 ,1,2
,1,2Development and Characterization of Whole Genome SSR in Tetraploid Wild Peanut (Arachis monticola)
WANG YuLong1,2, HUANG BingYan2, WANG SiYu2,3, DU Pei2, QI FeiYan2, FANG YuanJin2, SUN ZiQi2, ZHENG Zheng2, DONG WenZhao2, ZHANG XinYou,1,2通讯作者:
收稿日期:2019-03-17接受日期:2019-05-15网络出版日期:2019-08-01
| 基金资助: |
Received:2019-03-17Accepted:2019-05-15Online:2019-08-01
作者简介 About authors
王玉龙,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (1471KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
王玉龙, 黄冰艳, 王思雨, 杜培, 齐飞艳, 房元瑾, 孙子淇, 郑峥, 董文召, 张新友. 四倍体野生种花生A.monticola全基因组SSR的开发与特征分析[J]. 中国农业科学, 2019, 52(15): 2567-2580 doi:10.3864/j.issn.0578-1752.2019.15.002
WANG YuLong, HUANG BingYan, WANG SiYu, DU Pei, QI FeiYan, FANG YuanJin, SUN ZiQi, ZHENG Zheng, DONG WenZhao, ZHANG XinYou.
0 引言
【研究意义】花生是世界重要的油料作物和经济作物,是人类食用植物油和植物蛋白的重要来源,主要种植在亚洲、非洲和美洲等100多个国家和地区。中国花生年产量居世界首位,花生在国民经济中具有重要地位。栽培花生的2个二倍体祖先野生种Arachis duranensis(AA)和Arachis ipaensis(BB)[1]及其近缘四倍体野生种Arachis monticola(AABB)[2]全基因组序列已公布,对其基因组序列信息的深入挖掘和有效利用,开展全基因组范围内SSR(simple sequence repeats)的研究,探究其分布规律,并进行引物开发,将对花生属植物遗传进化分析及分子标记开发起到重要作用。【前人研究进展】SSR是指由1—6个核苷酸组成的不同类型基序多次重复而形成的相对较短的DNA序列[3]。在DNA复制、修复以及重组过程中DNA聚合酶滑动错配及后续的错误产生[4],其重复次数高度可变且侧翼序列相对保守,广泛分布于原核生物和真核生物基因组中[5,6,7]。研究表明SSR能够影响基因调控、转录、进化、蛋白质功能、基因组结构、DNA复制以及细胞循环等[8,9]。SSR分子标记是建立在PCR基础上的DNA分子标记,与其他分子标记相比,SSR具有多态性高、可重复性好、操作简便、及共显性等优点,是目前最常用的分子标记技术之一。SSR分子标记在花生属植物中也得到了广泛的应用,包括F1真杂种的鉴定[10]、指纹图谱的构建[11,12,13]、品种遗传多样性分析[14,15,16,17,18]、连锁图谱构建及QTL定位[19,20,21,22,23]等。目前,花生SSR分子标记的开发主要通过以下6种方式:(1)通过构建基因组文库的方式(包括基因组文库富集法和筛选基因组文库法)进行SSR标记开发[24,25,26,27,28,29,30,31,32];(2)基于公共数据库及自己开发的EST(expressed sequence tags)序列进行SSR标记的开发[33,34,35,36,37,38,39,40];(3)通过同源转移法进行SSR标记的开发,利用近缘物种开发的SSR引物应用到花生上[41];(4)基于构建BAC文库进行SSR标记开发[42];(5)基于转录组数据的SSR标记开发[43,44];(6)基于全基因组序列的SSR标记开发[45,46]。以上研究中花生SSR标记的开发多以栽培种为研究对象,对野生种花生的研究相对较少。2012年,ZHAO等[47]整合了栽培花生和野生花生中SSR标记的总数为9 274个;Kazusa Marker数据库[48]收集了9 893个花生SSR标记,花生SSR标记逐渐丰富。【本研究切入点】花生SSR标记开发虽取得一些进展,但由于花生基因组庞大且复杂,遗传基础狭窄,SSR标记多态性较低,需要开发更多的SSR标记,以适应花生遗传研究。传统的SSR标记开发费时费力,目前,高通量测序技术高速发展,植物全基因组测序成本逐渐降低,为SSR序列的开发提供了极大便利。在拟南芥[49]、水稻[49]、黄瓜[50]、烟草[51]、短柄草[52]、葡萄[53]等植物中,已有大量的文献报道其基因组序列中SSR的信息,YU等[54]开发了用于研究植物(包含110种)中微卫星DNA和SSR标记的数据库。栽培花生二倍体野生种祖先和四倍体野生种A.monticola全基因组的测序完成,使花生的SSR高效开发成为可能。ZHAO等[45]对栽培花生的2个二倍体野生种祖先首先进行了全基因组序列SSR分析,探究了其分布规律,共开发了204 439个SSR标记。但目前对四倍体野生种A.monticola全基因组范围内SSR的研究,鲜见报道。【拟解决的关键问题】本研究采用生物信息学方法在A.monticola全基因组序列中开展SSR位点分析,对基因组中不同区域及各条染色体的SSR位点进行统计,探究其分布规律,并利用搜索的SSR两端的保守序列进行引物设计继而开发出单一位点SSR分子标记。为进一步研究花生基因组的进化,真假杂种F1的鉴定,品种多样性分析,遗传图谱的构建等提供便利和理论依据。1 材料与方法
1.1 植物材料
选用花生区组的4个不同种的花生材料为基因组DNA模板进行SSR引物的扩增验证。2个二倍体野生种花生分别为栽培花生的祖先种、1个四倍体野生种和1个栽培花生品种(表1)。Table 1
表1
表1供试材料的基本信息
Table 1
