 ,四川省南充市农业科学院大豆研究所/国家大豆产业技术体系南充综合试验站,四川南充 637000
,四川省南充市农业科学院大豆研究所/国家大豆产业技术体系南充综合试验站,四川南充 637000Development New Molecular Markers for Quantitative Trait Locus (QTL) Analysis of the Seed Protein Content Based on Whole Genome Re-Sequencing in Soybean
WANG Jia, ZENG ZhaoQiong, LIANG JianQiu, YU XiaoBo, WU HaiYing, ZHANG MingRong,Soybean Research Institute, Nanchong Academy of Agricultural Sciences/Nanchong comprehensive experimental station of National Soybean Industry Technology System, Nanchong 637000, Sichuan通讯作者:
收稿日期:2019-03-25接受日期:2019-05-7网络出版日期:2019-08-16
| 基金资助: |
Received:2019-03-25Accepted:2019-05-7Online:2019-08-16
作者简介 About authors
王嘉,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (4355KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
王嘉, 曾召琼, 梁建秋, 于晓波, 吴海英, 张明荣. 基于全基因组重测序的大豆分子标记开发及籽粒蛋白质含量QTL定位[J]. 中国农业科学, 2019, 52(16): 2743-2757 doi:10.3864/j.issn.0578-1752.2019.16.001
WANG Jia, ZENG ZhaoQiong, LIANG JianQiu, YU XiaoBo, WU HaiYing, ZHANG MingRong.
0 引言
【研究意义】大豆是中国重要的粮油饲兼用作物,在国民经济中占有重要地位。南方地区是中国大豆第三大主产区,同时也是重要的高蛋白生态区,常年种植面积200万hm2,种植方式以与玉米、果树等高大作物套作为主[1,2,3]。玉米-大豆带状复合种植模式作为南方旱地农业的一种主体模式,在不影响玉米产量的同时增加大豆种植面积和产量,对保障区域及国家粮食安全具有重要意义[4,5]。在玉米-大豆套作模式中,玉豆共生期50—60 d,大豆作为矮秆作物,共生期间会受到玉米的严重荫蔽影响,这就要求大豆耐荫性好、抗倒力强[6]。因此,结合高蛋白生态优势,培育高产、耐荫、抗倒伏等优良特性的大豆品种,提高大豆生产潜力,提升大豆品种竞争力对改善中国大豆产业现状至关重要。然而传统的育种方法需要10—15年才能培育出一个大豆新品种,既费时又费力[7]。近年来,随着现代分子生物学的迅猛发展对作物遗传育种产生了极其深远的影响,特别是分子标记辅助选择技术克服了常规育种周期长、预见性差、选择效率低的局限性。随着大豆基因组测序的完成以及高通量测序技术的快速发展,使得基因组范围内进行遗传分析成为可能,为功能性分子标记的开发提供了支撑。【前人研究进展】基于高通量重测序技术进行遗传分析已经在玉米[8,9,10]、水稻[11,12,13]、谷子[14,15]、油菜[16]、番茄[17]、辣椒[18]等作物中得到了广泛应用。束永俊等[19]基于大豆基因组重测序数据,共设计163对引物,其中73对引物的PCR产物具有酶切多态性,开发出CAPS标记73个,这73个CAPS标记靶向的基因与大豆重要农艺性状的形成相关。SONG等[20]对荷豆12进行全基因组重测序,共检测到49 276个小片段插入/缺失位点(insertion/deletion,InDel)和242 059单核苷酸多态位点(single nucleotide polymorphisms,SNP)。基于突变位点,开发了243个InDel标记,其中165个在荷豆12和Williams 82之间存在多态性,多态性率68%。为检测这些InDel标记的效能,构建了一个皱叶突变体的遗传图谱,并成功将CRINKLY LEAF位点定位到第7染色体上一个360 kb的区域内。在大豆籽粒蛋白质含量QTL定位方面,自1992年DIERS等[21]发展关于大豆蛋白含量QTL定位研究后,大量大豆蛋白含量相关QTL位点被报道。截止目前,美国农业部大豆种质资源数据库(SoyBase,http://www.soybase.org)收录了多达303个蛋白含量相关的QTL,其中,利用连锁分析检测到241个,利用关联分析检测到62个。对这些QTL分析发现,大豆蛋白质含量在遗传背景不同的群体中检测到的QTL数量有差异,QTL所在的连锁群或连锁区域也存在差异。连锁分析和关联分析均显示在第20染色体(I连锁群)检测到大豆籽粒蛋白质含量QTL的频率最高,其中,A688-Satt239在多个环境和多个遗传群体中均检测到,可能是一个比较稳定的大豆籽粒蛋白质含量QTL区段,且在该区段上预测了一些大豆籽粒蛋白质含量相关的候选基因[22]。其次,第15染色体(E连锁群)和第6染色体(C2连锁群)也分别存在一个多次检测到的QTL区段。【本研究切入点】目前,针对大豆籽粒蛋白质含量等性状的遗传规律,前人已开展了大量研究并取得较大进展。但前人研究所用的高蛋白亲本材料蛋白质含量普遍集中在45%—50%,鲜有超过50%的育成品种作亲本,且构建遗传群体的亲本表型差异较小。【拟解决的关键问题】本研究通过南豆12进行全基因组重测序,深度挖掘南豆12全基因组突变类型。针对其超高蛋白、耐荫、抗倒伏等优良特性分析相关变异基因及其表达模式,开发特异性分子标记,对籽粒蛋白质进行QTL定位分析,为后续高蛋白、耐荫、抗倒育种研究提供参考和分子标记资源。1 材料与方法
1.1 供试材料及性状考察
母本南豆12是南充市农业科学院育成的大豆品种,该品种籽粒蛋白质含量为51.79%,蛋脂和为69.42%,具有耐荫性好、抗倒力强、抗病毒病等优良特性,适宜与玉米间、套种植[23,24,25]。该品种于2009—2015连续7年被列为农业部主导品种和四川省主导品种,成为川渝地区大面积间套作当家品种和重要骨干亲本。父本为地方品种十月黄,该品种籽粒蛋白质质含量为40.50%,耐荫性差,极易倒伏。两亲本杂交获得F1植株,F1自交获得672个单株组成的F2分离群体。栽培大豆测序品种Williams 82由南京农业大学馈赠,蛋白质含量为39.5%,套作下重倒。所有材料均种植于南充市农业科学院潆溪试验基地,田间管理同常规生产,确保所有样本的外部生长环境一致。成熟后单株收获、脱粒,自然风干后使用近红外品质分析仪测定籽粒蛋白质含量。1.2 DNA提取、重测序及基因组变异检测与注释及变异基因分析
