 ,, 邱丽娟中国农业科学院作物科学研究所/国家农作物基因资源与遗传改良重大科学工程/农业部种质资源利用重点实验室,北京 100081
,, 邱丽娟中国农业科学院作物科学研究所/国家农作物基因资源与遗传改良重大科学工程/农业部种质资源利用重点实验室,北京 100081QTL Mapping of Hard Seededness in Wild Soybean Using BSA Method
CHEN JingJing, LIU XieXiang, YU LiLi, LU YiPeng, ZHANG SiTian, ZHANG HaoChen, GUAN RongXia,, QIU LiJuanInstitute of Crop Sciences, Chinese Academy of Agricultural Sciences/National Key Facility for Crop Gene Resources and Genetic Improvement (NFCRI)/Key Laboratory of Germplasm Utilization, Ministry of Agriculture, Beijing 100081通讯作者:
责任编辑: 李莉
收稿日期:2019-02-28接受日期:2019-04-14网络出版日期:2019-07-01
| 基金资助: |
Received:2019-02-28Accepted:2019-04-14Online:2019-07-01
作者简介 About authors
陈静静,E-mail:1061184187@qq.com。

摘要
关键词:
Abstract
Keywords:
PDF (3934KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
陈静静, 刘谢香, 于莉莉, 卢一鹏, 张嗣天, 张昊辰, 关荣霞, 邱丽娟. 利用BSA法发掘野生大豆种子硬实性相关QTL[J]. 中国农业科学, 2019, 52(13): 2208-2219 doi:10.3864/j.issn.0578-1752.2019.13.002
CHEN JingJing, LIU XieXiang, YU LiLi, LU YiPeng, ZHANG SiTian, ZHANG HaoChen, GUAN RongXia, QIU LiJuan.
0 引言
【研究意义】栽培大豆在大约5000年前由一年生野生大豆驯化而来[1,2],种子硬实性是与大豆驯化相关的一个重要性状。与非硬实种子相比,硬实大豆种子具有较高的种子活力与较长的种子寿命[3,4,5]。在热带和亚热带地区高温多雨条件下,硬实种子不易吸胀,延缓了种子的劣变,有利于种子的保存与运输[4]。野生大豆的硬实性有利于其在自然环境下的生存繁衍,但该性状却影响了野生大豆在栽培大豆杂交改良中的利用效率。因此,发掘硬实相关的重要QTL位点,对于硬实性状的遗传解析及野生大豆利用具有重要意义。【前人研究进展】遗传研究证明大豆种子硬实性由多基因控制。大豆硬实的形成受种皮和种脐影响最大,与种皮的角质层和栅栏层有关,正常吸胀的种子其背部角质层存在细小裂痕,硬实种子的角质层却无裂痕。在显微镜下观察硬实种子时,栅栏细胞外存在一条致密的透明带,该透明带可能与大豆种子硬实性高度相关[1,6-10]。KEIM等[11]检测到5个与野生大豆种子硬实性相关的RFLP(restrict fragment length polymorphism)标记,可解释71%的遗传变异,其中一个包括i位点的区间可解释32%的遗传变异。日本****在C2(第6染色体)、D1b(第2染色体)和I(第20染色体)连锁群发现大豆硬实性相关的QTL,并发现控制种皮色的I位点(第8染色体)和控制茸毛色的T位点(第6染色体)与大豆硬实紧密连锁[12,13,14]。SINGH等[15]检测到4个SSR标记(Satt434、Satt538、Satt281和Satt598)与种皮吸胀性紧密连锁,解释3.9%—4.5%的表型变异。