| 材料名称 Materials | 区组 Section | 种别 Species | 倍性 Ploidy | 基因组 Genomes |
|---|---|---|---|---|
| PI 219823 | 花生区组Section arachis | A.duranesis | 二倍体Diploid (2n=2x=20) | A |
| PI 468322 | 花生区组Section arachis | A.ipaensis | 二倍体Diploid (2n=2x=20) | B |
| PI 219824 | 花生区组Section arachis | A.moticola | 四倍体Tetraploid (2n=2x=40) | AB |
| Tifrunner | 花生区组Section arachis | A.hypogaea | 四倍体Tetraploid (2n=2x=40) | AB |
新窗口打开|下载CSV
1.2 基因组序列
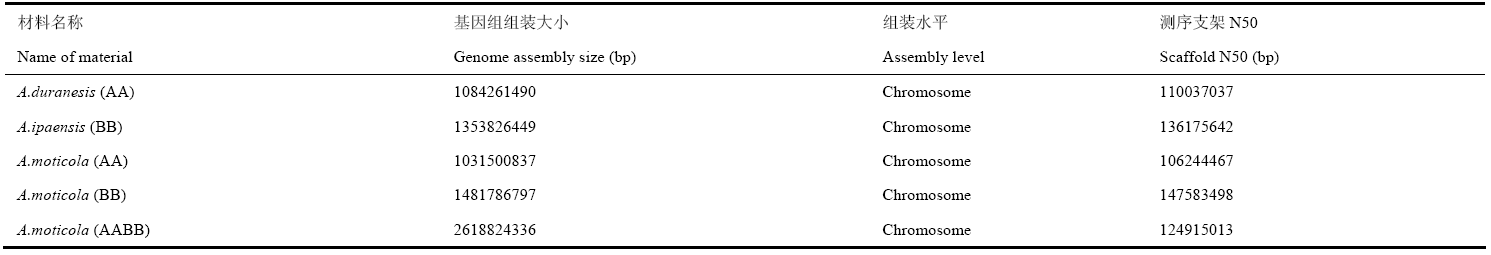
A.monticola全基因组序列及基因注释文件下载自华大基因GigaScience数据库,A. duranensis 和A. ipaensis基因组序列来自花生基因组数据库(PeanutBase)。基因组大小及组装水平见电子附表1。Supplementary Table 1
附表1
附表1供试材料基因组大小及组装水平
Supplementary Table 1
 |
新窗口打开|下载CSV
1.3 基因组序列中SSR的搜索
利用生物信息学SSR搜索工具MISA(http://pgrc. ipk-gatersleben.de/misa/)进行全基因组中满足定义的SSR位点检索。为方便与栽培花生的2个野生种祖先A. duranensis和A. ipaensis全基因SSR位点数量与分布特征比较,查找标准与ZHAO等[45]所用标准保持一致:单碱基重复≥12 bp(基序M=1;重复次数N≥12);2碱基重复≥12 bp(M=2;N≥6);3碱基重复≥15 bp(M=3;N≥5);4 碱基重复≥16 bp(M=4;N≥4);5 碱基重复≥15 bp(M=5;N≥3);6 碱基重复≥18 bp(M=6;N≥3)。混合微卫星中2个SSR距离小于100 bp。利用Excel 2016、Perl脚本和R语言对所得数据进行统计分析与绘图。1.4 A.monticola全基因组SSR引物设计
根据鉴定的四倍体野生种花生A.monticola全基因组SSR位点侧翼的保守序列,利用引物设计工具Primer 3(Linux版)进行引物设计,引物长度18—23 bp,退火温度50—65℃,GC含量为40%—60%,扩增产物长度为100—300 bp。1.5 电子PCR进行单位点SSR标记分析
利用获得的SSR引物在A. monticola全基因组进行电子PCR扩增及电子定位,去除有多处扩增的引物,保证引物扩增的特异性。1.6 SSR引物通用性验证
随机选取100对引物在通用生物系统(安徽)有限公司进行引物合成(电子附表2)。采用CTAB法提取4种供试材料的基因组DNA。PCR总反应体系为10 μL,包括20 ng·μL-1 DNA样品2 μL、10×PCR Reaction Buffer(Mg2+ plus)1 μL、dNTP Mixture 0.8 μL、ddH2O 5.75 μL、TaKaRa Taq 0.05 μL(5 U·μL-1)、正反向引物各0.2 μL。PCR反应程序为94℃ 5 min;94℃ 30 s,55—51℃ 45 s,72℃ 45 s,30个循环;72℃ 7 min,4℃保存。Supplementary Table 2
附表2
附表2100 对SSR 引物基本信息
Supplementary Table 2
 |
新窗口打开|下载CSV
完成PCR循环48 h内取1.5 μL产物在8%非变性聚丙烯酰胺凝胶上恒电压220 V电泳1 h 20 min,用NaOH银染方法进行染色显影。显影完成之后,拍照保存。
2 结果
2.1 A.monticola全基因组SSR位点数量和分布
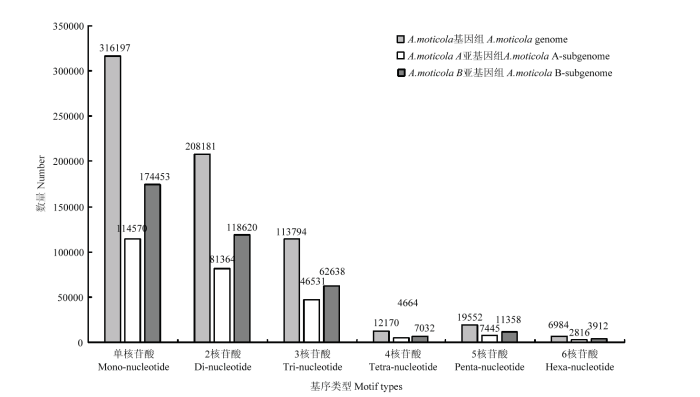
利用MISA软件,在全长分别为1 084 261 490、1 353 826 449和2 618 653 824 bp的花生二倍体祖先种A. duranensis、A. ipaensis和四倍体野生种A. monticola基因组内分别搜寻到满足条件的SSR位点,A. monticola基因组中共搜索到SSR位点676 878个,二倍体祖先种分别搜索到135 529和199 957个,与ZHAO等[45]的结果一致。A. monticola基因组检测到的SSR总长16 896 431 bp,占基因组的0.6452%,分布于5 127条scaffold序列中,平均3.8 kb出现一个SSR序列。A亚基因组平均4 kb出现一个SSR,B亚基因组平均3.9 kb出现一个SSR。SSR在四倍体野生种花生A. monticola基因组上种类丰富,单核苷酸至六核苷酸均有分布,且数量上差异较大(表2)。在全部的6种基序(单、二、三、四、五、六碱基)类型中,单一重复类型(单个重复基元)SSR有538 886个(79.61%),而复合型(多个重复基元)SSR则有137 992个(20.39%)。单一类型重复基元数量最多的是单碱基重复,达到316 197个,其次是二碱基重复,三碱基重复和五碱基重复。A.monticola基因组SSR位点主要集中在一、二、三碱基基序类型上,且三者约占总SSR位点数目的94.28%,分别为46.71%、30.76%和16.81%。五碱基基序类型的SSR位点所占的比例高于四、六碱基基序类型,六碱基基序类型在6种基序类型中出现频率最低,不同重复基元频数高低与核苷酸数量没有严格相关性(图1)。A、B染色体组各类型重复基元丰富度表现与全基因组相一致,且B基因组SSR位点数量更多。Table 2
表2
表2A. monticola、A. duranensis和A.ipaensis基因组SSR位点信息
Table 2