待材料第一片三出复叶全展时,分别采集所有材料中间小叶,采用改良的CTAB法[26]提取叶片总DNA。质检合格的南豆12和十月黄的DNA,分别随机选取5株混样送至华大基因研究院,在Illumina HiSeq 2500测序平台下,进行深度为40×的基因组重测序。其余材料DNA-20℃保存备用。以Glyma.Wm82.a2v1作为参考基因组[27],应用BWA软件将质控后的测序数据比对到参考基因组上[28]。GATK(https://www.broadinstitute.org/gatk/)被用于检测SNP和InDel[29],Breakdancer(http:// breakdancer.sourceforge.net/)被用于检测SV[30],基于SOAP比对结果,计算基因组上各个位置间测序深度,并对各个位置的深度进行标准化,从而计算出基因组上各个位置的拷贝数变异值。应用BGI自主开发的软件对检测结果进行注释和统计。
1.3 转录组数据分析及qRT-PCR
提取南豆12花后14、21、28和35 d的种子RNA,每个时期3株等量混匀,送至公司进行转录组测序,3个生物学重复。荫蔽处理转录组数据来自GONG等[31]研究结果,分别选取遮阴和对照处理的成熟叶和嫩叶进行转录组测序。选取10个重测序挖掘的高蛋白、耐荫等相关且差异表达的变异基因进行qRT-PCR验证,qRT-PCR所用引物见表1。qRT-PCR分析使用一步法qRT-PCR试剂盒(One Step qRT-PCR Kit),反应体系为10 μL 2×one step qRT-PCR Master Mix、0.4 μL Primer F(10 μmol·L-1)、0.4 μL Primer R(10 μmol·L-1)、0.65 μL RT enzyme Mix和0.1—100 ng RNA Template,添加ddH2O至20 μL。qRT-PCR反应程序为50℃反转录5 min,95℃ 3 min;95℃ 10 s,60℃ 30 s,40个循环;以ACT11为内参,以65—95℃作产物溶解曲线,每个qRT-PCR反应设3次生物学重复。Table 1
表1
表1qRT-PCR验证基因引物信息
Table 1
| 基因ID Gene ID | 基因名称 Gene name | 正向引物 Forward primer (5'-3') | 反向引物 Reverse primer (5'-3') |
|---|---|---|---|
| Glyma.18G176100 | ABI3 | CTCATCATGCAAATCCTACAGC | CTATCTGATGCAACTCGACTCT |
| Glyma.13G123500 | Gy5 | CAGATATAGAGCACCCAGAGAC | GGTGTTGAAGGATTGTGCTAAG |
| Glyma.18G175500 | MAG2 | CAAATTTGGGGCATGGCTAATA | CTTGAAAATTGTTTCGGTTGCG |
| Glyma.18G226500 | CRA1 | CTGCAAGAAGTGGTCGTTTTTA | ATTTCAGTTGTTCCATGTTCCG |
| Glyma.02g248300 | PAT2 | AAAGGCGTTGAAGCTGGATATA | AATCTCCTATCTGGATCTCCGA |
| Glyma.11g112800 | BBX21 | CACAACAGGTTTCTTCTCACTG | TGTTCTTTCTTCAATGGCAGTG |
| Glyma.14g062100 | COI1 | GTACATACACGACTCCAAGGAC | TTTCCCTTCAGGTTTAACGACT |
| Glyma.05g062700 | SAV1 | GTGGGTCTCCAATGTTTTGTAC | GCAAATTGTTTCAATGGGTGTG |
| Glyma.18g043000 | KAN | TTGGAGTTCACATTAGGGAGAC | AAGTTTGTCACATGTTACCGTG |
| Glyma.03g227300 | PHYA | CAAAGTCTGATAGAGCAGCAAC | CTCACTGTAAGCGATGACCTTA |
| ACT11 | CGGTGGTTCTATCTTGGCATC | GTCTTTCGCTTCAATAACCCTA |
新窗口打开|下载CSV
1.4 CAPS标记和InDel标记开发
根据比对结果,提取非同义突变SNP位点两端各1 000 bp序列,利用NEBcutter V2.0(http://nc2.neb.com/ NEBcutter2/index.php)进行限制性内切酶识别位点分析,选取对酶切位点产生影响的SNP,利用Primer 5在该SNP的旁侧序列上设计引物开发CAPS标记。若SNP处无酶切位点,利用dCAPS Finder 2.0开发dCAPS标记[32]。参照New England Biolabs(NEB)公司的限制性内切酶操作指南,用限制性内切酶酶切PCR扩增的目标基因片段。酶切反应体系包含2 μL限制性内切酶(10 U·μL-1)、2 μL buffer和10 μL PCR扩增产物,18 μL无核酸酶水。37℃恒温水浴2 h,用2%琼脂糖凝胶电泳检测标记多态性。InDel标记选取发生在CDS区域上的InDel位点进行引物设计,为后续检测方便,以差异碱基数10个以上,且两侧高度保守为理想的InDel设计区。提取InDel位点两端各500 bp序列,使用Primer 5软件设计引物。所有引物由生工生物工程(上海)股份有限公司合成。
1.5 遗传连锁图谱构建及QTL分析
采用JoinMap 4.0软件构建遗传连锁图谱,在重组率r<0.1情况下对标记进行分群,采用极大似然函数值作为标记排序的目标函数,用Kosambi作图函数将重组率转换为遗传距离。采用QTL分析软件Windows QTL Cartographer 2.5及复合区间作图(composite interval mapping,CIM)法对籽粒蛋白质含量进行QTL定位及效应检测[33]。LOD≥2.0时,即认为该区间可能存在一个QTL。运行软件后可同时给出各QTL的加性效应和解释的表型变异。2 结果
2.1 南豆12全基因组重测序结果及变异基因分析
