 ,华中农业大学作物遗传改良国家重点实验室,武汉 430070
,华中农业大学作物遗传改良国家重点实验室,武汉 430070Development of an Efficient Editing System in Arabidopsis by CRISPR-Cas9
ZHANG Cheng, HE MingLiang, WANG Wei, XU FangSen,National Key Laboratory of Crop Genetic Improvement, Huazhong Agricultural University, Wuhan 430070通讯作者:
责任编辑: 李莉
收稿日期:2019-10-9网络出版日期:2020-06-16
| 基金资助: |
Received:2019-10-9Online:2020-06-16
作者简介 About authors
张成,E-mail:zcheng93@webmail.hzau.edu.cn。

摘要
关键词:
Abstract
Keywords:
PDF (3127KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
张成, 何明亮, 汪威, 徐芳森. 一种CRISPR-Cas9介导的拟南芥高效基因编辑系统的构建与应用[J]. 中国农业科学, 2020, 53(12): 2340-2348 doi:10.3864/j.issn.0578-1752.2020.12.003
ZHANG Cheng, HE MingLiang, WANG Wei, XU FangSen.
0 引言
【研究意义】通过基因编辑手段获得基因敲除材料,对于研究基因在植物中的生物学功能具有重要的意义。而获得高编辑效率的CRISPR-Cas9系统能加速获得基因敲除材料的进程。【前人研究进展】在植物基因功能研究中,靶向基因编辑技术有着十分重要的作用,特别是在目标基因与其同源基因同源性较高的情况下很难通过其他途径获得想要的突变体。近些年,基于细菌免疫系统中CRISPR(clustered regularly interspaced short palindromic repeats)-Cas(CRISPR associated)的靶向基因编辑技术席卷了整个生物基因组研究领域[1,2]。常用的CRISPR-Cas9系统由两部分组成:带有核定位信号的Cas9和由21 bp靶序列(tracrRNA)的CRISPR(crRNA)构成的sgRNA(synthetic guide RNA)。其中,由sgRNA转录而成的非编码RNA与带有核定位信号的Cas9蛋白结合形成复合体,在PAM(protospacer-adjacent motif,Cas9系统中为NGG)位点上游3 bp位置切割DNA产生DNA双链切割(double-stranded breaks,DSBs),继而发生同源重组修复(homology-directed repair,HDR)或非同源重组修复(nonhomologous end joining,NHEJ),而容易出错的NHRJ修复方式导致在切割位点附近发生核苷酸的随机缺失,插入或者替换[1,3]。目前,CRISPR-Cas9系统已经成功在多种植物中得以应用,包括拟南芥、水稻、玉米、番茄、小麦、油菜和烟草等[4,5,6,7,8,9,10,11]。目前,在拟南芥中主要是利用农杆菌蘸花的方法,通过T-DNA插入到拟南芥卵细胞或者胚胎干细胞中的基因组获得拟南芥转基因植株。在之前的拟南芥CRISPR-Cas9研究中,主要是利用花椰菜花叶病毒(cauliflower mosaic virus,CaMV)35s启动子启动Cas9的表达,但发现拟南芥中的编辑效率较水稻下降很多,且发生基因编辑的T1代大部分为嵌合体。这主要由于35s启动子在植物的胚胎形成过程中表达量不高,而是在营养器官中高表达量的原因[8,11]。因此,为了提高拟南芥中基因编辑的效率以及遗传稳定性,选用合适的启动子启动Cas9的表达显得尤为重要。目前已经有一些研究人员使用了在拟南芥卵细胞中表达水平高的启动子来启动Cas9的表达,比如EC1.1/EC1.2启动子[12]、DD45和SPL启动子[13]、YAO启动子[14]、INCURVATA2启动子[15]、APETALA1启动子[16]以及RPS5A和WOX2启动子[17]。【本研究切入点】拟南芥CRISPR载体的编辑效率大多较低,且从T1代编辑株系的鉴定到T3代纯合突变且无Cas9材料的获得过程耗时费力,造成严重的时间、人力和资源的浪费。创建拟南芥高效的CRISPR编辑系统有利于加速基因敲除材料的获得,且节省时间和成本。【拟解决的关键问题】本研究通过构建一个高效CRISPR-Cas9系统用于拟南芥的基因编辑,并用拟南芥木葡聚糖内糖基转移/水解酶基因TOUCH 4(TCH4)对该基因编辑系统进行验证。采用DsRed2红色荧光筛选标记筛选阳性植株及无Cas9植株。通过对测序峰图的分析对编辑结果进行解码,以便降低获得稳定遗传的无Cas9突变体材料的时间、人力和财务成本。1 材料与方法
1.1 材料
拟南芥为Columbia-0(Col-0)生态型,生长条件为长日照(16 h光照/8 h黑暗),环境温度为22℃。1.2 拟南芥CRISPR-Cas9载体改造
拟南芥RPS5A编码一个核糖体蛋白,从胚胎发育早期到后期所有的生长发育阶段都表现为持续的高效表达[18,19]。使用的原始载体为pKSE401(由陈其军教授课题组提供),为了构建pRSE-WH载体,首先使用引物pKSE-F:5′-GCGGGACTCTGGGGTTCG-3′和pKSE-R:5′-GCGGGACTCTGGGGTTCG-3′,用宝生物公司的primer STAR MAX高保真酶扩增不包含35S启动子和潮霉素抗性的pKSE载体片段,同时以pBinGlyRed3的质粒为模板,用引物RED-F:5′-GCCAACATGGTGGAGGAGGAGTCCACCATGGTAGATCTG-3′和RED-R:5′-ACCCCAGAGTCCCGCT CAGGGTACCAGGAACAGGTG-3′扩增红光蛋白片段,利用Infusion(TaKaRa)将2个片段连接,转化后得到pRSE401载体。以pRSE401载体为模板,使用引物pRSE-F:5′-CTCGACCTCAACACAACATATA C-3′和pRSE-R:5′-GACCTGCAGGCATGCAAGC-3′,扩增得到不包含35S启动子的pRSE401载体片段。利用拟南芥基因组DNA为模板,使用引物AtRPS5A-F:5′-GCATGCCTGCAGGTCCTCAACTTTTGATTCGCTATTTGC-3′和AtRPS5A-R:5′-TGTGTTGAGGTCGAG GGCTGTGGTGAGAGAAACAG-3′扩增得到AtRPS5A启动子片段,使用Infusion连接pRSE401载体片段和AtRPS5A启动子片段,得到pRSE-WH载体。1.3 TCH4的CRISPR载体构建