利用硬实大豆PI 594619与大豆品种PI 587982A杂交,发现PI 594619的硬实性状受1个单显性基因控制,并将该基因定位于第2染色体(D1b连锁群)[16]。最近,利用冀豆12与硬实地方品种黑豆杂交构建分离群体,除前期定位的第2和第6染色体外,在第14染色体(B2连锁群)上也定位了硬实相关QTL[17]。JANG等[18]通过精细定位,将第2染色体一个控制大豆种子硬实性的QTL(qHS1)定位于93 kb区间,在10个注释基因中推测编码1,4-β-葡聚糖内切酶的Glyma02g43680为候选基因,将来自硬实近等基因系的qHS1基因组片段转入吸胀品种中,发现转基因促进了栅栏细胞外层1,4-β-葡聚糖的积累,导致种子产生硬实。而SUN等[19]研究发现野生大豆PI 468916和PI 479752的硬实性受单显性基因GmHs1-1控制,并将该基因锚定到第2染色体22 kb区间,排除了Glyma02g43680,最后确定编码钙调磷酸跨膜蛋白的Glyma02g43700.1为候选基因,并通过转基因试验证明该基因是控制大豆种子硬实的关键位点。上述研究说明了控制大豆种子硬实性遗传机制的复杂性,同时也说明不同遗传背景下可能存在不同的硬实性相关QTL位点。【本研究切入点】前人通过传统的群体遗传图谱构建方法进行大豆种子硬实性基因/QTL定位的研究,往往费时费力。BSA法作为一种快速检测与目标性状连锁标记的方法[20],在大豆株高、抗逆和品质性状的基因/QTL定位研究中被广泛应用[21,22,23,24,25,26,27,28,29,30,31]。但迄今为止,在大豆硬实性重要基因/QTL定位中的研究尚未见BSA法应用的报道。【拟解决的关键问题】本研究利用中国栽培大豆中黄39与野生大豆NY27-38杂交构建的分离群体,采用BSA法开展大豆种子硬实性QTL定位研究,通过与传统遗传作图方法比较,明确BSA法发掘大豆种子硬实相关主效QTL位点的效率,促进野生大豆种子硬实性遗传机制研究,为野生大豆在大豆遗传改良中的合理利用奠定基础。1 材料与方法
1.1 供试材料
以中国农业科学院作物科学研究所选育的中黄39为母本,以野生大豆NY27-38(从国家种质资源库保存的河北省承德市的野生大豆ZYD2738中选择的单株)为父本配制杂交组合。2014年在北京顺义种植来自一个F1单株的F2分离群体,F3代开始每个世代都采用单粒传繁殖,2017年在北京顺义种植重组自交系群体(recombinant inbred lines,RIL)(F7,203个家系);2015年在北京顺义种植来自另一个F1单株的F2分离群体(301个单株)。行长3 m,株距0.5 m,行距1.1 m。田间采集单株叶片,液氮冷冻后保存在-80℃冰箱备用。在植株完熟时单株收获,为避免种子的机械损伤,采用手工脱粒。1.2 种子吸胀性鉴定
从每个单株上选取整齐一致的种子90粒,在铺有一层滤纸的培养皿中放置30粒,加入30 mL蒸馏水,3次重复。将培养皿放置于25℃培养箱中暗处理4 h,体积增加的种子,为正常吸胀种子,大小不变的种子为硬实种子。统计每个培养皿中正常吸胀种子数,以3次重复的吸胀百分率平均值作为单株的吸胀率。吸胀率=正常吸胀种子粒数/测定粒数×100%,吸胀率≤20%的为硬实,吸胀率介于20%—80%的为中间类型,吸胀率≥80%的为吸胀[19]。1.3 基因组DNA提取与DNA池构建(BSA法)
从每个单株上取壹元硬币大小的嫩叶,用DNA纯化试剂盒(genomic DNA purification kit,Thermo Scientific)提取单株叶片基因组DNA。在F2群体中,共取样38个单株,包括22个种子正常吸胀单株(吸胀率>90%)和16个硬实单株(吸胀率<10%);在F7群体中,共取样40个单株,包括20个完全吸胀单株(吸胀率=100%)和20个完全硬实单株(吸胀率=0%),单株DNA等量混合,分别构建吸胀和硬实DNA池。1.4 分子标记的选择及分析