测序结果显示,DNA文库共获得199.35 M个原始reads。去除带接头的或低质量的reads后得到169 072 750(84.82%)个Clean reads,定位到参考基因组的Clean reads数占比95.73%,正确识别率大于Q30的碱基占比89.52%以上,基因组GC含量39.27%,样品平均覆盖深度55×。根据Clean reads在参考基因组的定位结果进行变异检测,共得到2 453 344个SNP,476 953个Small Indel,20 085个SV以及12 605个CNV(图1-a)。通过寻找参考基因组与南豆12基因组间的各种变异,发现与Williams 82相比,南豆12基因组共存在38 896个基因变异,其中14 491个基因存在2种或2种以上类型的突变,318个基因同时存在4种类型的变异(图1-b)。KOG分析结果表明,信号传导机制,功能预测,翻译后修饰、蛋白折叠和伴侣蛋白,转录,碳水化合物转运与代谢,氨基酸转运与代谢以及次生代谢产物的生物合成、转运和分解等7个功能类存在较多的变异基因,其中,信号传导机制类变异基因2 630个,氨基酸转运与代谢类变异基因713个,次生代谢产物的生物合成、转运和分解类变异基因807个(图1-c)。KEGG分析结果表明,植物激素信号传导存在1 301个变异基因,内质网蛋白质加工存在839个变异基因,其他次生代谢产物的生物合成存在485个变异基因,环境适应存在375个变异基因(图1-d)。图1
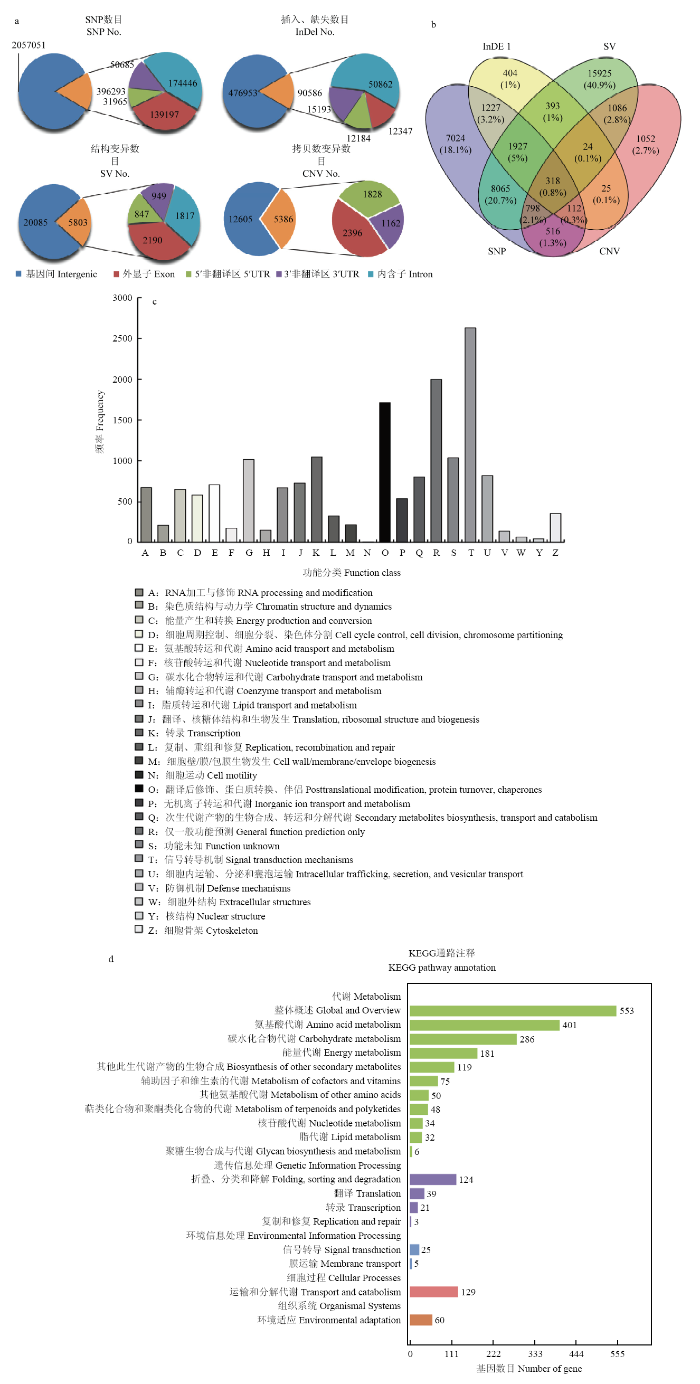 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1南豆12全基因组注释结果
a:南豆12全基因组注释结果;b:4种变异类型的基因韦恩图;c:变异基因COG注释分类图;d:变异基因KEGG注释图
Fig. 1Whole genome annotation results of Nandou 12
a: Whole genome annotation results of Nandou 12; b: Diagram of gene variations identified in four variations on Nandou 12 coding region; c: Classification of gene variations compared with COG database by blast; d: Classification of gene variations compared with KEGG database by blast
2.2 南豆12和十月黄相关性状等位基因的比较分析
为了鉴定南豆12和十月黄中存在的关于高蛋白、耐荫、抗倒伏等性状丰富的等位基因信息,对其基因型进行了比较。基于全基因组重测序数据,两亲本间共检测到9 705个SNP位点和796个InDel位点。进一步分析位于基因区域内可能介导功能变异的SNP发现,1 658个SNP出现在编码区,其中,595个出现在CDS区的SNP,导致263个基因因非同义替换事件而出现蛋白序列差异。根据发生非同义替换的SNP数目,分析发生非同义替换最多的前10个基因发现(表2),Glyma.18G176100存在的非同义替换SNP位点最多,其基因内存在72个SNP位点,31个位于CDS区域的SNP中有20个为非同义替换。Glyma.18G176100属于ABI3-like转录因子家族成员,且大豆ABI3-like转录因子通过与贮藏蛋白基因启动子中的RY重复元件结合并促进贮藏蛋白基因的表达[34],ABI3-like转录因子对大豆蛋白质的积累具有重要作用。Glyma.13G123500(Gy5)为大豆球蛋白基因家族成员之一,编码一个11S球蛋白亚基,在种子形成的胚乳期大量表达,其变异会影响大豆种子贮藏蛋白的积累[35]。比较发现,Glyma.13G123500存在46个SNP,33个位于CDS区域的SNP中18个属于非同义替换。其余含有较多非同义替换SNP的基因功能主要涉及荫蔽反应、储藏蛋白合成及运输以及形态建成等。Table 2
表2
表2南豆12和十月黄之间出现非同义替换最多的前10个基因
Table 2
| 基因ID Gene ID | 基因名称 Gene name | 注释 Annotation | 基因区域内的SNP数目 Genic SNP | CDS中的SNP SNP in CDS | 非同义替换SNP Non-syn SNP |
|---|---|---|---|---|---|
| Glyma.18G176100 | ABI3 | ABI3-like转录因子家族成员,促进种子储藏蛋白的积累 A member of ABI3-like transcription factor family, promoting the accumulation of seed storage proteins | 72 | 31 | 20 |
| Glyma.13G123500 | Gy5 | 大豆球蛋白基因家族成员之一,编码一个11S球蛋白亚基 Encodes an 11S globulin subunit, a member of the soybean globulin gene family | 46 | 33 | 18 |
| Glyma.11G112800 | BBX21 | 编码B-box锌指转录因子bbx21,与cop1基因相互作用调节避荫 Encodes a B-box zinc finger transcription factor BBX21, genetically interacts with COP1 to regulate shade avoidance. | 30 | 29 | 16 |
| Glyma.18G175500 | MAG2 | 参与种子贮藏蛋白从内质网到液泡的运输 Involved in transportation of seed storage proteins from the ER to the vacuole | 36 | 27 | 14 |
| Glyma.13G335200 | OBP3 | 编码一个主要在根中表达的包含转录因子的核定位Dof域 Encodes a nuclear localized Dof domain containing transcription factor expressed primarily in roots. | 35 | 35 | 14 |
| Glyma.02G057500 | SAV1 | 编码一个油菜素类固醇生物合成途径的限速酶22α羟化酶 Encodes a 22α hydroxylase whose reaction is a rate-limiting step in brassinosteroid biosynthetic pathway | 51 | 29 | 12 |
| Glyma.08G171800 | GA2OX4 | 编码作用于赤霉素C19的赤霉素2-氧化酶 Encodes a gibberellin 2-oxidase that acts on C19 gibberellins | 88 | 26 | 11 |
| Glyma.20G048500 | VPS26B | 细胞内蛋白转运 Intracellular protein transport | 32 | 25 | 11 |
