量根据2-ΔΔCt算法计算得出。利用Graph Prism 7进行数据计算并绘图。Table 1
表1
表1本研究使用的引物
Table 1
| 引物ID Primer ID | 引物序列 Primer sequence |
|---|---|
| miR-2478-y | Loop: GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGACTGGTGTC F: GCGCGCGTCGAATCCCACT |
| novel-m0017-5p | Loop: GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGACGGGCATA F: GCGCGCGTGGACCAGTGGC |
| novel-m0010-5p | Loop: GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGACGCTCGCT F: GCGCGCGTTTAGTTTGGCT |
| miR-3968-y | Loop: GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGACTGGTGTC F: GCGCGCGTCGAATCCCACT |
| miR-146-x | Loop: GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGACAACCCAT R: GCGCGCGTGAGAACTGAAT |
| miR-24-y | Loop: GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGACGTTCCTG F: GCGCGCGTGGCTCAGTTCA |
| miR-2965-y | Loop: GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGACTGCTCTC F: GCGCCGGAGGACTGCT |
| miR-318-y | Loop: GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGACGACCAAC F: CGGCCGCTCACTGGGC |
| R: CAGTGCGTGTCGTGGAGT | |
| 5S rRNA | F: CGAGCGGTTTCCCATCTCAGTA R: AAAACACCGGAACTCGTCAGCT |
新窗口打开|下载CSV
2 结果
2.1 sRNA-seq的数据质控与过滤
东方蜜蜂微孢子虫孢子(NcCK)测序得到 44 055 635条raw reads,经质控和过滤得到35 566 428条clean reads;经筛滤得到NcT1和NcT2的raw reads分别为40 126 400和42 145 184条,经质控和过滤得到的clean reads分别为21 525 444和30 316 756条。NcCK、NcT1和NcT2比对上参考基因组的平均clean reads数分别为7 188 407、2 103 266和4 926 712,平均比对率分别达到60.91%、29.26%和48.67%(表2)。Table 2
表2
表2sRNA-seq 数据总览
Table 2
| 样品Sample | 原始读段Raw reads | 有效标签Clean tags | 映射读段Mapped reads | 映射率Mapping ratio (%) |
|---|---|---|---|---|
| NcCK1 | 16506662 | 13466818 | 7560765 | 56.14 |
| NcCK2 | 15368310 | 11938692 | 7655068 | 64.12 |
| NcCK3 | 12180663 | 10160918 | 6349388 | 62.49 |
| NcT1-1 | 13345686 | 6950844 | 1865571 | 26.84 |
| NcT1-2 | 12837034 | 7324633 | 2510215 | 34.27 |
| NcT1-3 | 13943680 | 7249967 | 1934012 | 26.68 |
| NcT2-1 | 13820326 | 10605224 | 5503089 | 51.89 |
| NcT2-2 | 14311818 | 9988951 | 4763103 | 47.68 |
| NcT2-3 | 14013040 | 9722581 | 4513943 | 46.43 |
新窗口打开|下载CSV
2.2 东方蜜蜂微孢子虫的miRNA预测与结构特征
基于质控后的高质量测序数据,共预测出545个miRNA,包括510个保守miRNA和35个新miRNA。结构特征分析结果显示,上述miRNA的长度分布介于18—26 nt,分布在22 nt的miRNA数量最多(28.25%),其次为分布在18 nt和23 nt的miRNA(20.58%和14.77%)(图1-A);此外,miRNA的首位碱基偏向为U,其次为A和C(图1-B)。图1
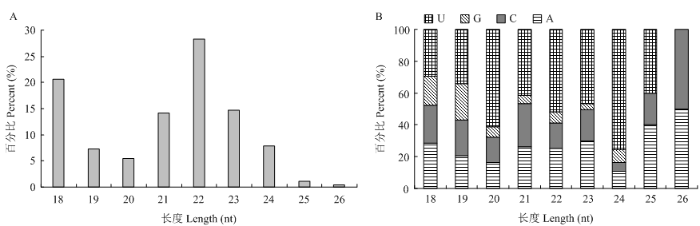 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1东方蜜蜂微孢子虫miRNA的结构特征
A:MiRNA长度分布Length distribution of miRNAs;B:MiRNA的首位碱基First nucleotide bias of miRNA
Fig. 1Structural characteristics of N. ceranae miRNAs
2.3 东方蜜蜂微孢子虫的miRNA表达谱
表达量聚类分析结果显示,miRNA的表达量在东方蜜蜂微孢子虫孢子和侵染过程的东方蜜蜂微孢子虫中差异明显,一些miRNA(miR-44-x、miR-8797-x和miR-9277-y等)在孢子中的表达量较低,但随着病原增殖表达量逐渐提高;另一些miRNA(miR-451-x、miR-486-x和miR-122-x等)在孢子中的表达量较高,但在病原的侵染过程呈下降趋势;部分miRNA(miR-8-y、miR-12-z和miR-281-y等)在孢子中保持较低的表达水平,在侵染7 d的意蜂工蜂中肠中表达量较高,但在侵染10 d的宿主中肠中又降至较低的表达水平(图2-A)。Venn分析结果显示,有147个miRNA在3组样品中共同表达,分别有86、139和41个仅在NcCK、NcT1和NcT2中特异性表达(图2-B)。图2
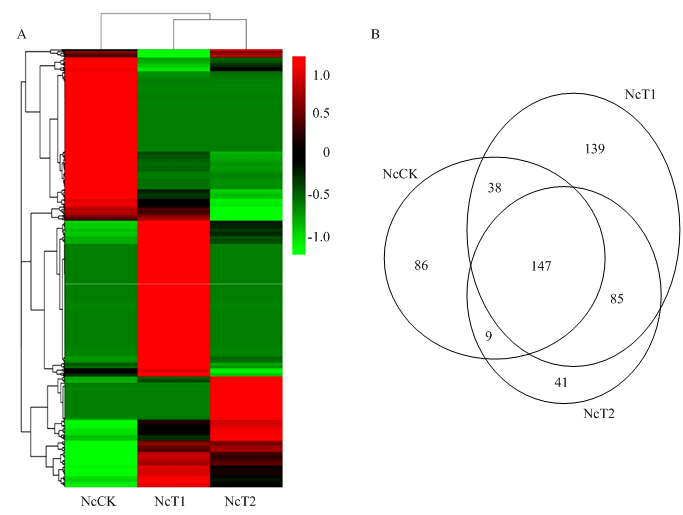 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2各个样品中miRNA的表达分析
A:各个样品中miRNA表达量的热图分析Heat map analysis of miRNA expression in each sample;B:各个样品中miRNA的Venn分析Venn analysis of miRNAs in each sample
Fig. 2Expression analysis of miRNAs in each sample
上述共有miRNA的靶mRNA可注释到27个功能条目,包括代谢过程(340)、催化活性(295)和细胞进程(292);以及84条通路如代谢途径(122)、核糖体(55)和次级代谢产物生物合成(46)。NcCK、NcT1和NcT2特有miRNA的靶mRNA可分别注释到27、27和24个功能条目,注释数目最多的条目均为代谢进程(333、329和242)、催化活性(294、291和212)和细胞进程(291、282和208);这些靶mRNA还可注释到84、84和83条通路,注释数目最多的条目均为代谢途径(121、119和87)、次级代谢产物生物合成(49、50和35)和核糖体(47、53和33)。括号内的数字代表注释到该条目(或通路)的靶mRNA数。