拟南芥TCH4(AT3G11940,XTH22)属于木葡聚糖内糖基转移/水解酶(xyloglucan endotransglucosylase/ hydrolase,XTH)家族中的一员[20],具有2种催化活性:1)内切木葡聚糖;2)将内切产生的木葡聚糖还原端连接到另一个木葡聚糖的非还原端。XTH一方面可以剪切细胞壁中的木葡聚糖交联网络引起细胞壁的松弛,一方面可以参与细胞壁新组分的组装,通过对细胞壁结构的调节参与对植物生长发育的调节[21,22,23,24,25,26]。利用CRISPR-P 2.0(http://crispr.hzau.edu.cn)对拟南芥TCH4设计2个靶点,靶点一序列为:5′-CCTTTCACTGCTTCTTACCG-3′,靶点二序列为:5′-GGATTGGAATCCAGAACCAG-3′。在引物的上游添加接头:5′-ATTG-3′,在引物的下游添加接头:5′-AAAC-3′。用于基因编辑第一个靶点的引物为:F:5′-ATTGCCTTTCACTGCTTCTTACCG-3′,R:5′-AAA CCGGTAAGAAGCAGTGAAAGG-3′。用于基因编辑第二个靶点的引物为:F:5′-ATTGGGATTGGAATCCA GAACCAG-3′;R:5′-AAACCTGGTTCTGGATTCCAA TCC-3′。采用25 μL退火体系(5 μL 10 mmol·L-1的上游引物,5 μL 10 mmol·L-1的下游引物,15 μL去离子水),退火程序为95℃ 3 min,95℃—16℃每20 s下降1℃,16℃保存,得到带有黏性末端的靶序列二聚体。酶切连接体系为靶序列二聚体2 μL、pRSE-WH 2 μL、10×NEB T4 buffer 1.5 μL、BsaⅠ(NEB)1 μL、10×BSA 1.5 μL、T4连接酶(NEB,高浓度)1 μL,去离子水补充至15 μL。反应程序为37℃ 5 h,50℃ 5 min,80℃ 10 min。将连接产物转化大肠杆菌,送公司测序鉴定正确后,提取质粒转化农杆菌。1.4 拟南芥转化以及阳性植株获得和筛选
采用沾花法转化拟南芥[27],收获T1代种子之后,筛选标记为红色荧光蛋白DsRed2,在绿色激发光下发射光为红色,使用体式荧光显微镜筛选阳性植株。1.5 编辑鉴定
采用CTAB法[28]提取T1代阳性植株的叶片基因组DNA,使用引物:TCR-F:5′-ATGGCGATCACTTAC TTGCTTC-3′和TCR-R:5′-TACTCTCTCTATGCAGCT AAGCAC-3′进行PCR扩增,测序检测T1代植株编辑情况。2 结果
2.1 拟南芥高效CRISPR-Cas9载体改造
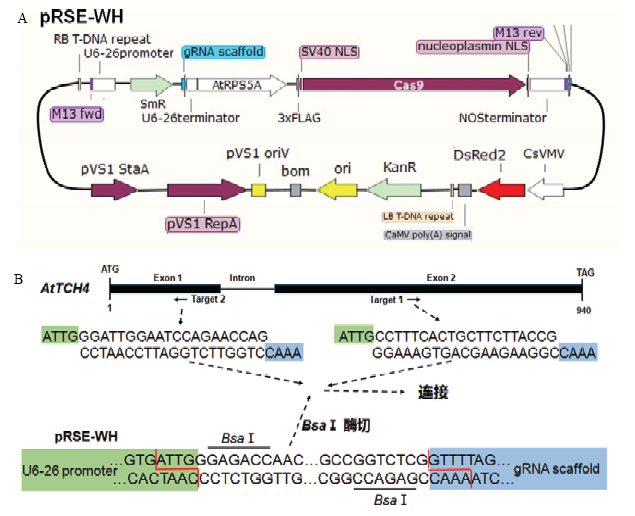
首先,用DsRed2红色荧光蛋白替换了原始的潮霉素抗性基因(图1-A)。然后,利用拟南芥中RPS5A的启动子替换pKSE401载体中的CaMV35s启动子,从而达到在拟南芥胚胎发育早期进行基因编辑的目的。从拟南芥Col-0基因组DNA中扩增得到长度为1 659 bp的RPS5A的启动子,将其整合到pRSE-WH载体中(图1-A)。在pRSE-WH中,sgRNA由AtU6-26启动子启动,而在sgRNA和U6-26启动子中间包含2个BsaⅠ酶切位点,可直接一步将20 bp的靶序列连入载体。替换后的红色荧光蛋白可直接用于阳性植株的筛选以及后期无Cas9植株的筛选。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1CRISPR/Cas9载体图谱和TCH4的靶点示意图
A:改造后的CRISPR/Cas9载体pRSE-WH;B:2个靶位点在TCH4中的位置
Fig. 1CRISPR/Cas9 vectors and target mapping of TCH4 gene
A: Reconstructed CRISPR/Cas9 vector pRSE-WH; B: Position of two targets in TCH4 gene locus
2.2 TCH4的CRISPR载体构建
TCH4的CDS全长为855 bp,基因结构包括2个外显子和1个内含子。使用CRISPR-P 2.0(http://crispr. hzau.edu.cn)对拟南芥TCH4设计2个靶点。第一个靶点位于第二个外显子上,第二个靶点位于第一个外显子上(图1-B)。在20 bp靶序列正向序列及反向互补序列的5′端添加接头序列,通过变性-复性形成带有黏性末端的二聚体,而pRSE-WH载体经过BsaⅠ酶切形成带有粘性末端的线性化载体片段,利用T4连接酶连接即可将靶序列整合到pRSE-WH载体中。将带有靶点的2个载体分别命名为TCR1和TCR2。2.3 红光筛选
使用DsRed2作为筛选标记,用于阳性筛选。组成型表达的DsRed2蛋白,使得拟南芥种皮在绿光激发下发出红光,区别于其他阴性种子(图2)。将T1代红色的阳性种子播种(图2-A),收获得到T2代种子,大部分的T2代种子中,红光种子与非红光种子的比例近似3﹕1(红光种子与非红光种子分别为65和28颗,图2-B),符合分离定律。T2代挑选红色种子繁种得到T3代不同植株种子中,三分之一的几率得到纯合红色种子(图2-C)。而T2代非红光种子后代全部为非红光种子(图2-D)。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2阳性植株的筛选
A:T1代种子;B:T2代种子;C:T3代红光种子;D:T3代无红光种子
Fig. 2Screening of positive plants