| Glyma.18G226500 | CRA1 | 编码一个12S种子储存蛋白 Encodes a 12S seed storage protein | 72 | 26 | 10 |
| Glyma.14G062100 | COI1 | 避荫 Shade avoidance | 28 | 25 | 10 |
新窗口打开|下载CSV
2.3 变异基因表达模式分析及qRT-PCR验证
基于南豆12和十月黄重测序结果,结合转录数据对发生变异的储藏蛋白合成及运输、荫蔽反应及形态建成和抗倒伏相关性状的基因进行表达模式分析。结果发现,在大豆籽粒发育过程中,相关基因呈现不同的表达模式,跟储藏蛋白合成相关的基因如Gy5(Glyma.13G123500)、ABI3(Glyma.18G176100)、CAR1(Glyma.03G163500)等基因在花后14—28 d一直处于较低表达水平,到花后35 d时表达量急剧上升(图2-a)。在荫蔽调节方面,参与荫蔽调节的相关基因在成熟叶与嫩叶中的表达水平呈现明显的差异,例如参与叶片发育的基因SAV1(Glyma.11g067700)在成熟叶中,对照和遮阴处理下表达水平均较低,而在嫩叶中表达水平升高。此外,部分基因如Glyma. 05g017800、Glyma.10g138800、Glyma.10g138800等无论在成熟叶还是嫩叶中,遮阴处理下的表达水平均高于对照,表明这些基因可能参与荫蔽条件的响应(图2-b)。图2
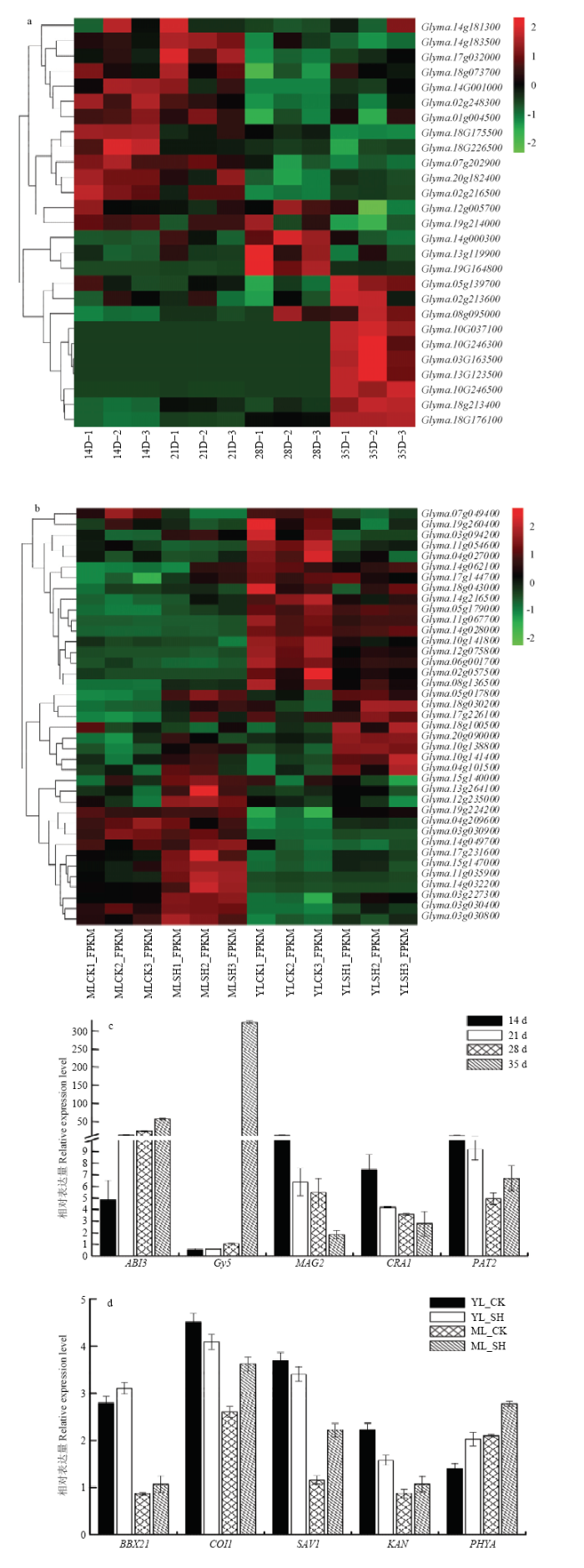 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2变异基因表达热图及转录组测序数据的qRT-PCR验证
a:籽粒发育时期蛋白合成与积累相关基因表达热图;b:遮阴处理下荫蔽调节相关基因在成熟叶和嫩叶中的表达模式图;c、d:随机挑选的变异基因的qRT-PCR验证
Fig. 2Expression heatmap of mutant genes and validation of transcriptome sequencing data by qRT-PCR
a: Expression heatmap of related to protein synthesis and accumulation gene during seed development; b: Expression pattern of shade regulation related genes in mature and young leaves under shading treatment; c, d: Validation of transcriptome sequencing data by qRT-PCR
随机挑选其中10个差异表达的变异基因进行qRT-PCR验证(5个储藏蛋白相关基因,5个荫蔽调节相关基因)。qRT-PCR分析结果显示5个上调表达基因的表达趋势均与转录组测序结果相符合,5个下调表达基因也与转录组测序结果基本一致(图2-c和图2-d),说明转录组测序的结果具有较高的准确性。
2.4 CAPS标记和InDel标记开发及多态性分析
在重测序基础上,经10种限制性内切酶(AluⅠ、BanⅡ、BstNⅠ、XbaⅠ、EcoRⅠ、BamHⅠ、HindⅢ、PstⅠ、HinfⅠ和XhoⅠ)酶切位点分析后,在两亲本材料中共获得540个均匀分布在全基因组上的CAPS/dCAPS标记位点。利用亲本和F2群体进行CAPS/dCAPS引物多态性筛选,获得332对多态引物,多态性率61.48%。其中,有7对多态性引物分别靶向Gy1、Gy5、CG-1、SAV3、AP4M等功能基因(图3-a和图3-b)。通过搜索InDel位点,根据其在基因组上的分布,筛选并设计300对代表性InDel引物。利用南豆12、十月黄和F2群体对InDel引物多态性进行筛选,获得201对多态引物,占总设计数比例为67.0%。其中,GmIn113靶向调节避荫反应的基因BBX21(图3-c和图3-d)。图3
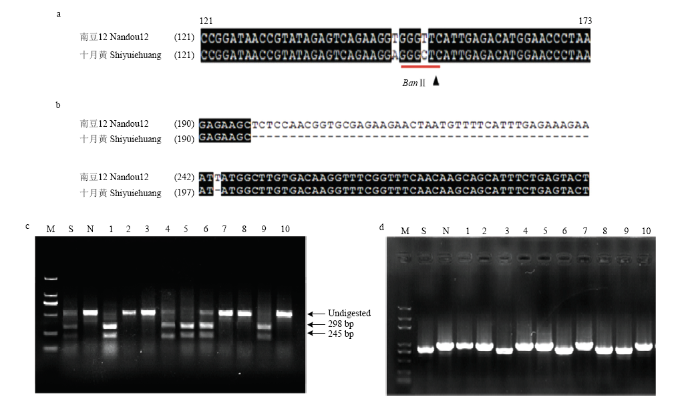 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3CAPS标记和InDel标记开发及多态性分析
a:靶向Gy1含限制性酶切位点BanⅡ的SNP部分序列比对结果。线条代表标注出的限制性内切酶识别位点,箭头代表酶切位点;b:南豆12和十月黄中BBX21包含InDel的部分序列比对结果;c:靶向Gy1的CAPS标记酶切检测结果;d:靶向BBX21的InDel标记的琼脂糖凝胶电泳结果。S:Shiyuehuang,N:南豆12,1—10:部分F2材料
Fig. 3Development and polymorphism analysis of CAPS and InDel markers
a: Alignment of the partial sequence of containing the SNP which resulted in the creation of the restriction sites (BanⅡ) between Nandou 12 and Shiyuehuang in Gy1. Lines represent the resriction sites, while arrows indicate the position of digesting site; b: Sequence alignment results of BBX21 gene containing InDel in Nandou 12 and Shiyuehuang; c: Detection of CAPS labeled enzyme digestion targeting Gy1 gene; d: The results of InDel markers amplification for targeting BBX21 gene were analyzed by agarose gel electrophoresis. S: Shiyuehuang, N: Nandou 12, Figure exhibited partial verifying lines (code number: 1-10)
2.5 遗传图谱构建及QTL分析
采用JoinMap 4.0软件对533个多态性标记进行连锁分析,构建了一张包含332个CAPS/dCAPS标记、201个InDel标记、20个连锁群(分别对应于大豆20条染色体)的大豆遗传连锁图谱,覆盖基因组长度2 973.87 cM,标记间的平均遗传距离为5.58 cM,各连锁群长度从83.69 cM(C2)到194.50 cM(C1)(数据未发表)。对亲本及F2的籽粒蛋白质含量分析发现,粗蛋白含量在F2群体中均呈连续分布和双向超亲分离,基本符合正态分布,可以进行QTL分析(表3)。利用Windows QTL CartographerV 2.5复合区间作图法对F2群体的籽粒蛋白质含量进行QTL分析,共检测到与籽粒蛋白质含量相关的QTL位点6个,分别分布在C2、E、I和M4条连锁群上(表4和图4),其中在I连锁群上(第20染色体)检测到2个QTL位点,位于35.61 cM处的QTL加性效应为-0.47,可解释9.32%的表型变异。位于103.41 cM处的QTL加性效应为-0.68,可解释18.25%的表型变异,为主效QTL位点。