| 材料 Materials | 统计量 Statistic | 单核苷酸Mono-nucleotide | 2核苷酸Di-nucleotide | 3核苷酸Tri-nucleotide | 4核苷酸Tetra-nucleotide | 5核苷酸Penta-nucleotide | 6核苷酸Hexa-nucleotide | 总计 Total |
|---|---|---|---|---|---|---|---|---|
| A.duranesis (AA) | 数目Amount | 29689 | 47805 | 42529 | 4988 | 7658 | 2860 | 135529 |
| 类型比例 Type ratio (%) | 21.91 | 35.27 | 31.38 | 3.68 | 5.65 | 2.11 | 100.00 | |
| 平均距离 Average distance (kb) | 36.5 | 22.7 | 25.5 | 217.4 | 141.6 | 379.1 | 8.0 | |
| A.ipaensis (BB) | 数目Amount | 57589 | 75334 | 45717 | 6736 | 11092 | 3489 | 199957 |
| 类型比例 Type ratio (%) | 28.80 | 37.68 | 22.86 | 3.37 | 5.55 | 1.74 | 100 | |
| 平均距离 Average distance (kb) | 23.5 | 18.0 | 29.6 | 201.0 | 122.1 | 388.0 | 6.7 | |
| A.moticola (AABB) | 数目Amount | 316197 | 208181 | 113794 | 12170 | 19552 | 6984 | 676878 |
| 类型比例 Type ratio (%) | 46.71 | 30.76 | 16.81 | 1.80 | 2.89 | 1.03 | 100.00 | |
| 平均距离 Average distance (kb) | 8.3 | 12.6 | 23.0 | 215.2 | 133.9 | 375.0 | 3.8 | |
| A.moticola (A-subgenome) | 数目Amount | 114570 | 81364 | 46531 | 4664 | 7445 | 2816 | 257390 |
| 类型比例 Type ratio (%) | 44.51 | 31.61 | 18.08 | 1.81 | 2.89 | 1.09 | 100.00 | |
| 平均距离 Average distance (kb) | 9.0 | 12.7 | 22.2 | 221.2 | 138.5 | 366.3 | 4.0 | |
| A.moticola (B-subgenome) | 数目Amount | 174453 | 118620 | 62638 | 7032 | 11358 | 3912 | 378013 |
| 类型比例 Type ratio (%) | 46.15 | 31.38 | 16.57 | 1.86 | 3.00 | 1.03 | 100.00 | |
| 平均距离 Average distance (kb) | 8.5 | 12.5 | 23.7 | 210.7 | 130.5 | 378.8 | 3.9 |
新窗口打开|下载CSV
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1A. monticola基因组SSR不同基序类型(1—6 bp)的数量分布
Fig. 1Quantitative distribution of different motif types (1-6 bp) of SSR in A. monticola genome
SSR位点在A.monticola基因组上的数量分布如表3所示,基因间区SSR数量最多,58 755个SSR位点分布在基因区。在基因区,内含子区域SSR数量最多,其次是CDS区域、UTR区域。
Table 3
表3
表3SSR位点在A. monticola基因组上不同区域的数量分布
Table 3
| 总计 Total | 基因区 Genetic region | 外显子区 Exon region | CDS区 CDS region | 5′-UTR区 5′-UTR region | 3′-UTR区 3′-UTR region | 内含子区 Intron region | 基因间区 Intergenic region |
|---|---|---|---|---|---|---|---|
| 538886 | 58755 | 14429 | 7344 | 4566 | 2519 | 44326 | 480131 |
新窗口打开|下载CSV
2.2 A.monticola全基因组SSR基序类型和结构
A.monticola全基因组重复基元非常丰富,从A.monticola基因组中鉴定出395个不同的重复基元(SSR motifs)。其中,在A. monticola基因组中,单核苷酸有2种,二核苷酸有4种,三核苷酸有10种,四、五、六核苷酸分别有28、93和258种;A. monticola的A基因组共鉴定出342种不同的重复基元,B基因组共搜索到356种不同的重复基元,其中,A、B亚基因组单核苷酸、二核苷酸、三核苷酸重复基元类型数目相同,分别为2、4和10种;四、五、六核苷酸分别存在特异的重复基元类型(表4)。四核苷酸重复基元类型:A基因组26种,特异性重复基元3种;B基因组25种,特异性重复基元2种。五核苷酸重复基元类型:A基因组81种,特异性重复基元6种;B基因组85种,特异性重复基元10种。六核苷酸重复基元:A基因组219种,特异性重复基元25种;B基因组230种,特异性重复基元36种。A/T是最丰富的重复基元,占基因组SSR的45.36%,其次是AT/AT、AAT/ATT、AG/CT、AAG/CTT和AC/GT,分别占基因组的19.26%、8.81%、8.71%、3.91%和2.41%。A.monticola全基因组数量最多的20种SSR的重复基元如图2所示。Table 4
表4
表4A.monticola A、B亚基因组特异性重复基元类型
Table 4