2.4 东方蜜蜂微孢子虫的miRNA差异表达谱
差异表达分析结果显示,NcCK vs NcT1、NcCK vs NcT2和NcT1 vs NcT2比较组中分别含有164、122和60个DEmiRNA,其中上调miRNA数分别为90、55和16个,下调miRNA数分别为74、67和44个。Venn分析结果显示,上述3个比较组中共同上调表达的miRNA有5个,分别为miR-5106-y(log2 FC= 15.76、19.69和3.93)、miR-301-x(log2FC=12.80、15.44和2.64)、novel-m0017-5p(log2FC=1.09、3.28和2.18)、miR-5108-y(log2FC =17.59、19.55和1.96)和miR-7465-x(log2FC=14.14、15.36和1.22);共同下调表达的miRNA有6个,分别为miR-378-y(log2 FC =-1.35、-2.61和-1.26)、miR-128-y(log2FC=-1.87、-3.87和-2.00)、miR-183-x(log2FC =-3.12、-16.80和-13.67)、miR-16-y(log2FC =-3.05、-16.85和-13.81)、miR-27-x(log2FC=-1.90、-16.05和-14.16)和miR-1307-y(log2FC =-1.46、-16.36和-14.90)。2.5 东方蜜蜂微孢子虫DEmiRNA的靶mRNA预测及功能和代谢通路注释
NcCK vs NcT1、NcCK vs NcT2和NcT1 vs NcT2比较组中DEmiRNA分别预测出1 885、1 733和1 524个靶mRNA。GO数据库注释结果显示,上述靶mRNA可分别注释到27、25和26个功能条目,均涉及生物学调控、细胞组分和分子功能3类(图3)。3个比较组中DEmiRNA的靶mRNA注释数量最多的均为代谢进程、催化活性、细胞进程、结合和细胞。但每个比较组在这些功能条目上富集的靶mRNA数并不相同,NcCK vs NcT1组的功能条目上富集的靶mRNA最多,NcCK vs NcT2组次之,NcT1 vs NcT2组的功能条目上富集的靶mRNA数最少。3个比较组中相差的功能条目为病毒、病毒组分和分子功能调节,但富集的靶mRNA数均仅为1个。NcCK vs NcT1组包含这3个功能条目,NcCK vs NcT2组仅有分子功能调节,NcT1 vs NcT2组包含病毒和病毒组分2个功能条目。图3
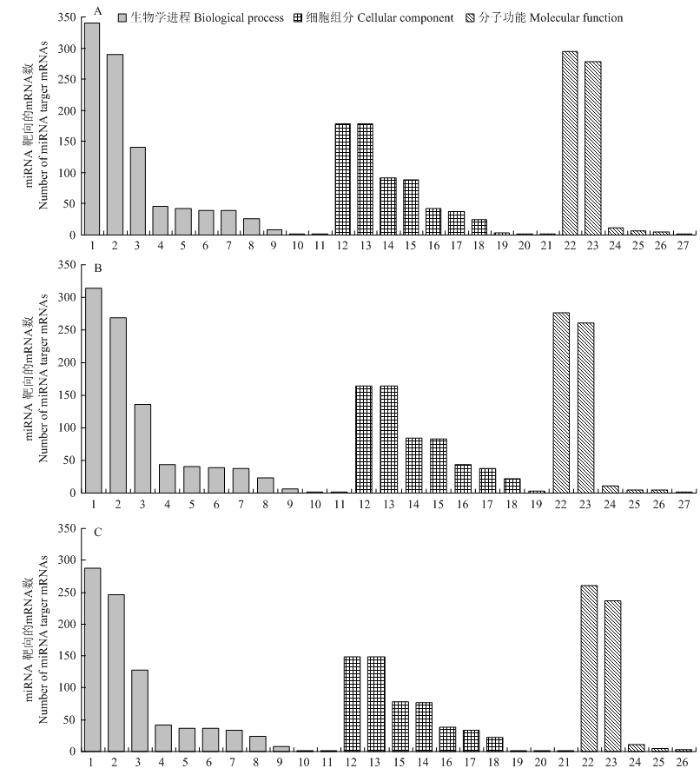 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3各个比较组中DEmiRNA靶基因的GO数据库注释
A:NcCK vs NcT1靶向的mRNA Target mRNA in NcCK vs NcT1;B:NcCK vs NcT2靶向的mRNA Target mRNA in NcCK vs NcT2;C:NcT1 vs NcT2靶向的mRNA Target mRNA in NcT1 vs NcT2。1:代谢进程Metabolic process;2:细胞进程Cellular process;3:单一有机体进程Single-organism process;4:定位Localization;5:生物学调控Biological regulation;6:生物进程调控Regulation of biological process;7:细胞成分组织或生物合成Cellular component organization or biogenesis;8:应激反应Response to stimulus;9:信号Signaling;10:复制Reproduction;11:生长Growth;12:细胞Cell;13:细胞组分Cell part;14:细胞器Organelle;15:大分子复合物Macromolecular complex;16:细胞膜Membrane;17:细胞膜组分Membrane part;18:细胞器组分Organelle part;19:膜封闭腔Membrane-enclosed lumen;20:病毒Virion;21:病毒组分Virion part;22:催化活性Catalytic activity;23:结合Binding;24:运输活性Transporter activity;25:分子结构活性Structural molecule activity;26:核苷酸结合转录因子活性Nucleic acid binding transcription factor activity;27:分子功能调节Molecular function regulator
Fig. 3GO database annotation of DEmiRNA’s target genes in each comparison group
KEGG数据库注释结果显示,上述NcCK vs NcT1、NcCK vs NcT2和NcT1 vs NcT2比较组中DEmiRNA的靶mRNA可分别注释到84、84和84条通路。NcCK vs NcT1中DEmiRNA的靶mRNA注释到最多的通路分别为代谢途径、核糖体、次级代谢产物生物合成、真核生物核糖体生物合成以及抗生素的生物合成;NcCK vs NcT2中DEmiRNA的靶mRNA注释到最多的通路分别为代谢途径、核糖体、次级代谢产物生物合成、真核生物核糖体生物合成以及抗生素的生物合成;NcT1 vs NcT2中DEmiRNA的靶mRNA注释到最多的通路分别为代谢途径、核糖体、次级代谢产物生物合成、抗生素的生物合成以及内质网蛋白质加工(表3)。
Table 3
表3
表3各比较组中DEmiRNA的靶mRNA富集的前15条通路
Table 3
| 通路 Pathway | NcCK vs NcT1比较组 NcCK vs NcT1 group | NcCK vs NcT2比较组 NcCK vs NcT2 group | NcT1 vs NcT2比较组 NcT1 vs NcT2 group |
|---|---|---|---|
| 新陈代谢途径 Metabolic pathways | 119 | 113 | 103 |
| 核糖体 Ribosome | 56 | 50 | 41 |
| 次生代谢产物合成 Biosynthesis of secondary metabolites | 48 | 47 | 42 |
| 真菌生物核糖体的生物合成 Ribosome biogenesis in eukaryotes | 37 | 38 | 29 |
| 抗生素的生物合成 Biosynthesis of antibiotics | 36 | 35 | 31 |
| 内质网蛋白质加工 Protein processing in endoplasmic reticulum | 35 | 32 | 29 |
| 嘌呤代谢 Purine metabolism | 33 | 28 | 26 |
| 嘧啶代谢 Pyrimidine metabolism | 32 | 28 | 25 |
| 细胞周期 Cell cycle | 31 | 30 | 28 |
| RNA转运 RNA transport | 30 | 26 | 24 |
| 蛋白酶体 Proteasome | 28 | 25 | 24 |
| 减数分裂 Meiosis | 26 | 24 | 24 |
| mRNA监督通路 mRNA surveillance pathway | 24 | 21 | 22 |
| DNA复制 DNA replication | 24 | 24 | 22 |
| 泛素蛋白水解 Ubiquitin mediated proteolysis | 24 | 24 | 20 |
新窗口打开|下载CSV
2.6 东方蜜蜂微孢子虫DEmiRNA的调控网络构建及分析