Table 3
表3
表3亲本及F2群体籽粒蛋白质含量表型分析
Table 3
| 性状 Trait | 亲本 Parent | F2群体 F2 population | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 南豆12 Nandou 12 | 十月黄 Shiyuehuang | 范围 Range | 均值 Mean | 方差 Variance | 标准差 SD | 变异系数 CV | 偏度 Skewness | 峰度 Kurtosis | ||
| 蛋白质含量 Protein contents | 50.96 | 41.35 | 40.34—51.72 | 45.37 | 3.47 | 1.86 | 4.10 | 0.16 | -0.09 | |
新窗口打开|下载CSV
Table 4
表4
表4利用复合区间作图法检测到的籽粒蛋白质含量QTL
Table 4
| 数量性状座位 QTL | 染色体 Chromosome | 标记区间 Position (cM) | 置信区间 Confidence interval | LOD | 加性效应 Additive | 贡献率 R2 (%) |
|---|---|---|---|---|---|---|
| qSPC-1 | Gm06(C2) | 34.01 | GmIn063—GmCA130 | 8.09 | -0.23 | 5.39 |
| qSPC-2 | Gm07(M) | 46.71 | GmCA168—GmCA171 | 5.26 | 0.22 | 5.36 |
| qSPC-3 | Gm15(E) | 26.51 | GmIn149—GmIn150 | 4.53 | 0.21 | 4.68 |
| qSPC-4 | Gm15(E) | 72.21 | GmIn152—GmIn163 | 9.93 | -0.29 | 9.32 |
| qSPC-5 | Gm20(I) | 35.61 | GmCA493—GmIn285 | 8.40 | -0.47 | 12.04 |
| qSPC-6 | Gm20(I) | 103.41 | GmdCA522—GmCA537 | 15.06 | -0.68 | 18.86 |
新窗口打开|下载CSV
图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4大豆籽粒蛋白质含量QTL在连锁群上的分布图
SPC:籽粒蛋白含量
Fig. 4Putative QTL locations of seed protein content on the genetic map
Seed protein content
3 讨论
3.1 大豆籽粒蛋白质含量的QTL分析
由于大豆种子含有约40%的蛋白质,全世界71%的膳食消费依赖于大豆,且这一比例还在逐年上升[36]。因此,通过遗传改良提高大豆籽粒蛋白质含量是大豆重要的育种目标之一。研究表明,大豆蛋白质含量是以加性效应为主的数量性状,受微效多基因控制,且存在基因型与环境互作现象[37]。随着分子标记技术和统计方法的发展,基于连锁作图的QTL定位分析方法被广泛应用于解析动植物的复杂数量性状。近20年来,美国农业部大豆种质资源数据库(SoyBase, http://www.soybase.org)报道了大量与蛋白质含量相关的QTL。然而,这些QTL中仅有57个被验证[38]。虽然各个染色体上均检测到大量大豆蛋白含量相关的QTL,但是绝大多数位点检测的频率不高。这些QTL中大多数是利用F2、重组自交系(RIL)和回交自交系(BIL)群体检测到的,然而这些遗传群体大多来自表型差异较小的2个亲本,因此很难有效地检测影响相关性状表型差异显著比例的微效QTL[38]。除了受分析方法、遗传背景、群体大小、标记数目以及环境影响外,等位基因的分散导致不可能用单个群体检测到影响特定性状的所有QTL。因此,利用表现型差异较大的大豆品种构建相关性状的图谱群体是提高QTL鉴定准确性的前提[21]。此外,需要通过不同的试验来检测不同的QTL和QTL等位基因。在本研究中,以高蛋白品种南豆12为亲本,结合全基因组重测序和转录组测序,利用开发出的分子标记构建的遗传连锁图谱共检测到6个与大豆蛋白含量相关的QTL。其中,位于I连锁群上的主效QTL与前人利用多群体多环境检测到大豆籽粒蛋白质含量QTL置信区间重叠(数据来自SoyBase,http://www.soybase.org),进一步印证了该处可能是一个比较稳定的大豆籽粒蛋白质含量QTL区段,为下一步分子设计育种提供了有价值的参考。3.2 可靠的标记资源是开展分子标记辅助选择育种的必要前提
随着现代分子生物学的快速发展,分子标记辅助选择日益成为作物改良过程中的一种重要辅助手段。分子标记也由过去基于基因组DNA随机开发的、位置不确定的DNA标记,向代表编码区序列的分子标记以及具有相应功能的功能标记转变[39]。同时,覆盖全基因组的丰富的分子标记是鉴定大豆相关性状主效QTL和候选基因的必要前提。目前,在植物育种中应用最广泛的是基于PCR技术的分子标记,如RAPD、SSR、SCAR以及ISSR等。ZENG等[40]利用SSR标记以及基于亲本间差异开发的分子标记,采取杂交、回交与分子标记定向选择等技术策略,成功将优质目标基因的优异等位聚合到受体材料,在高产的基础上,使稻米外观品质、蒸煮食味品质、口感和风味等方面均有显著改良。成功实现了“籼稻的产量,粳稻的品质”的理想目标。ZHANG等[41]利用3个大豆蛋白质含量主效QTL的侧翼标记SSR和PAV标记,通过标记辅助亲本选择和标记辅助子代选择2个阶段进行标记辅助育种,在F2:5:6代获得了最高比原始亲本蛋白质含量高54.15%的材料。本研究开发的部分CAPS标记是由基因上的SNP位点转化来的,InDel标记更是直接来自基因编码区的插入/缺失变异,2种标记都保留了与目标基因直接连锁的特性,具有基因特异性。代表Gy1、CG1和AP4M序列变异的CAPS标记与大豆种子储藏蛋白含量紧密连锁,代表BBX21序列变异的InDel标记则与耐荫密切相关。PCR扩增结果显示,这些标记在F2分离群体中存在明显的多态性。利用构建的遗传连锁图谱,结合籽粒蛋白质含量数据对这些标记的效能进行了验证,检测到的QTL位点与前人定位结果置信区间重叠。上述结果表明,本研究开发的部分标记,可为下一步分子设计育种提供可靠的标记资源。3.3 中国大豆生产面临的现状及探讨
