The Potential Role of MicroRNAs and MicroRNA-Mediated Competing Endogenous Networks During the Developmental Process of Apis mellifera ligustica Worker’s Midgut
DU Yu, FAN XiaoXue, JIANG HaiBin, WANG Jie, FAN YuanChan, ZHU ZhiWei, ZHOU DingDing, WAN JieQi, LU JiaXuan, XIONG CuiLing, ZHENG YanZhen, CHEN DaFu, GUO RuiCollege of Animal Sciences (College of Bee Science), Fujian Agriculture and Forestry University, Fuzhou 350002通讯作者:
责任编辑: 杨鑫浩
收稿日期:2019-10-27网络出版日期:2020-06-16
| 基金资助: |
Received:2019-10-27Online:2020-06-16
作者简介 About authors
杜宇,E-mail:m18505700830@163.com。
范小雪,E-mail:imfanxx@163.com。

摘要
关键词:
Abstract
Keywords:
PDF (3681KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
杜宇, 范小雪, 蒋海宾, 王杰, 范元婵, 祝智威, 周丁丁, 万洁琦, 卢家轩, 熊翠玲, 郑燕珍, 陈大福, 郭睿. 微小RNA及其介导的竞争性内源RNA调控网络在意大利蜜蜂工蜂中肠发育过程中的潜在作用[J]. 中国农业科学, 2020, 53(12): 2512-2526 doi:10.3864/j.issn.0578-1752.2020.12.017
DU Yu, FAN XiaoXue, JIANG HaiBin, WANG Jie, FAN YuanChan, ZHU ZhiWei, ZHOU DingDing, WAN JieQi, LU JiaXuan, XIONG CuiLing, ZHENG YanZhen, CHEN DaFu, GUO Rui.
0 引言
【研究意义】意大利蜜蜂(Apis mellifera ligustica,简称意蜂)是具有重要生态、经济和科研价值的社会性昆虫,广泛应用于我国和其他养蜂国家的养蜂生产[1]。成年蜜蜂的肠道分为前肠、中肠和后肠,其中中肠是食物消化、营养吸收以及免疫防御的主要部位,其内壁富含几丁质的围食膜具有分子筛功能,可保护中肠抵御病原侵染以及促进营养成分吸收[2,3]。微小RNA(microRNA,miRNA)是一类高度保守的长度约为18—25 nt的内源性单链非编码RNA(non-coding RNA,ncRNA)[4],主要通过特异性结合靶mRNA的3′ UTR,导致靶mRNA的翻译抑制或降解,从而在转录后水平行使基因表达调控功能[5]。较多的研究结果表明miRNA广泛参与昆虫生长发育和免疫防御等生物学过程[6,7]。目前,蜜蜂肠道发育的分子机理尚不明确,ncRNA在蜜蜂肠道发育过程中的作用研究还很有限。对意蜂工蜂中肠发育过程的miRNA差异表达谱、调控网络及其差异表达miRNA(differentially expressed miRNA,DEmiRNA)的潜在作用进行全面分析和探讨,可在miRNA组学层面进一步揭示意蜂工蜂中肠发育的分子机理。【前人研究进展】1993年,LEE等[8]首次报道lin-4在秀丽隐杆线虫(Caenorhabditis elegans)体内具有调节生长发育的作用。近十年来,随着高通量测序技术的飞速发展,越来越多的miRNA在动物、植物和微生物中被鉴定出来[9,10,11]。但相比于果蝇(Drosophila melanogaster)[12]等模式生物,蜜蜂的miRNA研究相对滞后,涉及miRNA参与调控蜜蜂肠道发育的研究尤为缺乏。LIU等[13]研究发现与神经发育相关的miR-31a、ame-miR-210和ame-miR-278等9个miRNA在意蜂哺育蜂与采集蜂头部的差异表达,并推测它们可能影响蜜蜂的劳动分工过程;SHI等[14]研究发现ame-let-7、ame-miR-13b和ame-miR-279在意蜂工蜂脑部的表达具有显著的时空特异性,且长度为22 nt的miRNA在蜂王与工蜂体内的表达量存在显著差异,作者推测这些miRNA参与调控与蜜蜂级型分化相关的信号通路;LOUREN?O等[15]研究发现,意蜂工蜂被革兰氏阳性菌藤黄微球菌(Micrococcus luteus)和革兰氏阴性菌粘质沙雷氏菌(Serratia marcescens)感染后miR-137等38个miRNA差异表达,并与Toll、Imd、JNK和Jak-STAT通路相关的25个mRNA之间存在潜在的调控关系,影响抗菌肽合成和黑化作用激活等免疫防御过程。2011年,SALMENA等[16]首次提出竞争性内源RNA(competing endogenous RNA,ceRNA)假说,认为含有miRNA应答元件(miRNA response element,MRE)的RNA,如长链非编码RNA(long non-coding RNA,lncRNA)、环状RNA(circular RNA,circRNA)和假基因转录本等,可作为ceRNA竞争性结合miRNA,从而间接影响mRNA的表达。此后,该假说已被越来越多的研究结果[17,18]所证实,WANG等[18]研究发现自噬细胞自噬促进因子lncRNA-APF可直接结合miR-188-3p抑制其活性,从而间接调节ATG7的表达以调控自噬程序和自噬细胞死亡。前期研究中,笔者所在课题组运用二代测序技术及生物信息学方法系统解析了意蜂幼虫肠道发育过程的DEmiRNA表达谱及调控网络[19],揭示了miR-342-y、ame-miR-6052、miR-iab-4-x、miR-281-x、novel-m0031-3p等DEmiRNA可能在幼虫肠道发育中发挥重要的调控作用;此外,还相继解析了意蜂工蜂中肠发育过程的差异表达lncRNA(differentially expressed lncRNA,DElncRNA)表达谱、调控网络及潜在作用[20],差异表达circRNA(differentially expressed circRNA,DEcircRNA)表达谱、调控网络及潜在功能[21],DEmRNA表达谱及ceRNA网络[22],在组学层面深入细致探讨了mRNA和ncRNA介导的中肠发育机理,并筛选出若干功能研究的候选分子。【本研究切入点】根据ceRNA机制,miRNA作为联系mRNA与lncRNA和circRNA等ncRNA的桥梁,在调控网络中具有核心地位。笔者所在课题组前期已在mRNA组学、lncRNA组学和circRNA组学层面对意蜂工蜂中肠发育机理进行了探究,本研究在此基础上进一步对中肠发育过程miRNA差异表达谱、DEmiRNA介导的ceRNA调控网络,以及DEmiRNA的潜在功能进行深入分析,从而将mRNA和ncRNA联系起来,展示意蜂工蜂中肠发育过程的ceRNA调控网络全貌。【拟解决的关键问题】利用small RNA-seq(sRNA-seq)技术、生物信息学方法和分子生物学手段对意蜂工蜂中肠发育过程DEmiRNA介导的ceRNA调控网络及潜在作用进行分析和验证,进而与前期在DEmRNA、DElncRNA和DEcircRNA组学层面的研究结果进行比较分析和探讨,以期在组学水平揭示DEmiRNA及复杂调控网络介导的中肠发育机理,为在分子水平阐明意蜂工蜂中肠的发育机理打下基础。1 材料与方法
试验于2017年9月至2019年10月在福建农林大学动物科学学院(蜂学学院)蜜蜂保护实验室完成。1.1 供试生物材料
供试意蜂工蜂取自福建农林大学动物科学学院(蜂学学院)教学蜂场。1.2 中肠样品制备与Illumina测序
按照笔者实验室前期已建立的方法[20,21,22]进行意蜂的人工饲养。(1)从群势较强且显微镜检无东方蜜蜂微孢子虫的3个蜂群中提取老熟封盖子脾至实验室恒温培养箱(34±0.5)℃;(2)每30 min将刚出房的工蜂(记为0 d)放入四周打孔且已消毒的干净塑料盒(35只/盒),每个塑料盒上方插入一支装有50%(w/v)无菌糖水的饲喂器;每日检查工蜂存活情况,及时清理死亡工蜂;(3)待工蜂出房7 d和10 d时,在超净台用干净镊子拉取工蜂中肠,放入灭菌后的RNA-Free的EP管,经液氮速冻后迅速转移至-80℃超低温冰箱保存备用,每只中肠的取样时间严格控制在15 s以内。进行3次生物学重复,每个生物学重复包含3只中肠样品。7 d工蜂中肠样品(Am7)的3个生物学重复:Am7-1、Am7-2和Am7-3;10 d工蜂中肠样品(Am10)的3个生物学重复:Am10-1、Am10-2和Am10-3。文库构建方法如下:用Trizol法从上述样本中提取total RNA,琼脂糖凝胶电泳切胶选择18—30 nt的片段。然后连接3′接头,连接产物以15%变性PAGE胶电泳分离,切胶选择36—44 nt目的条带。回收切胶产物,连接5′接头,然后对连接了两侧接头的小RNA样本进行反转录PCR。反转录产物以3.5%琼脂糖凝胶电泳分离,切胶选择140—160 bp区域条带,胶回收产物即为终文库。建好的文库委托广州基迪奥生物技术有限公司进行测序,测序平台为Illumina MiSeq。测序数据已上传NCBI SRA数据库,BioProject号:PRJNA408312。1.3 测序数据质控、参考基因组比对及miRNA的差异表达分析
对于下机的原始读段(raw reads),按照前期已建立的方法[19,23]步骤进行质量控制:(1)过滤掉质量值<20的碱基数超过1个的reads;(2)过滤除掉含有未知碱基(N)的reads;(3)过滤3′或5′接头的reads,并去除长度<18 bp的reads;(4)过滤包含poly A的reads。过滤得到的clean reads用于后续分析。利用Bowit软件将非注释序列标签(tags)与西方蜜蜂基因组(assembly Amel 4.5)比对,得到相关tags对应的位置信息,即mapped tags。使用miRDeep2软件将mapped tags与miRBase数据库中已知的miRNA前体序列进行比对,鉴定已知miRNA的表达。利用每百万标签序列(tags per million,TPM)算法公式(TPM=T×106/N,T表示miRNA的tags,N表示总miRNA的tags)对表达量进行归一化处理。使用R软件计算意蜂工蜂中肠样品不同生物学重复之间的Pearson相关性系数。显著性DEmiRNA的筛选标准为|log2fold change|≥1且P≤0.05。
1.4 DEmiRNA的靶mRNA预测及分析
通过TargetFinder软件对DEmiRNA进行靶向预测。利用Blast软件将上述靶mRNA序列分别映射GO(Gene Ontology)和KEGG(Kyoto Encyclopedia of Genes and Genomes)数据库。结合本课题组前期研究结果[20,21,22],得到DEmiRNA与DEmRNA、DElncRNA和DEcircRNA的靶向结合关系,并根据DEmiRNA与DEmRNA的靶向结合关系筛选自由能≤-20 kcal·mol-1的靶mRNA,进行KEGG数据库注释,再通过比对前期在差异基因方面的研究结果[22]找到相同的代谢通路,利用Cytoscape软件对DEmiRNA靶向的DElncRNA、DEcircRNA以及通路中的DEmRNA进行调控网络的构建及可视化。1.5 DEmiRNA的茎环反转录实时荧光定量PCR(Stem- loop RT-qPCR)验证
参考前期已建立的方法[19,23-24],通过Stem-loop RT-qPCR检测4个随机挑选的DEmiRNA(miR-210-z、miR-342-y、miR-7975-y和miR-155-x)在Am7和Am10中的表达情况,以验证数据的可靠性。通过DNAMAN软件设计特异性引物和通用反向引物,委托上海生工生物工程有限公司合成引物,引物信息详见表1。以snRNA U6作为内参,使用M5 microRNA抽提试剂盒(Mei5bio公司,中国)分别提取Am7和Am10的miRNA作为模板,利用Stem-loop引物进行反转录得到相应的cDNA后,以cDNA作为模板进行qPCR。qPCR反应按照SYBR Green Dye试剂盒(Vazyme公司,中国)操作说明书进行。反应体系20 μL包含SYBR Green Dye 10 μL,cDNA模板1.3 μL,正、反向引物各1 μL,DEPC水6.7 μL。qRT-PCR反应在ABI QuantStudio 3荧光定量PCR仪(ABI,美国)上进行,程序设置为95℃预变性5 min;95℃变性10 s,60℃退火延伸30 s,共40个循环。每个反应进行3次生物学重复和技术重复。利用2-ΔΔCt法计算miRNA相对表达量。利用Graph Prism 7软件处理数据和绘图,通过t检验计算组间显著性。Table 1
表1
表1RT-qPCR引物信息
Table 1
| 引物名称Primer name | 引物序列Primer sequence (5′-3′) |
|---|---|
| Loop-miR-210-z | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGACAGCCGC |
| F-miR-210-z | ACACTCCAGCTGGGCTGTGCGTGTGACA |
| Loop-miR-342-y | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGACGGGTGC |
| F-miR-342-y | ACACTCCAGCTGGGTCTCACACAGAAATC |
| Loop-miR-7975-y | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTGGTGCCG |
| F-miR-7975-y | ACACTCCAGCTGGGATCCTGGTCA |
| Loop-miR-155-x | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGAACCCCTA |
| F-miR-155-x | ACACTCCAGCTGGGTTAATGCTAATTGTGA |
| R | CTCAACTGGTGTCGTGGA |
| U6-F | GTTAGGCTTTGACGATTTCG |
| U6-R | GGCATTTCTCCACCAGGTA |
新窗口打开|下载CSV
2 结果
2.1 意蜂工蜂中肠测序数据的质控与评估
Am7和Am10的sRNA-seq分别得到13 511 614条和13 448 175条raw reads,严格过滤后分别得到13 119 002条和13 010 751条clean reads,占raw reads的比例均≥96.13%(表2)。此外,Am7和Am10的组内各生物学重复之间的Pearson相关性系数均达到0.9870和0.9988以上(图1)。上述结果说明本研究中样本重复性和数据质量较好,可用于进一步分析。Table 2
表2
表2sRNA-seq数据总览
Table 2
| 样品 Sample | 原始读段 Raw reads | 有效读段 Clean reads |
|---|---|---|
| Am7-1 | 14021728 | 13619226 (97.13%) |
| Am7-2 | 13479635 | 13081146 (97.04%) |
| Am7-3 | 13033478 | 12656635 (97.11%) |
| Am10-1 | 13083648 | 12577875 (96.13%) |
| Am10-2 | 13544596 | 13137501 (96.99%) |
| Am10-3 | 13716280 | 13316876 (97.09%) |
新窗口打开|下载CSV
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1各意蜂工蜂中肠样品不同生物学重复间的Pearson相关性系数
Fig. 1Pearson correlation coefficients among different biological replicas within each A. m. ligustica worker’s midgut sample group
2.2 意蜂工蜂中肠发育过程的miRNA差异表达谱
差异表达分析结果显示,Am7 vs Am10比较组共含有112个显著性DEmiRNA,包括38个显著上调miRNA和74个显著下调miRNA。其中,上调倍数最高的是miR-7132-y,其次是miR-2188-x和miR-1388-x(表3);而下调倍数最高的是miR-8503-x,其次是miR-298-x和miR-291-y(表4)。Table 3
表3
表3意蜂工蜂中肠发育过程中显著上调前10个miRNA
Table 3
| 差异表达miRNA ID DEmiRNA ID | 以2为底miRNA的相对变化倍数的对数值 log2fold change | P值 P value |
|---|---|---|
| miR-7132-y | 13.27 | 8.68E-09 |
| miR-2188-x | 12.72 | 2.31E-06 |
| miR-1388-x | 12.43 | 6.78E-06 |
| miR-1338-x | 12.38 | 7.39E-06 |
| miR-1388-y | 11.76 | 4.20E-04 |
| miR-1338-y | 11.67 | 9.18E-04 |
| miR-7132-x | 11.16 | 9.01E-03 |
| miR-3964-y | 11.09 | 3.37E-03 |
| miR-7935-y | 11.01 | 4.31E-03 |
| miR-6537-x | 10.46 | 0.023 |
新窗口打开|下载CSV
Table 4
表4
表4意蜂工蜂中肠发育过程中显著下调前10个miRNA
Table 4
| 差异表达miRNA ID DEmiRNA ID | 以2为底miRNA的相对变化倍数的对数值 log2fold change | P值 P value |
|---|---|---|
| miR-8503-x | -14.09 | 3.09E-04 |
| miR-298-x | -13.64 | 2.65E-08 |