因各比较组中DEmiRNA及其靶mRNA数量较多,形成的调控网络十分复杂,故根据靶mRNA的KEGG数据库注释信息,选择与东方蜜蜂微孢子虫的增殖、环境应答和能量代谢密切相关的MAPK信号通路和糖酵解/糖异生通路富集靶mRNA及相应DEmiRNA构建调控网络,分析结果显示NcCK vs NcT1中35个DEmiRNA靶向结合10个MAPK信号通路相关靶mRNA,49个DEmiRNA靶向结合13个糖酵解/糖异生通路相关靶mRNA(图4-A、4-B);NcCK vs NcT2中26个DEmiRNA靶向结合8个MAPK信号通路相关靶mRNA,40个DEmiRNA靶向结合13个糖酵解/糖异生通路相关靶mRNA(图4-C、4-D);NcT1 vs NcT2中12个DEmiRNA靶向结合9个MAPK信号通路相关靶mRNA,17个DEmiRNA靶向结合10个糖酵解/糖异生通路相关靶mRNA(图4-E、4-F)。图4
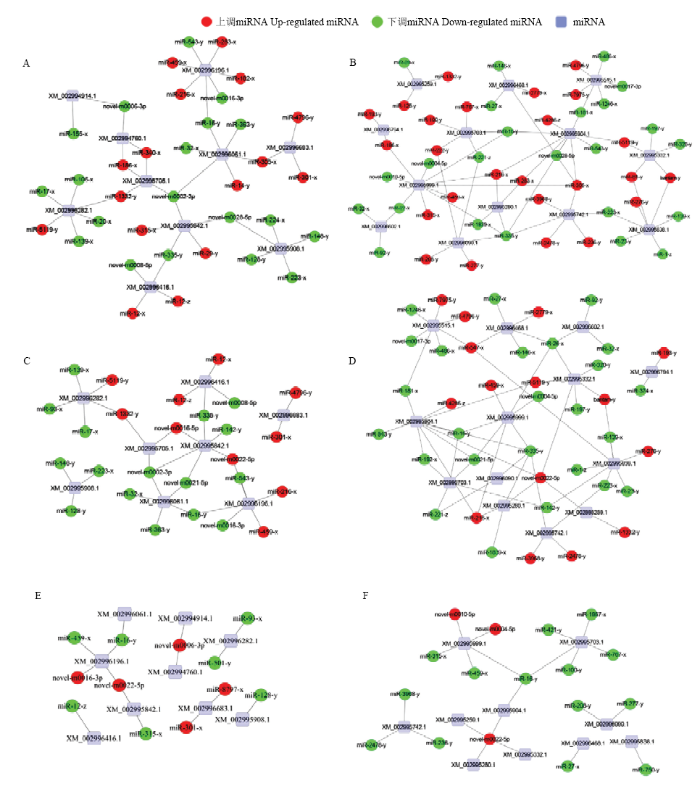 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4MAPK信号通路和糖酵解/糖异生通路相关靶mRNA及相应DEmiRNA的调控网络
Fig. 4Regulatory networks of DEmiRNAs and corresponding target mRNAs associated with MAPK signaling pathway and glycolysis/gluconeogenesis pathway
进一步筛选出蓖麻毒素B凝集素、孢壁蛋白、孢壁蛋白前体、孢壁和锚定吸盘复合蛋白和极管蛋白等毒力因子以及ATP/ADP移位酶、ABC转运蛋白、己糖激酶、丙酮酸激酶和6-磷酸果糖激酶等侵染相关因子相关mRNA存在靶向结合关系的DEmiRNA,并构建DEmiRNA-靶mRNA的调控网络。3个比较组中,共有15个DEmiRNA靶向结合糖酵解途径3个关键酶基因,其中有10个DEmiRNA靶向己糖激酶编码基因(图5-A);2个DEmiRNA可靶向结合抑制细胞凋亡蛋白编码基因(图5-B);78个DEmiRNA可靶向结合4个ATP/ADP移位酶编码基因和7个ABC转运蛋白编码基因(图5-C);61个DEmiRNA可靶向结合3个极管蛋白编码基因、5个孢壁蛋白编码基因、孢壁蛋白前体编码基因、孢壁和锚定吸盘复合蛋白编码基因(图5-D);27个DEmiRNA可靶向结合7个蓖麻毒素B凝集素的蛋白编码基因(图5-E)。
图5
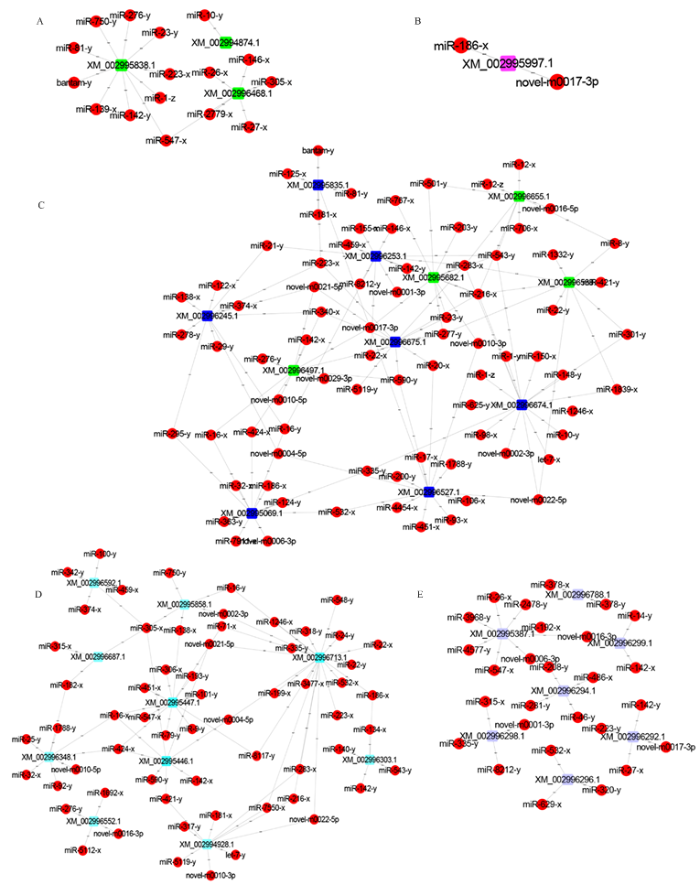 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5东方蜜蜂微孢子虫毒力因子和侵染相关因子相关mRNA及相应DEmiRNA的调控网络
Fig. 5Regulatory networks of DEmiRNAs and corresponding target mRNAs related to virulence factors and infection-associated factors of N. ceranae
2.7 东方蜜蜂微孢子虫DEmiRNA的RT-qPCR验证
从NcCK vs NcT1、NcCK vs NcT2和NcT1 vs NcT2比较组中分别随机选取5、3和5个显著性DEmiRNA进行RT-qPCR验证,结果显示其表达趋势变化与测序数据中的变化趋势一致,证实测序数据和miRNA差异表达趋势的可靠性(图6)。图6
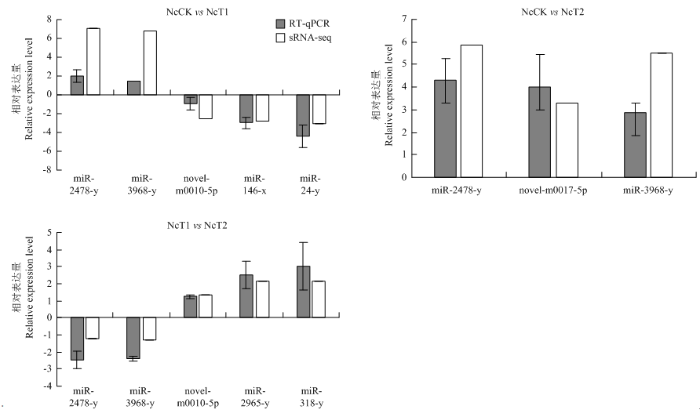 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6DEmiRNA的RT-qPCR验证
Fig. 6RT-qPCR confirmation of DEmiRNAs
3 讨论
3.1 东方蜜蜂微孢子虫 miRNA的数量和结构特征
HUANG等[35]利用二代测序技术对东方蜜蜂微孢子虫感染1—6 d的西方蜜蜂中肠组织进行转录组测序,分析发现东方蜜蜂微孢子虫的1 122个差异表达基因可归入4种表达模式。随后,该团队研究报道东方蜜蜂微孢子虫的5个miRNA不仅能靶向结合病原自身的mRNA,还可以靶向结合宿主的mRNA[20],暗示miRNA可能介导西方蜜蜂对东方蜜蜂微孢子虫的跨界调控。本研究通过对东方蜜蜂微孢子虫感染7 d和10 d的意蜂工蜂中肠以及纯化孢子进行测序,利用生物信息学软件预测出545个miRNA,其长度主要分布在21—24 nt,首位碱基主要偏向U,上述结构特征类似于其他已报道的真菌miRNA[17]。本研究鉴定到的miRNA为东方蜜蜂微孢子虫的miRNA信息提供了很好的补充。笔者团队前期利用高通量测序技术和生物信息学手段从东方蜜蜂微孢子虫孢子中分别鉴定出83个长链非编码RNA(long non-coding RNA,lncRNA)和204个环状RNA(circular RNA,circRNA)[24,25]。本研究鉴定出的miRNA和前期鉴定出的lncRNA、circRNA丰富了东方蜜蜂微孢子虫的ncRNA信息,也为其他微孢子虫的ncRNA研究提供了重要参考。3.2 侵染意蜂工蜂的东方蜜蜂微孢子虫的miRNA表达谱差异
本研究发现,共有147个miRNA在NcCK、NcT1和NcT2中共同表达,推测这些miRNA在东方蜜蜂微孢子虫孢子和侵染状态均具有一定的生物学功能。miRNA的表达具有时空特异性。东方蜜蜂微孢子虫的生活史约为6 d[35],感染意蜂后7 d和10 d的东方蜜蜂微孢子虫理论上处于第二生活史时期,其中感染7 d的病原孢子可能处于弹射极丝侵染宿主中肠上皮细胞阶段,而感染10 d的病原可能处于裂殖体阶段。在本研究中分别有86、139和41个miRNA在NcCK、NcT1和NcT2中特异性表达,推测这些miRNA在东方蜜蜂微孢子虫孢子感染工蜂的不同时期发挥特定的调控作用。从NcCK vs NcT1、NcCK vs NcT2和NcT1 vs NcT2比较组中分别筛选出164、122和60个DEmiRNA,表明东方蜜蜂微孢子虫的侵染和增殖过程伴随着miRNA表达量的动态变化,上述DEmiRNA与病原的侵染和增殖具有密切的相关性。此外,miR-5106-y、miR-301-x、novel-m0017-5p、miR-5108-y和miR-7465-x 5个miRNA在各比较组中共同上调表达,而miR-378-y、miR-128-y、miR-183-x、miR-16-y、miR-27-x和miR-1307-y 6个miRNA共同下调表达,暗示这些DEmiRNA在东方蜜蜂微孢子虫增殖和侵染过程具有特殊功能,值得进一步的功能研究。3.3 东方蜜蜂微孢子虫DEmiRNA通过调控MAPK信号通路调节病原的环境应答和增殖
MAPK、cAMP和双组分信号传导系统是真菌应答外界环境的3个关键信号通路,均涉及调控真菌生长发育和毒力因子[42]。真菌的MAPK级联是由外界刺激触发,调节细胞周期、繁殖、形态发生、应激反应和毒性等一系列过程[43]。MAPK信号通路参与调节酿酒酵母(Saccharomyces cerevisia)的细胞壁完整性、配合、菌丝生长、孢子形成以及细胞应激反应[43]。笔者团队前期研究发现,球囊菌可能通过激活MAPK信号通路促进对意蜂幼虫的侵染[44];但对于侵染中蜂幼虫的球囊菌,该信号通路受到明显抑制[45];此外还发现在侵染意蜂工蜂的过程中,东方蜜蜂微孢子虫的MAPK信号通路富集基因多数上调表达[23]。本研究中,NcCK vs NcT1、NcCK vs NcT2和NcT1 vs NcT2比较组中DEmiRNA的靶mRNA分别有10、8和9个注释到MAPK信号通路,表明相应的DEmiRNA可能调控MAPK信号通路相关基因的表达水平,从而调节东方蜜蜂微孢子虫对外界环境的应答以及病原的增殖;进一步分析发现,上述DEmiRNA包含的下调miRNA(20、16和7个)多于上调DEmiRNA(15、10和5个),体现出意蜂工蜂与东方蜜蜂微孢子虫互作的复杂性,宿主可能通过跨界调控等机制对病原的MAPK信号通路进行调节。转录因子ste12基因(ste12)是MAPK信号通路下游的关键基因,有研究报道灰霉菌(Botrytis cinerea)、白色念球菌(Candida albicans)、新型隐球菌(Cryptococcus neoformans)、粗糙脉胞菌和构巢曲霉(Aspergillus nidulans)等真菌如缺少ste12将导致生殖周期产生缺陷[46]。前期研究发现,东方蜜蜂微孢子虫的ste12在NcCK vs NcT1和NcCK vs NcT2比较组中均上调表达[23]。本研究中,靶向ste 12 mRNA的miR-223-x(log2FC=-17.58和-17.58)、miR-148-y(log2FC=-4.54和-5.29)、miR-224-x(log2 FC=-1.98和-3.53)和Novel-m0028-5p(log2FC=-14.19和-1.95)在NcCK vs NcT1、NcCK vs NcT2中表达量均为下调,表明东方蜜蜂微孢子虫在侵染过程中通过下调上述miRNA的表达水平减弱对ste 12 mRNA的抑制,从而上调ste 12的表达量,以促进自身的增殖和侵染。3.4 东方蜜蜂微孢子虫DEmiRNA通过调控糖酵解/糖异生、ATP/ADP移位酶和ABC转运蛋白满足增殖过程的能量需求