从SoyBase数据库(https://soybase.org/)中选择分布于大豆20条染色体的543个SSR标记(每条染色体的标记数在17—40个,平均每条染色体上27个标记),由博迈德生物技术有限公司合成,用中黄39和NY27-38进行多态性引物筛选。根据发表文章合成CAPS标记Hs1AseI的2条引物(F:5′-TTTAACCTCAAGTGGAGTTAC-3′;R:5′-CTAAC TTCAAGAGTGCAATGT-3′)[19]。PCR总反应体系为20 μL,包括10×Easy Taq Buffer(Mg2+)2.0 μL、2.5 mmol·L-1 dNTPs 1.5 μL、无菌水9.3 μL、Taq 酶(5 U·μL-1)0.2 μL、引物(2 μmol·L-1)2.0 μL和DNA(20 ng μL-1)5.0 μL。反应程序为95℃ 5 min;95℃ 30 s,55℃ 30 s,72℃ 30 s,34个循环;72℃ 5 min,4℃保存。SSR标记的扩增产物进行6%变性聚丙烯酰胺凝胶电泳。CAPS标记Hs1AseI的PCR产物用内切酶AseⅠ酶切,酶切体系为10 μL:PCR产物5.0 μL、无菌水3.4 μL、AseⅠ内切酶(NEB, USA)0.6 μL和10×NEB Buffer 1.0 μL,混匀并置于37℃酶切1 h,酶切产物用1.4%琼脂糖电泳检测。
1.5 BSA法和遗传群体作图法定位硬实性相关QTL
BSA法:利用亲本间有多态性的259个多态性SSR标记对F2和F7群体的吸胀池和硬实池进行检测,筛选在硬实和吸胀DNA池间表现多态性的SSR标记(其中一个DNA池扩增的条带粗细为另一个DNA池相应条带的1/2或更低时,判定该标记在2个DNA池间存在多态性)。用池间有多态性的标记检测F2群体中所有单株的DNA,利用QTL IciMapping4.0(http://www.isbreeding.net)软件绘制遗传连锁图谱,并进行QTL定位(临界阈值LOD=2.5)。遗传群体作图法:利用分布于大豆20条染色体的192个SSR标记检测F7群体203个单株,利用QTL IciMapping4.0软件绘制遗传连锁图谱,并进行QTL定位(临界阈值LOD=2.5)。
2 结果
2.1 大豆种子硬实性鉴定及遗传分析
2.1.1 大豆种子硬实性鉴定 从中黄39、NY27-38,以及F1单株收获的种子(F1:2)中各取50粒完整种子,用蒸馏水处理。处理2 h,中黄39的种子开始吸胀,其中26%的种子完全吸胀,74%的种子表现种皮皱缩,未达到完全吸胀;NY27-38有2粒种子吸胀;F1:2有4粒种子吸胀。处理4 h,中黄39的种子100%吸胀;NY27-38种子有3粒吸胀;F1:2种子有4粒吸胀。根据亲本表现将种子吸胀鉴定时间确定为4 h(图1-A和图1-B)。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1中黄39、NY27-38、F1:2种子及分离群体(F2和F7)的表型鉴定
A:中黄39、NY27-38、F1:2种子处理0、2和4 h的吸胀表型;B:处理12 h,中黄39、NY27-38、F1:2种子在不同时间点的吸胀比例;C:F2群体吸胀频率分布;D:F7群体吸胀频率分布
Fig. 1Hard seededness of Zhonghuang39, NY27-38, F1:2 seeds and F2, F7 populations
A: Phenotypic illustration of Zhonghuang39, NY27-38 and F1:2 seeds from a F1 plant (Zhonghong39 × NY27-38) at 0, 2 and 4 h; B: Permeable proportion of seeds from Zhonghuang39, NY27-38 and their F1:2 progeny at multiple time points over 12 h; C: Frequency distribution for seed-coat permeability of F2 population; D: Frequency distribution for seed-coat permeability of F7 population
