 ,福建农林大学动物科学学院(蜂学学院),福州 350002
,福建农林大学动物科学学院(蜂学学院),福州 350002Investigation of Competing Endogenous RNA Regulatory Network and Putative Function of Long Non-Coding RNAs in Nosema ceranae Spore
ZHOU DingDing, SHI XiaoYu, WANG Jie, FAN YuanChan, ZHU ZhiWei, JIANG HaiBin, FAN XiaoXue, XIONG CuiLing, ZHENG YanZhen, FU ZhongMin, XU GuoJun, CHEN DaFu, GUO Rui,College of Animal Sciences (College of Bee Science), Fujian Agriculture and Forestry University, Fuzhou 350002通讯作者:
责任编辑: 岳梅
收稿日期:2019-10-23接受日期:2019-11-26网络出版日期:2020-05-16
| 基金资助: |
Received:2019-10-23Accepted:2019-11-26Online:2020-05-16
作者简介 About authors
周丁丁,E-mail:zdd03569981@163.com。
史小玉,E-mail:2395965008@qq.com。

摘要
关键词:
Abstract
Keywords:
PDF (2655KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
周丁丁, 史小玉, 王杰, 范元婵, 祝智威, 蒋海宾, 范小雪, 熊翠玲, 郑燕珍, 付中民, 徐国钧, 陈大福, 郭睿. 东方蜜蜂微孢子虫孢子中长链非编码RNA的竞争性内源RNA调控网络及潜在功能[J]. 中国农业科学, 2020, 53(10): 2122-2136 doi:10.3864/j.issn.0578-1752.2020.10.018
ZHOU DingDing, SHI XiaoYu, WANG Jie, FAN YuanChan, ZHU ZhiWei, JIANG HaiBin, FAN XiaoXue, XIONG CuiLing, ZHENG YanZhen, FU ZhongMin, XU GuoJun, CHEN DaFu, GUO Rui.
0 引言
【研究意义】东方蜜蜂微孢子虫(Nosema ceranae)是一种常见的蜜蜂真菌病原,特异性侵染成年蜜蜂的中肠上皮细胞,导致宿主产生诸多生理和病理变化[1,2,3]。目前,有关东方蜜蜂微孢子虫非编码RNA(non-coding RNA,ncRNA)的信息尤为有限[4,5]。利用组学技术探究东方蜜蜂微孢子虫长链非编码RNA(long non-coding RNA,lncRNA)的不同作用方式及潜在功能,可为探明东方蜜蜂微孢子虫孢子发芽以及在宿主细胞内的增殖相关机制提供依据,筛选出的关键lncRNA有望为蜜蜂微孢子虫病的诊断和治疗提供新型分子标记和靶点。【前人研究进展】真核细胞中存在大量的ncRNA,其中lncRNA是一类长度>200 nt,没有长阅读框,由RNA聚合酶Ⅱ转录,同时5′端加帽与3′端加尾而形成的转录本,具有复杂而特定的二级结构,通过支架、向导和诱饵等方式参与调节细胞的增殖、分化、凋亡及代谢等生命活动[4,5,6]。有研究表明lncRNA可通过顺式(cis)作用调控同一染色体上邻近蛋白编码基因的表达,或通过反式(trans)作用调控距离较远基因的转录激活及表达[6,7,8,9]。此外,含有微小RNA反应元件(microRNA response element,MRE)的lncRNA还能够作为“分子海绵”吸附结合miRNA,通过发挥竞争性内源RNA(competing endogenous RNA,ceRNA)调控作用间接影响miRNA与mRNA的靶向结合[6,10-11]。基于链特异性建库策略的二代测序技术的发展与成熟为在全基因组水平鉴定lncRNA提供了强大工具[6,12]。目前,已在人类[13]、小鼠[14]、拟南芥[15]、果蝇[16]等模式生物中鉴定出大量lncRNA。但相比较而言,真菌的lncRNA研究起步较晚且发展滞后。FAUQUENOY等[9]运用生物信息学方法在裂殖酵母(Schizosaccharomyces pombe)中筛选出与mRNA邻近的68个lncRNA,进一步研究发现lncRNA对邻近蛋白编码基因具有调控作用;LIU等[17]利用RNA-seq技术对未发芽和发芽的家蚕微孢子虫(Nosema bombycis)进行转录组测序,基于测序数据分别组装出2 756和2 690个unigenes,进而通过比较分析筛选出66个显著差异表达基因。然而对于东方蜜蜂微孢子虫等蜜蜂病原,lncRNA的相关信息极其匮乏。GUO等[18]通过基于链特异性cDNA文库的lncRNA-seq技术对蜜蜂白垩病病原蜜蜂球囊菌(Ascosphaera apis,简称球囊菌)的菌丝和孢子混合样品进行测序,通过生物信息学方法鉴定出379个lncRNA,进一步分析发现这些lncRNA具有类似于其他物种已报道lncRNA的结构特征;ATKINSON[19]利用RNA-seq技术在裂殖酵母中鉴定出5 775个lnRNA,进而通过结构优化发现其中的1 557个lnRNA已被注释,多数lncRNA表达与mRNA表达呈正相关,这些lncRNA的cis作用及核内的共转录功能介导了裂殖酵母的调控机制;GUO等[20]利用lncRNA的cDNA芯片技术对人的慢性骨髓白血病细胞的lncRNA进行了全面分析,发现lncRNA-BGL3作为一种ceRNA靶向结合miR-17家族的多个miRNA,进而调控肿瘤抑制因子PTEN相关蛋白编码基因的表达。【本研究切入点】目前,东方蜜蜂微孢子虫的lncRNA研究非常滞后,相关信息匮乏。笔者团队前期已利用基于cDNA链特异性文库的lncRNA-seq技术对东方蜜蜂微孢子虫纯化孢子进行深度测序,联用CPC、CNCI、CPAT和Pfam软件鉴定出83个lncRNA,进而对上述lncRNA的种类和结构特征进行了初步分析,并利用RT-PCR验证了其中13个lncRNA的真实表达[21]。然而,这些lncRNA在东方蜜蜂微孢子虫孢子中具有怎样的调控功能以及如何发挥作用仍未可知。【拟解决的关键问题】利用small RNA-seq(sRNA-seq)技术对东方蜜蜂微孢子虫纯化孢子进行测序,结合高质量的sRNA数据和前期获得的lncRNA数据进一步深入分析东方蜜蜂微孢子虫lncRNA的cis作用和ceRNA作用,为东方蜜蜂微孢子虫的lncRNA信息提供有益补充,并揭示lncRNA在东方蜜蜂微孢子虫孢子中的潜在功能。1 材料与方法
试验于2018年12月至2019年4月在福建农林大学完成。1.1 生物材料
供试东方蜜蜂微孢子虫纯化孢子由福建农林大学动物科学学院(蜂学学院)蜜蜂保护实验室制备和保存[18,21]。1.2 LncRNA组学数据来源