作为重要的植物蛋白质和食用油来源,中国对大豆的需求量逐年提高。2017年中国大豆总产量为1 450万t,而进口大豆高达9 554万t,自给率仅13.2%。受国内外环境影响,2018年中国大豆种植面积创下新高,但2018年大豆进口量仍高达8 803万t,国产大豆的发展形势依然严峻。西南、华南间套作食用大豆生产区被农业部《十二五种植业发展规划》列为全国大豆优势主产区。发展间套作大豆,不仅科学的解决了作物间争地、争肥、争劳动力的矛盾,同时具有显著的增产节肥增效增收优势,能够一定程度的缓解中国大豆供不应求的紧张局面。如何提高大豆耐荫抗倒性,筛选出适合玉豆带状套作种植的材料,是当下玉米大豆套作大豆研究的重点[42]。利用已有骨干亲本,解析其优异遗传背景,开发相关性状紧密连锁的分子标记,结合杂交、回交与分子标记定向选择等技术,将优质目标基因的优异等位聚合到受体材料,并充分保留原品种的优良特性。在保留原有优异特性的基础上,使大豆蛋白质含量、耐荫性、抗倒伏能力等方面均有显著改良。通过高效、精准、定向的分子设计育种是加快选育优良品种的有效途径。本研究对间套作育种的重要亲本南豆12进行遗传解析,发现大量储藏蛋白、环境适应相关的重要基因或同源基因发生突变,这些变异可能导致了相关基因的功能分化或不等量表达,进而塑造了南豆12的优良特性,变异基因的具体功能还需要下一步验证。但针对这些突变位点,开发在高产优质大豆品种选育中更有功效的标记,使基因选择与表型选择真正结合,加快育种进程,选育出适合南方地区间套作的优良品种,有助于加强中国大豆的市场竞争力。4 结论
基于亲本间的变异位点,开发了533个分子标记,构建了一张长度为2 973.87 cM包含20个连锁群的遗传连锁图谱,QTL分析检测到大豆籽粒蛋白质含量QTL位点6个,其中主效QTL位点1个。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.7505/j.issn.1007-9084.2016.02.003Magsci [本文引用: 1]

<p>为全面揭示大豆优良品种和重要亲本的全基因组突变类型,采用高通量重测序技术,对大豆品种齐黄34进行全基因组重测序,测序深度13×,共检测到1 519 494个单核苷酸多态位点,357 549个小片段插入缺失位点,4 506个结构变异;这些变异共导致了17 748基因变异;大量光周期相关的重要基因发生突变,其中包括CRYPTOCHROME 2、GIGANTEA、Timing of CAB expression 1、E1、PHYTOCHROME A、Flowering Locus T、CONSTANS、Terminal Flower like等;齐黄34为E1基因型,为分子标记辅助选择提供了重要的标记资源。</p>
DOI:10.7505/j.issn.1007-9084.2016.02.003Magsci [本文引用: 1]
<p>为全面揭示大豆优良品种和重要亲本的全基因组突变类型,采用高通量重测序技术,对大豆品种齐黄34进行全基因组重测序,测序深度13×,共检测到1 519 494个单核苷酸多态位点,357 549个小片段插入缺失位点,4 506个结构变异;这些变异共导致了17 748基因变异;大量光周期相关的重要基因发生突变,其中包括CRYPTOCHROME 2、GIGANTEA、Timing of CAB expression 1、E1、PHYTOCHROME A、Flowering Locus T、CONSTANS、Terminal Flower like等;齐黄34为E1基因型,为分子标记辅助选择提供了重要的标记资源。</p>
[本文引用: 1]
DOI:10.3864/j.issn.0578-1752.2018.08.001Magsci [本文引用: 1]
【目的】降低果皮纤维素是甜玉米品质改良的重要目标,然而玉米果皮纤维素含量调控的研究甚少,相关调控基因尚未定位。利用纤维素含量差异的重组自交系(RILs)群体,通过特异位点扩增片段(specific-locus amplified fragment-sequencing,SLAF)测序和分离混池分析(bulked segregant analysis,BSA)定位控制玉米果皮纤维素含量的染色体区段,鉴定调控玉米果皮纤维素含量的候选基因。【方法】以果皮纤维素含量显著差异的E327和G5-1为亲本,构建重组自交系(RILs)。对RILs群体进行果皮纤维素含量的测定,并根据纤维素含量的结果选择纤维素含量高、低的样本进行混池,用于SLAF标签的鉴定和BSA分析。在BSA分析中,首先对两混池和2个亲本DNA用HaeⅢ和Hpy166Ⅱ进行酶切,回收414—464 bp的酶切片段进行Illumina建库,并进行SLAF测序,然后根据多态性SLAF标签开发SNP标记,利用SNP标记对玉米果皮纤维素含量进行关联分析,鉴定调控甜玉米果皮纤维素含量的染色体区段。分析这些区段所包含的玉米基因,并找到它们对应的拟南芥同源基因,通过查阅拟南芥相关基因功能研究的文献,进一步鉴定控制玉米果皮纤维素含量的候选基因。【结果】两亲本和2个混池SLAF建库测序得到的SLAF符合预期,基于SLAF测序数据,鉴定了73 786个多态性SLAF标签,这些SLAF标签均匀分布在玉米的10条染色体上。在这些多态性标签中得到了523 395 SNP位点信息。通过关联分析,调控果皮纤维素变异的基因被定位到玉米基因组的6个染色区段,都位于玉米的第5染色体上。在这些区段上,一共有47个玉米基因。通过进一步的研究分析,在这些关联的染色体区段最终确定了9个候选基因。【结论】定位到调控玉米果皮纤维素的含量的基因,表明此方法可以用于基因定位。
DOI:10.3864/j.issn.0578-1752.2018.08.001Magsci [本文引用: 1]
【目的】降低果皮纤维素是甜玉米品质改良的重要目标,然而玉米果皮纤维素含量调控的研究甚少,相关调控基因尚未定位。利用纤维素含量差异的重组自交系(RILs)群体,通过特异位点扩增片段(specific-locus amplified fragment-sequencing,SLAF)测序和分离混池分析(bulked segregant analysis,BSA)定位控制玉米果皮纤维素含量的染色体区段,鉴定调控玉米果皮纤维素含量的候选基因。【方法】以果皮纤维素含量显著差异的E327和G5-1为亲本,构建重组自交系(RILs)。对RILs群体进行果皮纤维素含量的测定,并根据纤维素含量的结果选择纤维素含量高、低的样本进行混池,用于SLAF标签的鉴定和BSA分析。在BSA分析中,首先对两混池和2个亲本DNA用HaeⅢ和Hpy166Ⅱ进行酶切,回收414—464 bp的酶切片段进行Illumina建库,并进行SLAF测序,然后根据多态性SLAF标签开发SNP标记,利用SNP标记对玉米果皮纤维素含量进行关联分析,鉴定调控甜玉米果皮纤维素含量的染色体区段。分析这些区段所包含的玉米基因,并找到它们对应的拟南芥同源基因,通过查阅拟南芥相关基因功能研究的文献,进一步鉴定控制玉米果皮纤维素含量的候选基因。【结果】两亲本和2个混池SLAF建库测序得到的SLAF符合预期,基于SLAF测序数据,鉴定了73 786个多态性SLAF标签,这些SLAF标签均匀分布在玉米的10条染色体上。在这些多态性标签中得到了523 395 SNP位点信息。通过关联分析,调控果皮纤维素变异的基因被定位到玉米基因组的6个染色区段,都位于玉米的第5染色体上。在这些区段上,一共有47个玉米基因。通过进一步的研究分析,在这些关联的染色体区段最终确定了9个候选基因。【结论】定位到调控玉米果皮纤维素的含量的基因,表明此方法可以用于基因定位。