中黄39×NY27-38的F1:2种子处理4 h时,92%的种子表现为硬实,说明硬实为显性,吸胀为隐性。F2群体301个单株中,吸胀率≤20%的有53株,吸胀率介于20%—80%的为155株,吸胀率≥80%的为93株(图1-C)。F7群体203个家系,吸胀率≤20%的为105个,吸胀率介于20%—80%的为45个,吸胀率≥80%的为53个(图1-D)。
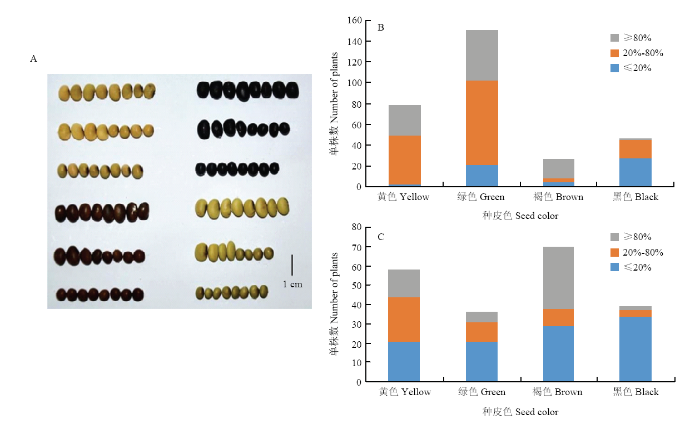
2.1.2 种子硬实性与种皮色的关系 前人研究发现大豆种子硬实性与种皮色相关位点I和T连锁[12]。本研究中,F2分离群体包括78个黄种皮、151个绿种皮、26个褐种皮和46个黑种皮单株。吸胀和硬实大豆中都包括不同种皮色材料,但比例各不相同(图2-A)。硬实单株中黑种皮材料最多(占50.0%),中间类型和吸胀单株主要为绿种皮(在中间类型中占54.3%,在吸胀单株中占50.0%)(图2-B)。F7群体包括58个黄种皮、36个绿种皮、70个褐种皮和39个黑种皮单株,硬实单株中黑种皮材料最多(占32.4%),中间类型主要是黄种皮(占51.1%),吸胀单株中褐种皮最多(占60.4%)(图2-C)。
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2F2与F7群体不同种皮色大豆的吸胀表型分布
A:F2群体不同种皮色大豆处理4 h吸胀性;B:F2群体不同种皮色大豆的表型分布;C:F7群体不同种皮色大豆的表型分布
Fig. 2Phenotypic distribution for seed-coat permeability with different seed coat colors of F2 and F7 populations
A: Photographic illustration of seed-coat permeability with different seed-coat colors of F2 population at 4 h; B: Relationship of seed-coat colors and frequency distribution of permeability in F2 population; C: Relationship of seed-coat colors and frequency distribution of permeability in F7 population
2.2 大豆种子硬实性QTL定位
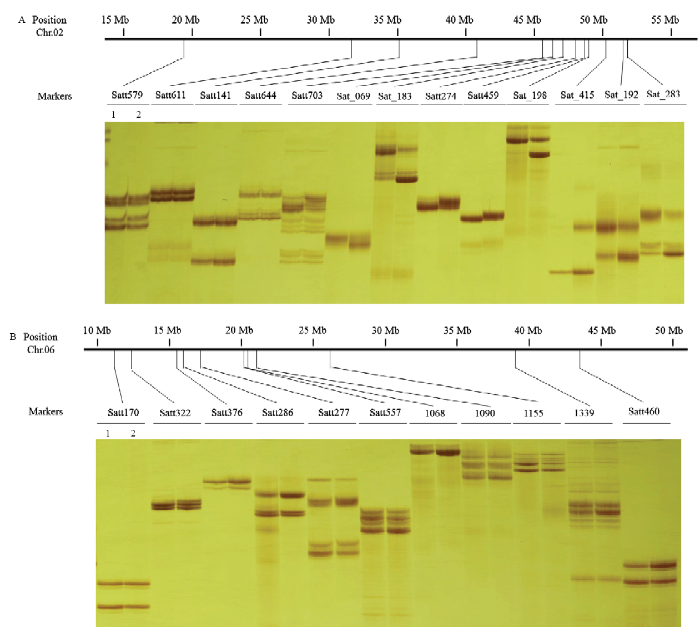
2.2.1 BSA法定位种子硬实相关QTL 用亲本间有多态性的259个SSR标记分析F2群体吸胀和硬实DNA池,共筛选到18个与大豆种子硬实相关的SSR标记,其余的标记在2个DNA池间无差别。在第2染色体上16.3 Mb区间有10个多态性SSR标记,包括Satt141、Satt703、Sat_069、Sat_183、Satt274、Satt459、Sat_198、Sat_415、Sat_192和Sat_283,其中Satt274、Satt459在DNA池间显示带型有/无的差异,即在吸胀DNA池中只有吸胀亲本中黄39的条带,硬实DNA池中只有硬实亲本NY27-38的条带,Sat_198和Sat_415在吸胀DNA池中只有吸胀亲本(中黄39)带型,硬实池中是双亲带型。其余位点在2个DNA池均扩增出双亲带型,但不同亲本来源的条带粗细有差别。说明在该染色体16.3 Mb区间的标记在吸胀和硬实DNA池间都显示多态性(图3-A)。