| 重复基元类型 Motif types | A亚基因组特异重复基元 Specific motifs in A-subgenome | B亚基因组特异重复基元 Specific motifs in B-subgenome |
|---|---|---|
| 四核苷酸 Tetra-nucleotide | AACG/CGTT AGCC/CTGG AGCG/CGCT | AAGC/CTTG AGGC/CCTG |
| 五核苷酸 Penta-nucleotide | ACCTG/AGGTC AGCCT/AGGCT ACGGG/CCCGT AGCCC/CTGGG ACTGG/AGTCC ACGTC/ACGTG | AACGC/CGTTG AACTT/AAGTT AAGGC/CCTTG ACAGT/ACTGT ACATG/ATGTC ACCGT/ACGGT ACTAG/AGTCT AGCGC/CGCTG AGCGG/CCGCT ATGCC/ATGGC |
| 六核苷酸 Hexa-nucleotide | AAACCC/GGGTTT AAAGCT/AGCTTT AACAGG/CCTGTT AACCGT/ACGGTT AACGGT/ACCGTT AACGTC/ACGTTG AACGTT/AACGTT AACTCC/AGTTGG AAGTGT/ACACTT AATACC/ATTGGT AATTCG/AATTCG ACACCG/CGGTGT ACATCG/ATGTCG ACCCAG/CTGGGT ACCCAT/ATGGGT ACCTCG/AGGTCG ACGCCG/CGGCGT ACGGAG/CCGTCT ACTCAT/AGTATG ACTCGG/AGTCCG ACTGCC/AGTGGC AGCATG/ATGCTC AGCCCG/CGGGCT AGCCCT/AGGGCT AGCTAT/AGCTAT | AAACGC/CGTTTG AAAGGT/ACCTTT AACGAC/CGTTGT AACGAT/ATCGTT AACTCT/AGAGTT AACTGG/AGTTCC AAGACC/CTTGGT AAGCAT/ATGCTT AAGGCG/CCTTCG AAGGGC/CCCTTG AAGTAC/ACTTGT AATCCG/ATTCGG AATCGC/ATTGCG AATGAC/ATTGTC AATGCG/ATTCGC AATGGT/ACCATT ACAGAT/ATCTGT ACAGGC/CCTGTG ACAGGG/CCCTGT ACATAG/ATGTCT ACATGC/ATGTGC ACCATG/ATGGTC ACCCTG/AGGGTC ACCGAG/CGGTCT ACCTAG/AGGTCT ACGCGG/CCGCGT ACGCTC/AGCGTG ACGTCC/ACGTGG ACTAGG/AGTCCT ACTCTG/AGAGTC ACTGGC/AGTGCC ACTGGG/AGTCCC AGCCAT/ATGGCT AGGCCG/CCTCGG AGGGAT/ATCCCT ATCGGC/ATGCCG |
新窗口打开|下载CSV
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2A.monticola全基因组出现数量最多的20种SSR重复基元类型
Fig. 2Twenty SSR motif types with the largest number in A. monticola genome
2.3 A.monticola全基因组SSR碱基组成
在1—6个核苷酸的motifs中,单核苷酸重复基元数量最多的是A/T,二核苷酸最多的是AT/AT,三核苷酸最多的是AAT/ATT,四核苷酸最多的是AAAT/ATTT,其次是AAAAT/ATTTT和AAAAAT/ATTTTT,均为AT富集的重复基元,而GC富集的重复基元相对较少。2.4 A.monticola全基因组SSR基序重复次数分布
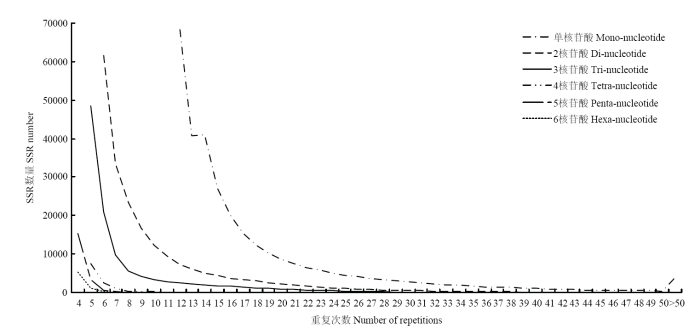
从图3可以看出,重复基元的重复次数多集中在50次以内;不同类型的motif的重复次数差异很大;同一种类型motif的SSR位点,随着motif重复次数增加,SSR的数量逐渐降低,SSR重复次数越少,SSR数量越多。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3不同类型的重复基元的重复次数分布
Fig. 3Distribution of repetition times of different motif types
2.5 A.monticola全基因组SSR在各染色体的分布
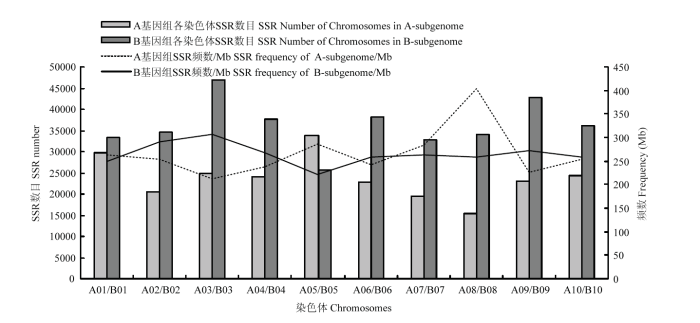
通过对A. monticola各个染色体SSR数目及SSR频数进行统计(图4),各条染色体上均搜索到大量SSR。在20条染色体中,B03染色体上SSR数量最多,达到47 033个,平均307.18个SSR/Mb,平均每3.33 kb就会出现一个SSR;A08染色体中SSR密度最高,共15 257个SSR,平均403.19个SSR/Mb,平均2.54 kb就会出现一个SSR。其中A基因组A05染色体中SSR数量最多,其次是A01、A03、A10、A04、A09、A06、A02、A07和A08,A08染色体中SSR数量最少,但是密度最高,A03染色体SSR密度最低。B染色体组B03染色体SSR数量最多,其次是B09、B06、B04、B10、B02、B08、B01、B07和B05,且SSR密度最高,B05染色体中SSR数量最少,且SSR密度最低。B染色体组SSR数量远高于A染色体组。图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4A. monticola各条染色体SSR数目及SSR频数分布
Fig. 4SSR number and SSR frequency distribution of chromosomes in A. monticola genome
2.6 A.monticola全基因组SSR引物开发,及单点SSR分析