| miR-291-y | -12.94 | 8.67E-05 |
| miR-292-y | -12.75 | 3.40E-04 |
| miR-4700-x | -12.55 | 0.015 |
| miR-293-y | -12.27 | 6.77E-04 |
| miR-2965-y | -12.13 | 0.030 |
| miR-2518-y | -12.13 | 6.13E-03 |
| miR-300-y | -12.03 | 2.96E-04 |
| miR-4577-y | -12.01 | 0.038 |
新窗口打开|下载CSV
2.3 意蜂工蜂中肠发育过程中DEmiRNA的靶mRNA预测及分析
利用TargetFinder软件对上述显著上调和显著下调miRNA进行靶向预测,分别预测出7 434和9 559个靶mRNA,并分别涉及47和51个功能条目。其中注释靶mRNA数前15位的条目均为结合(2 082和2 632)、细胞进程(2 074和2 526)、代谢进程(1 767和2 270)、单组织进程(1 588和1 957)、催化活性(1 288和1 720)、细胞(916和1 130)、细胞组件(916和1130)、细胞膜(913和1 098)、细胞膜组件(893和1 076)、细胞器(681和863)、生物学调控(655和775)、生物进程调控(601和729)、定位(565和713)、应激反应(556和679)、信号(494和615)(图2)。括号内数字代表注释在该条目的显著上调(下调)miRNA的靶mRNA数。图2
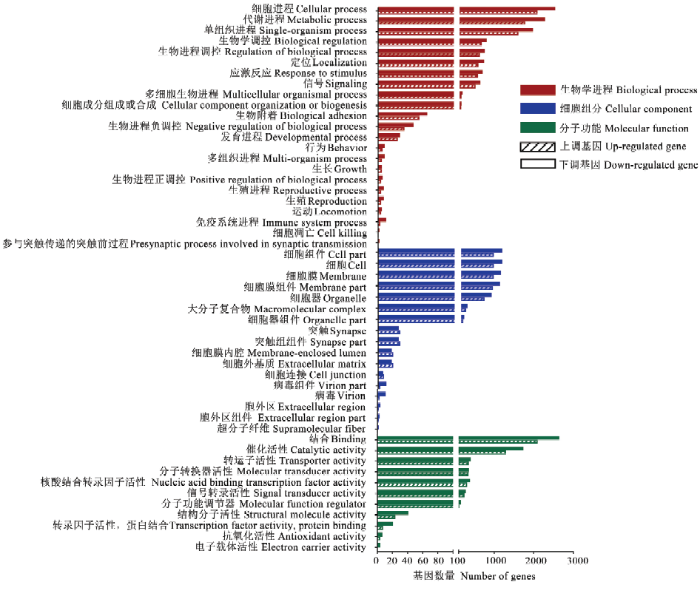 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2意蜂工蜂中肠发育过程DEmiRNA的GO数据库注释
Fig. 2GO database annotation of DEmiRNA target mRNAs involved in the developmental process of A. m. ligustica worker’s midgut
进一步对上述DEmiRNA的靶mRNA进行KEGG数据库注释,结果显示显著上调miRNA的靶mRNA可注释到315条通路,显著下调miRNA的靶mRNA可注释到324条通路。对于显著上调miRNA和显著下调miRNA,分别有118和134个靶mRNA注释到Hippo信号通路,155和190个靶mRNA注释到Wnt信号通路,58和63个靶mRNA注释到FoxO信号通路,30和41个靶mRNA注释到Notch通路;分别有98和109个靶mRNA注释到内吞作用,74和70个靶mRNA注释到泛素介导的蛋白水解,37和32个靶mRNA注释到溶酶体,111和117个靶mRNA注释到黑色素生成等细胞免疫通路;分别有17和22个靶mRNA注释到Jak-STAT信号通路,8和10个靶mRNA注释到NF-κB信号通路,8和18个靶mRNA注释到Toll/Imd信号通路,25和53个靶mRNA注释到MAPK信号通路等体液免疫通路。表5和表6分别展示了显著上调miRNA和显著下调miRNA显著富集的前15位通路。
Table 5
表5
表5意蜂工蜂中肠上调miRNA的靶mRNA显著富集的前15位通路
Table 5
| 通路 Pathway | 通路ID Pathway ID | 靶mRNA富集数 Number of target mRNAs | P值 P value |
|---|---|---|---|
| 钙信号通道 Calcium signaling pathway | ko04020 | 77 | 1.30E-14 |
| 催产素信号通路 Oxytocin signaling pathway | ko04921 | 82 | 1.07E-14 |
| 坏死性凋亡 Necroptosis | ko04217 | 58 | 5.72E-16 |
| 嗅觉传导 Olfactory transduction | ko04740 | 50 | 3.01E-17 |
| 长时程增强 Long-term potentiation | ko04720 | 70 | 3.98E-18 |
| Hippo信号通路 Hippo signaling pathway | ko04390 | 118 | 1.29E-18 |
| 促性腺激素释放激素信号通路 GnRH signaling pathway | ko04912 | 79 | 4.76E-19 |
| 神经营养因子信号通路 Neurotrophin signaling pathway | ko04722 | 89 | 3.17E-21 |
| 胆碱能突触 Cholinergic synapse | ko04725 | 79 | 2.90E-21 |
| 胃酸分泌 Gastric acid secretion | ko04971 | 69 | 2.05E-21 |
| 醛固酮合成与分泌 Aldosterone synthesis and secretion | ko04925 | 74 | 1.94E-21 |
| cAMP信号通路 cAMP signaling pathway | ko04024 | 113 | 1.73E-21 |
| 昼夜节律调节 Circadian entrainment | ko04713 | 75 | 2.93E-23 |
| 胰岛素分泌 Insulin secretion | ko04911 | 78 | 2.53E-23 |
| ErbB信号通路 ErbB signaling pathway | ko04012 | 80 | 2.43E-25 |
新窗口打开|下载CSV
Table 6
表6
表6意大利蜜蜂工蜂中肠下调miRNA的靶mRNA显著富集的前15位通路
Table 6
| 通路 Pathway | 通路ID Pathway ID | 靶mRNA富集数 Number of target mRNAs | P值 P value |
|---|---|---|---|
| Hippo信号通路 Hippo signaling pathway | ko04390 | 134 | 6.20E-17 |
| 醛固酮合成与分泌 Aldosterone synthesis and secretion | ko04925 | 78 | 1.79E-17 |
| 胆碱能突触 Cholinergic synapse | ko04725 | 84 | 1.71E-17 |
| 卵母细胞减数分裂 Oocyte meiosis | ko04114 | 103 | 8.04E-19 |
| 昼夜节律调节 Circadian entrainment | ko04713 | 79 | 3.12E-19 |
| 胃酸分泌 Gastric acid secretion | ko04971 | 75 | 1.66E-19 |
| 神经营养因子信号通路 Neurotrophin signaling pathway | ko04722 | 99 | 1.20E-19 |
| 胰岛素分泌 Insulin secretion | ko04911 | 83 | 9.94E-20 |
| 促性腺激素释放激素信号通路 GnRH signaling pathway | ko04912 | 91 | 9.12E-20 |
| 长时程增强 Long-term potentiation | ko04720 | 82 | 4.72E-20 |
| 钙信号通道 Calcium signaling pathway | ko04020 | 101 | 3.09E-22 |
| ErbB信号通路 ErbB signaling pathway | ko04012 | 87 | 3.11E-23 |
| cAMP信号通路 cAMP signaling pathway | ko04024 | 134 | 1.19E-23 |
| 炎症介质对TRP通道的调节Inflammatory mediator regulation of TRP channels | ko04750 | 97 | 9.4E-26 |
| 黑色素生成 Melanogenesis | ko04916 | 117 | 9.23E-28 |
新窗口打开|下载CSV
2.4 意蜂工蜂中肠发育过程中DEmiRNA调控网络的构建及分析
前期研究中,为深入分析意蜂工蜂中肠的发育机理,笔者所在课题组选取13条信号通路(AMPK、PI3K-Akt、Wnt、cAMP、FoxO、Hippo、mTOR、Jak-STAT、Toll-like受体、TGF-beta、Notch、MAPK和NF-κB)及各通路富集DEmRNA构建富集关系网络[22]。在此基础上,根据序列匹配原则,利用软件预测DEmiRNA与前期研究中DElncRNA[20]、DEcircRNA[21]、DEmRNA[22]的靶向结合关系,构建9条通路相关的调控网络,分析结果显示DEmiRNA与DEmRNA、DEcircRNA和DElncRNA之间存在复杂的调控关系,DEmiRNA居于网络的中心位置,而DElncRNA、DEcircRNA和DEmRNA处于网络外周;此外miR-5106-y能够靶向结合3个DElncRNA和2个DEcircRNA(图3)。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3意蜂工蜂中肠发育过程中9条信号通路相关的DElncRNA/DEcircRNA-DEmiRNA-DEmRNA关系网络
Fig. 3Relationship network of DElncRNA/DEcircRNA-DEmiRNA-DEmRNA associated with 9 signaling pathways in the midgut of A. m. ligustica worker
进一步分析发现,一个DEmiRNA靶向的DEmRNA可注释到多条信号通路,例如miR-291-y靶向的糖原合成酶激酶-3β亚型编码基因(XM_006567354.2)可注释到Wnt、PI3K-Akt和mTOR信号通路,miR- 376-y靶向的环盒子亚型编码基因(XM_016910739.1)可注释到Wnt和TGF-beta信号通路,miR-148-y靶向的非特性蛋白LOC411962编码基因(XM_006567604.2)可注释到MAPK和Toll/Imd信号通路,miR-5106-y靶向的丝氨酸/苏氨酸-蛋白磷酸酶-β亚基-ε编码基因(XM_016912389.1)可注释到PI3K-Akt和AMPK信号通路。此外,同一个DEmiRNA靶向的多个DEmRNA富集在同一条信号通路,例如ame-miR-6001-3p靶向的3个DEmRNA(XM_006562526.2、XM_016914568.1和XM_016914811.1)可注释到Hippo信号通路;miR-126-x靶向的2个DEmRNA(XM_006557903.2和XM_006557904.2)均注释到cAMP信号通路(图3)。
miR-342-y在意蜂工蜂中肠发育过程中呈显著性上调表达,与其在意蜂幼虫肠道发育过程的表达趋势[19]相反。进一步对miR-342-y及与其存在靶向结合关系的DElncRNA、DEcircRNA和DEmRNA进行预测和分析,结果显示miR-342-y可靶向结合3个DEcircRNA、4个DElncRNA和327个靶mRNA,上述靶mRNA可注释到松弛素信号通路(15)、趋化因子信号通路(13)、HIF-1信号通路(12)、神经活性配体-受体相互作用(10)、内吞作用(10)、碳水化合物的消化吸收(8)、雌激素信号通路(8)、腹背轴形成(8)和TRP通道的炎性介质调节(8)等162条通路(图4)。
图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4意蜂工蜂中肠的miR-342-y靶向结合的10条信号通路相关DElncRNA、DEcircRNA、DEmRNA关系网络
六边形代表信号通路 Hexagons indicate signaling pathways;红色三角形代表miRNA Red triangle indicates miRNA;黄色圆形代表DEcircRNA Yellow circles indicate DEcircRNAs;绿色平行四边形代表DElncRNA Green parallelograms indicate DElncRNAs;蓝色菱形代表mRNAs Blue diamonds indicate mRNAs;六边形和菱形的大小代表连接mRNA和信号通路的数量 The size of hexagon and diamond indicates the number of connected DEmRNAs or signaling pathways
Fig. 4Relationship network of miR-342-y and its target DElncRNAs, target DEcircRNAs, target DEmRNAs associated with 10 signaling pathways in the midgut of A. m. ligustica worker
2.5 意蜂工蜂中肠发育过程中DEmiRNA的Stem-loop RT-qPCR验证
利用Stem-loop RT-qPCR对miR-210-z、miR-342-y、miR-7975-y和miR-155-x进行表达量检测,结果显示它们的差异变化趋势与转录组测序中相应的表达量变化趋势一致(图5),证明了本研究中miRNA差异表达及测序数据的真实可靠性。图5
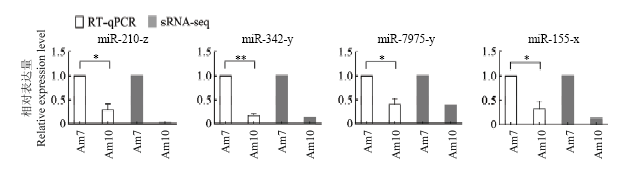 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5DEmiRNA的RT-qPCR验证
Fig. 5RT-qPCR validation of DEmiRNAs
3 讨论
昆虫肠道不仅是消化食物和吸收营养的主要场所,也是阻挡病原入侵血淋巴及其他器官的重要防线[25]。目前,有关成年蜜蜂肠道的研究主要集中在肠道微生物的结构特征和功能预测方面[26,27],肠道发育机理的相关研究极其有限。以Illumina为代表的二代测序技术近年来发展迅速,在miRNA等ncRNA的高通量挖掘方面表现出突出的优越性[28,29,30]。前期研究中,笔者所在课题组对意蜂工蜂响应微孢子虫胁迫的应答机制进行了lncRNA、miRNA和mRNA层面的全面解析[28,30-31];并在lncRNA、circRNA和mRNA组学层面探究了意蜂工蜂中肠的发育机理,系统解析了DElncRNA、DEcircRNA和DEmRNA表达谱及三者涉及的调控网络[20,21,22]。miRNA作为基因表达调控的关键因子,能够作为桥梁将编码RNA与非编码RNA联系起来。为进一步深入探究意蜂工蜂中肠发育的分子机理,本研究结合测序得到的意蜂工蜂中肠sRNA组学数据和前期获得的中肠circRNA、lncRNA和mRNA组学数据,对miRNA的差异表达谱及调控网络进行了深入分析和探讨。蜜蜂是一种完全变态昆虫,历经卵期、幼虫期、蛹期和成虫期4个阶段,其中,1—2日龄幼虫太小,直接剖取肠道技术上无法满足。前期研究中,笔者所在课题组从自然蜂群中移取2日龄意蜂幼虫至48孔培养板,经24 h的环境适应后,于3日龄时处理组幼虫饲喂接种球囊菌孢子,对照组幼虫饲喂不含球囊菌孢子的饲料,然后于4、5和6日龄分别剖取处理组和对照组幼虫肠道用于sRNA-seq;基于高质量的sRNA组学数据全基因组鉴定和分析了意蜂幼虫肠道miRNA及其结构特征[9],并解析了意蜂幼虫肠道发育过程的miRNA差异表达谱、调控网络及潜在功能[19]。蜜蜂预蛹期,内部的组织经历分解和重构,逐渐形成新的组织,此阶段无法获取完整肠道。本研究仅选取意蜂7日龄和10日龄工蜂中肠进行sRNA-seq,因为这两个时间点均处于意蜂成虫期,且本研究的分析和讨论未涉及变态发育。从Am7 vs Am10比较组中筛选出38个显著上调miRNA和74个显著下调miRNA,可分别靶向7 434和9 559个mRNA,通过与circRNA、lncRNA和mRNA的相互作用参与调节意蜂中肠发育过程中的各类生命活动。
3.1 DEmiRNA参与调控意蜂中肠发育过程的新陈代谢、信号传导和免疫应答
蜜蜂肠道在消化和吸收等正常生命活动中伴随着活跃的物质和能量代谢。本研究发现,显著上调和下调miRNA靶向结合的mRNA均可注释到嘌呤代谢(81和104个靶mRNA)、甘油磷脂代谢(49和52个靶mRNA)、醚脂质代谢(13和22个靶mRNA)等脂代谢相关通路;果糖和甘露糖代谢(23和23个靶mRNA)、磷酸肌醇代谢(43和64个靶mRNA)等碳水化合物代谢相关通路;赖氨酸降解(13和21个靶mRNA)、甘氨酸、丝氨酸和苏氨酸代谢(11和17个靶mRNA)等氨基酸代谢相关通路。此外,显著上调和下调miRNA靶向结合的mRNA还能注释到氮代谢(2和6个靶mRNA)和氧化磷酸化(22和25个靶mRNA)等多条能量代谢相关通路。上述结果表明意蜂工蜂中肠发育过程中可通过DEmiRNA调节部分物质代谢和能量代谢通路及相关基因的表达,以满足中肠生长和发育的物质和能量需求。昆虫肠道在生长发育过程中受到Hippo、Wnt、FoxO和Notch等信号通路的共同调节[32]。中肠干细胞能够分化为具有吸收功能的肠上皮细胞以及分泌功能的肠内分泌细胞,保证肠组织的自我更新和损伤修复,从而维持正常的形状和功能,而Notch、Hippo和Wnt信号通路在中肠干细胞的稳态平衡、更新分化过程中发挥重要作用[33,34]。Notch信号通路在果蝇胚胎期中肠发育过程中调节成体中肠祖细胞[35],在成年期调控中肠干细胞以维持中肠内环境稳态[36,37]。Wnt信号通路通过控制果蝇成年个体肠道的腔室内干细胞增殖以及调控腔室边界细胞正常发展,从而在内环境平衡和发育过程中发挥重要作用[38]。上皮细胞Hippo信号的缺失会增加Upd家族细胞因子的产生,并且刺激EGFR配体,进而激活Jak-STAT和EGFR信号通路并促进果蝇中肠干细胞的增殖[39]。昆虫蜕皮是生长发育的重要生理过程,而FoxO信号通路中的FoxO通过调控CPA的表达参与调控昆虫的蜕皮行为[40]。本研究中,对于显著上调和显著下调miRNA,分别有118和134个靶mRNA注释到Hippo信号通路,155和190个靶mRNA注释到Wnt信号通路,58和63个靶mRNA注释到FoxO信号通路,30和41个靶mRNA注释到Notch通路。相应的DEmiRNA通过上调或下调部分基因的表达水平对上述发育相关信号通路进行调控,从而影响意蜂工蜂中肠的生长和发育以及细胞分化。