A: T1 seeds; B: T2 seeds; C: T3 seeds with red fluorescence; D: T3 seeds without red fluorescence
2.4 编辑检测
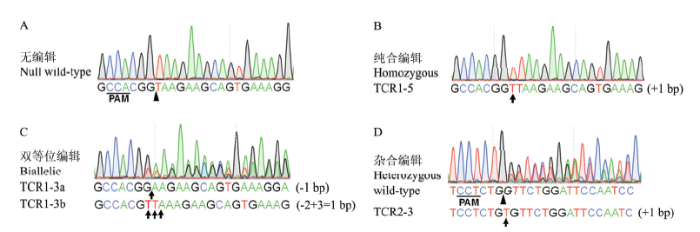
从T1代植株中挑选40株TCR1和19株TCR2,提取植株叶片DNA,对TCH4基因组片段进行PCR扩增,测序检测编辑结果。根据测序峰图手动解码编辑结果,结果发现,编辑类型可以分为4种:无编辑、纯合编辑、杂合编辑以及双等位编辑(图3)。在40株TCR1植株中,发生编辑的有32株,编辑效率达到80%,19株TCR2植株中,全部发生编辑,100%编辑效率。通过对不同的编辑类型进行统计,发现59株T1代阳性植株中,无编辑8株(13.56%)、纯合编辑9株(15.25%)、双等位编辑40株(67.80%)和杂合编辑2株(3.39%)(图4)。将每条染色体的编辑事件单独分析,118次编辑事件中,18次没有编辑,占比15.25%;45次单碱基插入,占比38.14%;1次多碱基插入,占比0.85%;11次单碱基缺失,占比9.32%;43次多碱基缺失,占比36.44%(表1)。其中多碱基缺失中,从几bp到几十bp均有发生,甚至几百bp缺失。在单碱基插入事件中,TCR1中只存在2种事件,4次A碱基插入和28次T碱基插入,而TCR2中4种碱基插入均有发生,5次A碱基插入,2次T碱基插入,1次C碱基插入和5次G碱基插入。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3T1代植株的编辑类型
Fig. 3Genotypes of the edited T1 plants
图4
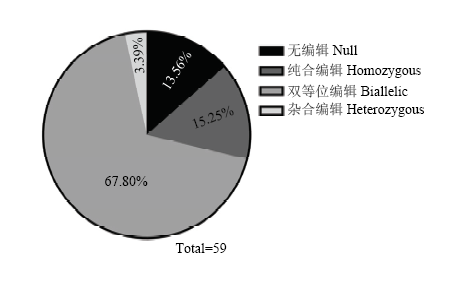 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4T1代植株各编辑类型所占比例
Fig. 4Proportion of editing types in T1 plants
Table 1
表1
表1TCH4 CRISPR T1代植株的编辑类型及比例
Table 1
| T1代植株染色体的编辑类型 Mutation patterns of chromosome in T1 plants | 发生次数 No. of edited lines | 突变比例 Mutation rate (%) |
|---|---|---|
| 无None | 18 | 15.25 |
| +1 | 45 | 38.14 |
| +32 | 1 | 0.85 |
| -1 | 11 | 9.32 |
| -2 | 4 | 3.39 |
| -4 | 1 | 0.85 |
| -5 | 2 | 1.69 |
| -6 | 2 | 1.69 |
| -7 | 4 | 3.39 |
| -10 | 4 | 3.39 |
| -11 | 1 | 0.85 |
| -12 | 1 | 0.85 |
| -13 | 3 | 2.54 |
| -14 | 1 | 0.85 |
| -16 | 1 | 0.85 |
| -17 | 1 | 0.85 |
| -19 | 1 | 0.85 |
| -20 | 6 | 5.08 |
| -22 | 1 | 0.85 |
| -23 | 1 | 0.85 |
| -31 | 1 | 0.85 |
| -34 | 2 | 1.69 |
| -49 | 1 | 0.85 |
| -55 | 1 | 0.85 |
| -218 | 1 | 0.85 |
| -313 | 1 | 0.85 |
| -503 | 1 | 0.85 |
| -577 | 1 | 0.85 |
| 合计Total | 118 | 100.00 |
新窗口打开|下载CSV
2.5 T2代编辑分析
在T1代发生纯合编辑以及双等位编辑的株系中选择了无红光种子进行繁种,并对T2代植株编辑情况进行测序检测。结果显示,TCR1-27株系T1代检测结果为T碱基插入的纯合编辑,所测序的6个T2代单株均为纯合的T碱基插入。同样,TCR1-10和TCR1-11的T1代均为双等位编辑,所测序的6个T2代单株均显示2条染色体中的突变都成功地遗传到后代中(图5)。图5
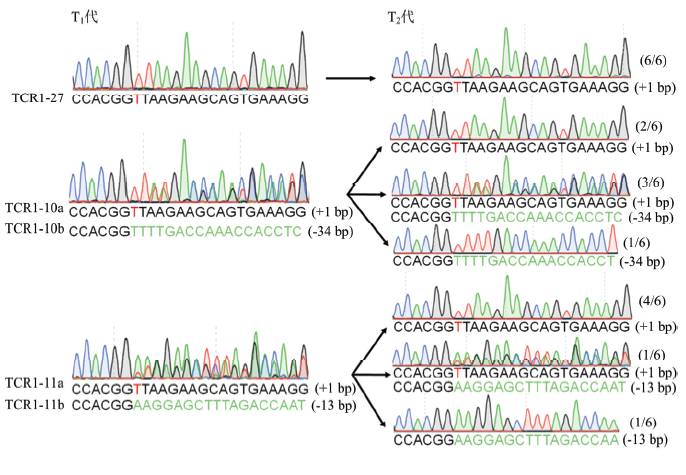 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5T1和T2代株系间的突变遗传
Fig. 5Mutation inheritance in T1 and T2 generation
3 讨论
以CRISPR为代表的基因编辑技术已广泛应用于农作物基因功能验证和农作物遗传改良。目前,已研发多种CRISPR载体运用于单子叶和双子叶植物的基因编辑[11-14,17,29]。植物营养遗传实验室尝试了使用载体pKSE401在拟南芥和油菜中运用CRISPR技术对拟研究的候选基因进行基因编辑。但是发现在CRISPR实际操作中存在的两个问题,一编辑效率低;二从T1代阳性植株筛选到T3代稳定遗传株系的获得,工作量大,成本高。而前者是主要的问题,因为一个高效的编辑系统能够很大的降低后期的工作量和成本。植物营养遗传实验室前期的工作中使用载体pKSE401进行甘蓝型油菜和拟南芥的编辑,其编辑效率较低,造成严重的时间、人力、资源的浪费。于是尝试了对实验室原有的CRISPR载体进行改造,试图提高编辑效率。与一些植物采用外植体侵染-再生转化植物不同的是,拟南芥遗传转化采用的是沾花法,农杆菌侵染的是雌性生殖器官中的胚囊。常用的35S启动子在胚囊中或者在整个胚胎发育过程中表达量并不高,而这很有可能导致了拟南芥中编辑效率的下降。因此,本研究采用了一个在胚胎发育过程中持续高表达的RPS5A启动子替换35S启动子。对TCH4的编辑结果显示,RPS5A启动子启动Cas9的系统确实有着很高的效率,测序的59株转基因阳性植株中编辑效率达到86%。而不同的靶点序列的编辑效率也有一定的差异,其中靶点一的编辑效率为80%,而靶点二的则是100%编辑。目前,CRISPR在水稻中的运用较好,编辑效率可以超过81%[11],在拟南芥中的编辑效率大多不超过40%[7,11,16]。而本研究改造的CRISPR编辑效率可高达86%。高的编辑效率可以很容易得到各种不同的突变类型,发生最多的是单碱基插入,占38.14%;其次是多碱基缺失,占36.44%;再次是单碱基缺失,占9.32%;而多碱基插入发生概率较低,118次编辑事件中只发生了一次。在这些不同的突变类型中,发现不同的靶点序列似乎对不同的突变类型有一定的偏好性。在TCR1的单碱基插入中,只发现了A/T插入,而且T插入占88%。但是TCR2的单碱基插入则4种碱基插入均有发现。导致这样的结果可能与产生DNA双链切割位点的碱基类型有关。TCR1的DNA双链切割位点碱基为G-T,而TCR2的DNA双链切割位点碱基为G-G。