前期研究中,笔者团队通过Percoll不连续密度梯度离心获得了东方蜜蜂微孢子虫的纯化孢子,利用lncRNA-seq技术对纯化孢子(NcL-1、NcL-2、NcL-3)进行了深度测序,获得了高质量的lncRNA组学数据[21]。链特异性cDNA文库构建及高通量测序委托北京百迈克生物科技有限公司完成,测序平台为Illumina HiSeq Xten。LncRNA-seq的原始数据已上传NCBI SRA数据库,BioProject号:PRJNA395264。1.3 sRNA-seq及数据质控
在东方蜜蜂微孢子虫的纯化孢子中加入适量液氮充分研磨至粉末,利用Trizol法分别抽提3份纯化孢子样品(NcS-1、NcS-2、NcS-3)的总RNA,通过Nanodrop超微量测定仪(Thermo公司,美国)检测上述RNA的浓度,通过Qubit 2.0(Life公司,美国)和Agilent 2100 bioanalyzer(Agilent公司,美国)分别检测上述RNA的纯度和完整性。对于检测合格的RNA,以1.5 μL的量作为RNA样本起始量,用无菌水补充体积至6 μL,使用small RNA Sample Pre Kit试剂盒(Illumina公司,美国)进行文库构建。由于sRNA的5′端含磷酸基团,3′端含羟基,利用T4 RNA Ligase 1和T4 RNA Ligase 2分别在sRNA 的3′端和5′端连接上接头序列,反转录合成cDNA,再进行PCR扩增,采用胶分离技术筛选目的片段,切胶回收得到的片段即为sRNA文库。文库构建完成后,使用Qubit 2.0对文库的浓度进行检测,将文库浓度稀释至1 ng·μL-1,使用Agilent 2100 bioanalyzer(Agilent公司,美国)对Insert Size进行检测,使用qPCR方法对文库的有效浓度进行准确定量,以保证文库质量。委托北京百迈克生物科技有限公司进行sRNA-seq,测序平台为Illumina MiSeq,sRNA的测序读长为single-end (SE) 50 nt。sRNA-seq的原始数据已上传NCBI SRA数据库,BioProject号:PRJNA395137。对于下机的原始数据,因含有接头序列或低质量序列,首先以以下流程进行数据质控:(1)去除低质量值的读段(reads);(2)去除未知碱基(N)含量≥10%的reads;(3)去除没有3′接头序列的reads;(4)剪切掉3′接头序列;(5)去除长度<18 nt和>30 nt的序列。经严格质控后得到的数据即为有效读段(clean reads)。
进一步通过Bowtie软件将clean reads分别与Silva数据库(
1.4 LncRNA上下游基因的功能注释
基于东方蜜蜂微孢子虫纯化孢子的lncRNA-seq数据,笔者团队前期已联用CPC、CNCI、CPAT和Pfam软件分别预测出162、295、342和206条lncRNA,四者的交集为83条lncRNA[21]。位于编码蛋白基因上下游的lncRNA可能在转录或转录后水平对其邻近蛋白编码基因的表达具有调控作用[8,9]。因此,将lncRNA上下游100 kb范围内的邻近基因作为其调控的潜在靶基因。利用Blast软件对lncRNA的上下游基因进行GO数据库(1.5 LncRNA的靶miRNA预测与ceRNA调控网络构建及分析
参照郭睿等[22]的方法,利用Target Finder软件[23]预测东方蜜蜂微孢子虫孢子中lncRNA 靶向结合的miRNA及miRNA靶向结合的mRNA,软件参数设置为:自由能≤-20 kcal·mol-1,空位开度=-0.9,空位扩展=-4.0。利用三者之间的靶向结合关系构建lncRNA- miRNA及lncRNA-miRNA-mRNA调控网络,并通过Cytoscape v3.7.2软件[24]对上述调控网络进行可视化。1.6 LncRNA和靶mRNA的RT-PCR验证
根据1.5中靶向预测的结果,随机选取2个lncRNA(MSTRG.3636.1和MSTRG.4883.1)和8个靶mRNA(gene565、gene1719、gene1695、gene1563、gene1439、gene1366、gene1360和gene1134)进行RT-PCR验证。根据上述lncRNA和mRNA的核酸序列利用DNAMAN软件设计相应的特异性引物,委托上海生工生物工程有限公司合成引物(表1)。参照GUO等[21]的方法,利用RNA抽提试剂盒(TaKaRa,中国)提取东方蜜蜂微孢子虫孢子的总RNA,作为模板进行反转录,得到的cDNA作为模板进行PCR。PCR反应体系(20 μL):cDNA 模板1 μL,上游引物1 μL,下游引物1 μL,PCR mix 10 μL,无菌水7 μL。将无菌水设为阴性对照,反应体系(20 μL):无菌水模板1 μL,上述2个lncRNA和8个靶mRNA的混合上游引物1 μL(每种0.1 μL)、混合下游引物 1 μL(每种0.1 μL),PCR mix 10 μL,无菌水7 μL。PCR程序:95℃预变性5 min;95℃变性50 s,55℃退火30 s,72℃延伸50 s,共35个循环;72℃再延伸10 min。PCR产物经1.6%琼脂糖凝胶电泳和凝胶成像仪(上海培清,中国)检测。Table 1
表1
表1本研究使用的引物
Table 1
| 核酸ID Nucleotide ID | 引物名称 Primer name | 引物序列 Primer sequence (5′-3′) | 预期目的片段 Product size (bp) |
|---|---|---|---|
| gene1439 | 1-F | ATGTATCTTATGCCAACGC | 382 |
| 1-R | TGAATCTTTCCTACACCCT | ||
| gene565 | 2-F | TCAAGCCTGTTAAGATGAC | 499 |
| 2-R | ACTACTTCTGGCGGTTTAT | ||
| gene1719 | 3-F | ATAGGAGGTAAACATCAG | 438 |
| 3-R | ATAGTAAAGTTCAGTGCC | ||
| gene1366 | 4-F | TTGAAGAAGGTGTCGCAGAAC | 182 |
| 4-R | AGCAGAGCCACATAAACAAGC | ||
| gene1360 | 5-F | AAGAAGCCGAAACAACCC | 101 |
| 5-R | GTTAGCCGCATCCATAGC | ||
| gene1134 | 6-F | TGTACGGTCTGGCTGCTTC | 352 |
| 6-R | GGACTTGGCTTTCCACTAA | ||
| gene1695 | 7-F | GATACAGCCTCTTTGG | 138 |
| 7-R | GGAACAATAACCCTAAA | ||
| gene1563 | 8-F | TGCCTACAATCAACTCTAAT | 384 |
| 8-R | CACCCAATACTTCAAATAAC | ||