昆虫中肠作为与外源病原体互作的重要场所,可通过分泌活性氧、抗菌肽以及各类蛋白酶协助宿主抵御病原侵染[25]。蜜蜂在与病原长期协同进化过程中,形成了以内吞作用、吞噬作用、黑化作用和蛋白酶促水解为代表的细胞免疫,以及Jak-STAT、NF-κB、Imd/Toll、MAPK和JNK等信号通路和抗菌肽释放为代表的体液免疫[41]。前人研究发现Jak-STAT信号通路作为昆虫先天免疫系统的重要组成部分,在病原侵染的过程中可被显著激活并广泛参与细胞凋亡、免疫调节等过程[42]。Toll/Imd信号通路和NF-κB信号通路共同介导了抗菌肽的合成与释放过程,并协同吞噬和包埋作用参与昆虫的应激免疫反应[43,44]。JNK途径在内的MAPK级联反应的激活,能够促进昆虫免疫的调节过程[45,46]。本研究发现,对于显著上调和下调的miRNA,分别有98和109个靶mRNA注释到内吞作用,74和70个靶mRNA注释到泛素蛋白水解,37和32个靶mRNA注释到溶酶体,111和117个靶mRNA注释到黑色素生成等细胞免疫通路;此外,分别有17和22个靶mRNA注释到Jak-STAT信号通路,8和10个靶mRNA注释到NF-κB信号通路,8和18个靶mRNA注释到Toll/Imd信号通路,25和53个靶mRNA注释到MAPK信号通路等体液免疫通路。上述结果共同表明,意蜂中肠发育过程中的DEmiRNA具有调节免疫应激反应的潜在作用。
3.2 DEmiRNA参与意蜂工蜂中肠发育过程的ceRNA调控网络
LncRNA和circRNA可作为ceRNA,通过MRE竞争性结合miRNA,以减轻miRNA对靶mRNA的抑制或降解作用,间接调控各类生物学过程[17,18]。笔者所在课题组前期研究发现,意蜂工蜂中肠的发育过程中,DEcircRNA和DElncRNA可分别结合多个miRNA,如lncRNA XR_001702484.1可靶向ame-miR-6001-3p、miR-376-y和miR-182-x等10个DEmiRNA[20];circRNA novel_circ_010719可靶向ame-miR-6001-3p、miR-148-y和miR-291-y等43个DEmiRNA[21]。经比较分析发现,本研究有112个DEmiRNA能够被201个DEcircRNA、112个DElncRNA和283个DEmRNA竞争性结合,其中靶向16个DEmRNA的miR-126-x(log2fold change=1.96, P= 4.54E-04)可被11个DEcircRNA和9个DElncRNA竞争性结合。上述结果表明,miRNA作为ceRNA调控网络的核心因子,通过介导circRNA和lncRNA对基因表达的调控,在意蜂工蜂中肠发育过程中发挥关键调控作用。一个mRNA可被多个miRNA靶向结合,并参与对多条信号通路的调控作用。前期研究发现,意蜂工蜂中肠发育过程中的57个DEmRNA涉及cAMP、AMPK、Hippo、Toll-like受体、MAPK、mTOR、Wnt、PI3K-Akt、FoxO、Jak-STAT、TGF-beta、Notch和NF-κB 13条发育和免疫相关信号通路[22]。结合前期研究结果进行比较分析,本研究发现miR-291-y、miR-376-y、miR-148-y、miR-5106-y、ame-miR-6001-3p、miR-182-x、miR-126-x、miR-3223-x 8个DEmiRNA靶向结合的12个DEmRNA主要富集在cAMP、AMPK、Hippo、Toll/Imd、TGF-beta、MAPK、mTOR、Wnt和PI3K-Akt 9条信号通路(图3)。XM_016912389.1编码的丝氨酸/苏氨酸-蛋白磷酸酶-β亚基-ε在细胞增殖、转化、凋亡及代谢等多种细胞事件中发挥重要调控作用[47]。本研究中,miR-5106-y的靶mRNA XM_016912389.1富集在PI3K-Akt和AMPK信号通路,说明miR-5106-y在意蜂工蜂中肠发育过程涉及这两条信号通路的调控。此外,分析结果显示同一DEmiRNA的靶向结合的多个mRNA富集在同一条信号通路,例如ame-miR-6001-3p靶向结合的4个DEmRNA中有3个富集在Hippo信号通路。前期研究发现多达29个DElncRNA[20]和14个DEcircRNA[21]能够靶向结合ame-miR-6001-3p,推测ame-miR-6001-3p在意蜂工蜂中肠发育过程除了能够直接调控细胞的增殖与凋亡、组织生长、器官大小,以及组织稳态维持,还能作为DElncRNA和DEcircRNA参与调控发育的媒介分子,值得进一步深入研究。cAMP信号通路在细胞中发挥信使的作用,细胞内cAMP浓度的改变影响多种细胞内信号转导途径,从而调控基因表达、蛋白活性以及细胞功能,对细胞的代谢、生长、分化和凋亡发生影响[48]。本研究中,miR-126-x靶向结合的2个DEmRNA都富集在cAMP信号通路,推测可能通过细胞内外信息传递调控细胞活动。ZHOU等[49]探究了lncRNA- miRNA-mRNA网络在舌鳞状细胞癌(SCCT)中的作用,与本研究中miR-148-y同家族的hsa-miR-148b-3p和hsa-miR-148a-3p可以靶向结合lncRNA KCNQ1OT1参与诱导SCCT细胞生长,推测本研究中的miR-148-y能够通过ceRNA网络在意蜂发育过程中参与调节中肠细胞的生长和发育。酪蛋白激酶在围食膜形成的过程中发挥作用[50],本研究中,ame-miR-6001-3p等28个DEmiRNA能够靶向XM_006569003.2在内的25个编码酪蛋白激酶相关蛋白的基因,暗示以上DEmiRNA参与调控围食膜的形成。几丁质是昆虫表皮和围食膜的重要组成成分之一,几丁质代谢随着昆虫不同生长发育阶段而变化[51],本研究发现ame-miR-6001-3p、miR-142-x、miR-3964-y等10个miRNA能够靶向结合XM_396925.6等4个能够编码几丁质酶的相关基因,推测此10个DEmiRNA参与肠道几丁质的形成过程。
昆虫肠道的发育过程伴随着复杂而广泛的mRNA与ncRNA的相互作用。前期研究发现miR-8503-x、miR-342-y、miR-4792-x和miR-5106-y等miRNA在意蜂幼虫肠道发育过程中发生显著性差异表达[19];本研究中,上述4个miRNA在意蜂工蜂中肠发育过程也呈现出显著性差异表达,差异变化倍数分别达到-14.08(P=3.09E-04)、-3.18(P=4.11E-06)、-3.31(P=8.04E-03)和-1.82(P=0.038),以上4个miRNA在意蜂的两种虫态均发生不同幅度的差异表达,暗示它们与意蜂不同虫态的肠道发育存在潜在关联。此外,还发现miR-342-y在意蜂幼虫肠道发育过程显著性下调表达[19];本研究中,miR-342-y在意蜂工蜂中肠发育中显著性上调表达,与其在幼虫肠道发育过程表达趋势相反,说明该miRNA在意蜂不同虫态的肠道发育过程扮演着不同角色。进一步分析发现,miR-342-y在ceRNA调控网络中可靶向结合3个circRNA、4个lncRNA和327个mRNA,进一步对上述靶mRNA进行代谢通路注释,结果显示参与对神经活性配体-受体相互作用、吞噬作用以及背腹轴形成等162条通路的调控。推测以miR-342-y为核心的ceRNA网络在意蜂不同虫态的肠道发育过程中发挥重要的调控作用,但在意蜂幼虫肠道和工蜂中肠相反的表达趋势暗示miR-342-y具有多面功能,值得进一步深入研究。
针对本研究筛选出的关键DEmiRNA,下一步将通过人工合成miRNA的类似物和抑制物,对意蜂工蜂和幼虫进行miRNA的过表达或敲减,同时检测对其靶向结合lncRNA、circRNA和mRNA表达水平的影响,进而揭示ceRNA介导意蜂工蜂中肠发育的分子机理。
4 结论
DEmiRNA可能通过参与调控的物质和能量代谢通路、Hippo和Wnt等信号通路以及细胞和体液免疫通路相关基因的表达影响意蜂中肠的生长和发育;miR-182-x、miR-291-y、miR-342-y、ame-miR-6001-3p等关键DEmiRNA介导的ceRNA调控网络可能在意蜂中肠发育过程发挥重要的调控作用。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.3864/j.issn.0578-1752.2011.24.018URL [本文引用: 1]

【Objective】 Through studying on the relationship between bee pollination and agricultural production, the economic value of bee pollination to agriculture was estimated for making clear the situation of apiculture in agriculture and providing a theoretical support for development of apicultural support polices in China. 【Method】 A bee pollination dependence valuation method was used to assess the economic value of bee as pollinators of 36 crops during 2006-2008, and the honeybee pollination demand of agricultural production in China was also discussed. 【Result】 There was a significant role in promoting the development of agriculture in China. The average economic value of 2006-2008, contributed by bee pollination, was estimated at¥304.22 billion, which was equivalent to 76 times the value of apicultural production, 12.30% of the gross output value of agriculture in China. There was a great demand for honeybee pollination in agriculture production, only vegetables, fruits, cotton and other crops required 60-87.95 million colonies(15 frames honeybees colony) pollination in 2008. 【Conclusion】 Beekeeping industry is an important component of modern agriculture, bee pollination is essential for agricultural production and there is also a huge demand. Therefore we should pay attention to beekeeping industry, not only to improve the level of social cognition in the value of bee pollination, but also provide powerful policy measures to support the development of apiculture in China.
DOI:10.3864/j.issn.0578-1752.2011.24.018URL [本文引用: 1]
【Objective】 Through studying on the relationship between bee pollination and agricultural production, the economic value of bee pollination to agriculture was estimated for making clear the situation of apiculture in agriculture and providing a theoretical support for development of apicultural support polices in China. 【Method】 A bee pollination dependence valuation method was used to assess the economic value of bee as pollinators of 36 crops during 2006-2008, and the honeybee pollination demand of agricultural production in China was also discussed. 【Result】 There was a significant role in promoting the development of agriculture in China. The average economic value of 2006-2008, contributed by bee pollination, was estimated at¥304.22 billion, which was equivalent to 76 times the value of apicultural production, 12.30% of the gross output value of agriculture in China. There was a great demand for honeybee pollination in agriculture production, only vegetables, fruits, cotton and other crops required 60-87.95 million colonies(15 frames honeybees colony) pollination in 2008. 【Conclusion】 Beekeeping industry is an important component of modern agriculture, bee pollination is essential for agricultural production and there is also a huge demand. Therefore we should pay attention to beekeeping industry, not only to improve the level of social cognition in the value of bee pollination, but also provide powerful policy measures to support the development of apiculture in China.
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s00253-015-6634-xURLPMID:25957492 [本文引用: 1]
High-throughput paired-end RNA sequencing (RNA-Seq) was performed to investigate the gene expression profile of a susceptible Bombyx mori strain, Lan5, and a resistant B. mori strain, Ou17, which were both orally infected with B. mori cypovirus (BmCPV) in the midgut. There were 330 and 218 up-regulated genes, while there were 147 and 260 down-regulated genes in the Lan5 and Ou17 strains, respectively. Gene ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment for differentially expressed genes (DEGs) were carried out. Moreover, gene interaction network (STRING) analyses were performed to analyze the relationships among the shared DEGs. Some of these genes were related and formed a large network, in which the genes for B. mori cuticular protein RR-2 motif 123 (BmCPR123) and the gene for B. mori DNA replication licensing factor Mcm2-like (BmMCM2) were key genes among the common up-regulated DEGs, whereas the gene for B. mori heat shock protein 20.1 (Bmhsp20.1) was the central gene among the shared down-regulated DEGs between Lan5 vs Lan5-CPV and Ou17 vs Ou17-CPV. These findings established a comprehensive database of genes that are differentially expressed in response to BmCPV infection between silkworm strains that differed in resistance to BmCPV and implied that these DEGs might be involved in B. mori immune responses against BmCPV infection.