利用SSR位点侧翼的保守序列,共设计出SSR引物192 303对,并利用这些标记进行电子定位,得到单位点标记96 828个(基因组中只有一个位点有扩增产物),检出率50.35%,单位点SSR在20条染色体上的分布密度呈现两端密集,中间稀疏的特点(图5和图6)。图5
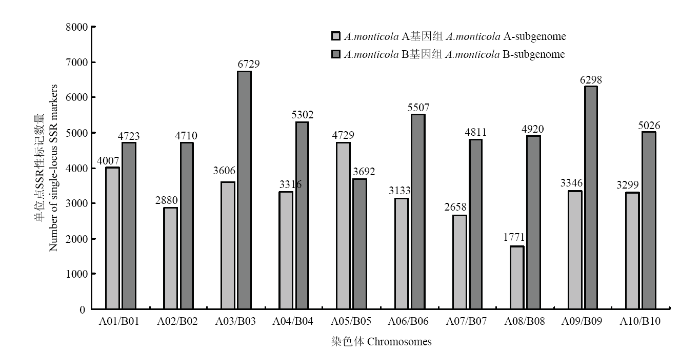 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5单位点SSR标记染色体上的数量分布
Fig. 5Quantitative distribution of single-locus SSR markers on chromosomes
图6
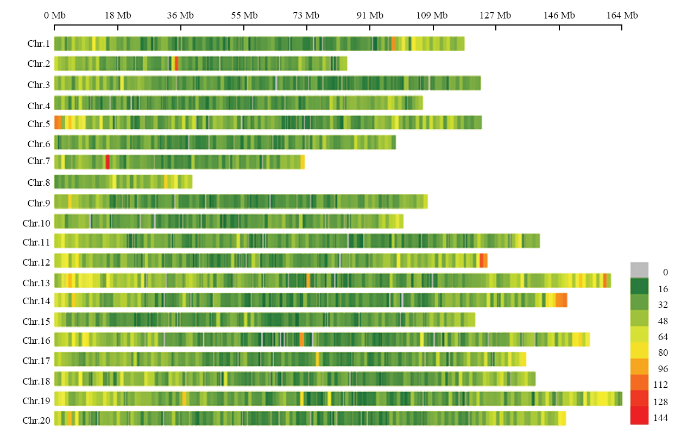 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6SSR单位点标记在各染色体上的密度分布(窗口为1M)
Fig. 6Density distribution of single-locus SSR markers on chromosomes(1M window size)
2.7 SSR引物的验证及通用性分析
在开发的单点SSR引物中,随机选取100对引物进行合成并实际扩增验证,在A. monticola中能扩增出稳定清晰条带的有90对,用于验证的SSR引物在参试的4份花生材料中扩增条带表现出较好的通用性。如图7所示,目标条带在参试的4份花生材料中表现出不同的特点:引物A02-7和引物A04-20在4份材料中均能扩增出目标条带;引物A02-8和引物A03-12只能在A. monticola和Tifrunner扩增出目的条带;引物A07-31、A07-32、A09-42、A09-43和A10-50能在A.monticola、A.ipaensis和Tifrunner中扩增出目的条带,而在A.duranensis不能扩增出来;引物B07-82只能在A. monticola、A .duranensis和Tifrunner中扩增出目的条带。引物A04-18只能在A.monticola扩增出目的条带;引物B10-100只能在A.duranensis中扩增出目的条带。图7
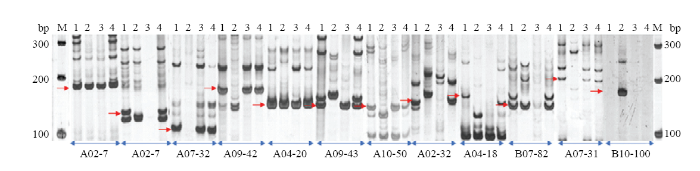 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图712种不同引物在4份材料基因组上扩增结果
M:DL 1000分子标记;1-4分别表示 A. moticola、A. duranesis、A. ipaensis、Tifrunner;红色箭头表示目标条带
Fig. 7Amplification results of 12 different primers on genomes of four materials
M: DL 1000 marker; 1-4 represent A. moticola, A. duranesis, A. ipaensis, Tifrunner; the red arrow indicates the target strip
3 讨论
3.1 A.monticola全基因组SSR分布特征
四倍体野生种花生A.monticola基因组上SSR种类丰富,单核苷酸至六核苷酸均有分布,且数量上差异较大,A.monticola全基因组SSR在基因组各区域的分布数量表明,大多数SSR分布在基因间区,然后是内含子区域,其次是外显子区域,最后是UTR区域,且5’UTR区域的SSR数量要高于3’UTR区域;在SSR重复碱基类型上,单碱基重复最为丰富,这与A. duranensis[45]、A. ipaensis[45]、烟草[51]SSR位点以二核苷酸出现频率最高结果不一致,然后是二碱基重复,其次是三碱基重复,五碱基重复,四碱基重复,六碱基重复。SSR主要集中在一、二、三碱基重复,四、五、六碱基重复较少,这与拟南芥[49]、水稻[49]、玉米[55]、棉花[56]的结果相一致,表明A.monticola进化水平相对较高。在SSR重复基序结构方面,单核苷酸A/T是最丰富的重复基元,占基因组SSR的45.36%,其次是AT/AT、AAT/ATT、AG/CT、AAG/CTT和AC/GT,分别占基因组的19.26%、8.81%、8.71%、3.91%和2.41%;二核苷酸最多的是AT/AT,此结果与A.duranensis[45]、A.ipaensis[45]、烟草[51]基因组SSR研究结果相同,水稻[49]、玉米[55]、大豆[57]、小麦[58]中以AT/GC出现的频数最高有所不同;三核苷酸最多的是AAT/ATT,与拟南芥[50]重复基序AAG/TTC出现频数最高,水稻[49]、玉米[55]等CCG/GCC重复基序最丰富,ATT/TAA最少的研究结果不同。四核苷酸、五核苷酸、六核苷酸最多的依次是AAAT/ATTT、AAAAT/ATTTT、AAAAAT/ ATTTTT;对于SSR在A.monticola各染色体上分布的数量和密度来看,在20条染色体中,B03染色体上SSR数量最多,达到47 033个,平均307.18个SSR/Mb,平均每3.33 kb就会出现一个SSR;A08染色体在20条染色体中最短,但SSR密度最高,共15 257个SSR,平均403.19个SSR/Mb,平均每2.54 kb就会出现一个SSR。3.2 A. monticola与A. duranensis、A. ipaensis基因组SSR分布比较