| MSTRG.3636.1 | 9-F | TAACTAGCCTACTCTATCCC | 257 |
| 9-R | TAAAAGCACTAACTACAACC | ||
| MSTRG.4883.1 | 10-F | CTGACAGACCATAGTCGGCATA | 128 |
| 10-R | TCTACAATGATGGGCACAGGA | ||
| nce-miR-8565 | 11-L | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCCTAAAGA | 67 |
| 11-F | CGCGCCTTTAATTGTGAAACATG | ||
| nce-miR-7502 | 12-L | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGAACTTGTC | 66 |
| 12-F | CGCGCCTAAAAACAAAAATGTA | ||
| nce-miR-7729 | 13-L | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTCTAGGCC | 62 |
| 13-F | CGCGCCTAGTGATGGCTGT | ||
| - | R | CTCAACTGGTGTCGTGGA |
新窗口打开|下载CSV
1.7 MiRNA的Stem-loop RT-PCR验证
通过Stem-loop RT-PCR验证对ceRNA调控网络中3个靶miRNA(nce-miR-7502、nce-miR-7729和nce-miR-8565)的表达进行验证。利用RNA抽提试剂盒(TaKaRa公司,日本)提取东方蜜蜂微孢子虫孢子的总RNA。参照郭睿等[25]和杜宇等[26]的方法,利用 DNAMAN软件(LynnonBiosoft公司,美国)设计 miRNA的Stem-loop引物、上游和下游引物,委托上海生工生物工程有限公司合成引物(表1)。利用Stem-loop引物经反转录得到cDNA,作为模板进行PCR扩增。PCR体系(20 μL):上下游引物(1.67 μmol·L-1)和cDNA模板各1 μL,PCR mix 10 μL,无菌水7 μL。将DEPC水设为阴性对照,反应体系(20 μL):DEPC水模板1 μL,上述3个靶miRNA的混合上游引物0.9 μL(每种0.3 μL)、混合下游引物0.9 μL(每种0.3 μL),PCR mix 10 μL,无菌水7.2 μL。PCR 程序:95℃ 5 min;95℃ 30 s,46℃ 30 s,72℃ 1 min,35个循环;72℃ 10 min。PCR产物经1.6%琼脂糖凝胶电泳检测。2 结果
2.1 LncRNA-seq及sRNA-seq的数据质控
前期研究中,东方蜜蜂微孢子虫纯化孢子样品(NcL-1、NcL-2、NcL-3)的lncRNA-seq得到的clean reads数分别为107 123 113、117 688 499和116 776 007条,占raw reads的比例分别为79.75%、78.84%和76.15%;Q30分别达到90.39%、90.49%和89.79%[21]。高质量的lncRNA组学数据可用于本研究中lncRNA的生物信息学分析。东方蜜蜂微孢子虫纯化孢子样品的sRNA-seq分别得到16 597 883、15 451 791和12 248 316条 raw reads,经过严格过滤得到的clean reads数分别为15 608 370(94.04%)、14 249 255(92.22%)和11 440 684(93.41%),Q30分别为98.61%、98.65%和98.58%(表2),说明本研究得到的sRNA数据质量良好,可进行下一步分析。将各孢子样品的unannotated reads比对到东方蜜蜂微孢子虫的参考基因组,比对上的reads总数为22 000 837条,比对率≥52.87%(表3)。Table 2
表2
表2sRNA-seq测序数据信息统计
Table 2
| 样品 Sample | 原始读段 Raw reads | 有效读段 Clean reads | 99.9%的碱基正确率 Q30 (%) |
|---|---|---|---|
| NcS-1 | 16597883 | 15608370 | 98.61 |
| NcS-2 | 15451791 | 14249255 | 98.65 |
| NcS-2 | 12248316 | 11440684 | 98.58 |
新窗口打开|下载CSV
Table 3
表3
表3sRNA-seq数据比对东方蜜蜂微孢子虫参考基因组的信息统计
Table 3
| 样品 Sample | 未注释的有效读段 Unannotated clean reads | 比对上的有效读段 Mapped clean reads | 比对率 Mapping ratio (%) |
|---|---|---|---|
| NcS-1 | 14596118 | 7717280 | 52.87 |
| NcS-2 | 12924265 | 7801135 | 60.36 |
| NcS-3 | 10842971 | 6482422 | 59.78 |
新窗口打开|下载CSV
2.2 东方蜜蜂微孢子虫孢子中lncRNA上下游基因的GO数据库注释
东方蜜蜂微孢子虫孢子中56个lncRNA共预测出310个上下游基因。对这些上下游基因进行GO数据库注释,结果显示它们可注释到生物学进程、分子功能和细胞组分相关的35个功能条目;在生物学进程中,注释基因数最多的条目分别为代谢进程(105)、细胞进程(100)、单组织进程(45)、生物学调控(19)和生物学进程调控(18);在分子功能中,注释基因数最多的分别为催化活性(96)、结合(95)、结构分子活性(9)、核酸结合转录因子活性(3)和转运器活性(3);在细胞组分中,注释基因数最多的分别为细胞(64)、细胞组件(64)、细胞器(46)、大分子复合物(34)和细胞器组件(13)(图1)。括号内的数字代表注释到该功能条目的上下游基因数。图1
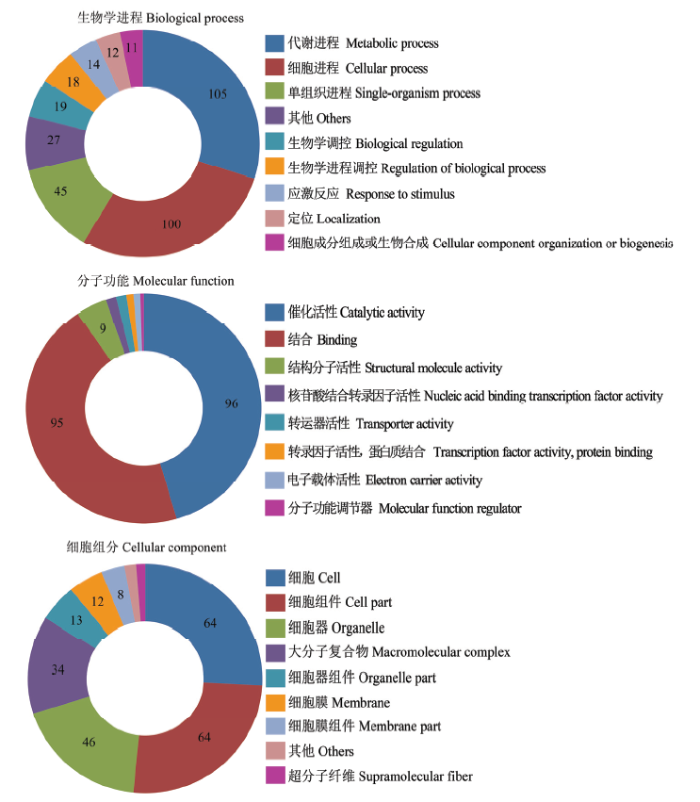 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1东方蜜蜂微孢子虫孢子中lncRNA的上下游基因的GO数据库注释
Fig. 1GO database annotation of upstream and downstream genes of lncRNAs in N. ceranae spore
2.3 东方蜜蜂微孢子虫孢子中lncRNA上下游基因的KEGG数据库注释
进一步对lncRNA的上下游基因进行KEGG数据库注释,结果显示310个上下游基因可注释到56条通路,注释基因数最多的前10位分别是新陈代谢途径(25)、抗生素的生物合成(12)、次生代谢产物的生物合成(11)、核糖体(10)、嘌呤代谢(7)、蛋白酶体(7)、RNA转运(7)、真核生物中核糖体的生物发生(7)、碳代谢(6)和嘧啶代谢(6)(图2)。括号内的数字代表注释到该通路的上下游基因数。图2