DOI:10.1016/s0092-8674(04)00045-5URLPMID:14744438 [本文引用: 1]
MicroRNAs (miRNAs) are endogenous approximately 22 nt RNAs that can play important regulatory roles in animals and plants by targeting mRNAs for cleavage or translational repression. Although they escaped notice until relatively recently, miRNAs comprise one of the more abundant classes of gene regulatory molecules in multicellular organisms and likely influence the output of many protein-coding genes.
DOI:10.1126/science.1115079URLPMID:16081698 [本文引用: 1]
MicroRNAs (miRNAs) are approximately 21-nucleotide-long RNA molecules regulating gene expression in multicellular eukaryotes. In metazoa, miRNAs act by imperfectly base-pairing with the 3' untranslated region of target messenger RNAs (mRNAs) and repressing protein accumulation by an unknown mechanism. We demonstrate that endogenous let-7 microribonucleoproteins (miRNPs) or the tethering of Argonaute (Ago) proteins to reporter mRNAs in human cells inhibit translation initiation. M(7)G-cap-independent translation is not subject to repression, suggesting that miRNPs interfere with recognition of the cap. Repressed mRNAs, Ago proteins, and miRNAs were all found to accumulate in processing bodies. We propose that localization of mRNAs to these structures is a consequence of translational repression.
DOI:10.1016/s0092-8674(03)00231-9URLPMID:12679032 [本文引用: 1]
Cell proliferation, cell death, and pattern formation are coordinated in animal development. Although many proteins that control cell proliferation and apoptosis have been identified, the means by which these effectors are linked to the patterning machinery remain poorly understood. Here, we report that the bantam gene of Drosophila encodes a 21 nucleotide microRNA that promotes tissue growth. bantam expression is temporally and spatially regulated in response to patterning cues. bantam microRNA simultaneously stimulates cell proliferation and prevents apoptosis. We identify the pro-apoptotic gene hid as a target for regulation by bantam miRNA, providing an explanation for bantam's anti-apoptotic activity.
DOI:10.1016/S0960-9822(03)00250-1URL [本文引用: 1]
Abstract
MicroRNAs (miRNAs) are small regulatory RNAs that are between 21 and 25 nucleotides in length and repress gene function through interactions with target mRNAs [1] and [2]. The genomes of metazoans encode on the order of several hundred miRNAs [3], but the processes they regulate have been defined for only two in C. elegans [4] and [5]. We searched for new inhibitors of apoptotic cell death by testing existing collections of P element insertion lines for their ability to enhance a small-eye phenotype associated with eye-specific expression of the Drosophila cell death activator Reaper. Here we report the identification of the Drosophila miRNA mir-14 as a cell death suppressor. Loss of mir-14 enhances Reaper-dependent cell death, whereas ectopic expression suppresses cell death induced by multiple stimuli. Animals lacking mir-14 are viable. However, they are stress sensitive and have a reduced lifespan. Mir-14 mutants have elevated levels of the apoptotic effector caspase Drice, suggesting one potential site of action. Mir-14 also regulates fat metabolism. Deletion of mir-14 results in animals with increased levels of triacylglycerol and diacylglycerol, whereas increases in mir-14 copy number have the converse effect. We discuss possible relationships between these phenotypes.DOI:10.1016/0092-8674(93)90529-yURLPMID:8252621 [本文引用: 1]
lin-4 is essential for the normal temporal control of diverse postembryonic developmental events in C. elegans. lin-4 acts by negatively regulating the level of LIN-14 protein, creating a temporal decrease in LIN-14 protein starting in the first larval stage (L1). We have cloned the C. elegans lin-4 locus by chromosomal walking and transformation rescue. We used the C. elegans clone to isolate the gene from three other Caenorhabditis species; all four Caenorhabditis clones functionally rescue the lin-4 null allele of C. elegans. Comparison of the lin-4 genomic sequence from these four species and site-directed mutagenesis of potential open reading frames indicated that lin-4 does not encode a protein. Two small lin-4 transcripts of approximately 22 and 61 nt were identified in C. elegans and found to contain sequences complementary to a repeated sequence element in the 3' untranslated region (UTR) of lin-14 mRNA, suggesting that lin-4 regulates lin-14 translation via an antisense RNA-RNA interaction.
[本文引用: 2]
[本文引用: 2]
DOI:10.1016/j.gene.2012.04.002URL [本文引用: 1]
Tobacco is one of the most important economic and agricultural crops worldwide. miRNAs have been increasingly acknowledged for their important roles in different biological processes of tobacco. However, few miRNAs have been identified so far in tobacco impeding the development of new tobacco strains with better properties. In this study, high-throughput sequencing technology was employed to identify novel tobacco miRNAs. A total of 84 potential miRNAs were obtained in tobacco, including 33 conserved and 51 novel miRNAs. Tissue-specific and topping-related miRNAs were identified. A tobacco miRNA microarray was also constructed to investigate miRNA expression patterns in different tissues, and their expression patterns were further validated by qRT-PCR and Northern Blot. Finally, the potential targets of these miRNAs were predicted based on a sequence homology search. Thus, in the current study, we have performed the comprehensive analysis of tobacco miRNAs, including their identification, expression pattern and target prediction. Our study opens a new avenue for further elucidation for their roles underlying the regulation of diversity of physiological processes. (C) 2012 Elsevier B.V.
DOI:10.13343/j.cnki.wsxb.20170535URL [本文引用: 1]
[Objective] In this study, pure culture of A. apis was sequenced using sRNA-seq technology, followed by prediction, identification and analysis of A. apis microRNAs. The microRNAs-mRNAs regulation network was further constructed. [Methods] Illumina Hiseq Xten platform was used to sequence mycelium and spores of A. apis, and corresponding softwares were used to predict and analyze A. apis microRNAs, some of which were identified via Stem-loop PCR. Cytoskype software was used to construct A. apis microRNAs-mRNAs regulation network. [Results] A total of 48268696 clean reads were obtained, and 118 miRNAs of A. apis were predicted, whose length was distributed between 18 nt and 25 nt. The preference of the first base of miRNAs with different length was obviously various. Stem-loop PCR result showed target fragments with expected sizes were amplified from 10 microRNAs, implying most of the predicted microRNAs' true existence. In total, 6529 target genes of A. apis microRNAs were predicted, and among them 5725 could be annotated in Nr, Swissprot, KOG, GO and KEGG databases. Further investigation demonstrated 24 target genes were annotated in MAPK signaling pathway. Cytoskype software analysis suggested complicated regulation networks exist between microRNAs and mRNAs in A. apis, and majority of miRNAs inside the networks bind to several mRNAs. [Conclusion] Our findings enrich the understanding of A. apis microRNAs, provide beneficial supplement for basic biology information of A. apis, and lay some foundation for illustrating the molecular mechanism regulating the pathogenesis of this widespread fungal pathogen.
DOI:10.13343/j.cnki.wsxb.20170535URL [本文引用: 1]
[Objective] In this study, pure culture of A. apis was sequenced using sRNA-seq technology, followed by prediction, identification and analysis of A. apis microRNAs. The microRNAs-mRNAs regulation network was further constructed. [Methods] Illumina Hiseq Xten platform was used to sequence mycelium and spores of A. apis, and corresponding softwares were used to predict and analyze A. apis microRNAs, some of which were identified via Stem-loop PCR. Cytoskype software was used to construct A. apis microRNAs-mRNAs regulation network. [Results] A total of 48268696 clean reads were obtained, and 118 miRNAs of A. apis were predicted, whose length was distributed between 18 nt and 25 nt. The preference of the first base of miRNAs with different length was obviously various. Stem-loop PCR result showed target fragments with expected sizes were amplified from 10 microRNAs, implying most of the predicted microRNAs' true existence. In total, 6529 target genes of A. apis microRNAs were predicted, and among them 5725 could be annotated in Nr, Swissprot, KOG, GO and KEGG databases. Further investigation demonstrated 24 target genes were annotated in MAPK signaling pathway. Cytoskype software analysis suggested complicated regulation networks exist between microRNAs and mRNAs in A. apis, and majority of miRNAs inside the networks bind to several mRNAs. [Conclusion] Our findings enrich the understanding of A. apis microRNAs, provide beneficial supplement for basic biology information of A. apis, and lay some foundation for illustrating the molecular mechanism regulating the pathogenesis of this widespread fungal pathogen.
DOI:10.1186/gb-2003-4-11-p8URL [本文引用: 1]
DOI:10.1111/j.1365-2583.2012.01135.xURL [本文引用: 1]
MicroRNAs (miRNAs) are endogenous small non-coding RNAs regulating gene expression in animals and plants. To find some differentially expressed miRNAs that may be associated with age-dependent behavioural changes in honey bees (Apis mellifera), we applied next-generation high-throughput sequencing technology to detect small RNAs in nurses and foragers. Our results showed that both nurses and foragers had a complicated small RNA population, and the length of small RNAs varied, 22 nucleotides being the predominant length. Combining deep sequencing and bioinformatic analysis, we discovered that nine known miRNAs were significantly different between nurses and foragers (P < 0.01; absolute value of fold-change =1). Some of their target genes were related to neural function. Moreover, 67 novel miRNAs were identified in nurses and foragers. Ame-miR-31a and ame-miR-13b were further validated using quantitative reverse-transcription PCR assays. The present study provides new information on the miRNA abundance of honey bees, and enhances our understanding of miRNA function in the regulation of honey bee development.
DOI:10.1007/s13592-014-0299-9URL [本文引用: 1]
DOI:10.1016/j.ibmb.2013.03.001URL [本文引用: 1]
In insects, a rapid and massive synthesis of antimicrobial peptides (AMPs) is activated through signaling pathways (Toll and Imd) to combat invading microbial pathogens. However, it is still unclear whether different types of bacteria provoke specific responses. Immune response mechanisms and the activation of specific genes were investigated by challenging Apis mellifera workers with the Gram-negative bacterium Serratia marcescens or the Gram-positive bacterium Micrococcus luteus. The immune system responded by activating most genes of the Toll and Imd pathways, particularly AMP genes. However, genes specifically regulated by M. luteus or S. marcescens were not detected, suggesting an interaction between the signaling pathways that lead to immune effectors synthesis. Despite this finding, kappaB motifs in the 5'-UTRs of selected genes suggest a pathway-specific control of AMP and transferrin-1 gene expression. Regulation by miRNAs was also investigated and revealed a number of candidates for the post-transcriptional regulation of immune genes in bees. (C) 2013 Elsevier Ltd.
DOI:10.1016/j.cell.2011.07.014URL [本文引用: 1]
Here, we present a unifying hypothesis about how messenger RNAs, transcribed pseudogenes, and long noncoding RNAs "talk" to each other using microRNA response elements (MREs) as letters of a new language. We propose that this "competing endogenous RNA" (ceRNA) activity forms a large-scale regulatory network across the transcriptome, greatly expanding the functional genetic information in the human genome and playing important roles in pathological conditions, such as cancer.
DOI:10.1038/nature12986URL [本文引用: 2]
Recent reports have described an intricate interplay among diverse RNA species, including protein-coding messenger RNAs and non-coding RNAs such as long non-coding RNAs, pseudogenes and circular RNAs. These RNA transcripts act as competing endogenous RNAs (ceRNAs) or natural microRNA sponges - they communicate with and co-regulate each other by competing for binding to shared microRNAs, a family of small non-coding RNAs that are important post-transcriptional regulators of gene expression. Understanding this novel RNA crosstalk will lead to significant insight into gene regulatory networks and have implications in human development and disease.
DOI:10.1038/ncomms7779URLPMID:25858075 [本文引用: 3]
The abnormal autophagy is associated with a variety of cardiovascular diseases. Long noncoding RNAs (lncRNAs) are emerging as new factors in gene regulation, but how lncRNAs operate in the regulation of autophagy in the heart is unclear. Here we report that a long noncoding RNA, named autophagy promoting factor (APF), can regulate autophagic cell death by targeting miR-188-3p and ATG7. The results show that miR-188-3p suppresses autophagy and myocardial infarction by targeting ATG7. Further, we find that APF lncRNA regulates miR-188-3p, and thus affects ATG7 expression, autophagic cell death and myocardial infarction. Our present study reveals a novel regulating model of autophagic programme, which comprises APF, miR-188-3p and ATG7 in the heart. Modulation of their levels may serve as potential targets and diagnostic tools for novel therapeutic strategies of myocardial infarction and heart failure.
DOI:10.3864/j.issn.0578-1752.2018.21.018URL [本文引用: 7]
【Objective】MicroRNA (miRNA) is a kind of key regulator for negative regulation of mRNA at post-transcriptional level. The objective of this study is to provide miRNA expression patterns and differential expression information, illuminate the function of differentially expressed miRNA (DEmiRNA) in the development of larval gut by comprehensively investigating the DEmiRNAs and their regulation networks during the developmental process of Apis mellifera ligustica larval gut. 【Method】Deep sequencing of the 4-, 5- and 6-day-old larval guts of A. m. ligustica was conducted using small RNA-seq (sRNA-seq) technology, followed by mapping of the data after quality-control with the reference genome of Apis mellifera, and the mapped tags were then compared to miRBase database. The miRNA expression level was normalized by TPM algorithm, and the expression clustering, prediction of secondary structure of precursor and differential expression analysis were performed using related bioinformatic softwares. TargetFinder software was used to predict target gene of DEmiRNA, which was annotated to GO and KEGG databases using Blast, furthermore, miRNA-mRNA regulation networks were constructed using Cytoscape software. Stem-loop RT-qPCR was used to verify the sequencing data in this study.【Result】High-throughput sequencing of larval gut samples produced 10 841 644, 12 037 678 and 9 230 496 clean tags, respectively. In Am4 vs Am5 comparison group, there were16 up-regulated and 10 down-regulated miRNAs, while Am5 vs Am6 comparison group included 5 up-regulated and 7 down-regulated miRNAs, respectively. Among them, Novel-m0031-3p was shared by both Am4 vs Am5 and Am5 vs Am6, binding 5 target genes associated with ecdysone inducible protein, 25 and 11 DEmiRNAs were specific for the above-mentioned two comparison groups. DEmiRNA in Am4 vs Am5 could bind 5 742 target genes, among them 2 725 targets could be annotated to 46 GO terms in GO database, and the largest ones were binding, cellular process, metabolic process and single-organism process. Similarly, 12 DEmiRNAs in Am5 vs Am6 could link 3 733 target genes, among them 2 725 targets could be annotated to 41 GO terms, and mostly enriched terms were binding, cellular process, single-organism process and metabolic process. In addition, 1 046 and 676 target genes of two comparison groups were related to 116 and 92 KEGG pathways, and the number of DEmiRNA target genes in Am4 vs Am5 was more than that in Am5 vs Am6, which annotated to Wnt signaling pathway, Hippo signaling pathway, purine metabolism and endocytosis. Further analysis demonstrated that up-regulated and down-regulated miRNAs in Am4 vs Am5 could bind 611 and 85 target genes, and ame-miR-6052 linked the most target genes and participated in regulating cytochrome P450 via 5 target genes. miR-281-x could bind 49 target genes and indirectly regulate histidine metabolism, TGF-β signaling pathway and Hippo signaling pathway. In Am5 vs Am6 comparison group, up-regulated and down-regulated miRNAs could bind 43 and 431 target genes, respectively, among them miR-iab-4-x linked the most target genes, and it could participate in regulating growth and development related pathways, such as dorso-ventral axis formation, Hippo signaling pathway, Wnt signaling pathway, FoxO signaling pathway, Notch signaling pathway and mTOR signaling pathway. Regulation network analysis indicated that complex networks formed between DEmiRNAs and target genes, and DEmiRNAs lied in the center while target genes lied in the periphery. Finally, Stem-loop RT-qPCR was carried out to validate the randomly selected three DEmiRNAs, and the result confirmed the reliability of sequencing data. 【Conclusion】The DEmiRNA and corresponding target genes in the A. m. ligustica larval gut were predicted and analyzed at genome-wide level, it was found that A. m. ligustica are capable of regulating the expression of many miRNAs such as ame-miR-6052, miR-iab-4-x, miR-281-x and novel-m0031-3p. The results not only offer the expression pattern and differential expression information of miRNA during the developmental process of A. m. ligustica larval gut, but also lay a foundation for clarifying the molecular mechanisms underlying the larval gut’s development.