本研究对四倍体野生种花生A. monticola全基因组进行了SSR查找,共得到SSR位点676 878个。其中A基因组257 390个,密度为1个/4 kb,B基因组378 013个,密度为1个/3.9 kb,而A. duranensis密度为1个/8 kb,A. ipaensis密度为1个/6.7 kb。总体水平上A. monticola全基因组SSR数量比A. duranensis(A基因组)、A. ipaensis(B基因组)SSR多,且密度更高。在重复基元数量分布方面,A.monticola单碱基、二碱基、三碱基重复在A亚基因组和B亚基因组分别高于A. duranensis(A基因组)、A. ipaensis(B基因组)。A. monticola四碱基、五碱基、六碱基重复在A亚基因组数量高于A. duranensis,在B亚基因组数量低于A. ipaensis;在重复基元分布比例方面,A. monticola A、B亚基因组除单碱基比例分布高于A. duranensis、A. ipaensis外,二碱基、三碱基、四碱基、五碱基、六碱基比例均低于A.duranensis、A.ipaensis;重复基元分布密度方面,在A. duranensis与A. ipaensis基因组中SSR二核苷酸重复数量最多且密度最高,而A. monticola基因组中单核苷酸重复数量最多且密度最高。出现这情况的原因有2种:(1)进化过程中SSR出现较大变异;(2)基因组序列组装存在问题。A. monticola全基因组SSR均为AT富集的重复基元,而GC富集的重复基元相对较少,这与A. duranensis和A. ipaensis基因组中SSR分布规律一致,这可能与打破GC键所需的能量要高于AT键所需的能量有关。基因组中较为丰富的重复序列的存在是导致花生基因组组装瓶颈的原因之一。3.3 A.monticola基因组SSR标记的应用
前人研究表明,SSR引物在植物种属间具有极高的通用性[41,45,59]。花生全基因组序列的公布加速了基因组进化研究。本研究从A. monticola基因组开发了SSR引物192 303对,其中96 828个单位点SSR标记,并在野生种花生A.duranensis、A.ipaensis和栽培花生Tifrunner中初步验证了其通用性。所使用材料A.monticola(AABB,2n=4x=40)是花生区组中唯一的异源四倍体野生种,被认为是栽培种花生亲缘关系最近的野生种[2];Tifrunner为栽培种;二倍体野生种A.duranensis和A.ipaensis被认为是栽培花生的2个祖先,栽培花生A、B基因组的供体[1]。在漫长的进化过程中4份材料基因组序列会存在差异,材料的特殊性导致引物在4个类型基因组的品种中扩增条带存在较大差异。经比对,本研究开发的SSR引物与ZHAO等[45]开发的引物有39 509对是重复序列,占比20.54%;其余152 794对为本研究新开发的SSR引物。由于在PCR过程中退火温度设置了不同的梯度,在扩增过程中会出现部分杂带(非特异性扩增)。本研究中SSR标记的开发对进一步研究花生属近缘种遗传多样性、基因组分子进化、染色体细胞学鉴定、品种真实性检测以及分子标记辅助选择育种等方面提供了重要的SSR分子标记库支撑。4 结论
A.monticola全基因组SSR位点各重复基元的数量、丰度、基序类型、重复次数以及碱基偏好性以及在各染色体上的分布均具有规律性。A.monticola基因组上SSR种类丰富,单核苷酸至六核苷酸均有分布,且数量上差异较大,其中,单核苷酸重复基元数量最多,且最密,六核苷酸重复基元数量最少,出现频率最低,不同重复基元频数高低与核苷酸数量没有严格相关性。SSR多分布在基因间区,基因区内含子区域SSR数量最多;A.monticola基因组中鉴定出395个不同的重复基元,A、B亚基因组具有其各自特异的重复基元类型;1—6个核苷酸的重复基元中,单个类型重复基元数量最多的均为AT富集的重复基元,而GC富集的重复基元相对较少;重复基元的重复次数多集中在50次以内,同一种类型重复基元的SSR位点,随着motif重复次数增加,SSR的数量逐渐降低;A. monticola B亚基因组SSR数量远高于A亚基因组。A. monticola全基因组SSR比A. duranensis、A. ipaensis基因组SSR数量多,密度也更高,A. monticola单核苷酸重复最丰富,与2个野生种二核苷酸数量最多的分布规律不同,单位点SSR在20条染色体上的分布密度呈现两端密集,中间稀疏的特点。基于A. monticola全基因组SSR位点开发的引物,用于花生属的其他3个种表现出部分通用性,可用于花生近缘种的相关遗传分析。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.3724/SP.J.1006.2016.01448URLMagsci [本文引用: 1]

<p>利用14个SSR标记构建了河南省2015年之前选育并审定的90个花生品种的DNA指纹图谱,用14个SSR标记产生的95个多态性位点可将90个花生品种完全区分开,其中84个品种间有≥2个位点的差异,在剩余的3对品种中,每对仅有1个差异位点。聚类分析结果表明,在遗传相似系数0.98处,90个花生品种被聚集成88类,有2对品种分别聚集在一起,是由于它们每一个品种分别以另一个品种作亲本选育而成,仅有1个差异SSR位点,表明所构建的指纹图谱是有效的。以遗传相似系数0.95为划分标准,有74.4%的品种具有特异性,与其他作物相比,河南省育成花生品种总体上亲缘关系相对较近。根据60个SSR标记的群体结构分析,90个花生品种可以分为3个亚群,与根据分枝开花习性和荚果类型的分类相吻合,亚群划分情况与聚类分析结果基本一致。</p>
DOI:10.3724/SP.J.1006.2016.01448URLMagsci [本文引用: 1]
<p>利用14个SSR标记构建了河南省2015年之前选育并审定的90个花生品种的DNA指纹图谱,用14个SSR标记产生的95个多态性位点可将90个花生品种完全区分开,其中84个品种间有≥2个位点的差异,在剩余的3对品种中,每对仅有1个差异位点。聚类分析结果表明,在遗传相似系数0.98处,90个花生品种被聚集成88类,有2对品种分别聚集在一起,是由于它们每一个品种分别以另一个品种作亲本选育而成,仅有1个差异SSR位点,表明所构建的指纹图谱是有效的。以遗传相似系数0.95为划分标准,有74.4%的品种具有特异性,与其他作物相比,河南省育成花生品种总体上亲缘关系相对较近。根据60个SSR标记的群体结构分析,90个花生品种可以分为3个亚群,与根据分枝开花习性和荚果类型的分类相吻合,亚群划分情况与聚类分析结果基本一致。</p>
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLMagsci [本文引用: 1]