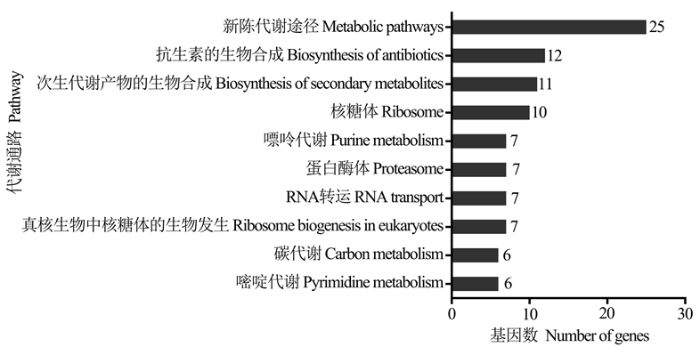 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2东方蜜蜂微孢子虫孢子中lncRNA的上下游基因的KEGG数据库注释
Fig. 2KEGG database annotation of upstream and downstream genes of lncRNAs in N. ceranae spore
2.4 东方蜜蜂微孢子虫孢子中lncRNA-miRNA调控网络的构建及分析
对东方蜜蜂微孢子虫孢子中lncRNA进行靶miRNA预测,发现3个lncRNA与4个miRNA存在靶向结合关系,其中MSTRG.3636.1靶向nce-miR- 8565,MSTRG.4498.1靶向nce-miR-7502和nce-miR- 8639,MSTRG.4883.1靶向nce-miR-7729(图3)。图3
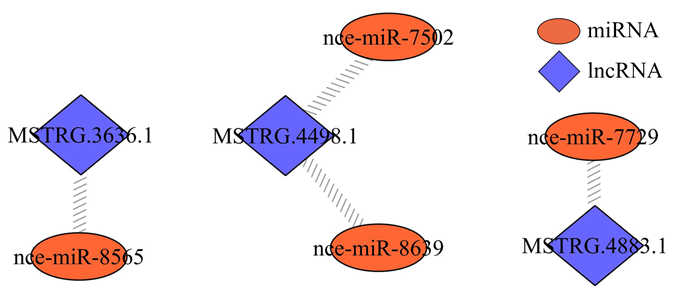 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3东方蜜蜂微孢子虫孢子中的lncRNA-miRNA调控网络
Fig. 3LncRNA-miRNA regulatory network in N. ceranae spore
2.5 东方蜜蜂微孢子虫孢子中lncRNA-miRNA-mRNA调控网络的构建及分析
利用软件预测lncRNA靶向结合miRNA的靶mRNA,综合lncRNA、miRNA与mRNA的靶向结合关系构建调控网络,分析发现MSTRG.4883.1靶向的nce-miR-7729共结合16个mRNA,MSTRG.3636.1靶向的nce-miR-8565共结合15个mRNA,lncRNA、miRNA和mRNA之间形成密切的调控关系;3个lncRNA的4个靶miRNA可结合59个mRNA,其中MSTRG.4498.1靶向的nce-miR-7502和nce-miR-8639共结合28个mRNA,三者形成更为复杂的调控关系(图4)。图4
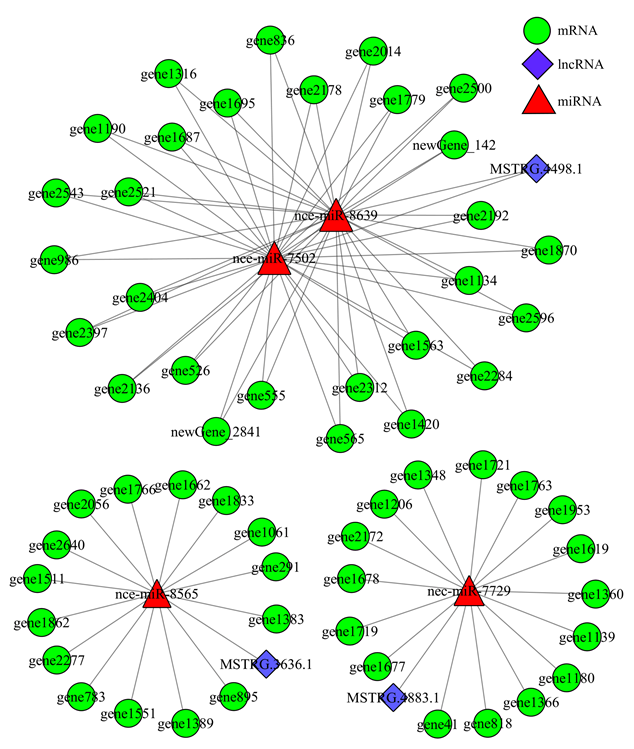 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4东方蜜蜂微孢子虫孢子中3个lncRNA的竞争性内源RNA调控网络
Fig. 4Competing endogenous network of three lncRNAs in N. ceranae spore
进一步分析发现,上述靶mRNA在Nr数据库中注释皆为假定蛋白;但在Swissprot数据库中均有功能注释信息,如gene2014、gene1662和gene1383分别注释为转录相关蛋白1、微管蛋白γ链和V型质子ATP酶催化亚基A(表4)。
Table 4
表4
表4东方蜜蜂微孢子虫孢子中lncRNA靶向miRNA的靶mRNA的Swissprot和Nr数据库注释
Table 4
| 核酸ID Nucleotide ID | Swissprot 数据库注释 Swissprot database annotation | Nr数据库注释 Nr database annotation |
|---|---|---|
| gene986 | 液泡氨基酸转运体5 Vacuolar amino acid transporter 5 | 假定蛋白NCER_101697 Hypothetical protein NCER_101697 |
| gene783 | 负辅因子2复合亚基 Negative cofactor 2 complex subunit beta | 假定蛋白NCER_101896 Hypothetical protein NCER_101896 |
| gene565 | 丝氨酸/苏氨酸多聚蛋白激酶1 Serine/threonine-protein kinase plo1 | 假定蛋白NCER__102113 Hypothetical protein NCER_102113 |
| gene2596 | 含C1778.09蛋白的TBC结构域 TBC domain-containing protein C1778.09 | 假定蛋白NCER_100108 Hypothetical protein NCER_100108 |
| gene2543 | TATA结合蛋白相关因子MOT1 TATA-binding protein-associated factor MOT1 | 假定蛋白NCER_100152 Hypothetical protein NCER_100152 |
| gene2397 | 丝氨酸/苏氨酸蛋白激酶CDC5同源物 Cell cycle serine/threonine-protein kinase CDC5 homolog | 假定蛋白NCER_100270 Hypothetical protein NCER_100270 |
| gene2312 | ERAD-相关E3泛素蛋白连接酶doa10 ERAD-associated E3 ubiquitin-protein ligase doa10 | 假定蛋白NCER_100375 Hypothetical protein NCER_100375 |
| gene2178 | 转运蛋白sec24 Transport protein sec24 | 假定蛋白NCER_100510 Hypothetical protein NCER_100510 |
| gene2014 | 转录相关蛋白1 Transcription-associated protein 1 | 假定蛋白NCER_100665 Hypothetical protein NCER_100665 |
| gene1870 | ERAD-相关E3泛素蛋白连接酶doa10 ERAD-associated E3 ubiquitin-protein ligase doa10 | 假定蛋白NCER_100805 Hypothetical protein NCER_100805 |
| gene1766 | 赖氨酸- tRNA连接酶 Lysine-tRNA ligase | 假定蛋白NCER_100909 Hypothetical protein NCER_100909 |
| gene1719 | T-复合体蛋白1亚基 T-complex protein 1 subunit alpha | 假定蛋白NCER_100958 Hypothetical protein NCER_100958 |
| gene1687 | DNA-锚定 RNA 聚合酶 II 亚基RPB2 DNA-directed RNA polymerase II subunit RPB2 | 假定蛋白NCER_100983 Hypothetical protein NCER_100983 |