DOI:10.3864/j.issn.0578-1752.2018.21.018URL [本文引用: 7]
【Objective】MicroRNA (miRNA) is a kind of key regulator for negative regulation of mRNA at post-transcriptional level. The objective of this study is to provide miRNA expression patterns and differential expression information, illuminate the function of differentially expressed miRNA (DEmiRNA) in the development of larval gut by comprehensively investigating the DEmiRNAs and their regulation networks during the developmental process of Apis mellifera ligustica larval gut. 【Method】Deep sequencing of the 4-, 5- and 6-day-old larval guts of A. m. ligustica was conducted using small RNA-seq (sRNA-seq) technology, followed by mapping of the data after quality-control with the reference genome of Apis mellifera, and the mapped tags were then compared to miRBase database. The miRNA expression level was normalized by TPM algorithm, and the expression clustering, prediction of secondary structure of precursor and differential expression analysis were performed using related bioinformatic softwares. TargetFinder software was used to predict target gene of DEmiRNA, which was annotated to GO and KEGG databases using Blast, furthermore, miRNA-mRNA regulation networks were constructed using Cytoscape software. Stem-loop RT-qPCR was used to verify the sequencing data in this study.【Result】High-throughput sequencing of larval gut samples produced 10 841 644, 12 037 678 and 9 230 496 clean tags, respectively. In Am4 vs Am5 comparison group, there were16 up-regulated and 10 down-regulated miRNAs, while Am5 vs Am6 comparison group included 5 up-regulated and 7 down-regulated miRNAs, respectively. Among them, Novel-m0031-3p was shared by both Am4 vs Am5 and Am5 vs Am6, binding 5 target genes associated with ecdysone inducible protein, 25 and 11 DEmiRNAs were specific for the above-mentioned two comparison groups. DEmiRNA in Am4 vs Am5 could bind 5 742 target genes, among them 2 725 targets could be annotated to 46 GO terms in GO database, and the largest ones were binding, cellular process, metabolic process and single-organism process. Similarly, 12 DEmiRNAs in Am5 vs Am6 could link 3 733 target genes, among them 2 725 targets could be annotated to 41 GO terms, and mostly enriched terms were binding, cellular process, single-organism process and metabolic process. In addition, 1 046 and 676 target genes of two comparison groups were related to 116 and 92 KEGG pathways, and the number of DEmiRNA target genes in Am4 vs Am5 was more than that in Am5 vs Am6, which annotated to Wnt signaling pathway, Hippo signaling pathway, purine metabolism and endocytosis. Further analysis demonstrated that up-regulated and down-regulated miRNAs in Am4 vs Am5 could bind 611 and 85 target genes, and ame-miR-6052 linked the most target genes and participated in regulating cytochrome P450 via 5 target genes. miR-281-x could bind 49 target genes and indirectly regulate histidine metabolism, TGF-β signaling pathway and Hippo signaling pathway. In Am5 vs Am6 comparison group, up-regulated and down-regulated miRNAs could bind 43 and 431 target genes, respectively, among them miR-iab-4-x linked the most target genes, and it could participate in regulating growth and development related pathways, such as dorso-ventral axis formation, Hippo signaling pathway, Wnt signaling pathway, FoxO signaling pathway, Notch signaling pathway and mTOR signaling pathway. Regulation network analysis indicated that complex networks formed between DEmiRNAs and target genes, and DEmiRNAs lied in the center while target genes lied in the periphery. Finally, Stem-loop RT-qPCR was carried out to validate the randomly selected three DEmiRNAs, and the result confirmed the reliability of sequencing data. 【Conclusion】The DEmiRNA and corresponding target genes in the A. m. ligustica larval gut were predicted and analyzed at genome-wide level, it was found that A. m. ligustica are capable of regulating the expression of many miRNAs such as ame-miR-6052, miR-iab-4-x, miR-281-x and novel-m0031-3p. The results not only offer the expression pattern and differential expression information of miRNA during the developmental process of A. m. ligustica larval gut, but also lay a foundation for clarifying the molecular mechanisms underlying the larval gut’s development.
DOI:10.3864/j.issn.0578-1752.2018.18.016URL [本文引用: 7]
【Objective】Long non-coding RNA (lncRNA) plays an important role in regulation of gene expression, epigenetics and cell cycle in eukaryotes. The objective of this study is to investigate the expression profile and role of lncRNAs in the developmental process of Apis mellifera ligustica worker’s midgut. 【Method】In this study, 7- and 10-day-old worker’s midguts of A. m. ligustica (Am7, Am10) were sequenced using RNA-seq technology and strand-specific library construction method. Using Perl script, raw reads were filtered to obtain clean reads with high-quality. Bowtie tool was used to compare clean reads to the ribosome database, and TopHat2 software was employed to compare unmapped clean reads to the reference genome. CPC and CNCI softwares were utilized to predict coding capacity of the transcripts. RT-PCR was performed to identify partial lncRNAs. Investigation of differentially expressed lncRNAs (DElncRNAs) was carried out with edgeR, followed by prediction of upstream and downstream genes, for which GO and KEGG pathway enrichment analyses were performed. RNAhybrid, Miranda and TargetScan softwares were utilized together to predict target miRNAs of DElncRNAs and target genes of miRNAs, and DElncRNAs-miRNAs-mRNAs regulation networks were visualized via Cytoscape. Finally, RT-qPCR was conducted to verify reliability of the sequencing data.【Result】134 802 058 and 147 051 470 raw reads were gained from deep sequencing of Am7 and Am10, respectively, and after stringent filtration, 134 166 157 and 146 293 288 were obtained. In total, 6 353 lncRNAs were predicted, and 3 890 DElncRNAs were obtained based on expression calculation, including 2 005 up-regulated lncRNAs and 1 885 down-regulated lncRNAs. The result of RT-PCR suggested the expected signal bands could be amplified from 8 lncRNAs, implying their true existence. There were 1 793 upstream and downstream genes of DElncRNAs, which were involved in 42 GO terms, including metabolic processes, developmental processes, cellular processes, stress responses, immune system processes and so forth. They were also associated with 251 KEGG pathways, including material metabolism pathways such as carbon metabolism, purine metabolism and fatty acid biosynthesis; energy metabolism pathways such as sulfur metabolism, methane metabolism and oxidative phosphorylation; signaling pathways such as Hippo, Wnt and Notch signaling pathways; cellular immune pathways such as lysosome, endocytosis and ubiquitin mediated proteolysis; humoral immune pathways such as MAPK, Jak-STAT and NF-kappa B pathways, these results demonstrated the DElncRNAs were involved in the material and energy metabolism, cell life activity and immunity regulation in the developmental process of A. m. ligustica worker’s midgut. Further analysis showed TCONS_00020918 might play a regulatory part in the nutrient absorption and caste differentiation in the worker’s midgut. Analysis of regulation networks demonstrated that complex networks existed between DElncRNAs and target miRNAs and mRNAs, partial DElncRNAs lie in the central of the networks and link many miRNAs, and partial miRNAs could be bound by many DElncRNAs, which indicated that these DElncRNAs might play an important role during the developmental process of the worker’s midgut. Finally, 5 DElncRNAs were randomly selected for RT-qPCR assay, and the result proved the reliability of sequencing data in this study.【Conclusion】DElncRNA is widely involved in the metabolism, cellular activity and immune regulation of A. m. ligustica worker’s midgut, and plays a role as a competitive endogenous RNA (ceRNA). The results provide the necessary data support for the screening and functional study of key lncRNA.
DOI:10.3864/j.issn.0578-1752.2018.18.016URL [本文引用: 7]
【Objective】Long non-coding RNA (lncRNA) plays an important role in regulation of gene expression, epigenetics and cell cycle in eukaryotes. The objective of this study is to investigate the expression profile and role of lncRNAs in the developmental process of Apis mellifera ligustica worker’s midgut. 【Method】In this study, 7- and 10-day-old worker’s midguts of A. m. ligustica (Am7, Am10) were sequenced using RNA-seq technology and strand-specific library construction method. Using Perl script, raw reads were filtered to obtain clean reads with high-quality. Bowtie tool was used to compare clean reads to the ribosome database, and TopHat2 software was employed to compare unmapped clean reads to the reference genome. CPC and CNCI softwares were utilized to predict coding capacity of the transcripts. RT-PCR was performed to identify partial lncRNAs. Investigation of differentially expressed lncRNAs (DElncRNAs) was carried out with edgeR, followed by prediction of upstream and downstream genes, for which GO and KEGG pathway enrichment analyses were performed. RNAhybrid, Miranda and TargetScan softwares were utilized together to predict target miRNAs of DElncRNAs and target genes of miRNAs, and DElncRNAs-miRNAs-mRNAs regulation networks were visualized via Cytoscape. Finally, RT-qPCR was conducted to verify reliability of the sequencing data.【Result】134 802 058 and 147 051 470 raw reads were gained from deep sequencing of Am7 and Am10, respectively, and after stringent filtration, 134 166 157 and 146 293 288 were obtained. In total, 6 353 lncRNAs were predicted, and 3 890 DElncRNAs were obtained based on expression calculation, including 2 005 up-regulated lncRNAs and 1 885 down-regulated lncRNAs. The result of RT-PCR suggested the expected signal bands could be amplified from 8 lncRNAs, implying their true existence. There were 1 793 upstream and downstream genes of DElncRNAs, which were involved in 42 GO terms, including metabolic processes, developmental processes, cellular processes, stress responses, immune system processes and so forth. They were also associated with 251 KEGG pathways, including material metabolism pathways such as carbon metabolism, purine metabolism and fatty acid biosynthesis; energy metabolism pathways such as sulfur metabolism, methane metabolism and oxidative phosphorylation; signaling pathways such as Hippo, Wnt and Notch signaling pathways; cellular immune pathways such as lysosome, endocytosis and ubiquitin mediated proteolysis; humoral immune pathways such as MAPK, Jak-STAT and NF-kappa B pathways, these results demonstrated the DElncRNAs were involved in the material and energy metabolism, cell life activity and immunity regulation in the developmental process of A. m. ligustica worker’s midgut. Further analysis showed TCONS_00020918 might play a regulatory part in the nutrient absorption and caste differentiation in the worker’s midgut. Analysis of regulation networks demonstrated that complex networks existed between DElncRNAs and target miRNAs and mRNAs, partial DElncRNAs lie in the central of the networks and link many miRNAs, and partial miRNAs could be bound by many DElncRNAs, which indicated that these DElncRNAs might play an important role during the developmental process of the worker’s midgut. Finally, 5 DElncRNAs were randomly selected for RT-qPCR assay, and the result proved the reliability of sequencing data in this study.【Conclusion】DElncRNA is widely involved in the metabolism, cellular activity and immune regulation of A. m. ligustica worker’s midgut, and plays a role as a competitive endogenous RNA (ceRNA). The results provide the necessary data support for the screening and functional study of key lncRNA.
DOI:10.3864/j.issn.0578-1752.2018.23.015URL [本文引用: 7]
【Objective】Circular RNA (circRNA) plays a primary role in alternative splicing, transcription regulation and expression regulation of parental gene. The objective of this study is to investigate the profile expression of circRNAs and differentially expressed circRNAs (DEcircRNAs) during the developmental process of the midguts of Apis mellifera ligustica workers, and to explore the role of DEcircRNAs in the development of midgut at the transcriptional level. 【Method】 On basis of the whole transcriptome data from 7- and 10-day-old worker’s midguts of A. m. ligustica (Am7 and Am10), find_circ software was used to predict circRNAs based on the filtered sequencing data. The circRNA expression level was normalized by RPM algorithm. Differential expression analysis for circRNAs was conducted via DEGseq software following standards fold change≥2.0, P<0.05 and false discovery rate (FDR) <0.05. Source genes of DEcircRNAs were annotated to GO and KEGG databases to gain function and pathway annotations by using BLAST. DEcircRNAs-miRNAs and DEcircRNAs-miRNAs-mRNAs networks were predicted with TargetFinder software and visualized using Cytoscape v.3.2.1 software. RT-qPCR was conducted to verify the reliability of sequencing data.【Result】 On average, 19 616 356 anchors reads were obtained from each A. m. ligustica worker’s midgut sample. Pearson correlations between different biological repeats within Am7 and Am10 groups were ≥0.950. In total, 256 DEcircRNAs including 105 up-regulated circRNAs and 151 down-regulated circRNAs were predicted. Novel_circ_009675 and novel_circ_013879 were highly expressed in Am7 and Am10, respectively. Source genes of DEcircRNAs could be annotated to 32 GO terms including binding, single-organism process and cellular process, among them 35, 35 and 7 source genes were involved in catalytic activity, metabolic process and stress response. Additionally, these source genes could be annotated to 35 KEGG pathways, in which 5, 5 and 4 source genes were associated with Hippo signaling pathway, endocytosis and phagosome, respectively; further investigation showed that 1, 2 and 2 source genes could be annotated to material metabolisms such as phosphoinositol metabolism, starch and sucrose metabolism and galactose metabolism; 5, 4, 3, 1 and 1 source genes could be annotated to immune signaling pathways including endocytosis, phagosome, lysosome, ubiquitin-mediated proteolysis and MAPK signaling pathway, respectively. These results suggested that the corresponding DEcircRNA was involved in the development, metabolism and immune defense of the midgut of A. m. ligustica. DEcircRNA-miRNA regulation network analysis showed that 141 DEcircRNAs could link to 107 miRNAs, most of these DEcircRNAs could only bind to 1-2 miRNAs, but novel_circ_011577 and novel_circ_010719 could respectively bind to 32 and 28 miRNAs. In addition, the number of DEcircRNAs combined with mir-136-y, ame-miR-6001-3p and mir-136-y was the highest (15, 14 and 14, respectively), which indicated that the corresponding DEcircRNA could play roles during the developmental process of A. m. ligustica worker’s midgut as competing endogenous RNAs. Furthermore, DEcircRNAs-ame-miR-6001-3p-mRNA network was constructed and analyzed, and the result indicated that 14 DEcircRNAs could jointly link to ame-miR-6001-3p, implying they were likely to indirectly regulate division and differentiation of stem cells in A. m. ligustica worker’s midgut via regulation of ame-miR-6001-3p. Six DEcircRNAs were randomly selected for RT-qPCR assay, the result showed the alteration trend of expression levels of 5 DEcircRNAs was consistent with that of the sequencing data, which proved the reliability of trancriptome data.【Conclusion】Through the deep investigation of DEcircRNAs during the developmental process of A. m. ligustica worker’s midgut, the expression profile and differential expression information of circRNAs in the development of midgut of worker bee were provided, and the role of DEcircRNAs in the development of midgut was revealed. It provides a basis for the screening and functional study of key circRNAs associated with the development of the midgut.