东方蜜蜂微孢子虫感染会对西方蜜蜂工蜂造成能量胁迫,引起后者较未感染工蜂摄取更多的糖水[6]。有研究表明受东方蜜蜂微孢子虫感染的西方蜜蜂的糖酵解相关基因表达量并无明显变化,但东方蜜蜂微孢子虫的催化糖酵解的核心酶基因在感染后24 h即开始表达,己糖激酶基因在感染后48 h也开始表达[35]。DOLGIKH等[47]研究发现东方蜜蜂微孢子虫的己糖激酶基因含有信号肽,合成并释放的己糖激酶在宿主的细胞核中积累。笔者团队前期发现,NcCK vs NcT1和NcCK vs NcT2比较组中分别有11和12个差异表达基因富集在糖酵解/糖异生通路,其中包括己糖激酶基因在内的3个关键酶基因在东方蜜蜂微孢子虫侵染意蜂工蜂的过程中均上调表达[23]。本研究中,NcCK vs NcT1和NcCK vs NcT2比较组中DEmiRNA的靶mRNA分别有13和13个注释到糖酵解/糖异生通路;共有10个DEmiRNA可靶向结合己糖激酶基因,其中miR-139-x(log2FC= -15.66和-15.66)、miR-1-z(log2FC=-15.16和-15.16)、miR-223-x(log2FC=-17.58和-17.58)、miR-23-y(log2 FC=-2.67和-2.67)在NcCK vs NcT1和NcCK vs NcT2中均为下调表达,推测东方蜜蜂微孢子虫在侵染过程中通过下调上述DEmiRNA的表达量减弱对己糖激酶基因的抑制,从而增强己糖激酶的合成,以调节宿主的能量代谢并满足自身增殖的能量需求。微孢子虫生长与繁殖所需的能量高度依赖宿主细胞提供。ATP/ADP移位酶和ABC转运蛋白参与微孢子虫对宿主物质和能量的窃取[37]。PALDI等研究发现通过RNAi敲减东方蜜蜂微孢子虫的ATP/ADP移位酶基因可导致病原的增殖水平下降[36]。前期研究发现3个ATP/ADP移位酶基因(XM_002996497.1、XM_002995682.1和XM_002996655.1)在NcCK vs NcT1和NcCK vs NcT2中表达量上调[23];本研究中,NcCK vs NcT1、NcCK vs NcT2和NcT1 vs NcT2中分别有4、5和2个DEmiRNA与上述3个基因的mRNA存在靶向结合关系且差异变化趋势相反,表明东方蜜蜂微孢子虫侵染过程中相应的DEmiRNA参与调节ATP/ADP移位酶基因的表达,通过加强ATP/ADP移位酶的合成满足自身能量需求并促进增殖。此外,另有1个ATP/ADP移位酶基因(XM_002996538.1)在NcCK vs NcT1和NcCK vs NcT2中下调表达[23];但与其mRNA靶向结合的miR-1332-y(log2FC=15.51和16.19)、miR-216-x(log2FC=16.33和15.48)、miR-283-x(log2FC=5.50和4.62)、miR-301-y(log2FC=15.58和14.83)、miR-8-y(log2FC=7.03和5.53)5个DEmiRNA的差异变化趋势与该基因的表达趋势恰好相反,体现出病原增殖过程能量代谢的活跃性和相关DEmiRNA参与调控能量代谢的广泛性。前期还发现东方蜜蜂微孢子虫的7个ABC转运蛋白编码基因在NcCK vs NcT1和NcCK vs NcT2中差异表达,其中2个ABC转运蛋白编码基因(XM_002995835.1和XM_002996245.1)上调表达,5个ABC转运蛋白基因(XM_002996253.1、XM_002995069.1、XM_ 002996674.1、TCONS_00003043和XM_002996675.1)下调表达[23],说明东方蜜蜂微孢子虫与意蜂工蜂之间存在复杂的互作,后者可能通过调节病原的部分ABC转运蛋白基因的表达水平限制其增殖。本研究中,NcCK vs NcT1和NcCK vs NcT2比较组中分别有6和12个DEmiRNA(包含3和7个下调miRNA)靶向结合XM_002995835.1和XM_ 002996245.1。NcCK vs NcT1和NcCK vs NcT2比较组中分别有11、17、12和12个DEmiRNA(包含3、5、6和2个上调miRNA)靶向结合XM_002996253.1、XM_002995069.1、XM_002996674.1、TCONS_00003043和XM_002996675.1。上述结果表明东方蜜蜂微孢子虫通过差异表达相应的miRNA调控ABC转运蛋白编码基因的表达。
3.5 东方蜜蜂微孢子虫DEmiRNA通过调控极管蛋白和孢壁蛋白基因的表达调节病原的侵染过程
东方蜜蜂微孢子虫的孢壁蛋白能够保护孢子抵御外界不良环境,同时在病原应答外界环境变化和附着宿主细胞等方面发挥关键作用[12]。东方蜜蜂微孢子虫孢子被蜜蜂摄入后,在中肠的碱性环境和离子浓度刺激下,内部极管快速弹射并刺入上皮细胞,然后经中空管道将有感染性的孢原质注入细胞[7,8,9]。在细胞识别和吸附以及极丝弹射的过程中,孢壁蛋白和极管蛋白发挥着非常重要的作用[48]。有研究表明家蚕微孢子虫的孢壁蛋白5与极管蛋白2、极管蛋白3存在相互作用,孢壁蛋白5对于极管蛋白锚定在孢壁上具有重要作用[48];孢壁蛋白9定位于极管上且能与极管蛋白1和极管蛋白2互作[49]。前期研究发现,东方蜜蜂微孢子虫的极管蛋白1、极管蛋白2和极管蛋白基因在NcCK、NcT1和NcT2中的表达量持续上调,印证了极管蛋白对东方蜜蜂微孢子虫侵染的重要性[23]。RODRíGUEZ-GARCíA等[38]研究报道干扰极管蛋白3基因的表达可显著降低东方蜜蜂微孢子虫孢子的数量。本研究发现,靶向结合极管蛋白1基因的miR-101-y(log2FC=-0.45和-2.75)、miR-451-x(log2FC=-7.03和-8.14)和miR-8117-y(log2FC=-4.63和-3.07)在NcCK vs NcT1和NcCK vs NcT2中下调表达;此外靶向结合极管蛋白2基因的novel-m0004-5p(log2FC=-3.81和-1.41)和novel-m0021-5p(log2FC=-4.41和-1.52)在NcCK vs NcT1和NcCK vs NcT2中也下调表达;还发现多达20个DEmiRNA靶向结合极管蛋白,其中有13个miRNA下调表达。以上结果表明东方蜜蜂微孢子虫在侵染过程中通过下调相关miRNA的表达量提高极管蛋白基因的表达水平,从而增强极管蛋白的合成,以促进病原对宿主细胞的侵染。前期研究还发现,孢壁蛋白8基因在NcCK vs NcT1中表达量下调,在NcCK vs NcT2中表达量未发生变化[23];本研究中,靶向结合孢壁蛋白8蛋白编码基因的miR-315-x在NcCK vs NcT1中上调表达(log2FC=12.80),在NcCK vs NcT2中没有发生差异表达(log2FC=0),说明miR-315-x通过调控孢壁蛋白8蛋白基因的表达参与东方蜜蜂微孢子虫对意蜂工蜂的侵染过程。此外,靶向结合孢壁蛋白9基因mRNA的miR-1788-y(log2FC=13.75和13.94)和miR-424-x(log2FC=13.19和13.86)在NcCK vs NcT1和NcCK vs NcT2比较组中上调表达,与该基因的差异变化趋势相反,推测这是病原和宿主之间的复杂互作的结果。3.6 东方蜜蜂微孢子虫DEmiRNA通过调控病原蓖麻毒素B凝集素和宿主细胞凋亡抑制基因促进侵染