| gene1678 | 锰-ATP转运酶4 Manganese-transporting ATPase 4 | 假定蛋白NCER_101005 Hypothetical protein NCER_101005 |
| gene1677 | 3型蛋白酶体亚基 Probable proteasome subunit beta type-3 | 假定蛋白NCER_101004 Hypothetical protein NCER_101004 |
| gene1662 | 微管蛋白γ链 Tubulin gamma chain | 假定蛋白NCER_101024 Hypothetical protein NCER_101024 |
| gene1563 | 核输出蛋白BRL1 Nucleus export protein BRL1 | 假定蛋白NCER_101114 Hypothetical protein NCER_101114 |
| gene1383 | V型质子ATP酶催化亚基A V-type proton ATPase catalytic subunit A | 假定蛋白NCER_101294 Hypothetical protein NCER_101294 |
| gene1366 | 长链脂肪酸CoA链2 Long chain fatty acid CoA ligase 2 | 假定蛋白NCER_101315 Hypothetical protein NCER_101315 |
| gene1360 | 热休克蛋白hsp98 Heat shock protein hsp98 | 假定蛋白NCER_101322 Hypothetical protein NCER_101322 |
| gene1348 | 丝氨酸/苏氨酸蛋白激酶MEC1同源物 Serine/threonine-protein kinase MEC1 homolog | 假定蛋白NCER_101332 Hypothetical protein NCER_101332 |
| gene1190 | 泛素羧基末端水解酶5 Ubiquitin carboxyl-terminal hydrolase 5 | 假定蛋白NCER_101487 Hypothetical protein NCER_101487 |
| gene1180 | 苯丙氨酸-tRNA连接酶亚基 Phenylalanine-tRNA ligase beta subunit | 假定蛋白NCER_101490 Hypothetical protein NCER_101490 |
| gene1134 | 二磷酸异戊基丁二醇异构酶 Isopentenyl-diphosphate Delta-isomerase | 假定蛋白NCER_101544 Hypothetical protein NCER_101544 |
| newGene_142 | 60S核糖体蛋白L40(前体) 60S ribosomal protein L40 (precursor) | 假定蛋白NCER_102067 Hypothetical protein NCER_102067 |
新窗口打开|下载CSV
2.6 东方蜜蜂微孢子虫孢子中lncRNA、靶miRNA和靶mRNA的分子验证
通过RT-PCR对上述ceRNA网络中lncRNA和靶mRNA的进行扩增,扩增产物的电泳结果显示8个靶mRNA均能扩增出预期大小的目的条带,阴性对照未扩增出片段;2个lncRNA也能扩增出预期片段,阴性对照未扩增出片段;利用Stem-loop RT-PCR对上述ceRNA网络中靶miRNA进行扩增,扩增产物的电泳结果显示3个靶miRNA均扩增出大小约100 bp的目的片段,阴性对照未扩增出片段(图5)。上述结果表明东方蜜蜂微孢子虫孢子中lncRNA、靶miRNA和靶mRNA真实表达。图5
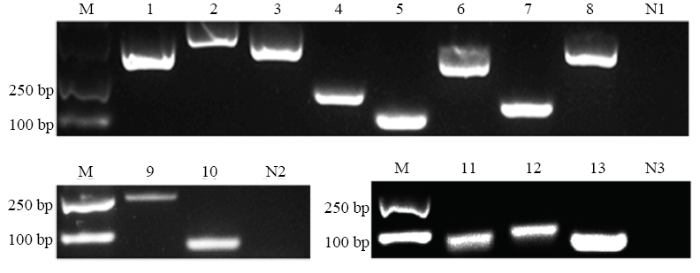 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5东方蜜蜂微孢子虫孢子中lncRNA、靶miRNA和靶mRNA的RT-PCR验证
M:DNA marker;1:假定蛋白NCER_101243编码基因Hypothetical protein NCER_101243 coding gene;2:假定蛋白NCER_102113编码基因Hypothetical protein NCER_102113 coding gene;3:假定蛋白NCER_100958编码基因Hypothetical protein NCER_100958 coding gene;4:假定蛋白NCER_101315编码基因Hypothetical protein NCER_101315 coding gene;5:假定蛋白NCER_101322编码基因Hypothetical protein NCER_101322 coding gene;6:假定蛋白NCER_101544编码基因Hypothetical protein NCER_101544 coding gene;7:假定蛋白NCER_100991编码基因Hypothetical protein NCER_100991 coding gene;8:假定蛋白NCER_1011114编码基因Hypothetical protein NCER_101114 coding gene;9:MSTRG.3636.1;10:MSTRG.4883.1;11:nce-miR-8565;12:nce-miR-7502;13:nce-miR-7729;N1、N2:阴性对照(无菌水)Negative control (sterile water);N3:阴性对照(DEPC水)Negative control (DEPC water)
Fig. 5RT-PCR verification of lncRNAs, target miRNAs and target mRNAs in N. ceranae spore
3 讨论