DOI:10.3864/j.issn.0578-1752.2018.23.015URL [本文引用: 7]
【Objective】Circular RNA (circRNA) plays a primary role in alternative splicing, transcription regulation and expression regulation of parental gene. The objective of this study is to investigate the profile expression of circRNAs and differentially expressed circRNAs (DEcircRNAs) during the developmental process of the midguts of Apis mellifera ligustica workers, and to explore the role of DEcircRNAs in the development of midgut at the transcriptional level. 【Method】 On basis of the whole transcriptome data from 7- and 10-day-old worker’s midguts of A. m. ligustica (Am7 and Am10), find_circ software was used to predict circRNAs based on the filtered sequencing data. The circRNA expression level was normalized by RPM algorithm. Differential expression analysis for circRNAs was conducted via DEGseq software following standards fold change≥2.0, P<0.05 and false discovery rate (FDR) <0.05. Source genes of DEcircRNAs were annotated to GO and KEGG databases to gain function and pathway annotations by using BLAST. DEcircRNAs-miRNAs and DEcircRNAs-miRNAs-mRNAs networks were predicted with TargetFinder software and visualized using Cytoscape v.3.2.1 software. RT-qPCR was conducted to verify the reliability of sequencing data.【Result】 On average, 19 616 356 anchors reads were obtained from each A. m. ligustica worker’s midgut sample. Pearson correlations between different biological repeats within Am7 and Am10 groups were ≥0.950. In total, 256 DEcircRNAs including 105 up-regulated circRNAs and 151 down-regulated circRNAs were predicted. Novel_circ_009675 and novel_circ_013879 were highly expressed in Am7 and Am10, respectively. Source genes of DEcircRNAs could be annotated to 32 GO terms including binding, single-organism process and cellular process, among them 35, 35 and 7 source genes were involved in catalytic activity, metabolic process and stress response. Additionally, these source genes could be annotated to 35 KEGG pathways, in which 5, 5 and 4 source genes were associated with Hippo signaling pathway, endocytosis and phagosome, respectively; further investigation showed that 1, 2 and 2 source genes could be annotated to material metabolisms such as phosphoinositol metabolism, starch and sucrose metabolism and galactose metabolism; 5, 4, 3, 1 and 1 source genes could be annotated to immune signaling pathways including endocytosis, phagosome, lysosome, ubiquitin-mediated proteolysis and MAPK signaling pathway, respectively. These results suggested that the corresponding DEcircRNA was involved in the development, metabolism and immune defense of the midgut of A. m. ligustica. DEcircRNA-miRNA regulation network analysis showed that 141 DEcircRNAs could link to 107 miRNAs, most of these DEcircRNAs could only bind to 1-2 miRNAs, but novel_circ_011577 and novel_circ_010719 could respectively bind to 32 and 28 miRNAs. In addition, the number of DEcircRNAs combined with mir-136-y, ame-miR-6001-3p and mir-136-y was the highest (15, 14 and 14, respectively), which indicated that the corresponding DEcircRNA could play roles during the developmental process of A. m. ligustica worker’s midgut as competing endogenous RNAs. Furthermore, DEcircRNAs-ame-miR-6001-3p-mRNA network was constructed and analyzed, and the result indicated that 14 DEcircRNAs could jointly link to ame-miR-6001-3p, implying they were likely to indirectly regulate division and differentiation of stem cells in A. m. ligustica worker’s midgut via regulation of ame-miR-6001-3p. Six DEcircRNAs were randomly selected for RT-qPCR assay, the result showed the alteration trend of expression levels of 5 DEcircRNAs was consistent with that of the sequencing data, which proved the reliability of trancriptome data.【Conclusion】Through the deep investigation of DEcircRNAs during the developmental process of A. m. ligustica worker’s midgut, the expression profile and differential expression information of circRNAs in the development of midgut of worker bee were provided, and the role of DEcircRNAs in the development of midgut was revealed. It provides a basis for the screening and functional study of key circRNAs associated with the development of the midgut.
DOI:10.3864/j.issn.0578-1752.2020.01.019URL [本文引用: 8]
【Objective】The whole transcriptome sequencing of Apis mellifera ligustica 7- and 10-day-old workers’ midguts (Am7 and Am10) was previously conducted. In this study, the differential expression profile and regulation network of genes were investigated to reveal the molecular mechanism underlying the midgut development.【Method】Gene expressions were calculated based on FPKM (fragments per kilobase of transcript per million mapped reads) algorithm, and differentially expressed genes (DEGs) were gained following the standard |log2 fold change|≥1 and P≤0.05. Target mRNAs of ame-miR-6001-3p were predicted utilizing TargetFinder. Annotations of all DEGs in GO and KEGG databases were performed using related software. In addition, DEGs enriched in 13 signaling pathways including AMPK, P13K-Akt, Wnt, cAMP, FoxO, Hippo, mTOR, Jak-STAT, Toll-like receptor, TGF-beta, Notch, MAPK and NF-κB, as well as DEGs targeted by ame-miR-6001-3p were screened out, followed by visualization of enrichment networks and regulation networks with Cytoscape. Finally, Stem loop RT-PCR and RT-qPCR were used to verify the expression and differential expression pattern of ame-miR-6001-3p and DEGs in Am7 and Am10.【Result】A total of 1 038 DEGs were identified in Am7 vs Am10 comparison group, including 515 up- and 523 down-regulated genes, respectively. These DEGs were associated with cellular process, metabolic process and catalytic activity, and significantly enriched in some material and energy metabolisms such as oxidative phosphorylation, amino sugar and nucleotide sugar metabolisms, fatty acid metabolism and purine metabolism, indicative of the active cellular and metabolic activities. Expression cluster analysis suggested that 20, 18, 15 and 14 DEGs were respectively enriched in AMPK signaling pathway, PI3K-Akt signaling pathway, endocytosis and Hippo signaling pathway. In addition, 57 DEGs were enriched in the aforementioned 13 signaling pathways associated with growth and development as well as immune defense, among them one DEG was enriched in several signaling pathways. Moreover, regulation network analysis showed that 54 up-regulated genes and 44 down-regulated genes were targets of ame-miR-6001-3p, respectively; these up-regulated genes were enriched in 43 pathways including inositol phosphate metabolism, Hippo signaling pathway, glutathione metabolism and insulin signaling pathway, while these down-regulated genes were enriched in 20 pathways including Hippo signaling pathway, metabolic pathways, glutathione metabolism and arachidonic acid metabolism. Moreover, RT-qPCR result showed that the variation trend of six randomly selected DEGs were consistent with that in sequencing data, confirming the reliability of DEGs. Finally, ame-miR-6001-3p was definitely expressed and significantly down-regulated in Am10.【Conclusion】In this work, the expression pattern of DEGs and the regulation network between DEGs and ame-miR-6001-3p as well as the potential role of DEGs during the developmental process of A. m. ligustica worker’s midgut were deeply analyzed. The results revealed that the DEGs may participate in various signaling pathways including TGF-beta, Wnt, Hippo, Notch, PI3K-Akt, mTOR, AMPK, NF-κB signaling pathways, thus affecting the growth and development as well as immune defense of the midgut; DEGs were likely to regulate several signaling pathways such as insulin signaling pathway during the midgut development via formation regulation networks with significantly down-regulated ame-miR-6001-3p.
DOI:10.3864/j.issn.0578-1752.2020.01.019URL [本文引用: 8]
【Objective】The whole transcriptome sequencing of Apis mellifera ligustica 7- and 10-day-old workers’ midguts (Am7 and Am10) was previously conducted. In this study, the differential expression profile and regulation network of genes were investigated to reveal the molecular mechanism underlying the midgut development.【Method】Gene expressions were calculated based on FPKM (fragments per kilobase of transcript per million mapped reads) algorithm, and differentially expressed genes (DEGs) were gained following the standard |log2 fold change|≥1 and P≤0.05. Target mRNAs of ame-miR-6001-3p were predicted utilizing TargetFinder. Annotations of all DEGs in GO and KEGG databases were performed using related software. In addition, DEGs enriched in 13 signaling pathways including AMPK, P13K-Akt, Wnt, cAMP, FoxO, Hippo, mTOR, Jak-STAT, Toll-like receptor, TGF-beta, Notch, MAPK and NF-κB, as well as DEGs targeted by ame-miR-6001-3p were screened out, followed by visualization of enrichment networks and regulation networks with Cytoscape. Finally, Stem loop RT-PCR and RT-qPCR were used to verify the expression and differential expression pattern of ame-miR-6001-3p and DEGs in Am7 and Am10.【Result】A total of 1 038 DEGs were identified in Am7 vs Am10 comparison group, including 515 up- and 523 down-regulated genes, respectively. These DEGs were associated with cellular process, metabolic process and catalytic activity, and significantly enriched in some material and energy metabolisms such as oxidative phosphorylation, amino sugar and nucleotide sugar metabolisms, fatty acid metabolism and purine metabolism, indicative of the active cellular and metabolic activities. Expression cluster analysis suggested that 20, 18, 15 and 14 DEGs were respectively enriched in AMPK signaling pathway, PI3K-Akt signaling pathway, endocytosis and Hippo signaling pathway. In addition, 57 DEGs were enriched in the aforementioned 13 signaling pathways associated with growth and development as well as immune defense, among them one DEG was enriched in several signaling pathways. Moreover, regulation network analysis showed that 54 up-regulated genes and 44 down-regulated genes were targets of ame-miR-6001-3p, respectively; these up-regulated genes were enriched in 43 pathways including inositol phosphate metabolism, Hippo signaling pathway, glutathione metabolism and insulin signaling pathway, while these down-regulated genes were enriched in 20 pathways including Hippo signaling pathway, metabolic pathways, glutathione metabolism and arachidonic acid metabolism. Moreover, RT-qPCR result showed that the variation trend of six randomly selected DEGs were consistent with that in sequencing data, confirming the reliability of DEGs. Finally, ame-miR-6001-3p was definitely expressed and significantly down-regulated in Am10.【Conclusion】In this work, the expression pattern of DEGs and the regulation network between DEGs and ame-miR-6001-3p as well as the potential role of DEGs during the developmental process of A. m. ligustica worker’s midgut were deeply analyzed. The results revealed that the DEGs may participate in various signaling pathways including TGF-beta, Wnt, Hippo, Notch, PI3K-Akt, mTOR, AMPK, NF-κB signaling pathways, thus affecting the growth and development as well as immune defense of the midgut; DEGs were likely to regulate several signaling pathways such as insulin signaling pathway during the midgut development via formation regulation networks with significantly down-regulated ame-miR-6001-3p.
[本文引用: 2]
[本文引用: 2]
DOI:10.3864/j.issn.0578-1752.2019.01.015URL [本文引用: 1]
【Objective】MicroRNA (miRNA) is a kind of key gene expression regulator, which can affect the interactions between host and pathogen. Ascosphaera apis is a lethal fungal pathogen that specifically infects honeybee larvae. The objective of this study is to analyze the differentially expressed miRNAs (DEmiRNAs) and their target genes in the Apis mellifera ligustica larval gut during the early infection stage of A. apis, reveal DEmiRNA’ roles in the stress responses of host at the miRNA omics level, and to screen the key miRNAs related to host response by constructing regulation networks of significant DEmiRNAs. 【Method】Normal and A. apis-infected 4-day-old larval gut of A. m. ligustica (AmCK and AmT) were deep-sequenced using small RNA-seq (sRNA-seq) technology, followed by quality-control of raw data and then mapping of the filtered data with the reference genome of Apis mellifera. The mapped tags were compared to the miRBase database to identify the expression of known miRNAs. The expression of miRNAs in each sample was normalized by TPM (tags per million) algorithm and significant DEmiRNAs were gained according to the standard |log2 fold change|≥1 and P≤0.05. Target genes of significant DEmiRNAs were predicted utilizing TargetFinder, and then annotated to the GO and KEGG databases. Cytoscape was used to visualize the regulation networks between significant DEmiRNAs and target mRNAs. Finally, Stem-loop RT-PCR and qPCR were conducted to verify the reliability of the sequencing data.【Result】sRNA-seq of AmCK and AmT produced 13 553 302 and 10 777 534 raw reads, and after strict filtration, 13 186 921 and 10 480 913 clean reads were obtained, respectively. The Pearson correlation coefficients among different biological replicates in each sample were above 0.9822 and 0.9508. There were 10 significant DEmiRNAs including 4 up-regulated miRNAs and 6 down-regulated miRNAs, and the overall expression level of DEmiRNAs in AmT was lower than that in AmCK. In total, 10 significant DEmiRNAs could link 3 788 target genes. The 1 240 target genes of up-regulated miRNAs could be annotated to 39 GO terms, and the mostly enriched terms were binding, cellular processes, metabolic processes, and response to stimulus. The 749 target genes of down-regulated miRNAs could be annotated to 34 GO terms, and the mostly enriched terms were cellular processes, binding, metabolic processes, and response to stimulus. The result of KEGG database annotation suggested that the target genes of up- and down-regulated miRNAs were respectively annotated in 95 and 66 pathways, the most abundant pathways were Wnt signaling pathway, Hippo signaling pathway, phototransduction and endocytosis, phosphatidylinositol signaling system, as well as purine metabolism. For up- and down-regulated miRNAs, there were 31 and 52 target genes could be annotated to endocytosis, 15 and 7 target genes could be annotated to ubiquitin-mediated proteolysis, 11 and 5 target genes could be annotated to Jak-STAT signaling pathway, 1 and 3 target genes could be annotated to the MAPK signaling pathway, respectively. Complex regulation networks existed between significant DEmiRNAs and their target mRNAs, among them 7 significant DEmiRNAs targeted 96 mRNAs associated with Wnt signaling pathway, and 8 significant DEmiRNAs targeted 55 mRNAs involved in endocytosis. Finally, the results of Stem-loop RT-PCR and qPCR verified the reliability of the sequencing data.【Conclusion】A. m. ligustica larval gut’s DEmiRNAs and their target genes during the early infection stage of A. apis were predicted and analyzed. DEmiRNA-mRNA regulation networks in the host were constructed and investigated. The results provide the expression profile and differential expression information of host miRNAs, and reveal that these DEmiRNAs likely participate in the stress responses of host via regulating biological processes such as cellular activity, metabolism, and immune defense. miR-4331-y, miR-4968-y, miR-8440-y, novel-m0023-5p and novel-m0025-3p jointly regulate Wnt signaling pathway and endocytosis of host and can be used as potential molecular targets for chalkbrood control.