蓖麻毒素B凝集素作为蓖麻毒素的B链,主要通过与细胞表面受体结合起到凝集素的作用,从而介导蓖麻毒素A链进入细胞发挥功能[50]。LIU等[41]对家蚕微孢子虫休眠状态和发芽状态的孢子进行转录组测序和分析,发现蓖麻毒素B凝集素在发芽孢子中的表达量下调;LIU等[51]发现在家蚕微孢子虫侵染家蚕卵巢BmN细胞的研究过程中加入蓖麻毒素B凝集素,能够增强病原对宿主细胞的黏附能力。前期研究发现,东方蜜蜂微孢子虫的7个蓖麻毒素凝集素基因在NcCK vs NcT1和NcCK vs NcT2比较组中表达量上调[23];本研究中,NcCK vs NcT1和NcCK vs NcT2比较组中的DEmiRNA可以靶向结合其中的6个蓖麻毒素凝集素基因的mRNA,其中有10个DEmiRNA在NcCK vs NcT1和NcCK vs NcT2比较组中的差异变化趋势与该基因相反,说明东方蜜蜂微孢子虫在侵染意蜂工蜂的过程中通过调节部分miRNA的表达水平对蓖麻毒素基因进行表达调控,从而增强对宿主细胞的黏附性和侵染,但宿主同样能够通过互作对病原的蓖麻毒素凝集素基因进行一定程度的抑制,再次体现出二者相互作用的复杂性;该基因或许能作为一个蜜蜂微孢子虫病治疗的有效靶点。东方蜜蜂微孢子虫能抑制西方蜜蜂的细胞凋亡[5]。细胞凋亡抑制基因通过结合活化半胱天冬酶抑制细胞的凋亡[35]。HUANG等[35]研究发现东方蜜蜂微孢子虫的细胞凋亡抑制基因在病原侵染西方蜜蜂后24 h即开始表达。前期研究发现,细胞凋亡抑制基因(XM_002995997.1)在NcCK vs NcT1和NcCK vs NcT2中表达量上调;本研究中,NcCK vs NcT1和NcCK vs NcT2比较组中有2个DEmiRNA可以靶向结合细胞凋亡抑制基因,其中novel-m0017-3p(log2FC =-14.05和-14.05)在NcCK vs NcT1和NcCK vs NcT2比较组中的差异变化趋势与该基因相反;而miR-186-x(log2FC=1.02和0.23)在NcCK vs NcT1和NcCK vs NcT2比较组中的差异变化趋势与该基因变化趋势一致,但差异变化倍数较小。以上结果表明东方蜜蜂微孢子虫主要通过下调novel-m0017-3p增强对宿主细胞凋亡抑制基因的抑制作用,从而延长宿主细胞的存活时间,更好地促进自身的繁殖。4 结论
在miRNA组学层面对侵染意蜂工蜂的东方蜜蜂微孢子虫的DEmiRNA、调控网络及潜在作用进行了深入解析,揭示了DEmiRNA在东方蜜蜂微孢子虫侵染过程中广泛参与调控病原的物质和能量代谢、细胞生命活动,部分DEmiRNA通过调节ste12、己糖激酶、孢壁蛋白、极管蛋白、ATP/ADP移位酶、ABC转运蛋白、蓖麻毒素等毒力及侵染相关因子基因的表达促进东方蜜蜂微孢子虫在宿主细胞内的增殖和侵染。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:22813931 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:29575513 [本文引用: 2]
[本文引用: 2]
[本文引用: 2]
URLPMID:16574143 [本文引用: 2]
[本文引用: 2]
DOI:10.1186/s12864-018-5316-3URLPMID:30547762 [本文引用: 1]

BACKGROUND: Trichophyton rubrum (T. rubrum) is an important model organism of dermatophytes, which are the most common fungal pathogens worldwide. Despite the severity and prevalence of the infection caused by these pathogens, current therapies are not sufficient. MicroRNA (miRNA) is a class of small noncoding RNAs that are key factors in the regulation of gene expression. These miRNAs are reported to be highly conserved in different organisms and are involved in various essential cellular processes. In this study, we performed an integrated analysis of microRNA-like RNAs (milRNAs) and mRNAs between conidial and mycelial stages to investigate the roles of milRNAs in regulating the expression of target genes in T. rubrum. RESULTS: A total of 158 conserved milRNAs and 12 novel milRNAs were identified in our study, corresponding to 5470 target genes, which were involved in various essential biological pathways. In addition, 137 target genes corresponding to 21 milRNAs were concurrent differentially expressed between the conidial and mycelial stages. Among these 137 target genes, 64 genes showed the opposite trend to their corresponding milRNAs in expression difference between the two stages, indicating possible negative regulation. Furthermore, 46% of differentially expressed target genes are involved in transcription, transcriptional and post-transcriptional regulation. Our results indicate that milRNAs might associate with other regulatory elements to control gene expression at both transcriptional and post-transcriptional level. CONCLUSIONS: This study provides the first analysis of milRNA expression profile in T. rubrum as well as dermatophytes in general. The results revealed the roles of milRNAs in regulating gene expression between the two major growth stages of this fungus. Our study deepens our understanding of T. rubrum and will serve as a foundation for further investigations to combat this fungus.