DsRed最早是从香菇珊瑚(Discosoma striata)中分离得到的一种红色荧光蛋白[30]。DsRed的发射光谱峰值为583 nm,激发光谱峰值为558 nm。DsRed在应用过程中被发现存在一些问题,包括成熟缓慢,易形成四聚体,有一定毒性。第二代DsRed2的氨基末端进行了一些突变改造,使得其组织蛋白凝集和毒性下降,荧光基团成熟时间变短。DsRed2不仅可以像GFP一样进行活体检测和连续观察,而且能消除植物本身的背景干扰,甚至在白光条件下也能检测到红光,因此,DsRed2可以作为一种可视标记应用于植物遗传转化[30,31]。在pRSE-WH中,采用DsRed2这种可视筛选标记。如果Cas9一直在植物中表达,它一方面可能会继续对靶位点进行切割、编辑;另一方面脱靶的可能性也一直存在。因此,最终希望得到无Cas9且稳定遗传的突变体。而Cas9基本上是与DsRed2连锁的,所以通过筛选无红光种子就能得到无Cas9的株系。在拟南芥CRISPR系统中,常用的抗生素筛选标记不适用于T2代无Cas9株系的筛选,而只能对T2代植株提取DNA,通过PCR扩增进行鉴定,理论上这会增加很大的工作量。而使用DsRed2可以很轻易的从T2代种子中筛选得到无Cas9的种子,并且能够保证T3代种子中无Cas9的稳定遗传。所以,在拟南芥CRISPR系统中DsRed2要明显优于常用的抗生素筛选。此外,常用的解码编辑结果的方法为对PCR产物进行克隆测序,并增加测序的单克隆数量获得编辑的结果,这无疑是最可靠的方式之一,但这会增加大量的时间、人力和物力的消耗。本研究在了解解码的机制后进行手动解码:对目标基因的基因组片段进行PCR扩增,测序检测编辑结果会出现2种峰型:单峰和套峰。其中,单峰表明扩增的PCR片段是单一的,可以很容易得到其中的DNA序列信息,而这种单峰的结果包括没有发生编辑和纯合编辑;套峰意味着PCR产物存在2个或者多个不同的DNA片段,会导致没有那么容易获得其中不同的DNA序列信息。可以通过以下几点去解码套峰中包含的信息:1)PCR过程中会导致不同DNA片段含量不一致,进而测序中表现出不同强度的信号值,可以通过这种信号强度的差异将不同的信息区分开;2)由于CRISPR-Cas9是基于DNA双链切割进而发生DNA修复产生的编辑,所以它的特点是编辑会发生在切割位点附近或者向外扩散,而未编辑的位点则应该与参考序列一致,因此,即使在套峰中2个信号强度相近的DNA片段,依旧可以通过碱基间的序列信息推断出其序列,解码出其中一条信息之后,套峰的信息减去其中已知的信息,就能得到另一条的序列信息。基于以上两点,对T1代阳性植株的测序信息进行解码,选择理想的编辑株系进行繁种,并在T2代中进行测序验证,其结果等同于在T1代中进行克隆测序,且节省了大量的时间、精力和成本。
4 结论
构建了一个适用于拟南芥中基因编辑的高效CRISPR载体pRSE-WH,能够简便地获得无Cas9且稳定遗传的T3代突变体。致谢:
中国农业大学陈其军教授提供了原始pKSE401载体;华中农业大学杨光圣教授课题组提供了体式荧光显微镜;华中农业大学汪社亮博士和英国The James Hutton Institute的Philip John White教授对文章的修改,在此表示感谢。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1126/science.1226740URL [本文引用: 2]
DOI:10.1126/science.1225829URLPMID:22745249 [本文引用: 1]

Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) systems provide bacteria and archaea with adaptive immunity against viruses and plasmids by using CRISPR RNAs (crRNAs) to guide the silencing of invading nucleic acids. We show here that in a subset of these systems, the mature crRNA that is base-paired to trans-activating crRNA (tracrRNA) forms a two-RNA structure that directs the CRISPR-associated protein Cas9 to introduce double-stranded (ds) breaks in target DNA. At sites complementary to the crRNA-guide sequence, the Cas9 HNH nuclease domain cleaves the complementary strand, whereas the Cas9 RuvC-like domain cleaves the noncomplementary strand. The dual-tracrRNA:crRNA, when engineered as a single RNA chimera, also directs sequence-specific Cas9 dsDNA cleavage. Our study reveals a family of endonucleases that use dual-RNAs for site-specific DNA cleavage and highlights the potential to exploit the system for RNA-programmable genome editing.