东方蜜蜂微孢子虫孢子处于休眠态,人们对于孢子中是否存在转录、翻译和新陈代谢等生命活动的认识十分有限。前期研究中,笔者团队证实东方蜜蜂微孢子虫孢子同样存在一定水平的转录,并基于高质量的lncRNA组学数据鉴定出83条lncRNA,同时对这些lncRNA的种类和结构特征进行了全面分析[21],为东方蜜蜂微孢子虫 lncRNA的深入研究提供了必要的数据基础。本研究结合前期获得的东方蜜蜂微孢子虫 lncRNA组学数据和测序得到的sRNA组学数据,进一步深入探究东方蜜蜂微孢子虫孢子中lncRNA的ceRNA调控网络及潜在功能。目前,动植物和微生物等物种的sRNA-seq数据在相应参考基因组上的比对率一般介于50%—70%[27,28,29],原因在于sRNA的长度很短,比对参考基因组时要求每个碱基均要匹配上,因此导致比对率偏低。本研究中,NcS-1、NcS-2和NcS-3的clean reads在东方蜜蜂微孢子虫参考基因组上的比对率介于52.87%—60.36%,与其他物种的研究报道类似,属于正常的比对率范围。目前,NONCODE[30]、LncRNAdb[31]和LncRNome[32]等lncRNA数据库中有限的功能注释信息局限于人类和其他少数模式物种(如小鼠、果蝇和拟南芥等)。GUO等[33]构建了家蚕微孢子虫感染家蚕(Bombyx mori)卵巢BmN细胞的连续感染系,进而利用脂质体介导将含gfp的转座子载体piggyBac和非转座子载体pIZT/V5-His转染感染细胞,通过分子生物学手段证实外源gfp被成功导入家蚕微孢子虫基因组;此外,GUO等[33]还通过饲喂家蚕微孢子虫孢子感染家蚕个体,然后通过无菌操作将蚕血加入正常BmN细胞,再进行脂质体转染的方法也成功将gfp导入了家蚕微孢子虫基因组;这是对微孢子虫直接进行转基因操作的首例报道,为东方蜜蜂微孢子虫等其他微孢子虫的转基因研究提供了重要的思路和方法借鉴。目前,东方蜜蜂微孢子虫的基因功能研究仍处于初步阶段。PALDI等[34]利用饲喂的方法将人工合成的靶向ADP/ATP转运器蛋白编码基因的双链RNA(dsRNA)导入被东方蜜蜂微孢子虫感染的西方蜜蜂体内,研究发现病原的ADP/ATP转运器蛋白转录本受到干扰而沉默,该沉默影响了病原增殖水平和宿主生理状态;HUANG等[35]通过饲喂小干扰RNA(siRNA)对东方蜜蜂微孢子虫感染的西方蜜蜂的Dicer进行敲减,发现病原孢子数显著下降,此外超过10%的病原蛋白编码基因呈现显著性差异表达。对于包括东方蜜蜂微孢子虫在内的绝大多数物种,通过分析和验证lncRNA的cis、trans和ceRNA作用,从而间接推测lncRNA的潜在功能仍是常用方法[6,7,8,9,10,11]。较多的研究表明lncRNA可以与一些较远基因上的增强子或启动子结合,通过trans作用行使其对编码蛋白基因表达的调控[7]。然而,本研究并没有预测到表达量存在正相关或负相关关系的lncRNA和mRNA,暗示lncRNA在东方蜜蜂微孢子虫的休眠态孢子中不行使trans作用。鉴于lncRNA的表达具有时空特异性[36],东方蜜蜂微孢子虫在孢子发芽和侵染宿主细胞的过程中是否存在trans作用或通过哪些方式发挥主要作用,值得进一步深入研究。利用链特异性建库的lncRNA-seq技术对东方蜜蜂微孢子虫感染的蜜蜂中肠组织进行测序,通过连续比对宿主蜜蜂参考基因组和东方蜜蜂微孢子虫参考基因组过滤得到病原的lncRNA组学数据,进而分析病原lncRNA的trans作用是具有可行性的方法。为探究东方蜜蜂微孢子虫孢子中lncRNA的潜在功能,本研究利用生物信息学方法对lncRNA的上下游基因和靶向结合的miRNA进行预测,进而对lncRNA的cis作用方式进行分析,发现东方蜜蜂微孢子虫孢子中56条lncRNA可能调控310个上下游基因,它们涉及代谢进程、细胞进程、催化活性、结合和细胞组件等35条功能条目,说明lncRNA可能广泛参与孢子休眠状态的基础生命活动,但其具体功能仍需要进一步实验验证。目前,lncRNA的相关研究主要集中在少数模式生物,如人类[13]、小鼠[14]、果蝇[16]、酵母[7,9]等;其中多数研究集中在lncRNA的生物信息学预测和分析,仅少数研究涉及lncRNA的功能验证,有限的技术手段是最大的限制因素。ZHAN等[37]研究发现山羊骨骼肌细胞中的1 153个lncRNA潜在调控1 455个上游及下游基因,部分上下游基因可能参与了骨骼肌细胞的发育,说明相应的lncRNA在山羊骨骼肌细胞发育过程中具有潜在的cis调控作用;FAUQUENOY等[9]通过对野生型和缺失一个拷贝rse1等位基因的杂合二倍体突变型裂殖酵母菌株进行研究,发现lncRNA通过抑制复合物的支架结构特异性抑制邻近的rse1等位基因表达,表明lncRNA通过cis作用抑制裂殖酵母的细胞分化。本研究中,对于东方蜜蜂微孢子虫的lncRNA,分别有105、100和45个上下游基因注释在代谢进程、细胞进程和单组织进程,说明相应的lncRNA通过cis作用维持东方蜜蜂微孢子虫孢子处于休眠态期间的基础新陈代谢。此外,分别有64、64和46个上下游基因涉及细胞组件、细胞和细胞器,表明lncRNA对东方蜜蜂微孢子虫孢子的细胞生命活动中具有潜在的调节功能。还发现有96个上下游基因涉及催化活性,进一步表明lncRNA可能参与东方蜜蜂微孢子虫孢子内新陈代谢过程的生化反应。LncRNA通过与DNA、RNA或蛋白质的相互作用而发挥调控作用[4,36]。东方蜜蜂微孢子虫在进行发芽和极丝弹射之前,首先需要识别和接触宿主细胞,进而与宿主细胞相互作用[38,39,40,41],上述过程必然也伴随着不同生物大分子之间的结合。本研究中,有95个上下游基因涉及分子功能中的结合,暗示lncRNA在东方蜜蜂微孢子虫孢子内可能通过与其他生物大分子的相互结合发挥特定的调控功能,也可能在病原识别和接触宿主细胞中扮演特定角色。微孢子虫具有一层坚硬的孢子壁,能够保护内部结构免受外界不良环境破坏[41]。东方蜜蜂微孢子虫在宿主细胞内增殖的过程需要不断适应外部环境并作出应激反应。本研究中,有14个上下游基因涉及应激反应,说明相应的lncRNA(MSTRG.4856.1、MSTRG.5126.1、MSTRG.5617.1、MSTRG.6243.1、MSTRG.6290.1、MSTRG.6328.1、MSTRG.6473.1、MSTRG.6633.1、MSTRG.6654.2、MSTRG.6688.1、MSTRG.6683.1、MSTRG.6692.1和MSTRG.6703.1)在孢子的环境应激方面发挥一定作用。
在糖酵解途径中,2-磷酸甘油酸可在烯醇化酶作用下转变为磷酸烯醇式丙酮酸,进而在丙酮酸激酶作用下转变为烯醇式丙酮酸,过程中产生两分子的H2O,同时两分子ADP转化为两分子ATP[42]。DOLGIKH等[43]检测了黄斑黑蟋蟀微孢子虫(Nosema grylli)孢子中6-磷酸葡萄糖脱氢酶、葡萄糖磷酸变位酶、磷酸葡萄糖异构酶、6-磷酸果糖激酶及3-磷酸甘油酸激酶等14种酶的活性,并分析了相关能量代谢途径,发现黄斑黑蟋蟀微孢子虫可能利用自身储备的碳水化合物而不是外源物质,推测糖酵解是黄斑黑蟋蟀微孢子虫碳水化合物分解代谢的主要途径。本研究发现,gene1402、gene2290、gene2665和gene926这4个上下游基因涉及糖酵解/糖异生途径,其中gene926和gene2290分别注释到Swissprot数据库中的胃瘤真菌的烯醇化酶基因和兔脑炎微孢子虫(Encephalitozoon cuniculi)的丙酮酸激酶基因,表明相应的lncRNA(MSTRG.6328.1和MSTRG.4700.1)具有调节糖酵解/糖异生途径的潜在功能,可能在东方蜜蜂微孢子虫孢子的能量代谢方面发挥作用。此外,共有85个上下游基因注释在10条碳水化合物代谢相关通路,包括代谢途径(25)、抗生素的生物合成(12)和次生代谢产物的生物合成(11)等,进一步说明相应的lncRNA参与调节东方蜜蜂微孢子虫孢子的能量代谢。核苷酸是组成DNA和RNA的重要部分,并作为电子信号和能量载体在生物体内发挥重要作用[44]。