URLPMID:24098464 [本文引用: 2]
[本文引用: 2]
DOI:10.1016/0092-8674(93)90529-yURLPMID:8252621 [本文引用: 1]
lin-4 is essential for the normal temporal control of diverse postembryonic developmental events in C. elegans. lin-4 acts by negatively regulating the level of LIN-14 protein, creating a temporal decrease in LIN-14 protein starting in the first larval stage (L1). We have cloned the C. elegans lin-4 locus by chromosomal walking and transformation rescue. We used the C. elegans clone to isolate the gene from three other Caenorhabditis species; all four Caenorhabditis clones functionally rescue the lin-4 null allele of C. elegans. Comparison of the lin-4 genomic sequence from these four species and site-directed mutagenesis of potential open reading frames indicated that lin-4 does not encode a protein. Two small lin-4 transcripts of approximately 22 and 61 nt were identified in C. elegans and found to contain sequences complementary to a repeated sequence element in the 3' untranslated region (UTR) of lin-14 mRNA, suggesting that lin-4 regulates lin-14 translation via an antisense RNA-RNA interaction.
DOI:10.1038/35002607URLPMID:10706289 [本文引用: 1]
The C. elegans heterochronic gene pathway consists of a cascade of regulatory genes that are temporally controlled to specify the timing of developmental events. Mutations in heterochronic genes cause temporal transformations in cell fates in which stage-specific events are omitted or reiterated. Here we show that let-7 is a heterochronic switch gene. Loss of let-7 gene activity causes reiteration of larval cell fates during the adult stage, whereas increased let-7 gene dosage causes precocious expression of adult fates during larval stages. let-7 encodes a temporally regulated 21-nucleotide RNA that is complementary to elements in the 3' untranslated regions of the heterochronic genes lin-14, lin-28, lin-41, lin-42 and daf-12, indicating that expression of these genes may be directly controlled by let-7. A reporter gene bearing the lin-41 3' untranslated region is temporally regulated in a let-7-dependent manner. A second regulatory RNA, lin-4, negatively regulates lin-14 and lin-28 through RNA-RNA interactions with their 3' untranslated regions. We propose that the sequential stage-specific expression of the lin-4 and let-7 regulatory RNAs triggers transitions in the complement of heterochronic regulatory proteins to coordinate developmental timing.
URLPMID:19167326 [本文引用: 2]
DOI:10.1093/nar/gkm952URLPMID:17991681 [本文引用: 2]
miRBase is the central online repository for microRNA (miRNA) nomenclature, sequence data, annotation and target prediction. The current release (10.0) contains 5071 miRNA loci from 58 species, expressing 5922 distinct mature miRNA sequences: a growth of over 2000 sequences in the past 2 years. miRBase provides a range of data to facilitate studies of miRNA genomics: all miRNAs are mapped to their genomic coordinates. Clusters of miRNA sequences in the genome are highlighted, and can be defined and retrieved with any inter-miRNA distance. The overlap of miRNA sequences with annotated transcripts, both protein- and non-coding, are described. Finally, graphical views of the locations of a wide range of genomic features in model organisms allow for the first time the prediction of the likely boundaries of many miRNA primary transcripts. miRBase is available at http://microrna.sanger.ac.uk/.
URLPMID:20417140 [本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:29692768 [本文引用: 2]
URLPMID:30142384 [本文引用: 1]
DOI:10.1111/imb.12534URLPMID:30171639 [本文引用: 2]
Nosema ceranae is a microsporidian parasite that infects the honeybee midgut epithelium. The protein-coding gene Dicer is lost in most microsporidian genomes but is present in N. ceranae. By feeding infected honeybees with small interfering RNA targeting the N. ceranae gene coding Dicer (siRNA-Dicer), we found that N. ceranae spore loads were significantly reduced. In addition, over 10% of total parasite protein-coding genes showed significantly divergent expression profiles after siRNA-Dicer treatment. Parasite genes for cell proliferation, ABC transporters and hexokinase were downregulated at 3 days postinfection, a key point in the middle of parasite replication cycles. In addition, genes involved in metabolic pathways of honeybees and N. ceranae showed significant co-expression. Furthermore, the siRNA-Dicer treatment partly reversed the expression patterns of honeybee genes. The honeybee gene mucin-2-like showed significantly upregulation in the siRNA-Dicer group compared with the infection group continually at 4, 5 and 6 days postinfection, suggesting that the siRNA-Dicer feeding promoted the strength of the mucus barrier resulted from interrupted parasite proliferation. As the gene Dicer broadly regulates N. ceranae proliferation and honeybee metabolism, our data suggest the RNA interference pathway is an important infection strategy for N. ceranae.
[本文引用: 10]
[本文引用: 10]
URLPMID:30269253 [本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
DOI:10.1128/AEM.67.5.2384-2387.2001URLPMID:11319129 [本文引用: 1]
A reverse transcriptase PCR (RT-PCR) assay was developed for the detection of acute bee paralysis virus (ABPV) and black queen cell virus (BQCV), two honeybee viruses. Complete genome sequences were used to design unique PCR primers within a 1-kb region from the 3' end of both genomes to amplify a fragment of 900 bp from ABPV and 700 bp from BQCV. The combined guanidinium thiocyanate and silica membrane method was used to extract total RNA from samples of healthy and laboratory-infected bee pupae. In a blind test, RT-PCR successfully identified the samples containing ABPV and BQCV. Sensitivities were approximately 1,600 genome equivalents of purified ABPV and 130 genome equivalents of BQCV.
URLPMID:21203504 [本文引用: 2]
[本文引用: 1]
DOI:10.1016/j.jip.2007.07.010URLPMID:17880997 [本文引用: 2]
Honey bee samples collected between 1995 and 2007 from 12 states were examined for the presence of Nosema infections. Our results showed that Nosema ceranae is a wide-spread infection of the European honey bee, Apis mellifera in the United States. The discovery of N. ceranae in bees collected a decade ago indicates that N. ceranae was transferred from its original host, Apis cerana to A. mellifera earlier than previously recognized. The spread of N. ceranae infection in A. mellifera warrants further epidemiological studies to identify conditions that resulted in such a widespread infection.