DOI:10.1126/science.1232033URL [本文引用: 1]
Bacteria and archaea have evolved adaptive immune defenses, termed clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) systems, that use short RNA to direct degradation of foreign nucleic acids. Here, we engineer the type II bacterial CRISPR system to function with custom guide RNA (gRNA) in human cells. For the endogenous AAVS1 locus, we obtained targeting rates of 10 to 25% in 293T cells, 13 to 8% in K562 cells, and 2 to 4% in induced pluripotent stem cells. We show that this process relies on CRISPR components; is sequence-specific; and, upon simultaneous introduction of multiple gRNAs, can effect multiplex editing of target loci. We also compute a genome-wide resource of similar to 190 K unique gRNAs targeting similar to 40.5% of human exons. Our results establish an RNA-guided editing tool for facile, robust, and multiplexable human genome engineering.
DOI:10.1073/pnas.1400822111URLPMID:24550464 [本文引用: 1]
The CRISPR (clustered regularly interspaced short palindromic repeat)/Cas (CRISPR-associated) system has emerged as a powerful tool for targeted gene editing in many organisms, including plants. However, all of the reported studies in plants focused on either transient systems or the first generation after the CRISPR/Cas system was stably transformed into plants. In this study we examined several plant generations with seven genes at 12 different target sites to determine the patterns, efficiency, specificity, and heritability of CRISPR/Cas-induced gene mutations or corrections in Arabidopsis. The proportion of plants bearing any mutations (chimeric, heterozygous, biallelic, or homozygous) was 71.2% at T1, 58.3% at T2, and 79.4% at T3 generations. CRISPR/Cas-induced mutations were predominantly 1 bp insertion and short deletions. Gene modifications detected in T1 plants occurred mostly in somatic cells, and consequently there were no T1 plants that were homozygous for a gene modification event. In contrast, approximately 22% of T2 plants were found to be homozygous for a modified gene. All homozygotes were stable to the next generation, without any new modifications at the target sites. There was no indication of any off-target mutations by examining the target sites and sequences highly homologous to the target sites and by in-depth whole-genome sequencing. Together our results show that the CRISPR/Cas system is a useful tool for generating versatile and heritable modifications specifically at target genes in plants.
DOI:10.1038/cr.2013.123URLPMID:23999856 [本文引用: 1]
DOI:10.1007/s11103-015-0342-xURLPMID:26188471 [本文引用: 1]
The CRISPR/Cas9 system is an efficient tool used for genome editing in a variety of organisms. Despite several recent reports of successful targeted mutagenesis using the CRISPR/Cas9 system in plants, in each case the target gene of interest, the Cas9 expression system and guide-RNA (gRNA) used, and the tissues used for transformation and subsequent mutagenesis differed, hence the reported frequencies of targeted mutagenesis cannot be compared directly. Here, we evaluated mutation frequency in rice using different Cas9 and/or gRNA expression cassettes under standardized experimental conditions. We introduced Cas9 and gRNA expression cassettes separately or sequentially into rice calli, and assessed the frequency of mutagenesis at the same endogenous targeted sequences. Mutation frequencies differed significantly depending on the Cas9 expression cassette used. In addition, a gRNA driven by the OsU6 promoter was superior to one driven by the OsU3 promoter. Using an all-in-one expression vector harboring the best combined Cas9/gRNA expression cassette resulted in a much improved frequency of targeted mutagenesis in rice calli, and bi-allelic mutant plants were produced in the T0 generation. The approach presented here could be adapted to optimize the construction of Cas9/gRNA cassettes for genome editing in a variety of plants.
DOI:10.1038/nbt.2654URLPMID:23929339 [本文引用: 2]
DOI:10.1038/cr.2013.114URLPMID:23958582 [本文引用: 2]
DOI:10.1093/mp/sst121URLPMID:23963532 [本文引用: 1]
DOI:10.1093/mp/ssu009URLPMID:24482433 [本文引用: 1]
DOI:10.1016/j.molp.2015.04.007URLPMID:25917172 [本文引用: 5]
CRISPR/Cas9 genome targeting systems have been applied to a variety of species. However, most CRISPR/Cas9 systems reported for plants can only modify one or a few target sites. Here, we report a robust CRISPR/Cas9 vector system, utilizing a plant codon optimized Cas9 gene, for convenient and high-efficiency multiplex genome editing in monocot and dicot plants. We designed PCR-based procedures to rapidly generate multiple sgRNA expression cassettes, which can be assembled into the binary CRISPR/Cas9 vectors in one round of cloning by Golden Gate ligation or Gibson Assembly. With this system, we edited 46 target sites in rice with an average 85.4% rate of mutation, mostly in biallelic and homozygous status. We reasoned that about 16% of the homozygous mutations in rice were generated through the non-homologous end-joining mechanism followed by homologous recombination-based repair. We also obtained uniform biallelic, heterozygous, homozygous, and chimeric mutations in Arabidopsis T1 plants. The targeted mutations in both rice and Arabidopsis were heritable. We provide examples of loss-of-function gene mutations in T0 rice and T1 Arabidopsis plants by simultaneous targeting of multiple (up to eight) members of a gene family, multiple genes in a biosynthetic pathway, or multiple sites in a single gene. This system has provided a versatile toolbox for studying functions of multiple genes and gene families in plants for basic research and genetic improvement.