第6染色体上23.4 Mb区间有8个SSR标记在2个池间条带粗细的差异,分别为Satt376、Satt286、Satt277、Satt557、BARCSOYSSR_06_1068、BARCSOYSSR_06_1090、BARCSOYSSR_06_1155和BARCSOYSSR_06_1339,未发现DNA池间带型有/无差异。利用这些SSR标记对F2群体所有单株进行检测,定位到2个主效QTL,硬实的增效性来自野生大豆NY27-38。第2染色体QTL位于标记Satt274与Sat_198之间,LOD值13.3,可解释17.2%的表型变异。第6染色体QTL位于标记BARCSOYSSR_06_0993与BARCSOYSSR_06_1068之间,LOD值为13.0,可解释17.8%的表型变异。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3F2群体DNA池间多态性SSR标记鉴定
A:第2染色体DNA池多态性SSR标记鉴定;B:第6染色体DNA池多态性SSR标记鉴定。1:吸胀池;2:硬实池
Fig. 3Identification of polymorphic SSR markers between two DNA bulks of F2 population
Polymorphic SSR markers between two DNA bulks on chromosome 2 (A) and chromosome 6 (B). 1: Permeable DNA bulk; 2: Impermeable DNA bulk
利用亲本间有多态性的259个SSR标记对F7群体的吸胀和硬实DNA池进行检测,共筛选到24个在吸胀和硬实池间存在差异的SSR标记,其余的标记在两池间无差别。第2染色体27.4 Mb区间上有11个,其中Satt274、Satt459显示带型有/无差异,其余标记在2个DNA池显示条带粗细差别(图4-A)。第6染色体27.8 Mb 区间上有9个标记,位于Satt376和Satt460之间(图4-B);第3染色体18.2 Mb 区间上有4个,分别为Satt125、Sat_280、Sat_266和Sat_236(图4-C)。在第3和第6染色体上,吸胀与硬实DNA池间的多态性SSR标记都是条带粗细差别。
图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4F7群体DNA池间多态性SSR标记鉴定
A:第2染色体DNA池多态性SSR标记鉴定;B:第6染色体DNA池多态性SSR标记鉴定;C:第3染色体DNA池多态性SSR标记鉴定。1:吸胀池;2:硬实池
Fig. 4Identification of polymorphic SSR markers between two DNA bulks of F7 population
Polymorphic SSR markers between two DNA bulks on chromosome 2 (A), chromosome 6 (B) and chromosome 3 (C). 1: Permeable DNA bulk; 2: Impermeable DNA bulk
2.2.2 遗传群体作图及硬实性相关QTL定位 为验证BSA方法检测硬实性相关QTL的效率,利用分布于大豆20条染色体的192个SSR标记检测F7群体203个家系,构建了长度为2 390.2 cM的遗传图谱(平均每条染色体上10个标记,标记间平均距离为12.4 cM),共定位到3个大豆种皮硬实性QTL,硬实的增效性来自野生大豆NY27-38。第2染色体的QTL位于标记Satt274与Sat_198之间,LOD值14.0,可解释23.3%的表型变异,该区间包括已克隆的GmHs1-1[19];第3染色体上的QTL位于标记Sat_266与Sat_236之间,LOD值2.7,可解释4.9%的表型变异;第6染色体上的QTL位于标记Sat_402与Satt557之间,LOD值11.5,可解释20.4%的表型变异(表1)。3个QTL的定位区间与BSA法筛选的池间差异SSR标记分布区间吻合(图4),说明BSA法可以筛选到所有F7群体用传统遗传作图法检测到的硬实相关主要QTL位点。