DOI:10.3864/j.issn.0578-1752.2019.01.015URL [本文引用: 1]
【Objective】MicroRNA (miRNA) is a kind of key gene expression regulator, which can affect the interactions between host and pathogen. Ascosphaera apis is a lethal fungal pathogen that specifically infects honeybee larvae. The objective of this study is to analyze the differentially expressed miRNAs (DEmiRNAs) and their target genes in the Apis mellifera ligustica larval gut during the early infection stage of A. apis, reveal DEmiRNA’ roles in the stress responses of host at the miRNA omics level, and to screen the key miRNAs related to host response by constructing regulation networks of significant DEmiRNAs. 【Method】Normal and A. apis-infected 4-day-old larval gut of A. m. ligustica (AmCK and AmT) were deep-sequenced using small RNA-seq (sRNA-seq) technology, followed by quality-control of raw data and then mapping of the filtered data with the reference genome of Apis mellifera. The mapped tags were compared to the miRBase database to identify the expression of known miRNAs. The expression of miRNAs in each sample was normalized by TPM (tags per million) algorithm and significant DEmiRNAs were gained according to the standard |log2 fold change|≥1 and P≤0.05. Target genes of significant DEmiRNAs were predicted utilizing TargetFinder, and then annotated to the GO and KEGG databases. Cytoscape was used to visualize the regulation networks between significant DEmiRNAs and target mRNAs. Finally, Stem-loop RT-PCR and qPCR were conducted to verify the reliability of the sequencing data.【Result】sRNA-seq of AmCK and AmT produced 13 553 302 and 10 777 534 raw reads, and after strict filtration, 13 186 921 and 10 480 913 clean reads were obtained, respectively. The Pearson correlation coefficients among different biological replicates in each sample were above 0.9822 and 0.9508. There were 10 significant DEmiRNAs including 4 up-regulated miRNAs and 6 down-regulated miRNAs, and the overall expression level of DEmiRNAs in AmT was lower than that in AmCK. In total, 10 significant DEmiRNAs could link 3 788 target genes. The 1 240 target genes of up-regulated miRNAs could be annotated to 39 GO terms, and the mostly enriched terms were binding, cellular processes, metabolic processes, and response to stimulus. The 749 target genes of down-regulated miRNAs could be annotated to 34 GO terms, and the mostly enriched terms were cellular processes, binding, metabolic processes, and response to stimulus. The result of KEGG database annotation suggested that the target genes of up- and down-regulated miRNAs were respectively annotated in 95 and 66 pathways, the most abundant pathways were Wnt signaling pathway, Hippo signaling pathway, phototransduction and endocytosis, phosphatidylinositol signaling system, as well as purine metabolism. For up- and down-regulated miRNAs, there were 31 and 52 target genes could be annotated to endocytosis, 15 and 7 target genes could be annotated to ubiquitin-mediated proteolysis, 11 and 5 target genes could be annotated to Jak-STAT signaling pathway, 1 and 3 target genes could be annotated to the MAPK signaling pathway, respectively. Complex regulation networks existed between significant DEmiRNAs and their target mRNAs, among them 7 significant DEmiRNAs targeted 96 mRNAs associated with Wnt signaling pathway, and 8 significant DEmiRNAs targeted 55 mRNAs involved in endocytosis. Finally, the results of Stem-loop RT-PCR and qPCR verified the reliability of the sequencing data.【Conclusion】A. m. ligustica larval gut’s DEmiRNAs and their target genes during the early infection stage of A. apis were predicted and analyzed. DEmiRNA-mRNA regulation networks in the host were constructed and investigated. The results provide the expression profile and differential expression information of host miRNAs, and reveal that these DEmiRNAs likely participate in the stress responses of host via regulating biological processes such as cellular activity, metabolism, and immune defense. miR-4331-y, miR-4968-y, miR-8440-y, novel-m0023-5p and novel-m0025-3p jointly regulate Wnt signaling pathway and endocytosis of host and can be used as potential molecular targets for chalkbrood control.
DOI:10.1038/nrmicro1870URLPMID:18327270 [本文引用: 2]
Recent genetic and molecular analyses have revealed how several strategies enable bacteria to persist and overcome insect immune defences. Genetic and genomic tools that can be used with Drosophila melanogaster have enabled the characterization of the pathways that are used by insects to detect bacterial invaders and combat infection. Conservation of bacterial virulence factors and insect immune repertoires indicates that there are common strategies of host invasion and pathogen eradication. Long-term interactions of bacteria with insects might ensure efficient dissemination of pathogens to other hosts, including humans.
DOI:10.1016/0040-8166(94)90098-1URLPMID:18621268 [本文引用: 1]
The midgut epithelium of the adult honeybee consists of columnar and endocrine cells, both originating from regenerative crypt cells. The regenerative crypt is composed of stem cells and differentiating cells. The stem cells generate two forms of endocrine cells along with differentiating enterocytes of two distinct stages. At first they can be seen as light crypt cells which are not secretory active; they then develop into more electron dense, active secretory crypt cells. The developing enterocytes are arranged as tetrads, each composed of cells with the same degree of differentiation. This can be explained by the occurrence of cytoplasmic bridges between the cells of each tetrad. These fusomes are probably responsible for the apparent intercellular coordination. This is a new example of intercellular bridges between somatic cells and, as far as we know the first description of fusomes in the insect midgut epithelium. The microvilli on top of the crypt cells develop within a spherical extracellular space where glycosaminoglycans are secreted. This occurs by the coordinated activity of the four surrounding electron dense crypt cells. Microvilli formation therefore seems to be the first in a series of successive functions of honeybee enterocytes during their ontogeny.
DOI:10.1007/s13592-012-0171-8URL [本文引用: 1]
Honeybees are subjected to direct contact with transgenic maize pollen due to their feeding activities on pollen. The potential side effects of transgenic cry1Ah-maize pollen on the midgut bacteria of the larvae and worker bees of Apis mellifera ligustica were investigated through denaturing gradient gel electrophoresis under both laboratory and field conditions. Newly emerged bees were fed transgenic cry1Ah-maize pollen, normal maize pollen, linear cry1Ah gene DNA, supercoiled plasmid DNA, and sugar syrup under the laboratory conditions. The results showed that there were no significant differences in the midgut bacterial community composition among the five treatments. No significant effects were observed in the midgut communities between larvae and adult honeybees fed transgenic cry1Ah-maize pollen and non-transgenic maize pollen in the field trials.
DOI:10.3390/insects10080245URL [本文引用: 2]
DOI:10.3390/insects10090258URL [本文引用: 1]
DOI:10.1007/s00284-018-1576-zURLPMID:30269253 [本文引用: 2]
Circular RNAs (circRNAs) are newly discovered endogenous non-coding RNAs (ncRNAs) that play key roles in microRNA function and transcriptional regulation. Though a large number of circRNAs had been identified in animals and plants, however, little is known regarding circRNAs in Nosema ceranae, a widespread fungal parasite of honeybee. In this study, using deep sequencing technology and bioinformatic analysis, we predicted 204 circRNAs from N. ceranae spore samples, including 174 exonic circRNAs and 30 intergenic circRNAs. In addition, the expression of seven N. ceranae circRNAs was confirmed by RT-PCR assay. Furthermore, regulation networks of circRNAs were constructed, and 15 circRNAs were found to act as sponges of the corresponding three miRNAs. GO categorization and pathway enrichment analysis suggested that the circRNAs are likely to play significant roles in N. ceranae spore. This is the first report of circRNAs generated by a microsporidia species. Our results provide novel insights into understanding the basic biology of N. ceranae.
DOI:10.3864/j.issn.0578-1752.2019.17.014URL [本文引用: 1]
【Objective】 The objective of this study is to reveal the cellular and humoral immune responses of Apis mellifera ligusitca worker’s midgut to Nosema ceranae stress, and to provide a basis for screening and functional study of key immune response genes by deep investigation of host differentially expressed genes (DEGs) and immune pathways.【Method】 Base on the previously obtained transcriptome datasets of normal and N. ceranae-stressed midguts of A. m. ligustica 7- and 10-day-old workers’ midgut samples (Am7CK, Am7T, Am10CK and Am10T), DEGs in each comparison group were filtered out following the standard FDR≤1, P≤0.05 and |log2 fold change|≥1. Additionally, Pearson correlations, Venn analysis, GO classification and KEGG pathway enrichment analysis were conducted by using related bioinformatic softwares. Further, DEGs enriched in immune pathways were summarized and analyzed. Finally, the reliability of transcriptome data was verified via real-time quantitative PCR (RT-qPCR).【Result】 Differential expression analysis showed that there were 472 up-regulated and 385 down-regulated genes in Am7CK vs Am7T comparison group, and 611 up-regulated and 360 down-regulated genes in Am10CK vs Am10T comparison group. Venn analysis suggested that the number of specific DEGs of the aforementioned two comparison groups was 739 and 853, respectively, and 118 DEGs were shared. GO classification indicated that the up- and down-regulated genes in Am7CK vs Am7T were respectively annotated to 23 and 29 functional terms; among them, the top five terms of up-regulated genes were binding, catalytic activity, metabolic process, cellular process and single tissue process; while the top five terms of down-regulated genes were metabolic process, single tissue process, catalytic activity, cellular process and binding. In addition, the up- and down-regulated genes in Am10CK vs Am10T were respectively involved in 36 and 26 functional terms; among them, the top five terms of up-regulated genes were single process, binding, cellular process, catalytic activity and metabolic activity; while the top five terms of down-regulated genes were binding, cellular process, catalytic activity, metabolic process and single process. KEGG metabolic pathway enrichment analysis showed that the up- and down-regulated genes in Am7CK vs Am7T were respectively enriched in 38 and 33 pathways, and among them the top five enriched pathways of up-regulated genes were bile secretion, endoplasmic reticulum protein processing, ubiquitin-mediated proteolysis, PI3K-Akt signaling pathway and neurotrophic factor signaling pathway; while the top five enriched pathways of down-regulated genes were cytoplasmic DNA-sensing pathway, purine metabolism, pyrimidine metabolism, RNA polymerase and ribosome; three cellular immune pathways including ubiquitin-mediated proteolysis, and seven humoral pathways including PI3K-Akt signaling pathway were detected. Additionally, the up- and down-regulated genes in Am10CK vs Am10T were respectively associated with 54 and 43 pathways, and among them the top five enriched pathways of up-regulated genes were Hippo signaling pathway, drug metabolism-cytochrome P450, metabolism of xenobiotics by cytochrome P450, ubiquitin-mediated proteolysis, and sphingolipid metabolism; while the top five enriched pathways of down-regulated genes were mRNA surveillance pathway, sphingolipid signaling pathway, fructose and mannose metabolism, galactose metabolism, and sphingolipid metabolism; seven cellular immune pathways including ubiquitin-mediated proteolysis, and two humoral pathways including NF-κB signaling pathway were found. The result of RT-qPCR verification demonstrated the expression trend of six randomly selected DEGs was consistent with that of the sequencing data, which confirmed the reliability of the transcriptome data. Further analysis noted that NF-κB signaling pathway was activated by N. ceranae in both 7- and 10-day-old workers’ midguts of A. m. ligustica, followed by immediate initiation of three antimicrobial peptides such as apidaecin, defensin-1 and hymenoptaecin, indicating their key importance in host defense against N. ceranae invasion.【Conclusion】 The immune responses of A. m. ligusitca worker’s midgut to N. ceranae were uncovered at transcriptomic level, and it was revealed that the host made both cellular and humoral immune responses at the early stage of N. ceranae stress. The host cellular immune responses may play a major role in resisting pathogen invasion. The host cellular immune continued to increase at the later stage of fungal stress, while the humoral immune drastically weakened. The ubiquitin mediated proteolysis and enriched DEGs, NF-κB signaling pathway and enriched DEGs, and antibacterial peptide-encoded genes such as apidaecin, defensin-1 and hymenoptaecin were likely to play pivotal roles in host immune response to N. ceranae stress.
DOI:10.3864/j.issn.0578-1752.2019.17.014URL [本文引用: 1]
【Objective】 The objective of this study is to reveal the cellular and humoral immune responses of Apis mellifera ligusitca worker’s midgut to Nosema ceranae stress, and to provide a basis for screening and functional study of key immune response genes by deep investigation of host differentially expressed genes (DEGs) and immune pathways.【Method】 Base on the previously obtained transcriptome datasets of normal and N. ceranae-stressed midguts of A. m. ligustica 7- and 10-day-old workers’ midgut samples (Am7CK, Am7T, Am10CK and Am10T), DEGs in each comparison group were filtered out following the standard FDR≤1, P≤0.05 and |log2 fold change|≥1. Additionally, Pearson correlations, Venn analysis, GO classification and KEGG pathway enrichment analysis were conducted by using related bioinformatic softwares. Further, DEGs enriched in immune pathways were summarized and analyzed. Finally, the reliability of transcriptome data was verified via real-time quantitative PCR (RT-qPCR).【Result】 Differential expression analysis showed that there were 472 up-regulated and 385 down-regulated genes in Am7CK vs Am7T comparison group, and 611 up-regulated and 360 down-regulated genes in Am10CK vs Am10T comparison group. Venn analysis suggested that the number of specific DEGs of the aforementioned two comparison groups was 739 and 853, respectively, and 118 DEGs were shared. GO classification indicated that the up- and down-regulated genes in Am7CK vs Am7T were respectively annotated to 23 and 29 functional terms; among them, the top five terms of up-regulated genes were binding, catalytic activity, metabolic process, cellular process and single tissue process; while the top five terms of down-regulated genes were metabolic process, single tissue process, catalytic activity, cellular process and binding. In addition, the up- and down-regulated genes in Am10CK vs Am10T were respectively involved in 36 and 26 functional terms; among them, the top five terms of up-regulated genes were single process, binding, cellular process, catalytic activity and metabolic activity; while the top five terms of down-regulated genes were binding, cellular process, catalytic activity, metabolic process and single process. KEGG metabolic pathway enrichment analysis showed that the up- and down-regulated genes in Am7CK vs Am7T were respectively enriched in 38 and 33 pathways, and among them the top five enriched pathways of up-regulated genes were bile secretion, endoplasmic reticulum protein processing, ubiquitin-mediated proteolysis, PI3K-Akt signaling pathway and neurotrophic factor signaling pathway; while the top five enriched pathways of down-regulated genes were cytoplasmic DNA-sensing pathway, purine metabolism, pyrimidine metabolism, RNA polymerase and ribosome; three cellular immune pathways including ubiquitin-mediated proteolysis, and seven humoral pathways including PI3K-Akt signaling pathway were detected. Additionally, the up- and down-regulated genes in Am10CK vs Am10T were respectively associated with 54 and 43 pathways, and among them the top five enriched pathways of up-regulated genes were Hippo signaling pathway, drug metabolism-cytochrome P450, metabolism of xenobiotics by cytochrome P450, ubiquitin-mediated proteolysis, and sphingolipid metabolism; while the top five enriched pathways of down-regulated genes were mRNA surveillance pathway, sphingolipid signaling pathway, fructose and mannose metabolism, galactose metabolism, and sphingolipid metabolism; seven cellular immune pathways including ubiquitin-mediated proteolysis, and two humoral pathways including NF-κB signaling pathway were found. The result of RT-qPCR verification demonstrated the expression trend of six randomly selected DEGs was consistent with that of the sequencing data, which confirmed the reliability of the transcriptome data. Further analysis noted that NF-κB signaling pathway was activated by N. ceranae in both 7- and 10-day-old workers’ midguts of A. m. ligustica, followed by immediate initiation of three antimicrobial peptides such as apidaecin, defensin-1 and hymenoptaecin, indicating their key importance in host defense against N. ceranae invasion.【Conclusion】 The immune responses of A. m. ligusitca worker’s midgut to N. ceranae were uncovered at transcriptomic level, and it was revealed that the host made both cellular and humoral immune responses at the early stage of N. ceranae stress. The host cellular immune responses may play a major role in resisting pathogen invasion. The host cellular immune continued to increase at the later stage of fungal stress, while the humoral immune drastically weakened. The ubiquitin mediated proteolysis and enriched DEGs, NF-κB signaling pathway and enriched DEGs, and antibacterial peptide-encoded genes such as apidaecin, defensin-1 and hymenoptaecin were likely to play pivotal roles in host immune response to N. ceranae stress.