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.3724/SP.J.1006.2009.02015Magsci [本文引用: 1]
<p><span >为了开发大豆基因靶向的功能分子标记,本研究采用生物信息学方法分析了大豆基因重测序数据,筛选出酶切位点突变的</span><span >SNP</span><span >位点,设计</span><span >PCR</span><span >引物</span><span >163</span><span >对,选用东北地区主栽品种绥农</span><span >14</span><span >的</span><span >DNA</span><span >为模板进行</span><span >PCR</span><span >扩增,其中</span><span >139</span><span >对引物获得大小为</span><span >400~1 200 bp</span><span >的特异片段。以大豆绥农</span><span >14</span><span >、合丰</span><span >25</span><span >、</span><span >Acher</span><span >、</span><span >Evans</span><span >、</span><span >Peking</span><span >、</span><span >PI209332</span><span >、固新野生大豆、科丰</span><span >1</span><span >号和南农</span><span >1138-2</span><span >的</span><span >DNA</span><span >为模板,采用筛选的</span><span >139</span><span >对引物进行</span><span >PCR</span><span >扩增,对扩增产物进行酶切分析,发现</span><span >73</span><span >对引物的</span><span >PCR</span><span >产物具有酶切多态性,开发出</span><span >CAPS</span><span >标记</span><span >73</span><span >个。通过功能注释分析发现,这</span><span >73</span><span >个</span><span >CAPS</span><span >标记靶向的基因主要参与细胞内亚细胞定位过程、蛋白质的结合与催化以及代谢过程等,与大豆重要农艺性状的形成相关,可以用于大豆品种的鉴定和分子系统进化的研究。</span></p>
DOI:10.3724/SP.J.1006.2009.02015Magsci [本文引用: 1]
<p><span >为了开发大豆基因靶向的功能分子标记,本研究采用生物信息学方法分析了大豆基因重测序数据,筛选出酶切位点突变的</span><span >SNP</span><span >位点,设计</span><span >PCR</span><span >引物</span><span >163</span><span >对,选用东北地区主栽品种绥农</span><span >14</span><span >的</span><span >DNA</span><span >为模板进行</span><span >PCR</span><span >扩增,其中</span><span >139</span><span >对引物获得大小为</span><span >400~1 200 bp</span><span >的特异片段。以大豆绥农</span><span >14</span><span >、合丰</span><span >25</span><span >、</span><span >Acher</span><span >、</span><span >Evans</span><span >、</span><span >Peking</span><span >、</span><span >PI209332</span><span >、固新野生大豆、科丰</span><span >1</span><span >号和南农</span><span >1138-2</span><span >的</span><span >DNA</span><span >为模板,采用筛选的</span><span >139</span><span >对引物进行</span><span >PCR</span><span >扩增,对扩增产物进行酶切分析,发现</span><span >73</span><span >对引物的</span><span >PCR</span><span >产物具有酶切多态性,开发出</span><span >CAPS</span><span >标记</span><span >73</span><span >个。通过功能注释分析发现,这</span><span >73</span><span >个</span><span >CAPS</span><span >标记靶向的基因主要参与细胞内亚细胞定位过程、蛋白质的结合与催化以及代谢过程等,与大豆重要农艺性状的形成相关,可以用于大豆品种的鉴定和分子系统进化的研究。</span></p>
[本文引用: 1]
[本文引用: 2]
DOI:10.7505/j.issn.1007-9084.2015.03.021Magsci [本文引用: 1]
<p>综述了近年来大豆籽粒蛋白质含量分子遗传方面的研究进展。由于遗传因素和环境因素对蛋白质含量均有很大的影响,因此,不同研究者的研究结果差异很大。仅利用表型数据的传统遗传研究表明,大豆籽粒蛋白质含量以加性效应为主,但不同材料之间遗传力差异很大,在31%~99%之间。在所有20个染色体上均发现有相关的QTL位点,共有298个QTL,贡献率介于0.002%~80%之间,其中20号(I连锁群)、15号(E连锁群)等染色体上均存在多次检测到的QTL区段。连锁分析和关联分析均显示20号染色体检测到大豆籽粒蛋白质含量QTL的频率均最高,且在该染色体上预测了一些大豆籽粒蛋白质含量相关的候选基因,但这些候选基因在染色体上的位置不一致,还需要进一步的验证。20号染色体有一个QTL区段(A688-Satt239)在多个环境和群体中均检测到,这为高蛋白分子标记辅助育种提供依据,并为后续的深入研究提供参考。</p>