Table 1
表1
表1不同群体检测到的大豆种子硬实性QTL
Table 1
| 群体 Population | 染色体 Chr. | 标记区间 Marker interval | 区间物理位置 Physical position of interval (bp,Wm82.a1.v1.1) | LOD值 LOD value | 贡献率 PVE (%) | 加性效应 Additive effect |
|---|---|---|---|---|---|---|
| F2 | 2 | Satt274-Sat_198 | 48345948—48621931 | 13.3 | 17.2 | 15.8 |
| 6 | 6-0993-6-1068 | 18697727—20742109 | 13.0 | 17.8 | 12.5 | |
| F7 | 2 | Satt274-Sat_198 | 48345948—48621931 | 14.0 | 23.3 | 20.2 |
| 3 | Sat_266-Sat_236 | 36046708—37622037 | 2.7 | 4.9 | 8.9 | |
| 6 | Sat_402-Satt557 | 16367687—20018845 | 11.5 | 20.4 | 19.2 |
新窗口打开|下载CSV
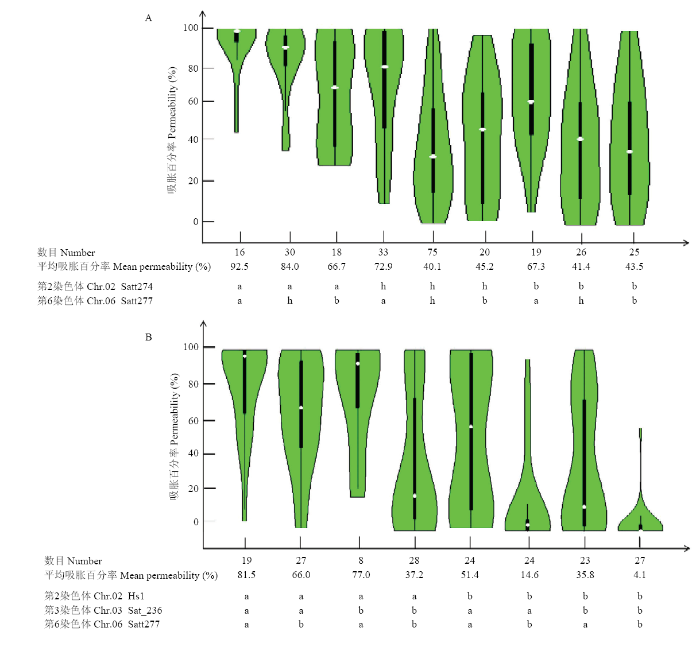
2.2.3 中黄39×NY27-38的F2和F7群体的QTL对吸胀性的影响 为分析不同QTL位点的效应,选择每个区间与QTL紧密连锁的标记,分析不同基因型材料的吸胀性。在F2群体中,当个体在第2染色体QTL位点的基因型为吸胀亲本中黄39基因型(标记为a),而第6染色体基因型分别为中黄39基因型(a)、杂合(h)、野生大豆基因型(b)的单株平均吸胀百分率为92.5%、84.0%和66.7%;当个体在第2染色体QTL位点的基因型为杂合(h),而第6染色体基因型分别为中黄39基因型(a)、杂合(h)、野生大豆基因型(b)的单株平均吸胀百分率为72.9%、40.1%和45.2%;当第2染色体基因型固定为b(来自野生大豆的基因型),而第6染色体基因型分别为a(吸胀等位变异)、h(杂合)、b(硬实等位变异)的平均吸胀百分率为67.3%、41.4%和43.5%(图5-A)。
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5小提琴图展示不同QTL对种子吸胀的影响
A:F2群体;B:F7群体
Fig. 5Violin plots showing the effect of different alleles of QTLs on seed permeability
A: F2 population; B: F7 population
在F7群体中,由于第2染色体定位区间包括已克隆的GmHs1-1,因此,该区间采用功能基因标记Hs1AseI的基因型[19]。当第2和第3染色体QTL位点的基因型为a,第6染色体基因型为b时的单株平均吸胀百分率为66.0%。当第2和第3染色体QTL位点基因型为b,第6染色体基因型为a时的单株平均吸胀百分率为35.8%。当第2和第6染色体基因型为a,第3染色体基因型为b时的单株平均吸胀百分率为77.0%。当第2和第6染色体基因型为b,第3染色体基因型为a时的单株平均吸胀百分率为14.6%。当3个定位区间基因型全为a时(n=19),平均吸胀百分率为81.5%;当3个定位区间基因型全为b时(n=27),平均吸胀百分率为4.1%(图5-B),说明3个位点存在加性效应,其中第2和第6染色体QTL效应更大。
3 讨论