DOI:10.1186/s13059-015-0715-0URLPMID:26193878 [本文引用: 1]
Arabidopsis mutants produced by constitutive overexpression of the CRISPR/Cas9 genome editing system are usually mosaics in the T1 generation. In this study, we used egg cell-specific promoters to drive the expression of Cas9 and obtained non-mosaic T1 mutants for multiple target genes with high efficiency. Comparisons of 12 combinations of eight promoters and two terminators found that the efficiency of the egg cell-specific promoter-controlled CRISPR/Cas9 system depended on the presence of a suitable terminator, and the composite promoter generated by fusing two egg cell-specific promoters resulted in much higher efficiency of mutation in the T1 generation compared with the single promoters.
DOI:10.1111/pbi.12468URLPMID:26360626 [本文引用: 1]
The Streptococcus-derived CRISPR/Cas9 system is being widely used to perform targeted gene modifications in plants. This customized endonuclease system has two components, the single-guide RNA (sgRNA) for target DNA recognition and the CRISPR-associated protein 9 (Cas9) for DNA cleavage. Ubiquitously expressed CRISPR/Cas9 systems (UC) generate targeted gene modifications with high efficiency but only those produced in reproductive cells are transmitted to the next generation. We report the design and characterization of a germ-line-specific Cas9 system (GSC) for Arabidopsis gene modification in male gametocytes, constructed using a SPOROCYTELESS (SPL) genomic expression cassette. Four loci in two endogenous genes were targeted by both systems for comparative analysis. Mutations generated by the GSC system were rare in T1 plants but were abundant (30%) in the T2 generation. The vast majority (70%) of the T2 mutant population generated using the UC system were chimeras while the newly developed GSC system produced only 29% chimeras, with 70% of the T2 mutants being heterozygous. Analysis of two loci in the T2 population showed that the abundance of heritable gene mutations was 37% higher in the GSC system compared to the UC system and the level of polymorphism of the mutations was also dramatically increased with the GSC system. Two additional systems based on germ-line-specific promoters (pDD45-GT and pLAT52-GT) were also tested, and one of them was capable of generating heritable homozygous T1 mutant plants. Our results suggest that future application of the described GSC system will facilitate the screening for targeted gene modifications, especially lethal mutations in the T2 population.
DOI:10.1016/j.molp.2015.10.004URLPMID:26524930 [本文引用: 2]
DOI:10.1007/s00425-014-2180-5URLPMID:25269397 [本文引用: 1]
MAIN CONCLUSION: Dividing tissue-targeted site-directed mutagenesis using RGEN of CRISPR/Cas system produces heritable mutations in Arabidopsis thaliana. Site-directed genome engineering in higher plants has great potential for basic research and molecular breeding. Here, we describe a method for site-directed mutagenesis of the Arabidopsis nuclear genome that efficiently generates heritable mutations using the RNA-guided endonuclease (RGEN) derived from bacterial clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 (CRISPR associated) protein system. To induce mutagenesis in proliferating tissues during embryogenesis and throughout the plant life cycle, the single guide RNA (sgRNA) and Cas9 DNA endonuclease were expressed from the U6 snRNA and INCURVATA2 promoters, respectively. After Agrobacterium-mediated introduction of T-DNAs encoding RGENs that targets FLOWERING LOCUS T (FT) and SQUAMOSA PROMOTER BINDING PROTEIN-LIKE 4 genes, somatic mutagenesis at the targeted loci was observed in T1 transformants. In the results of FT-RGEN, T1 plants often showed late flowering indicative of the presence of large somatic sectors in which the FT gene is mutated on both chromosomes. DNA sequencing analysis estimated that about 90 % of independent chromosomal DNA fragments carried mutations in the analyzed tissue of a T1 plant showing late flowering. The most frequently detected somatic polymorphism showed a high rate of inheritance in T2 plants, and inheritance of less frequent polymorphisms was also observed. As a result, late-flowering plants homozygous for novel, heritable null alleles of FT including a 1 bp insertion or short deletions were recovered in the following T2 and T3 generations. Our results demonstrate that dividing tissue-targeted mutagenesis using RGEN provides an efficient heritable genome engineering method in A. thaliana.
DOI:10.1104/pp.16.00663URLPMID:27208253 [本文引用: 2]
Mutations generated by CRISPR/Cas9 in Arabidopsis (Arabidopsis thaliana) are often somatic and are rarely heritable. Isolation of mutations in Cas9-free Arabidopsis plants can ensure the stable transmission of the identified mutations to next generations, but the process is laborious and inefficient. Here, we present a simple visual screen for Cas9-free T2 seeds, allowing us to quickly obtain Cas9-free Arabidopsis mutants in the T2 generation. To demonstrate this in principle, we targeted two sites in the AUXIN-BINDING PROTEIN1 (ABP1) gene, whose function as a membrane-associated auxin receptor has been challenged recently. We obtained many T1 plants with detectable mutations near the target sites, but only a small fraction of T1 plants yielded Cas9-free abp1 mutations in the T2 generation. Moreover, the mutations did not segregate in Mendelian fashion in the T2 generation. However, mutations identified in the Cas9-free T2 plants were stably transmitted to the T3 generation following Mendelian genetics. To further simplify the screening procedure, we simultaneously targeted two sites in ABP1 to generate large deletions, which can be easily identified by PCR. We successfully generated two abp1 alleles that contained 1,141- and 711-bp deletions in the ABP1 gene. All of the Cas9-free abp1 alleles we generated were stable and heritable. The method described here allows for effectively isolating Cas9-free heritable CRISPR mutants in Arabidopsis.