DOI:10.3864/j.issn.0578-1752.2019.01.015URL [本文引用: 1]
【目的】 微小RNA(microRNA,miRNA)是一类重要的基因表达调控因子,可影响宿主与病原间的互作过程。蜜蜂球囊菌(Ascosphaera apis)是一种特异性侵染蜜蜂幼虫的致死性真菌病原。本研究旨在对意大利蜜蜂(Apis mellifera ligustica,简称意蜂)幼虫肠道在球囊菌胁迫前期的差异表达miRNA(differentially expressed miRNA,DEmiRNA)及其靶基因进行深入分析,在miRNA组学水平探究意蜂幼虫在球囊菌侵染前期的胁迫应答,并通过构建显著DEmiRNA的调控网络筛选出与宿主应答相关的关键miRNA。【方法】 利用small RNA-seq(sRNA-seq)技术对正常及球囊菌侵染的意蜂4日龄幼虫肠道(AmCK和AmT)进行高通量测序,首先对原始数据进行质控和评估,随后将过滤后的数据与西方蜜蜂(Apis mellifera)参考基因组进行比对;将比对上的序列标签(tags)注释到miRBase数据库,得出已知miRNA的表达量;通过TPM(tags per million)算法对各样本中miRNA的表达量进行归一化处理,以|log2 fold change|≥1且P≤0.05作为标准筛选得到显著DEmiRNA;利用TargetFinder 软件预测显著DEmiRNA的靶基因,并对其进行GO和KEGG代谢通路(pathway)富集分析。利用Cytoscape软件对miRNA-mRNA调控网络进行可视化。最后,利用茎环反转录PCR(Stem-loop RT-PCR)和荧光定量PCR(qPCR)验证测序数据的可靠性。【结果】 AmCK和AmT样品的测序分别得到13 553 302和10 777 534条原始读段(raw reads),经严格过滤后得到的有效读段(clean reads)数分别为13 186 921和10 480 913条。各样品的生物学重复间的Pearson相关性系数分别在0.9822和0.9508以上。共有10个显著DEmiRNA,包括4个上调miRNA和6个下调miRNA。显著DEmiRNA在AmT的整体表达水平低于AmCK。10个显著DEmiRNA可靶向结合3 788个靶基因,其中上调miRNA的1 240个靶基因可注释到GO数据库中的39个GO条目,主要富集在结合、细胞进程、代谢进程和应激反应等;下调miRNA的749个靶基因可注释到34个GO条目,主要富集在细胞进程、结合、代谢进程和应激反应等。KEGG数据库注释结果显示,上调miRNA和下调miRNA的靶基因分别注释到95和66条代谢通路,富集基因数最多的分别是Wnt信号通路、Hippo信号通路、光传导以及内吞作用、磷脂酰肌醇信号系统、嘌呤代谢。对于上调和下调miRNA,分别有31和52个靶基因注释到内吞作用,15和7个靶基因注释到泛素介导的蛋白水解,11和5个靶基因注释到Jak-STAT信号通路,1和3个靶基因注释到MAPK信号通路。显著DEmiRNA与靶mRNA之间形成复杂的调控网络,7个显著DEmiRNA靶向结合96个与Wnt信号通路相关的mRNA,8个显著DEmiRNA靶向结合55个与内吞作用相关的mRNA。Stem-loop RT-PCR和qPCR结果验证了测序数据的可靠性。【结论】 对意蜂幼虫肠道在球囊菌侵染前期的DEmiRNA及其靶基因进行预测和分析,并构建和分析了DEmiRNA-mRNA调控网络,研究结果提供了宿主miRNA的表达谱和差异表达信息,揭示了DEmiRNA通过调控细胞生命活动、新陈代谢以及部分细胞和体液免疫等生物学过程参与宿主的胁迫应答。miR-4331-y、miR-4968-y、miR-8440-y、novel-m0023-5p和novel-m0025-3p共同参与了宿主的Wnt信号通路和内吞作用的调控,可作为白垩病治疗的潜在分子靶标。
DOI:10.3864/j.issn.0578-1752.2019.01.015URL [本文引用: 1]
【目的】 微小RNA(microRNA,miRNA)是一类重要的基因表达调控因子,可影响宿主与病原间的互作过程。蜜蜂球囊菌(Ascosphaera apis)是一种特异性侵染蜜蜂幼虫的致死性真菌病原。本研究旨在对意大利蜜蜂(Apis mellifera ligustica,简称意蜂)幼虫肠道在球囊菌胁迫前期的差异表达miRNA(differentially expressed miRNA,DEmiRNA)及其靶基因进行深入分析,在miRNA组学水平探究意蜂幼虫在球囊菌侵染前期的胁迫应答,并通过构建显著DEmiRNA的调控网络筛选出与宿主应答相关的关键miRNA。【方法】 利用small RNA-seq(sRNA-seq)技术对正常及球囊菌侵染的意蜂4日龄幼虫肠道(AmCK和AmT)进行高通量测序,首先对原始数据进行质控和评估,随后将过滤后的数据与西方蜜蜂(Apis mellifera)参考基因组进行比对;将比对上的序列标签(tags)注释到miRBase数据库,得出已知miRNA的表达量;通过TPM(tags per million)算法对各样本中miRNA的表达量进行归一化处理,以|log2 fold change|≥1且P≤0.05作为标准筛选得到显著DEmiRNA;利用TargetFinder 软件预测显著DEmiRNA的靶基因,并对其进行GO和KEGG代谢通路(pathway)富集分析。利用Cytoscape软件对miRNA-mRNA调控网络进行可视化。最后,利用茎环反转录PCR(Stem-loop RT-PCR)和荧光定量PCR(qPCR)验证测序数据的可靠性。【结果】 AmCK和AmT样品的测序分别得到13 553 302和10 777 534条原始读段(raw reads),经严格过滤后得到的有效读段(clean reads)数分别为13 186 921和10 480 913条。各样品的生物学重复间的Pearson相关性系数分别在0.9822和0.9508以上。共有10个显著DEmiRNA,包括4个上调miRNA和6个下调miRNA。显著DEmiRNA在AmT的整体表达水平低于AmCK。10个显著DEmiRNA可靶向结合3 788个靶基因,其中上调miRNA的1 240个靶基因可注释到GO数据库中的39个GO条目,主要富集在结合、细胞进程、代谢进程和应激反应等;下调miRNA的749个靶基因可注释到34个GO条目,主要富集在细胞进程、结合、代谢进程和应激反应等。KEGG数据库注释结果显示,上调miRNA和下调miRNA的靶基因分别注释到95和66条代谢通路,富集基因数最多的分别是Wnt信号通路、Hippo信号通路、光传导以及内吞作用、磷脂酰肌醇信号系统、嘌呤代谢。对于上调和下调miRNA,分别有31和52个靶基因注释到内吞作用,15和7个靶基因注释到泛素介导的蛋白水解,11和5个靶基因注释到Jak-STAT信号通路,1和3个靶基因注释到MAPK信号通路。显著DEmiRNA与靶mRNA之间形成复杂的调控网络,7个显著DEmiRNA靶向结合96个与Wnt信号通路相关的mRNA,8个显著DEmiRNA靶向结合55个与内吞作用相关的mRNA。Stem-loop RT-PCR和qPCR结果验证了测序数据的可靠性。【结论】 对意蜂幼虫肠道在球囊菌侵染前期的DEmiRNA及其靶基因进行预测和分析,并构建和分析了DEmiRNA-mRNA调控网络,研究结果提供了宿主miRNA的表达谱和差异表达信息,揭示了DEmiRNA通过调控细胞生命活动、新陈代谢以及部分细胞和体液免疫等生物学过程参与宿主的胁迫应答。miR-4331-y、miR-4968-y、miR-8440-y、novel-m0023-5p和novel-m0025-3p共同参与了宿主的Wnt信号通路和内吞作用的调控,可作为白垩病治疗的潜在分子靶标。
DOI:10.1371/journal.pone.0147549URLPMID:26840596 [本文引用: 6]
To clarify the mechanisms of Nosema ceranae parasitism, we deep-sequenced both honey bee host and parasite mRNAs throughout a complete 6-day infection cycle. By time-series analysis, 1122 parasite genes were significantly differently expressed during the reproduction cycle, clustering into 4 expression patterns. We found reactive mitochondrial oxygen species modulator 1 of the host to be significantly down regulated during the entire infection period. Our data support the hypothesis that apoptosis of honey bee cells was suppressed during infection. We further analyzed genome-wide genetic diversity of this parasite by comparing samples collected from the same site in 2007 and 2013. The number of SNP positions per gene and the proportion of non-synonymous substitutions per gene were significantly reduced over this time period, suggesting purifying selection on the parasite genome and supporting the hypothesis that a subset of N. ceranae strains might be dominating infection.
URLPMID:20622131 [本文引用: 1]
URLPMID:25914091 [本文引用: 1]
[本文引用: 2]
DOI:10.1038/35106579URLPMID:11719806 [本文引用: 1]
Microsporidia are obligate intracellular parasites infesting many animal groups. Lacking mitochondria and peroxysomes, these unicellular eukaryotes were first considered a deeply branching protist lineage that diverged before the endosymbiotic event that led to mitochondria. The discovery of a gene for a mitochondrial-type chaperone combined with molecular phylogenetic data later implied that microsporidia are atypical fungi that lost mitochondria during evolution. Here we report the DNA sequences of the 11 chromosomes of the approximately 2.9-megabase (Mb) genome of Encephalitozoon cuniculi (1,997 potential protein-coding genes). Genome compaction is reflected by reduced intergenic spacers and by the shortness of most putative proteins relative to their eukaryote orthologues. The strong host dependence is illustrated by the lack of genes for some biosynthetic pathways and for the tricarboxylic acid cycle. Phylogenetic analysis lends substantial credit to the fungal affiliation of microsporidia. Because the E. cuniculi genome contains genes related to some mitochondrial functions (for example, Fe-S cluster assembly), we hypothesize that microsporidia have retained a mitochondrion-derived organelle.