HEINZ等[45]研究了来自HIV/AIDS患者的人气管普孢子虫(Trachipleistophora hominis)中的4种核苷酸转运蛋白,验证了这些蛋白可以转运有放射性标记的2种嘌呤核苷酸,并未发现嘧啶核苷酸;作者进一步将人工合成酶与细胞内人气管普孢子虫、兔脑炎微孢子虫和东方蜜蜂微孢子虫酶比较,发现存在嘌呤和嘧啶核苷酸在磷酸化和氧化作用之间相互转换的一类核心酶,表明微孢子虫中的此类酶能够保持嘌呤和嘧啶处于不同的激活状态满足生命活动过程中的相关代谢途径。本研究中,分别有7和6个上下游基因注释到嘌呤代谢和嘧啶代谢,其中的共有基因为6个,分别为gene2676、 gene2395、gene1404、gene2450、gene1439和gene1058,说明相应的lncRNA(MSTRG.6703.1、MSTRG.6459.2、MSTRG.6459.1、MSTRG.5334.1、MSTRG.6512.1、MSTRG.5375.1、MSTRG.4883.1和MSTRG.4883.1)具有通过调控东方蜜蜂微孢子虫孢子中的嘌呤代谢和嘧啶代谢影响遗传信息传递、电子传递、信号通路及能量代谢的潜在作用。还发现分别有6、2、1、1和1个上下游基因分别注释到碳代谢、丙酮酸代谢、磷酸肌醇代谢、脂肪酸代谢和果糖甘露糖代谢等物质代谢通路;有 2、1和1个基因分别注释到甲烷代谢、氧化磷酸化和过氧化物酶体等能量代谢通路;上述结果进一步表明相应的lncRNA(MSTRG.4700.1、MSTRG.5292.1、MSTRG.5334.1、MSTRG.6328.1、MSTRG.6383.1、MSTRG.6688.1、MSTRG.6692.1和MSTRG.6703.1)通过cis作用参与东方蜜蜂微孢子虫孢子中的基本物质和能量代谢的调控。
SALMENA等[46]于2011年提出了“ceRNA假说”,该假说认为含有MRE的mRNA、假基因和lncRNA等RNA,能够竞争性结合miRNA,从而间接影响靶基因的表达量[46,47]。随后,越来越多的研究结果证实了这一主流假说[22,46-48]。WANG等[48]利用荧光素酶RNA免疫沉淀技术探究在低氧或炎症因子处理下lncRNA的ceRNA机制,结果显示lncRNA H19和HULC可作为ceRNA靶向结合let-7a/let-7b和miR-372/miR-373,从而参与炎症反应促进胆管癌细胞的迁移和侵袭。前期研究中,笔者团队曾对意大利蜜蜂(Apis mellifera ligustica)工蜂7 d和10 d中肠发育过程中lncRNA和circRNA差异表达谱及调控网络进行研究,分析发现此2种ncRNA均可作为潜在的ceRNA吸附miRNA,从而减少对靶mRNA的抑制或降解[22,49]。本研究发现,MSTRG.4498.1、MSTRG.4883.1和MSTRG.3636.1与4个miRNA(nce-miR-8565、nce-miR-7502、nce-miR-8639和nce-miR-7729)之间存在靶向结合关系;此外,上述4个miRNA与59个mRNA也存在靶向结合关系,表明lncRNA可能通过竞争性结合miRNA调控mRNA的表达量,进而影响东方蜜蜂微孢子虫孢子休眠态下的诸多生命活动。对于lncRNA-miRNA-mRNA调控网络,通过RT-PCR证实了2个lncRNA和8个靶mRNA的真实表达,通过Stem-loop RT-PCR证实了3个靶miRNA的真实表达(图5)。但本研究中的靶向结合关系是基于生物信息学软件预测出来的,上述靶向结合关系是否真实存在仍需通过双荧光素酶报告系统加以验证。对调控网络中的靶mRNA进行功能和代谢通路注释,结果显示它们涉及19条功能条目,包括代谢进程、细胞进程和催化活性等,进一步分析发现对于调控网络中间接调控靶mRNA的lncRNA,其中部分lncRNA也参与了对上下游基因的调控;此外,有24个靶mRNA富集在18条代谢通路,涉及嘌呤代谢、嘧啶代谢、脂肪酸代谢、过氧化物酶体及氧化磷酸化等。上述结果表明部分lncRNA能同时通过cis和ceRNA作用影响靶基因的表达水平,从而对东方蜜蜂微孢子虫孢子的新陈代谢和基本生命活动进行灵活调控。
4 结论
部分lncRNA可能通过cis作用调控上下游基因的表达,进而调节东方蜜蜂微孢子虫孢子中的物质代谢、能量代谢和环境应激等生物学过程;部分lncRNA可能作为ceRNA参与调节东方蜜蜂微孢子虫孢子休眠态下的诸多生命活动;此外,少数lncRNA能同时通过cis和ceRNA作用影响靶基因的表达水平,从而对东方蜜蜂微孢子虫孢子的新陈代谢和基本生命活动进行灵活调控。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 3]
[本文引用: 2]
[本文引用: 5]
[本文引用: 4]
[本文引用: 3]
[本文引用: 6]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 2]
[D].
[本文引用: 1]
[本文引用: 1]
[本文引用: 7]
[本文引用: 3]
[本文引用: 3]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
DOI:10.1111/imb.v28.1URL [本文引用: 1]
DOI:10.1016/j.molcel.2011.08.018URL [本文引用: 2]

Long noncoding RNAs (IncRNAs) are an important class of pervasive genes involved in a variety of biological functions. Here we discuss the emerging archetypes of molecular functions that IncRNAs execute as signals, decoys, guides, and scaffolds. For each archetype, examples from several disparate biological contexts illustrate the commonality of the molecular mechanisms, and these mechanistic views provide useful explanations and predictions of biological outcomes. These archetypes of IncRNA function may be a useful framework to consider how IncRNAs acquire properties as biological signal transducers and hint at their possible origins in evolution. As new IncRNAs are being discovered at a rapid pace, the molecular mechanisms of IncRNAs are likely to be enriched and diversified.