DOI:10.1093/pcp/pcw191URLPMID:27856772 [本文引用: 2]
The CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated 9) system is widely used as a tool for genome engineering in various organisms. A complex consisting of Cas9 and single guide RNA (sgRNA) induces a DNA double-strand break in a sequence-specific manner, resulting in knockout. Some binary vectors for CRISPR/Cas9 in plants have been reported, but there is a problem with low efficiency. Here, we present a newly developed, highly efficient CRISPR/Cas9 vector for Arabidopsis thaliana, pKAMA-ITACHI Red (pKIR), harboring the RIBOSOMAL PROTEIN S5 A (RPS5A) promoter to drive Cas9. The RPS5A promoter maintains high constitutive expression at all developmental stages starting from the egg cell and including meristematic cells. Even in the T1 generation, pKIR induced null phenotypes in some genes: PHYTOENE DESATURASE 3 (PDS3), AGAMOUS (AG) and DUO POLLEN 1 (DUO1). Mutations induced by pKIR were carried in the germ cell line of the T1 generation. Surprisingly, in some lines, 100% of the T2 plants had the adh1 (ALCOHOL DEHYDROGENASE 1) null phenotype, indicating that pKIR strongly induced heritable mutations. Cas9-free T2 mutant plants were obtained by removing T2 seeds expressing a fluorescent marker in pKIR. Our results suggest that the pKIR system is a powerful molecular tool for genome engineering in Arabidopsis.
URLPMID:11684664 [本文引用: 1]
Mutations in ribosomal protein (RP) genes in Drosophila lead to strong developmental phenotypes, expressed in the semi-dominant Minute syndrome. In plants, however, mutations in RP genes have so far only been reported to result in recessive developmental phenotypes. We present the analysis of an Arabidopsis promoter-trap line, in which a T-DNA insertion in an RPS5 gene (AtRPS5A) causes semi-dominant developmental phenotypes. Most cell-division processes are delayed or disturbed in the heterozygous mutant, and development is completely arrested at an early embryonic stage in the homozygous mutant. By analogy with Drosophila rp mutants, we have named this mutant Arabidopsis Minute-like 1 (aml1). As with other Arabidopsis RPs, RPS5 is represented by a small gene family, but in contrast to other described plant RPs, this family comprises only two members. The AtRPS5A gene (mutated in aml1) is strongly expressed in dividing cells, whereas expression of the second RPS5 gene, AtRPS5B, is lower than that of AtRPS5A, and is correlated with cell differentiation rather than cell division. From expression analyses we conclude that AtRPS5A is the most abundantly expressed RPS5 gene in Arabidopsis. The Minute-like defects in the aml1 mutant provide the first evidence that ribosome insufficiency leads to similar consequences in both plants and insects, and emphasize the general importance of efficient protein translation for cell proliferation in higher eukaryotes.
DOI:10.1016/j.devcel.2013.03.013URL [本文引用: 1]
In flowering plants, double fertilization is normally accomplished by the first pollen tube, with the fertilized ovule subsequently inhibiting the attraction of a second pollen tube. However, the mechanism of second-pollen-tube avoidance remains unknown. We discovered that failure to fertilize either the egg cell or the central cell compromised second-pollen-tube avoidance in Arabidopsis thaliana. A similar disturbance was caused by disrupting the fertilization-independent seed (FIS) class polycomb-repressive complex 2 (FIS-PRC2), a central cell- and endosperm-specific chromatin-modifying complex for gene silencing. Therefore, the two female gametes have evolved their own signaling pathways. Intriguingly, second-pollen-tube attraction induced by half-successful fertilization allowed the ovules to complete double fertilization, producing a genetically distinct embryo and endosperm. We thus propose that each female gamete independently determines second-pollen-tube avoidance to maximize reproductive fitness in flowering plants.
DOI:10.1093/pcp/pcf171URLPMID:12514239 [本文引用: 1]
The polysaccharide xyloglucan is thought to play an important structural role in the primary cell wall of dicotyledons. Accordingly, there is considerable interest in understanding the biochemical basis and regulation of xyloglucan metabolism, and research over the last 16 years has identified a large family of cell wall proteins that specifically catalyze xyloglucan endohydrolysis and/or endotransglucosylation. However, a confusing and contradictory series of nomenclatures has emerged in the literature, of which xyloglucan endotransglycosylases (XETs) and endoxyloglucan transferases (EXGTs) are just two examples, to describe members of essentially the same class of genes/proteins. The completion of the first plant genome sequencing projects has revealed the full extent of this gene family and so this is an opportune time to resolve the many discrepancies in the database that include different names being assigned to the same gene. Following consultation with members of the scientific community involved in plant cell wall research, we propose a new unifying nomenclature that conveys an accurate description of the spectrum of biochemical activities that cumulative research has shown are catalyzed by these enzymes. Thus, a member of this class of genes/proteins will be referred to as a xyloglucan endotransglucosylase/hydrolase (XTH). The two known activities of XTH proteins are referred to enzymologically as xyloglucan endotransglucosylase (XET, which is hereby re-defined) activity and xyloglucan endohydrolase (XEH) activity. This review provides a summary of the biochemical and functional diversity of XTHs, including an overview of the structure and organization of the Arabidopsis XTH gene family, and highlights the potentially important roles that XTHs appear to play in numerous examples of plant growth and development.
DOI:10.1046/j.1365-313x.1993.03050691.xURLPMID:8374618 [本文引用: 1]
The action on tamarind seed xyloglucan of the pure, xyloglucan-specific endo-(1-->4)-beta-D-glucanase from nasturtium (Tropaeolum majus L.) cotyledons has been compared with that of a pure endo-(1-->)-beta-D-glucanase ('cellulase') of fungal origin. The fungal enzyme hydrolysed the polysaccharide almost completely to a mixture of the four xyloglucan oligosaccharides: [formula: see text] Exhaustive digestion with the nasturtium enzyme gave the same four oligosaccharides plus large amounts of higher oligosaccharides and higher-polymeric material. Five of the product oligosaccharides (D, E, F, G, H) were purified and shown to be dimers of oligosaccharides A to C. D (glc8xyl6) had the structure A-->A, H (glc8xyl6 gal4) was C-->C, whereas E (glc8xyl6gal), F (glc8xyl6gal2) and G (glc8xyl6gal3) were mixtures of structural isomers with the appropriate composition. For example, F contained B2-->B2 (30%), A-->C (30%), C-->A (20%), B2-->B1 (15%) and others (about 5%). At moderate concentration (about 3 mM) oligosaccharides D to H were not further hydrolysed by the nasturtium enzyme, but underwent transglycosylation to give oligosaccharides from the group A, B, C, plus higher oligomeric structures. At lower substrate concentrations, hydrolysis was observed. Similarly, tamarind seed xyloglucan was hydrolysed to a greater extent at lower concentrations. It is concluded that the xyloglucan-specific nasturtium-seed endo-(1-->4)-beta-D-glucanase has a powerful xyloglucan-xyloglucan endo-transglycosylase activity in addition to its known xyloglucan-specific hydrolytic action. It would be more appropriately classified as a xyloglucan endo-transglycosylase. The action and specificity of the nasturtium enzyme are discussed in the context of xyloglucan metabolism in the cell walls of seeds and in other plant tissues.