3.1 不同遗传背景大豆种子硬实性QTL定位
大豆种子硬实是一个复杂的数量性状,不同遗传背景下,控制硬实的位点不完全相同。在本研究中,F2群体硬实与吸胀分离比不符合3﹕1(χ2=10.73,P=0.0043),说明该群体硬实性并非由单基因控制,与已有报道不同[18,19]。利用F2和RIL群体在第2染色体276.0 kb区间定位的主效QTL,包含前人在第2染色体的定位区间[12,16,18-19],与艾丽娟等[17]发现的第2染色体QTL(标记Sat_069与Sat_183)相距941.0 kb。说明第2染色体上的硬实相关位点是不同野生大豆中共有的重要QTL位点。F2与F7群体的第6染色体上与硬实相关的区间部分重叠,该区间包含在地方品种黑豆中检测到的26.9 MB区间(Sat_402—Satt460)内[17]。WATANABE等[13]发现的QTL RAS1(标记Satt489与Satt100之间)和SAKAMOTO等[12]定位的大豆种子硬实性QTL(标记Satt316和Satt489之间)都与本研究在第6染色体的定位区间相距3.1 Mb。SINGH等[15]检测到的与Satt281紧密连锁QTL与本研究第6染色体的定位区间相距9.8 Mb。这些研究表明,在第6染色体也存在不同材料间共有的硬实性相关QTL位点,可以作为进一步研究的重要位点。前人研究报道大豆种子硬实QTL与种皮色I位点和T位点位置相邻近或可能其本身是控制大豆种子硬实的一个QTL[11,12,13]。大豆种皮色相关的T位点(18534606—18541507,Wm82.a1. v1.1)位于本研究的第6染色体QTL区间内,这在一定程度上解释了不同种皮色的家系吸胀性存在差异的原因,尤其是黑种皮大豆吸胀率显著低于其他种皮色大豆。除此之外,本研究在第3染色体1.6 Mb区间定位到一个尚未报道的贡献率为4.9%的QTL,说明该位点可能为NY27-38特有的位点。3.2 BSA法可用于大豆硬实性QTL的高效发掘
BSA法在大豆基因/QTL定位中已有较多报道,MANSUR等[21]利用RFLP标记验证了BSA方法发掘大豆成熟期、株高、抗倒伏和产量性状相关QTL的有效性。之后BSA法逐步被应用于大豆抗病、抗虫和耐逆境胁迫性状的研究,在大豆离子含量和营养成分等相关的品质性状研究中也有报道[21,22,23,24,25,26,27,28,29,30,31]。为确定BSA法在大豆种子硬实性QTL定位中的应用效果,本研究利用F2和F7群体进行相关研究,分别检测到18个和24个在硬实和吸胀DNA池间有差异的SSR标记。利用F7群体构建包括192个SSR标记的遗传图谱,复合区间作图定位到3个硬实相关QTL,分别对应在第2、第6和第3染色体利用BSA法筛选的区间,表明利用BSA法挖掘大豆种子硬实QTL的高效性。值得关注的是,利用BSA法在F7群体中检测到一个位于第3染色体的微效QTL(LOD=2.7,贡献率4.9%),BSA法在F2群体中未检测到该位点。为确定是否由于该位点在F2群体中效应较小而不能被BSA法检测到,本研究利用该区间的3个分子标记(Satt125、Satt549、Satt312)对F2群体进行检测,遗传作图后也未检测到超过阈值的QTL,说明了不同分离世代QTL位点的贡献率不同。比如在第2染色体(已克隆的GmHs1-1)定位区间为中黄39的基因型(a)时,若其他位点基因型为野生大豆基因型(b),F2单株的平均吸胀率为66.7%,而在F7分离群体中这类单株的平均吸胀率只有37.2%(图5)。利用BSA法发掘农艺性状相关QTL位点时,如果极端个体数量过少,容易检测到假阳性位点。与基因型分离最丰富的F2 群体相比,RIL群体具有来自双亲的稳定重组,在本研究中利用极端个体进行DNA池构建时,RIL群体的极端家系数量更多(吸胀率为0和100%的家系分别有20个和20个),在F2群体中吸胀率为0和100%的家系分别有0个和8个,将极端个体表型值分别调整为10%和90%时才有足够的个体构建DNA池。