<FONT face=Verdana>【目的】评价ICRISAT花生微核心种质资源的遗传多样性水平,揭示ICRISAT花生微核心种质资源遗传多样性,验证传统植物学分类的可靠程度,为充分发掘、利用ICRISAT花生微核心种质资源提供必要信息。【方法】采用27对花生SSR引物,对ICRISAT微核心花生种质168份材料(来自世界五大洲42个国家)进行遗传多样性分析;利用NTSYS-pc V2.0软件进行主成分分析(PCA)并绘制三维空间聚类图;利用Popgene V1.32估算种质群间的Nei78遗传距离等参数并进行UPGMA聚类分析,采用MEGA 3.1绘制种质群间聚类图。【结果】27对SSR引物共扩增出115条多态性条带,每对引物平均扩增出4.2930个等位变异,其中有效等位变异数2.7931,有效等位变异所占比重为65.49%;PM137、16C6、14H6、8D9和7G02等引物最为有效,其Shannon’s信息指数均在1.5以上,等位变异数5个以上,有效变异数3.7个以上。在多粒型群体中,来源于南美洲和印度种质资源的遗传多样性较低,来源于南美洲和非洲种质资源的遗传多样性较高;在珍珠豆型群体中,来源于北美洲种质资源的遗传多样性较低,来源于南美洲和非洲种质资源的遗传多样性较高;在普通型群体中,来源于北美洲种质资源的遗传多样性较低,来源于南美洲、美国和非洲种质资源的遗传多样性较高。来自南美洲的花生种质资源具有较高的遗传多样性,与花生起源于南美洲的结论一致。PCA分析,发现栽培种花生种质资源由4个差异明显的基因源构成,“hypogaea”包括普通型种质资源,“vulgaris”包括珍珠豆型种质资源,“fastigiata 1”包括多粒型种质资源,“fastigiata 2”包括多粒型种质资源。植物学分类单位间的Nei78遗传距离介于16.336—23.607 cM,UPGMA聚类方法将花生属植物学分类单位聚成5个组群,“组群1”对应“hypogaea”基因源,“组群2”对应“vulgaris”基因源,“组群3”对应“fastigiata 1”、“fastigiata 2”基因源之和,“组群4”和“组群5”分别代表秘鲁型和赤道型基因源,聚类结果支持4个基因源的划分。【结论】ICRISAT花生微核心种质资源具有丰富的遗传多样性,不同来源的变种群间存在明显的遗传差异,并分化成4个基因源,研究结果部分支持栽培种花生传统的植物学分类体系。为拓宽花生育成品种的遗传基础,应充分发掘ICRISAT微核心种质各基因源的遗传潜力。<BR></FONT>
URLMagsci [本文引用: 1]
<FONT face=Verdana>【目的】评价ICRISAT花生微核心种质资源的遗传多样性水平,揭示ICRISAT花生微核心种质资源遗传多样性,验证传统植物学分类的可靠程度,为充分发掘、利用ICRISAT花生微核心种质资源提供必要信息。【方法】采用27对花生SSR引物,对ICRISAT微核心花生种质168份材料(来自世界五大洲42个国家)进行遗传多样性分析;利用NTSYS-pc V2.0软件进行主成分分析(PCA)并绘制三维空间聚类图;利用Popgene V1.32估算种质群间的Nei78遗传距离等参数并进行UPGMA聚类分析,采用MEGA 3.1绘制种质群间聚类图。【结果】27对SSR引物共扩增出115条多态性条带,每对引物平均扩增出4.2930个等位变异,其中有效等位变异数2.7931,有效等位变异所占比重为65.49%;PM137、16C6、14H6、8D9和7G02等引物最为有效,其Shannon’s信息指数均在1.5以上,等位变异数5个以上,有效变异数3.7个以上。在多粒型群体中,来源于南美洲和印度种质资源的遗传多样性较低,来源于南美洲和非洲种质资源的遗传多样性较高;在珍珠豆型群体中,来源于北美洲种质资源的遗传多样性较低,来源于南美洲和非洲种质资源的遗传多样性较高;在普通型群体中,来源于北美洲种质资源的遗传多样性较低,来源于南美洲、美国和非洲种质资源的遗传多样性较高。来自南美洲的花生种质资源具有较高的遗传多样性,与花生起源于南美洲的结论一致。PCA分析,发现栽培种花生种质资源由4个差异明显的基因源构成,“hypogaea”包括普通型种质资源,“vulgaris”包括珍珠豆型种质资源,“fastigiata 1”包括多粒型种质资源,“fastigiata 2”包括多粒型种质资源。植物学分类单位间的Nei78遗传距离介于16.336—23.607 cM,UPGMA聚类方法将花生属植物学分类单位聚成5个组群,“组群1”对应“hypogaea”基因源,“组群2”对应“vulgaris”基因源,“组群3”对应“fastigiata 1”、“fastigiata 2”基因源之和,“组群4”和“组群5”分别代表秘鲁型和赤道型基因源,聚类结果支持4个基因源的划分。【结论】ICRISAT花生微核心种质资源具有丰富的遗传多样性,不同来源的变种群间存在明显的遗传差异,并分化成4个基因源,研究结果部分支持栽培种花生传统的植物学分类体系。为拓宽花生育成品种的遗传基础,应充分发掘ICRISAT微核心种质各基因源的遗传潜力。<BR></FONT>
DOI:10.7505/j.issn.1007-9084.2014.02.020URLMagsci [本文引用: 1]
<p>本研究从212对SSR标记引物中筛选出48对引物对63份花生品种进行遗传多样性分析,共得到251个等位变异,变异范围为2~13个,平均每个标记位点有5.23个变异;48个SSR标记的多态性信息含量为0.252~0.873,平均为0.647;63份材料的遗传多样性指数为0.508~2.243,平均值为1.272;品种间的遗传相似系数在0.657~0.960之间,不同类型的花生品种间的遗传相似性较小,不同来源花生品种间的亲缘关系也较远;聚类分析结果表明,63个花生品种在遗传相似系数为0.74处分为4大类,聚类分析结果与传统的花生分类结果吻合。</p>
DOI:10.7505/j.issn.1007-9084.2014.02.020URLMagsci [本文引用: 1]