DOI:10.7505/j.issn.1007-9084.2015.03.021Magsci [本文引用: 1]
<p>综述了近年来大豆籽粒蛋白质含量分子遗传方面的研究进展。由于遗传因素和环境因素对蛋白质含量均有很大的影响,因此,不同研究者的研究结果差异很大。仅利用表型数据的传统遗传研究表明,大豆籽粒蛋白质含量以加性效应为主,但不同材料之间遗传力差异很大,在31%~99%之间。在所有20个染色体上均发现有相关的QTL位点,共有298个QTL,贡献率介于0.002%~80%之间,其中20号(I连锁群)、15号(E连锁群)等染色体上均存在多次检测到的QTL区段。连锁分析和关联分析均显示20号染色体检测到大豆籽粒蛋白质含量QTL的频率均最高,且在该染色体上预测了一些大豆籽粒蛋白质含量相关的候选基因,但这些候选基因在染色体上的位置不一致,还需要进一步的验证。20号染色体有一个QTL区段(A688-Satt239)在多个环境和群体中均检测到,这为高蛋白分子标记辅助育种提供依据,并为后续的深入研究提供参考。</p>
[本文引用: 1]
[本文引用: 1]
DOI:10.3864/j.issn.0578-1752.2015.13.005Magsci [本文引用: 1]
【目的】从内源赤霉素代谢角度,阐明玉米大豆套作环境下,大豆苗期茎秆发生藤蔓化倒伏的原因。【方法】在大豆单作和玉米大豆套作两种种植方式下,以耐荫抗倒型大豆南豆12和不耐荫抗倒型大豆南032-4为试验材料,采用石蜡切片、液相色谱质谱联用、SYBR<sup>®</sup>GreenⅡ荧光定量PCR等方法,对茎秆形态、解剖结构、赤霉素含量、赤霉素代谢相关基因表达等进行测定和分析。【结果】与大豆单作相比,玉米大豆套作下,由于受玉米荫蔽的影响,南豆12和南032-4两个大豆材料苗期均出现典型的避荫性反应,即主茎节数减少,节间长和株高显著增加,苗期出现藤蔓化和倒伏,但两大豆材料间反应程度不同,差异极显著。与南032-4比,南豆12受套作荫蔽的影响较小,茎秆藤蔓化程度低,倒伏率仅29.93%,极显著低于南032-4(93.94%)(<em>P</em><0.05);南032-4株高增加32.64 cm,与南豆12(22.95 cm)呈显著差异(<em>P</em><0.05);两材料主茎节数减少,材料间无差异;南032-4中上部节间长度显著长于南豆12,说明套作下植株株高的增加来源于节间的伸长,而非节数的增多,且中部及上部节间过度伸长易导致植株茎秆藤蔓化和倒伏。从茎秆纵切面的解剖结构可知,玉米大豆套作条件下,大豆主茎细胞都有不同程度的伸长,南032-4茎秆髓部、木质部、韧皮部细胞伸长明显,而南豆12在两种种植模式下各部分细胞无明显变化,说明大豆株高的增加源于细胞的伸长,非细胞的分裂。内源赤霉素代谢分析结果表明,套作条件下,两材料茎秆内源赤霉素含量均显著降低,南豆12茎秆内赤霉素A<sub>4</sub>(GA<sub>4</sub>)的含量显著低于南032-4;对编码赤霉素代谢途径中关键酶基因表达量的分析表明,除赤霉素合成酶基因<em>GmGA20ox5</em>、<em>GmGA3ox6</em>和赤霉素降解酶基因<em>GmGA2ox4</em>在不同种植方式和不同材料间均保持较低的水平外,茎秆中其他内源赤霉素合成酶GA-20氧化酶基因、GA3-氧化酶基因和赤霉素降解酶GA2-氧化酶基因表达量均高于单作,且南032-4的表达量高于南豆12,说明植株内源赤霉素的含量对编码赤霉素代谢途径中关键酶基因具有前馈和反馈作用,南豆12茎尖维持较低活性赤霉素GA<sub>4</sub>水平,抑制植株主茎过度伸长,最终表现出较强的耐荫抗倒性。【结论】大豆内源赤霉素GA<sub>4</sub>的合成受基因型和光环境的共同影响,从而影响大豆茎秆生长发育,最终导致不同材料在耐荫抗倒表型上的典型差异。
DOI:10.3864/j.issn.0578-1752.2015.13.005Magsci [本文引用: 1]
【目的】从内源赤霉素代谢角度,阐明玉米大豆套作环境下,大豆苗期茎秆发生藤蔓化倒伏的原因。【方法】在大豆单作和玉米大豆套作两种种植方式下,以耐荫抗倒型大豆南豆12和不耐荫抗倒型大豆南032-4为试验材料,采用石蜡切片、液相色谱质谱联用、SYBR<sup>®</sup>GreenⅡ荧光定量PCR等方法,对茎秆形态、解剖结构、赤霉素含量、赤霉素代谢相关基因表达等进行测定和分析。【结果】与大豆单作相比,玉米大豆套作下,由于受玉米荫蔽的影响,南豆12和南032-4两个大豆材料苗期均出现典型的避荫性反应,即主茎节数减少,节间长和株高显著增加,苗期出现藤蔓化和倒伏,但两大豆材料间反应程度不同,差异极显著。与南032-4比,南豆12受套作荫蔽的影响较小,茎秆藤蔓化程度低,倒伏率仅29.93%,极显著低于南032-4(93.94%)(<em>P</em><0.05);南032-4株高增加32.64 cm,与南豆12(22.95 cm)呈显著差异(<em>P</em><0.05);两材料主茎节数减少,材料间无差异;南032-4中上部节间长度显著长于南豆12,说明套作下植株株高的增加来源于节间的伸长,而非节数的增多,且中部及上部节间过度伸长易导致植株茎秆藤蔓化和倒伏。从茎秆纵切面的解剖结构可知,玉米大豆套作条件下,大豆主茎细胞都有不同程度的伸长,南032-4茎秆髓部、木质部、韧皮部细胞伸长明显,而南豆12在两种种植模式下各部分细胞无明显变化,说明大豆株高的增加源于细胞的伸长,非细胞的分裂。内源赤霉素代谢分析结果表明,套作条件下,两材料茎秆内源赤霉素含量均显著降低,南豆12茎秆内赤霉素A<sub>4</sub>(GA<sub>4</sub>)的含量显著低于南032-4;对编码赤霉素代谢途径中关键酶基因表达量的分析表明,除赤霉素合成酶基因<em>GmGA20ox5</em>、<em>GmGA3ox6</em>和赤霉素降解酶基因<em>GmGA2ox4</em>在不同种植方式和不同材料间均保持较低的水平外,茎秆中其他内源赤霉素合成酶GA-20氧化酶基因、GA3-氧化酶基因和赤霉素降解酶GA2-氧化酶基因表达量均高于单作,且南032-4的表达量高于南豆12,说明植株内源赤霉素的含量对编码赤霉素代谢途径中关键酶基因具有前馈和反馈作用,南豆12茎尖维持较低活性赤霉素GA<sub>4</sub>水平,抑制植株主茎过度伸长,最终表现出较强的耐荫抗倒性。【结论】大豆内源赤霉素GA<sub>4</sub>的合成受基因型和光环境的共同影响,从而影响大豆茎秆生长发育,最终导致不同材料在耐荫抗倒表型上的典型差异。
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}