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1126/science.1136606URLPMID:17303754 [本文引用: 1]
The adult Drosophila midgut contains multipotent intestinal stem cells (ISCs) scattered along its basement membrane that have been shown by lineage analysis to generate both enterocytes and enteroendocrine cells. ISCs containing high levels of cytoplasmic Delta-rich vesicles activate the canonical Notch pathway and down-regulate Delta within their daughters, a process that programs these daughters to become enterocytes. ISCs that express little vesiculate Delta, or are genetically impaired in Notch signaling, specify their daughters to become enteroendocrine cells. Thus, ISCs control daughter cell fate by modulating Notch signaling over time. Our studies suggest that ISCs actively coordinate cell production with local tissue requirements by this mechanism.
URLPMID:1295737 [本文引用: 1]
The complex embryonic phenotype of the six neurogenic mutations Notch, mastermind, big brain, Delta, Enhancer of split and neuralized was analyzed by using different antibodies and PlacZ markers, which allowed us to label most of the known embryonic tissues. Our results demonstrate that all of the neurogenic mutants show abnormalities in many different organs derived from all three germ layers. Defects caused by the neurogenic mutations in ectodermally derived tissues fell into two categories. First, all cell types that delaminate from the ectoderm (neuroblasts, sensory neurons, peripheral glia cells and oenocytes) are increased in number. Secondly, ectodermal tissues that in the wild type form epithelial structures lose their epithelial phenotype and dissociate (optic lobe, stomatogastric nervous system) or show significant differentiative abnormalities (trachea, Malpighian tubules and salivary gland). Abnormalities in tissues derived from the mesoderm were observed in all six neurogenic mutations. Most importantly, somatic myoblasts do not fuse and/or form an aberrant muscle pattern. Cardioblasts (which form the embryonic heart) are increased in number and show differentiative abnormalities; other mesodermal cell types (fat body, pericardial cells) are significantly decreased. The development of the endoderm (midgut rudiments) is disrupted in most of the neurogenic mutations (Notch, Delta, Enhancer of split and neuralized) during at least two stages. Defects occur as early as during gastrulation when the invaginating midgut rudiments prematurely lose their epithelial characteristics. Later, the transition of the midgut rudiments to form the midgut epithelium does not occur. In addition, the number of adult midgut precursor cells that segregate from the midgut rudiments is strongly increased. We propose that, at least in the ectodermally and endodermally derived tissues, neurogenic gene function is primarily involved in interactions among cells that need to acquire or to maintain an epithelial phenotype.
DOI:10.1038/nature04371URLPMID:16340959 [本文引用: 1]
Adult stem cells maintain organ systems throughout the course of life and facilitate repair after injury or disease. A fundamental property of stem and progenitor cell division is the capacity to retain a proliferative state or generate differentiated daughter cells; however, little is currently known about signals that regulate the balance between these processes. Here, we characterize a proliferating cellular compartment in the adult Drosophila midgut. Using genetic mosaic analysis we demonstrate that differentiated cells in the epithelium arise from a common lineage. Furthermore, we show that reduction of Notch signalling leads to an increase in the number of midgut progenitor cells, whereas activation of the Notch pathway leads to a decrease in proliferation. Thus, the midgut progenitor's default state is proliferation, which is inhibited through the Notch signalling pathway. The ability to identify, manipulate and genetically trace cell lineages in the midgut should lead to the discovery of additional genes that regulate stem and progenitor cell biology in the gastrointestinal tract.
DOI:10.1038/nature04333URLPMID:16340960 [本文引用: 1]
Vertebrate and invertebrate digestive systems show extensive similarities in their development, cellular makeup and genetic control. The Drosophila midgut is typical: enterocytes make up the majority of the intestinal epithelial monolayer, but are interspersed with hormone-producing enteroendocrine cells. Human (and mouse) intestinal cells are continuously replenished by stem cells, the misregulation of which may underlie some common digestive diseases and cancer. In contrast, stem cells have not been described in the intestines of flies, and Drosophila intestinal cells have been thought to be relatively stable. Here we use lineage labelling to show that adult Drosophila posterior midgut cells are continuously replenished by a distinctive population of intestinal stem cells (ISCs). As in vertebrates, ISCs are multipotent, and Notch signalling is required to produce an appropriate fraction of enteroendocrine cells. Notch is also required for the differentiation of ISC daughter cells, a role that has not been addressed in vertebrates. Unlike previously characterized stem cells, which reside in niches containing a specific partner stromal cell, ISCs adjoin only the basement membrane, differentiated enterocytes and their most recent daughters. The identification of Drosophila intestinal stem cells with striking similarities to their vertebrate counterparts will facilitate the genetic analysis of normal and abnormal intestinal function.
DOI:10.1371/journal.pgen.1005822URLPMID:26845150 [本文引用: 1]
Intestinal stem cell (ISC) self-renewal and proliferation are directed by Wnt/beta-catenin signaling in mammals, whereas aberrant Wnt pathway activation in ISCs triggers the development of human colorectal carcinoma. Herein, we have utilized the Drosophila midgut, a powerful model for ISC regulation, to elucidate the mechanisms by which Wingless (Wg)/Wnt regulates intestinal homeostasis and development. We provide evidence that the Wg signaling pathway, activation of which peaks at each of the major compartment boundaries of the adult intestine, has essential functions. Wg pathway activation in the intestinal epithelium is required not only to specify cell fate near compartment boundaries during development, but also to control ISC proliferation within compartments during homeostasis. Further, in contrast with the previous focus on Wg pathway activation within ISCs, we demonstrate that the primary mechanism by which Wg signaling regulates ISC proliferation during homeostasis is non-autonomous. Activation of the Wg pathway in absorptive enterocytes is required to suppress JAK-STAT signaling in neighboring ISCs, and thereby their proliferation. We conclude that Wg signaling gradients have essential roles during homeostasis and development of the adult intestine, non-autonomously controlling stem cell proliferation inside compartments, and autonomously specifying cell fate near compartment boundaries.
DOI:10.1073/pnas.1012759107URLPMID:21078993 [本文引用: 1]
Intestinal stem cells (ISCs) in the Drosophila adult midgut are essential for maintaining tissue homeostasis and replenishing lost cells in response to tissue damage. Here we demonstrate that the Hippo (Hpo) signaling pathway, an evolutionarily conserved pathway implicated in organ size control and tumorigenesis, plays an essential role in regulating ISC proliferation. Loss of Hpo signaling in either midgut precursor cells or epithelial cells stimulates ISC proliferation. We provide evidence that loss of Hpo signaling in epithelial cells increases the production of cytokines of the Upd family and multiple EGFR ligands that activate JAK-STAT and EGFR signaling pathways in ISCs to stimulate their proliferation, thus revealing a unique non-cell-autonomous role of Hpo signaling in blocking ISC proliferation. Finally, we show that the Hpo pathway mediator Yorkie (Yki) is also required in precursor cells for injury-induced ISC proliferation in response to tissue-damaging reagent DSS.
[D].
[本文引用: 1]
[D].
[本文引用: 1]
DOI:10.1186/1472-6785-6-4URLPMID:16551367 [本文引用: 1]
BACKGROUND: Honey bees, Apis mellifera, face many parasites and pathogens and consequently rely on a diverse set of individual and group-level defenses to prevent disease. One route by which honey bees and other insects might combat disease is through the shielding effects of their microbial symbionts. Bees carry a diverse assemblage of bacteria, very few of which appear to be pathogenic. Here we explore the inhibitory effects of these resident bacteria against the primary bacterial pathogen of honey bees, Paenibacillus larvae. RESULTS: Here we isolate, culture, and describe by 16S rRNA and protein-coding gene sequences 61 bacterial isolates from honey bee larvae, reflecting a total of 43 distinct bacterial taxa. We culture these bacteria alongside the primary larval pathogen of honey bees, Paenibacillus larvae, and show that many of these isolates severely inhibit the growth of this pathogen. Accordingly, symbiotic bacteria including those described here are plausible natural antagonists toward this widespread pathogen. CONCLUSION: The results suggest a tradeoff in social insect colonies between the maintenance of potentially beneficial bacterial symbionts and deterrence at the individual and colony level of pathogenic species. They also provide a novel mechanism for recently described social components behind disease resistance in insect colonies, and point toward a potential control strategy for an important bee disease.
DOI:10.1242/dev.02411URLPMID:16794031 [本文引用: 1]
High levels of interspecies conservation characterise all signal transduction cascades and demonstrate the significance of these pathways over evolutionary time. Here, we review advances in the field of JAK/STAT signalling, focusing on recent developments in Drosophila. In particular, recent results from genetic and genome-wide RNAi screens, as well as studies into the developmental roles played by this pathway, highlight striking levels of physical and functional conservation in processes such as cellular proliferation, immune responses and stem cell maintenance. These insights underscore the value of model organisms for improving our understanding of this human disease-relevant pathway.
DOI:10.5483/bmbrep.2005.38.2.121URLPMID:15826489 [本文引用: 1]
Drosophila protects itself from infection by microbial organisms by means of its pivotal defense, the so-called innate immunity system. This is its sole defense as it lacks an adaptive immunity system such as is found in mammals. The strong conservation of innate immunity systems in organisms from Drosophila to mammals, and the ease with which Drosophila can be manipulated genetically, makes this fly a good model system for investigating the mechanisms of virulence of a number of medically important pathogens. Potentially damaging endogenous and/or exogenous challenges sensed by specific receptors initiate signals via the Toll and/or Imd signaling pathways. These in turn activate the transcription factors Dorsal, Dorsal-related immune factor (Dif) and Relish, culminating in transcription of genes involved in the production of antimicrobial peptides, melanization, phagocytosis, and the cytoskeletal rearrangement required for appropriate responses. Clarifying the regulatory interactions between the various pathways involved is very important for understanding the specificity and termination mechanism of the immune response.
DOI:10.1111/hel.12152URLPMID:25495001 [本文引用: 1]
BACKGROUND: Helicobacter pylori (H. pylori) infection leads to acute induction of Sonic Hedgehog (Shh) in the stomach that is associated with the initiation of gastritis. The mechanism by which H. pylori induces Shh is unknown. Shh is a target gene of transcription factor Nuclear Factor-kappaB (NFkappaB). We hypothesize that NFkappaB mediates H. pylori-induced Shh. MATERIALS AND METHODS: To visualize Shh ligand expression in response to H. pylori infection in vivo, we used a mouse model that expresses Shh fused to green fluorescent protein (Shh::GFP mice) in place of wild-type Shh. In vitro, changes in Shh expression were measured in response to H. pylori infection using 3-dimensional epithelial cell cultures grown from whole dissociated gastric glands (organoids). Organoids were generated from stomachs collected from the fundic region of control and mice expressing a parietal cell-specific deletion of Shh (PC-Shh(KO) mice). RESULTS: Within 2 days of infection, H. pylori induced Shh expression within parietal cells of Shh::GFP mice. Organoids expressed all major gastric cell markers, including parietal cell marker H(+) ,K(+) -ATPase and Shh. H. pylori infection of gastric organoids induced Shh expression; a response that was blocked by inhibiting NFkappaB signaling and correlated with IkappaB degradation. H. pylori infection of PC-Shh(KO) mouse-derived organoids did not result in the induction of Shh expression. CONCLUSION: Gastric organoids allow for the study of the interaction between H. pylori and the differentiated gastric epithelium independent of the host immune response. H. pylori induces Shh expression from the parietal cells, a response mediated via activation of NFkappaB signaling.
URLPMID:7601337 [本文引用: 1]
The transmission of extracellular signals into their intracellular targets is mediated by a network of interacting proteins that regulate a large number of cellular processes. Cumulative efforts from many laboratories over the past decade have allowed the elucidation of one such signaling mechanism, which involves activations of several membranal signaling molecules followed by a sequential stimulation of several cytoplasmic protein kinases collectively known as mitogen-activated protein kinase (MAPK) signaling cascade. Up to six tiers in this cascade contribute to the amplification and specificity of the transmitted signals that eventually activate several regulatory molecules in the cytoplasm and in the nucleus to initiate cellular processes such as proliferation, differentiation, and development. Moreover, because many oncogenes have been shown to encode proteins that transmit mitogenic signals upstream of this cascade, the MAPK pathway provides a simple unifying explanation for the mechanism of action of most, if not all, nonnuclear oncogenes. The pattern of MAPK cascade is not restricted to growth factor signaling and it is now known that signaling pathways initiated by phorbol esters, ionophors, heat shock, and ligands for seven transmembrane receptors use distinct MAPK cascades with little or no cross-reactivity between them. In this review we emphasize primarily the first MAPK cascade to be discovered that uses the MEK and ERK isoforms and describe their involvement in different cellular processes.
DOI:10.4049/jimmunol.165.5.2671URL [本文引用: 1]
DOI:10.1042/bj3530417URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1186/s12885-019-5983-8URLPMID:31391008 [本文引用: 1]
BACKGROUND: Numerous studies have highlighted that long non-coding RNAs (lncRNAs) can bind to microRNA (miRNA) sites as competing endogenous RNAs (ceRNAs), thereby affecting and regulating the expression of mRNAs and target genes. These lncRNA-associated ceRNAs have been theorized to play a significant role in cancer initiation and progression. However, the roles and functions of the lncRNA-miRNA-mRNA ceRNA network in squamous cell carcinoma of the tongue (SCCT) are still unclear. METHODS: The miRNA, mRNA and lncRNA expression profiles from 138 patients with SCCT were downloaded from The Cancer Genome Atlas database. We identified the differential expression of miRNAs, mRNAs, and lncRNAs using the limma package of R software. We used the clusterProfiler package for GO and KEGG pathway annotations. The survival package was used to estimate survival analysis according to the Kaplan-Meier curve. Finally, the GDCRNATools package was used to construct the lncRNA-miRNA-mRNA ceRNA network. RESULTS: In total, 1943 SCCT-specific mRNAs, 107 lncRNAs and 100 miRNAs were explored. Ten mRNAs (CSRP2, CKS2, ADGRG6, MB21D1, GMNN, RIPOR3, RAD51, PCLAF, ORC1, NAGS), 9 lncRNAs (LINC02560, HOXC13 - AS, FOXD2 - AS1, AC105277.1, AC099850.3, STARD4 - AS1, SLC16A1 - AS1, MIR503HG, MIR100HG) and 8 miRNAs (miR - 654, miR - 503, miR - 450a, miR - 379, miR - 369, miR - 190a, miR - 101, and let-7c) were found to be significantly associated with overall survival (log-rank p < 0.05). Based on the analysis of the lncRNA-miRNA-mRNA ceRNA network, one differentially expressed (DE) lncRNA, five DEmiRNAs, and three DEmRNAs were demonstrated to be related to the pathogenesis of SCCT. CONCLUSIONS: In this study, we described the gene regulation by the lncRNA-miRNA-mRNA ceRNA network in the progression of SCCT. We propose a new lncRNA-associated ceRNA that could help in the diagnosis and treatment of SCCT.
[本文引用: 1]
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}