DOI:10.1016/0003-9861(92)90423-tURLPMID:1416968 [本文引用: 1]
Oligosaccharide subunits were prepared from xyloglucan (XG) by partial hydrolysis with cellulase and added back at micro- to millimolar concentrations to XG in the presence of nasturtium seed xyloglucanase (XG-ase). The oligosaccharides (0.2 mM) stimulated the capacity of this XG-ase to reduce the viscosity of XG solutions by 10- to 20-fold. Purification and fractionation of seed XG-ase activity by gel permeation fast protein liquid chromatography produced a single peak that was much more active in the presence than absence of added XG oligosaccharide. [14C]Fucose-labeled XG nonasaccharide was synthesized by pea fucosyltransferase and shown to be incorporated into polymeric XG in the presence of seed XG-ase without the net production of new reducing chain ends, even while the loss of XG viscosity and XG depolymerization were enhanced. It is concluded that in vitro seed XG-ase can transfer cleavage products of XG to XG oligosaccharides via endotransglycosylation reactions, thereby reducing XG M(r) without hydrolysis. Since this is the only XG-cleaving enzyme that develops in nasturtium seeds during germination, it may be that its transglycosylase and hydrolase capacities are both necessary to account for the rapid and complete depolymerization of XG that takes place.
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/BF02344317URL [本文引用: 1]
DOI:10.1042/bj2790529URL [本文引用: 1]
DOI:10.1046/j.1365-313x.1998.00343.xURLPMID:10069079 [本文引用: 1]
The Agrobacterium vacuum infiltration method has made it possible to transform Arabidopsis thaliana without plant tissue culture or regeneration. In the present study, this method was evaluated and a substantially modified transformation method was developed. The labor-intensive vacuum infiltration process was eliminated in favor of simple dipping of developing floral tissues into a solution containing Agrobacterium tumefaciens, 5% sucrose and 500 microliters per litre of surfactant Silwet L-77. Sucrose and surfactant were critical to the success of the floral dip method. Plants inoculated when numerous immature floral buds and few siliques were present produced transformed progeny at the highest rate. Plant tissue culture media, the hormone benzylamino purine and pH adjustment were unnecessary, and Agrobacterium could be applied to plants at a range of cell densities. Repeated application of Agrobacterium improved transformation rates and overall yield of transformants approximately twofold. Covering plants for 1 day to retain humidity after inoculation also raised transformation rates twofold. Multiple ecotypes were transformable by this method. The modified method should facilitate high-throughput transformation of Arabidopsis for efforts such as T-DNA gene tagging, positional cloning, or attempts at targeted gene replacement.
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.molp.2019.03.009URLPMID:30928636 [本文引用: 1]
CRISPR/Cas9 genome editing relies on sgRNA-target DNA base pairing and a short downstream PAM sequence to recognize target DNA. The strict protospacer adjacent motif (PAM) requirement hinders applications of the CRISPR/Cas9 system since it restricts the targetable sites in the genomes. xCas9 and SpCas9-NG are two recently engineered SpCas9 variants that can recognize more relaxed NG PAMs, implying a great potential in addressing the issue of PAM constraint. Here we use stable transgenic lines to evaluate the efficacies of xCas9 and SpCas9-NG in performing gene editing and base editing in rice. We found that xCas9 can efficiently induce mutations at target sites with NG and GAT PAM sequences in rice. However, base editors containing xCas9 failed to edit most of the tested target sites. SpCas9-NG exhibited a robust editing activity at sites with various NG PAMs without showing any preference for the third nucleotide after NG. Moreover, we showed that xCas9 and SpCas9-NG have higher specificity than SpCas9 at the CGG PAM site. We further demonstrated that different forms of cytosine or adenine base editors containing SpCas9-NG worked efficiently in rice with broadened PAM compatibility. Taken together, our work has yielded versatile genome-engineering tools that will significantly expand the target scope in rice and other crops.
DOI:10.1073/pnas.97.22.11984URLPMID:11050229 [本文引用: 2]
DsRed is a recently cloned 28-kDa fluorescent protein responsible for the red coloration around the oral disk of a coral of the Discosoma genus. DsRed has attracted tremendous interest as a potential expression tracer and fusion partner that would be complementary to the homologous green fluorescent protein from Aequorea, but very little is known of the biochemistry of DsRed. We now show that DsRed has a much higher extinction coefficient and quantum yield than previously reported, plus excellent resistance to pH extremes and photobleaching. In addition, its 583-nm emission maximum can be further shifted to 602 nm by mutation of Lys-83 to Met. However, DsRed has major drawbacks, such as strong oligomerization and slow maturation. Analytical ultracentrifugation proves DsRed to be an obligate tetramer in vitro, and fluorescence resonance energy transfer measurements and yeast two-hybrid assays verify oligomerization in live cells. Also, DsRed takes days to ripen fully from green to red in vitro or in vivo, and mutations such as Lys-83 to Arg prevent the color change. Many potential cell biological applications of DsRed will require suppression of the tetramerization and acceleration of the maturation.
DOI:10.1038/nbt0102-83URLPMID:11753367 [本文引用: 1]
The red fluorescent protein DsRed has spectral properties that are ideal for dual-color experiments with green fluorescent protein (GFP). But wild-type DsRed has several drawbacks, including slow chromophore maturation and poor solubility. To overcome the slow maturation, we used random and directed mutagenesis to create DsRed variants that mature 10-15 times faster than the wild-type protein. An asparagine-to-glutamine substitution at position 42 greatly accelerates the maturation of DsRed, but also increases the level of green emission. Additional amino acid substitutions suppress this green emission while further accelerating the maturation. To enhance the solubility of DsRed, we reduced the net charge near the N terminus of the protein. The optimized DsRed variants yield bright fluorescence even in rapidly growing organisms such as yeast.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}