URLPMID:27230544 [本文引用: 1]
DOI:10.1093/abbs/gmv140URLPMID:26837419 [本文引用: 2]
Nosema bombycis is an obligate intracellular parasitic fungus that utilizes a distinctive mechanism to infect Bombyx mori. Germination, an indispensible process through which microsporidia infect the host cells, is regarded as a key developmental turning point for microsporidia from dormant state to reproduction state. Thus, elucidating the transcriptome changes before and after germination is crucial for parasite control. However, the molecular basis of germination of microsporidia remains unknown. To investigate this germination process, the transcriptome of N. bombycis ungerminated spores and germinated spores were sequenced and analyzed. More than 60 million high-quality transcript reads were generated from these two groups using RNA-Seq technology. After assembly, 2756 and 2690 unigenes were identified, respectively, and subsequently annotated based on known proteins. After analysis of differentially expressed genes, 66 genes were identified to be differentially expressed (P
[本文引用: 1]
[本文引用: 1]
URLPMID:11104818 [本文引用: 2]
[本文引用: 1]
[本文引用: 1]
DOI:10.13343/j.cnki.wsxb.20160551URL [本文引用: 1]
[Objective] RNA-seq technology was used to sequence the larval guts of Apis cerana cerana under stress of Ascosphaera apis. Subsequently, trend was analyzed for differentially expressed genes (DEGs) to obtain significant gene expression patterns, followed by transcriptome analysis of A. apis stressing the larval gut.[Methods] Infected honeybee larval guts were sequenced at Illumina HiSeq 2500 platform and in-depth analyses were done using corresponding biological software. Finally, RT-qPCR was conducted to validate RNA-seq data.[Results] A total of 41133932 high-quality clean reads were obtained. Trend analysis result showed that 22865 DEGs were grouped into 8 gene expression patterns, among them 16769 DEGs were assigned to 4 significant expression patterns including 2 up-regulated trends and 2 down-regulated trends. GO enrichment analysis result showed that all DEGs within significant up-and down-regulated patterns were enriched in 40 and 37 GO terms, respectively, and the mostly enriched one is cellular process (2486 unigenes). KEGG enrichment analysis result displayed that the DEGs within significant up-and down-regulated trends were enriched in 119 and 112 pathways, respectively, and biosynthesis of amino acids (127 unigenes) and ribosome (98 unigenes) were mostly enriched. A. apis facilitated its proliferation through enhancing the biosynthesis and the host could fight A. apis by inhibiting the protein synthesis of the fungal pathogen during the stress process. Furthermore, expression levels of 11 DEGs enriched in the pathogen's MAPK signaling pathway decreased when the stressing time of A. apis was prolonged, suggesting that A. c. cerana larvae could constrain the pathogen's replication by disturbing this pathway.[Conclusion] This is the first report of transcriptome investigation of A. apis infecting A. c. cerana larvae. Our data provide gene expression profiles of A. apis stressing the larval gut of A. c. cerana, as well lay the foundation of unraveling molecular mechanisms regulating the pathogenesis of A. apis.
DOI:10.13343/j.cnki.wsxb.20160551URL [本文引用: 1]
[Objective] RNA-seq technology was used to sequence the larval guts of Apis cerana cerana under stress of Ascosphaera apis. Subsequently, trend was analyzed for differentially expressed genes (DEGs) to obtain significant gene expression patterns, followed by transcriptome analysis of A. apis stressing the larval gut.[Methods] Infected honeybee larval guts were sequenced at Illumina HiSeq 2500 platform and in-depth analyses were done using corresponding biological software. Finally, RT-qPCR was conducted to validate RNA-seq data.[Results] A total of 41133932 high-quality clean reads were obtained. Trend analysis result showed that 22865 DEGs were grouped into 8 gene expression patterns, among them 16769 DEGs were assigned to 4 significant expression patterns including 2 up-regulated trends and 2 down-regulated trends. GO enrichment analysis result showed that all DEGs within significant up-and down-regulated patterns were enriched in 40 and 37 GO terms, respectively, and the mostly enriched one is cellular process (2486 unigenes). KEGG enrichment analysis result displayed that the DEGs within significant up-and down-regulated trends were enriched in 119 and 112 pathways, respectively, and biosynthesis of amino acids (127 unigenes) and ribosome (98 unigenes) were mostly enriched. A. apis facilitated its proliferation through enhancing the biosynthesis and the host could fight A. apis by inhibiting the protein synthesis of the fungal pathogen during the stress process. Furthermore, expression levels of 11 DEGs enriched in the pathogen's MAPK signaling pathway decreased when the stressing time of A. apis was prolonged, suggesting that A. c. cerana larvae could constrain the pathogen's replication by disturbing this pathway.[Conclusion] This is the first report of transcriptome investigation of A. apis infecting A. c. cerana larvae. Our data provide gene expression profiles of A. apis stressing the larval gut of A. c. cerana, as well lay the foundation of unraveling molecular mechanisms regulating the pathogenesis of A. apis.
URLPMID:20798817 [本文引用: 1]
DOI:10.1007/s00436-019-06279-wURLPMID:30863897 [本文引用: 1]
The secretion of hexokinases (HKs) by microsporidia followed by their accumulation in insect host nuclei suggests that these enzymes play regulatory and catalytic roles in infected cells. To confirm whether HKs exert catalytic functions in insect cells, we expressed in E. coli the functionally active HKs of two entomopathogenic microsporidia, Nosema bombycis and Nosema ceranae, that cause silkworm and honey bee nosematoses. N. bombycis HK with C-terminal polyHis tag and N. ceranae enzyme with N-terminal polyHis tag were cloned into pOPE101 and pRSET vectors, respectively, and overexpressed. Specific activities of N. bombycis and N. ceranae enzymes isolated by metal chelate affinity chromatography were 29.2 +/- 0.5 and 60.2 +/- 1.2 U/mg protein at an optimal pH range of 8.5-9.5. The kinetic characteristics of the recombinant enzymes were similar to those of HKs from other parasitic and free-living organisms. N. bombycis HK demonstrated Km 0.07 +/- 0.01 mM and kcat 1726 min(-1) for glucose, and Km 0.39 +/- 0.05 mM and kcat 1976 min(-1) for ATP, at pH 8.8. N. ceranae HK showed Km 0.3 +/- 0.04 mM and kcat 3293 min(-1) for glucose, and Km 1.15 +/- 0.11 mM and kcat 3732 min(-1) for ATP, at the same pH value. These data demonstrate the capability of microsporidia-secreted HKs to phosphorylate glucose in infected cells, suggesting that they actively mediate the effects of the parasite on host metabolism. The present findings justify further study of the enzymes as targets to suppress the intracellular development of silkworm and honey bee pathogens.
URLPMID:17557882 [本文引用: 2]
URLPMID:29522765 [本文引用: 1]
URLPMID:3288622 [本文引用: 1]
Ricin A-chain cleaves the N-glycosidic bond at A-4324 in 28 S rRNA when intact rat ribosomes are the substrate. Cleavage occurs at a concentration of the toxin of 1 X 10(-10) M, and specificity for this single residue is retained when the concentration is as high as 3 X 10(-7) M. The apparent Michaelis constant (Km) for the reaction is 2.6 microM, and the turnover number (Kcat) is 1777 min-1. The same N-glycosidic bond is cleaved by ricin A-chain in naked 28 S rRNA, but at a greatly reduced rate. The Km value for this reaction is 5.8 microM. The results suggest that the A-chain has a similar affinity for 28 S rRNA in ribosomes and in the absence of ribosomal proteins. Ricin A-chain has no effect on 23 S rRNA in Escherichia coli ribosomes, however, the N-glycosidic bond at A-2600 in naked 23 S rRNA is cleaved by the toxin; this corresponds to the ricin site in eukaryotic 28 S rRNA. Since the Km value (3.3 microM) for the reaction with E. coli 23 S rRNA approximates that obtained with rat liver ribosomes, it is possible that E. coli ribosomal protein(s) protect this site against ricin attack in intact ribosomes. Ricin A-chain also acted on naked 16 S rRNA cleaving the N-glycosidic bond of adenine at position 1014. The results suggest that ricin A-chain recognizes a specific structure in rRNA, perhaps a loop and stem having the sequence GAGA in the loop.
DOI:10.1093/abbs/gmw093URLPMID:27649890 [本文引用: 1]
Nosema bombycis is an obligate intracellular parasitic fungus that utilizes a distinctive mechanism to infect Bombyx mori Spore germination can be used for host cell invasion; however, the detailed mechanism remains to be elucidated. The ricin-B-lectin (RBL) gene is significantly differentially regulated after N. bombycis spore germination, and NbRBL might play roles in spore germination and infection. In this study, the biological function of NbRBL was examined. Protein sequence analysis showed that NbRBL is a secreted protein that attaches to carbohydrates. The relative expression level of the NbRBL gene was low during the first 30 h post-infection (hpi) in BmN cells, and high expression was detected from 42 hpi. Gene cloning, prokaryotic expression, and antibody preparation for NbRBL were performed. NbRBL was detected in total and secreted proteins using western blot analysis. Subcellular localization analysis showed that NbRBL is an intracellular protein. Spore adherence and infection assays showed that NbRBL could enhance spore adhesion to BmN cells; the proliferative activities of BmN cells incubated with anti-NbRBL were higher than those in negative control groups after N. bombycis infection; and the treatment groups showed less damage from spore invasion. We therefore, propose that NbRBL is released during spore germination, enhances spore adhesion to BmN cells, and contributes to spore invasion.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}