这与WANG等[32]对F2、RIL和DH群体综合分析后的结论相同,即利用F2群体构建DNA池进行QTL位点发掘效果较差,RIL群体建池效果更好。但无论F2或RIL群体,均可利用BSA法快速发掘与硬实相关的主效QTL染色体区间(图3和图4)。目前,BSA法已与全基因组测序(bulked-segregant analysis sequencing,BSA-Seq)或转录组测序(bulked segregant RNA- sequencing,BSR-Seq)相结合,用于不同作物重要性状相关QTL或候选基因的发掘[33,34,35,36]。与传统遗传群体作图相比(本研究为203个家系,192个SSR标记),BSA法只需要用亲本间多态性的分子标记(本研究中为259个SSR标记)对极端个体混合获得的2个DNA池进行检测,一旦确定候选区间,也仅需要用候选区间的几个标记检测整个群体,通过连锁分析进行QTL准确定位,大大提高QTL发掘效率。同时,本研究发现不同染色体上两个DNA池间有多态性的标记分布在16.3—27.8Mb区间内,说明在用BSA法进行QTL区间筛选时,确保每5.0 Mb有一个多态性标记,在候选区间会检测到连续多个多态性位点,可有效排除假阳性位点。
4 结论
在F2(中黄39×野生大豆NY27-38)分离群体检测到2个与硬实性相关QTL位点,在F7分离群体检测到3个与硬实性相关QTL位点。利用192个SSR标记检测F7分离群体203个家系,构建遗传图谱,在第2、3和6染色体共定位到3个大豆种子硬实性QTL,与BSA法发掘的QTL位点一致。证明用分离群体中的16—22个极端个体构建DNA池,在染色体上每5.0 Mb物理距离选择一个亲本间多态性标记,即可有效发掘大豆种子硬实性主要QTL。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 2]
.
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/BF02857926URL [本文引用: 1]
DOI:10.2135/cropsci1978.0011183X001800020006xURL [本文引用: 2]
DOI:10.1016/0378-4290(94)00075-NURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1093/aob/mch133URL
[本文引用: 1]
.
[本文引用: 1]
[本文引用: 2]
DOI:10.1270/jsbbs.54.133URL [本文引用: 5]
DOI:10.1270/jsbbs.54.399URL [本文引用: 3]
[本文引用: 1]
DOI:10.1007/s12298-008-0016-0URL [本文引用: 2]
DOI:10.1007/s00122-014-2355-2URL [本文引用: 2]
[本文引用: 3]
[本文引用: 3]
DOI:10.1371/journal.pone.0128527URL [本文引用: 3]
[本文引用: 7]
DOI:10.1073/pnas.88.21.9828URL [本文引用: 1]
[本文引用: 3]
[本文引用: 2]
DOI:10.2135/cropsci2004.0560URL [本文引用: 2]
DOI:10.1007/s00122-008-0752-0URL [本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
.
[本文引用: 2]
DOI:10.2135/cropsci2005.05-0168URL [本文引用: 2]
.
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s11032-019-0948-9 [本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}