[本文引用: 1]
[本文引用: 1]
DOI:10.3390/insects10080245URL [本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
DOI:10.1111/jeu.1997.44.issue-3URL [本文引用: 1]
DOI:10.1007/s11302-015-9483-2URL [本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.cell.2011.07.014URL [本文引用: 3]
DOI:10.1038/nature09144URL [本文引用: 1]
DOI:10.1186/s13045-016-0348-0URL [本文引用: 2]
DOI:10.3864/j.issn.0578-1752.2018.23.015URL [本文引用: 1]
【Objective】Circular RNA (circRNA) plays a primary role in alternative splicing, transcription regulation and expression regulation of parental gene. The objective of this study is to investigate the profile expression of circRNAs and differentially expressed circRNAs (DEcircRNAs) during the developmental process of the midguts of Apis mellifera ligustica workers, and to explore the role of DEcircRNAs in the development of midgut at the transcriptional level. 【Method】 On basis of the whole transcriptome data from 7- and 10-day-old worker’s midguts of A. m. ligustica (Am7 and Am10), find_circ software was used to predict circRNAs based on the filtered sequencing data. The circRNA expression level was normalized by RPM algorithm. Differential expression analysis for circRNAs was conducted via DEGseq software following standards fold change≥2.0, P<0.05 and false discovery rate (FDR) <0.05. Source genes of DEcircRNAs were annotated to GO and KEGG databases to gain function and pathway annotations by using BLAST. DEcircRNAs-miRNAs and DEcircRNAs-miRNAs-mRNAs networks were predicted with TargetFinder software and visualized using Cytoscape v.3.2.1 software. RT-qPCR was conducted to verify the reliability of sequencing data.【Result】 On average, 19 616 356 anchors reads were obtained from each A. m. ligustica worker’s midgut sample. Pearson correlations between different biological repeats within Am7 and Am10 groups were ≥0.950. In total, 256 DEcircRNAs including 105 up-regulated circRNAs and 151 down-regulated circRNAs were predicted. Novel_circ_009675 and novel_circ_013879 were highly expressed in Am7 and Am10, respectively. Source genes of DEcircRNAs could be annotated to 32 GO terms including binding, single-organism process and cellular process, among them 35, 35 and 7 source genes were involved in catalytic activity, metabolic process and stress response. Additionally, these source genes could be annotated to 35 KEGG pathways, in which 5, 5 and 4 source genes were associated with Hippo signaling pathway, endocytosis and phagosome, respectively; further investigation showed that 1, 2 and 2 source genes could be annotated to material metabolisms such as phosphoinositol metabolism, starch and sucrose metabolism and galactose metabolism; 5, 4, 3, 1 and 1 source genes could be annotated to immune signaling pathways including endocytosis, phagosome, lysosome, ubiquitin-mediated proteolysis and MAPK signaling pathway, respectively. These results suggested that the corresponding DEcircRNA was involved in the development, metabolism and immune defense of the midgut of A. m. ligustica. DEcircRNA-miRNA regulation network analysis showed that 141 DEcircRNAs could link to 107 miRNAs, most of these DEcircRNAs could only bind to 1-2 miRNAs, but novel_circ_011577 and novel_circ_010719 could respectively bind to 32 and 28 miRNAs. In addition, the number of DEcircRNAs combined with mir-136-y, ame-miR-6001-3p and mir-136-y was the highest (15, 14 and 14, respectively), which indicated that the corresponding DEcircRNA could play roles during the developmental process of A. m. ligustica worker’s midgut as competing endogenous RNAs. Furthermore, DEcircRNAs-ame-miR-6001-3p-mRNA network was constructed and analyzed, and the result indicated that 14 DEcircRNAs could jointly link to ame-miR-6001-3p, implying they were likely to indirectly regulate division and differentiation of stem cells in A. m. ligustica worker’s midgut via regulation of ame-miR-6001-3p. Six DEcircRNAs were randomly selected for RT-qPCR assay, the result showed the alteration trend of expression levels of 5 DEcircRNAs was consistent with that of the sequencing data, which proved the reliability of trancriptome data.【Conclusion】Through the deep investigation of DEcircRNAs during the developmental process of A. m. ligustica worker’s midgut, the expression profile and differential expression information of circRNAs in the development of midgut of worker bee were provided, and the role of DEcircRNAs in the development of midgut was revealed. It provides a basis for the screening and functional study of key circRNAs associated with the development of the midgut.
DOI:10.3864/j.issn.0578-1752.2018.23.015URL [本文引用: 1]
【Objective】Circular RNA (circRNA) plays a primary role in alternative splicing, transcription regulation and expression regulation of parental gene. The objective of this study is to investigate the profile expression of circRNAs and differentially expressed circRNAs (DEcircRNAs) during the developmental process of the midguts of Apis mellifera ligustica workers, and to explore the role of DEcircRNAs in the development of midgut at the transcriptional level. 【Method】 On basis of the whole transcriptome data from 7- and 10-day-old worker’s midguts of A. m. ligustica (Am7 and Am10), find_circ software was used to predict circRNAs based on the filtered sequencing data. The circRNA expression level was normalized by RPM algorithm. Differential expression analysis for circRNAs was conducted via DEGseq software following standards fold change≥2.0, P<0.05 and false discovery rate (FDR) <0.05. Source genes of DEcircRNAs were annotated to GO and KEGG databases to gain function and pathway annotations by using BLAST. DEcircRNAs-miRNAs and DEcircRNAs-miRNAs-mRNAs networks were predicted with TargetFinder software and visualized using Cytoscape v.3.2.1 software. RT-qPCR was conducted to verify the reliability of sequencing data.【Result】 On average, 19 616 356 anchors reads were obtained from each A. m. ligustica worker’s midgut sample. Pearson correlations between different biological repeats within Am7 and Am10 groups were ≥0.950. In total, 256 DEcircRNAs including 105 up-regulated circRNAs and 151 down-regulated circRNAs were predicted. Novel_circ_009675 and novel_circ_013879 were highly expressed in Am7 and Am10, respectively. Source genes of DEcircRNAs could be annotated to 32 GO terms including binding, single-organism process and cellular process, among them 35, 35 and 7 source genes were involved in catalytic activity, metabolic process and stress response. Additionally, these source genes could be annotated to 35 KEGG pathways, in which 5, 5 and 4 source genes were associated with Hippo signaling pathway, endocytosis and phagosome, respectively; further investigation showed that 1, 2 and 2 source genes could be annotated to material metabolisms such as phosphoinositol metabolism, starch and sucrose metabolism and galactose metabolism; 5, 4, 3, 1 and 1 source genes could be annotated to immune signaling pathways including endocytosis, phagosome, lysosome, ubiquitin-mediated proteolysis and MAPK signaling pathway, respectively. These results suggested that the corresponding DEcircRNA was involved in the development, metabolism and immune defense of the midgut of A. m. ligustica. DEcircRNA-miRNA regulation network analysis showed that 141 DEcircRNAs could link to 107 miRNAs, most of these DEcircRNAs could only bind to 1-2 miRNAs, but novel_circ_011577 and novel_circ_010719 could respectively bind to 32 and 28 miRNAs. In addition, the number of DEcircRNAs combined with mir-136-y, ame-miR-6001-3p and mir-136-y was the highest (15, 14 and 14, respectively), which indicated that the corresponding DEcircRNA could play roles during the developmental process of A. m. ligustica worker’s midgut as competing endogenous RNAs. Furthermore, DEcircRNAs-ame-miR-6001-3p-mRNA network was constructed and analyzed, and the result indicated that 14 DEcircRNAs could jointly link to ame-miR-6001-3p, implying they were likely to indirectly regulate division and differentiation of stem cells in A. m. ligustica worker’s midgut via regulation of ame-miR-6001-3p. Six DEcircRNAs were randomly selected for RT-qPCR assay, the result showed the alteration trend of expression levels of 5 DEcircRNAs was consistent with that of the sequencing data, which proved the reliability of trancriptome data.【Conclusion】Through the deep investigation of DEcircRNAs during the developmental process of A. m. ligustica worker’s midgut, the expression profile and differential expression information of circRNAs in the development of midgut of worker bee were provided, and the role of DEcircRNAs in the development of midgut was revealed. It provides a basis for the screening and functional study of key circRNAs associated with the development of the midgut.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}