<p>本研究从212对SSR标记引物中筛选出48对引物对63份花生品种进行遗传多样性分析,共得到251个等位变异,变异范围为2~13个,平均每个标记位点有5.23个变异;48个SSR标记的多态性信息含量为0.252~0.873,平均为0.647;63份材料的遗传多样性指数为0.508~2.243,平均值为1.272;品种间的遗传相似系数在0.657~0.960之间,不同类型的花生品种间的遗传相似性较小,不同来源花生品种间的亲缘关系也较远;聚类分析结果表明,63个花生品种在遗传相似系数为0.74处分为4大类,聚类分析结果与传统的花生分类结果吻合。</p>
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 10]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 6]
[本文引用: 2]
URL [本文引用: 3]
URL [本文引用: 3]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[D].
[本文引用: 3]
[D].
[本文引用: 3]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.3724/SP.J.1006.2014.00924URLMagsci [本文引用: 1]
<div ><span >分子标记具种属间通用性可提高其利用效率,并降低标记开发成本。本研究基于</span><span >Roche 454</span><span >超高通量测序技术获得普通菜豆基因组测序结果,共开发了</span><span >560</span><span >个普通菜豆基因组</span><span >SSR</span><span >标记。利用</span><span >2</span><span >份普通菜豆品种对标记进行初步筛选,有</span><span >421</span><span >个标记能够有效扩增。用新开发的标记分析</span><span >16</span><span >份豇豆和</span><span >16</span><span >份小豆的通用性。结果显示,</span><span >185</span><span >个普通菜豆基因组</span><span >SSR</span><span >标记在豇豆中能有效扩增,通用性比率为</span><span >43.9%</span><span >;</span><span >161</span><span >个</span><span >SSR</span><span >标记在小豆中能有效扩增,通用性比率为</span><span >38.2%</span><span >;在豇豆和小豆中都能获得有效扩增条带的标记共</span><span >138</span><span >个;并且</span><span >普通菜豆基因序列</span><span >SSR</span><span >标记在豇豆和小豆中的通用性比率高于基因间序列</span><span >SSR</span><span >标记。</span><span >通用性标记的多态性分析表明,豇豆和小豆的多态性比率分别为</span><span >34.0%</span><span >和</span><span >24.8%</span><span >;且</span><span >豇豆和小豆中基因间标记的多态性都比基因内标记的多态性高</span><span >。上述通用性标记</span><span >为豇豆属作物的多样性评价、连锁图谱的构建及基因定位等方面的研究</span><span >提供了便利<span >。</span></span></div>
DOI:10.3724/SP.J.1006.2014.00924URLMagsci [本文引用: 1]
<div ><span >分子标记具种属间通用性可提高其利用效率,并降低标记开发成本。本研究基于</span><span >Roche 454</span><span >超高通量测序技术获得普通菜豆基因组测序结果,共开发了</span><span >560</span><span >个普通菜豆基因组</span><span >SSR</span><span >标记。利用</span><span >2</span><span >份普通菜豆品种对标记进行初步筛选,有</span><span >421</span><span >个标记能够有效扩增。用新开发的标记分析</span><span >16</span><span >份豇豆和</span><span >16</span><span >份小豆的通用性。结果显示,</span><span >185</span><span >个普通菜豆基因组</span><span >SSR</span><span >标记在豇豆中能有效扩增,通用性比率为</span><span >43.9%</span><span >;</span><span >161</span><span >个</span><span >SSR</span><span >标记在小豆中能有效扩增,通用性比率为</span><span >38.2%</span><span >;在豇豆和小豆中都能获得有效扩增条带的标记共</span><span >138</span><span >个;并且</span><span >普通菜豆基因序列</span><span >SSR</span><span >标记在豇豆和小豆中的通用性比率高于基因间序列</span><span >SSR</span><span >标记。</span><span >通用性标记的多态性分析表明,豇豆和小豆的多态性比率分别为</span><span >34.0%</span><span >和</span><span >24.8%</span><span >;且</span><span >豇豆和小豆中基因间标记的多态性都比基因内标记的多态性高</span><span >。上述通用性标记</span><span >为豇豆属作物的多样性评价、连锁图谱的构建及基因定位等方面的研究</span><span >提供了便利<span >。</span></span></div>

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}