 ,, 闫会, 高闰飞, 后猛, 唐维, 王欣, 张允刚, 李强,*中国农业科学院甘薯研究所 / 江苏徐淮地区徐州农业科学研究所 / 农业农村部甘薯生物学与遗传育种重点实验室, 江苏徐州 221131
,, 闫会, 高闰飞, 后猛, 唐维, 王欣, 张允刚, 李强,*中国农业科学院甘薯研究所 / 江苏徐淮地区徐州农业科学研究所 / 农业农村部甘薯生物学与遗传育种重点实验室, 江苏徐州 221131Construction linkage maps and identification of quantitative trait loci associated with important agronomic traits in purple-fleshed sweetpotato
MA Meng,, YAN Hui, GAO Run-Fei, KOU Meng, TANG Wei, WANG Xin, ZHANG Yun-Gang, LI Qiang,*Sweetpotato Research Institute, Chinese Academy of Agricultural Sciences / Xuzhou Institute of Agricultural Sciences in Jiangsu Xuhuai District / Key Laboratory of Biology and Genetic Breeding of Sweetpotato, Ministry of Agriculture and Rural Affairs, Xuzhou 221131, Jiangsu, China通讯作者: * 李强, E-mail:instrong@163.com
收稿日期:2020-12-13接受日期:2021-03-19网络出版日期:2021-04-12
| 基金资助: |
Corresponding authors: *E-mail:instrong@163.com
Received:2020-12-13Accepted:2021-03-19Published online:2021-04-12
| Fund supported: |
作者简介 About authors
E-mail:1325428037@qq.com

摘要
理想农艺性状是甘薯育种的重要目标, 而选择甘薯理想农艺性状的育种手段还很缺乏。本研究以分枝数多、蔓长中等、高产紫肉甘薯品种徐紫薯8号为母本, 分枝数少、长蔓、中等产量白肉甘薯品种美国红为父本, 以F1代分离群体的274个单株为作图群体, 利用SSR分子标记技术, 构建了甘薯分子连锁图谱, 能够加密已有的遗传图谱。其中母本图谱包含24个连锁群(linkage groups, LGs), 图谱总长1325.8 cM, 标记间平均距离9.2 cM; 父本图谱包含21个LGs, 图谱总长1088.6 cM, 标记间平均距离8.2 cM。通过复合区间作图法对甘薯地上部分枝数、茎蔓直径、最长蔓长、叶柄长度和节间长度5个重要农艺性状进行QTL分析, 检测到1个与分枝数相关的QTL, 解释表型变异的53.2%; 1个与茎蔓直径相关的QTL, 解释表型变异的16.7%; 2个与最长蔓长相关的QTL, 解释表型变异的9.5%和13.7%; 2个与叶柄长度相关定位的重要农艺性状QTL, 可以开发与其连锁的分子标记, 辅助室内早代苗期筛选具有理想农艺性状的株系, 从而提高田间选择效率的QTL, 解释表型变异的8.8%和11.3%; 5个与节间长度相关的QTL, 解释表型变异的9.6%~28.1%。利用定位的重要农艺性状QTL,可以开发与其连锁的分子标记,辅助室内早代苗期筛选具有理想农艺性状的株系,从而提高田间选择效率。。
关键词:
Abstract
Ideal agronomic traits are the important objectives in sweetpotato breeding, but the breeding methods are still lacking. We constructed linkage maps using a mapping population of 274 individuals derived from a cross between the female parent Xuzishu 8 (a purple-fleshed cultivar with many branches, medium vine, and high yield) and the male parent Meiguohong (a white-fleshed cultivar with few branches, long vine, and medium yield) by simple sequence repeats (SSR) markers in this study. The female parent map contained 24 linkage groups, and covered 1325.8 cM with an average marker interval of 9.2 cM. The male parent map contained 21 linkage groups, and covered 1088.6 cM with an average marker interval of 8.2 cM. The maps could increase the density of existing genetic maps. Using the composite interval mapping, we analyzed five important agronomic traits, including branch number, vine diameter, longest vine length, petiole length, and internode length in sweetpotato, thus identified one QTL related to branch number explaining the phenotypic variance of 53.2%, one QTL related to internode diameter explaining the phenotypic variance of 16.7%, two QTLs related to longest vine length explaining the phenotypic variance of 9.5% and 13.7%, two QTLs related to petiole length explaining the phenotypic variance of 8.8% and 11.3%, and five QTLs related to internode length explaining the phenotypic variance of 9.6%-28.1%. The QTLs can be used to develop molecular markers and assist the screening of plants with ideal agronomic traits at early seedling stage, thus improved the efficiency of field selection.
Keywords:
PDF (1173KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
马猛, 闫会, 高闰飞, 后猛, 唐维, 王欣, 张允刚, 李强. 紫甘薯SSR标记遗传图谱构建与重要农艺性状QTL定位. 作物学报, 2021, 47(11): 2147-2162 DOI:10.3724/SP.J.1006.2021.04271
MA Meng, YAN Hui, GAO Run-Fei, KOU Meng, TANG Wei, WANG Xin, ZHANG Yun-Gang, LI Qiang.
甘薯[Ipomoea batatas (L.) Lam.]是一种用途广泛的六倍体作物, 可作为粮食、饲料、工业原料及新型能源使用, 在我国占据重要的产业地位[1]。甘薯地下部块根产量及品质性状与地上部的生长有密切关系, 既相互促进又相互制约[2]。有研究表明, 甘薯茎越粗, 蔓长越短, 越有利于甘薯干物质积累[3]; 基部分枝数和最长蔓长可作为高产品种选育的重要参考。在甘薯育种过程中, 品种基部分枝数较多、茎直径较粗, 往往蔓长相对较短, 既有利于甘薯田间管理及收获, 又有利于提高薯块产量。而叶柄的长短对于甘薯长势及产量也有巨大影响, 叶柄较长会减少叶片叠加, 能够保持良好的叶面积和叶层结构, 以及提高叶片的光能利用率。因此, 加大对甘薯重要农艺性状的研究, 揭示甘薯重要性状遗传规律, 对于加快甘薯品种选育, 具有重要意义。
甘薯很多重要的农艺性状如分枝数、最长蔓长等都是多基因控制的数量性状, 其表现型是基因型与环境共同作用的结果[4]。随着分子生物学的发展, 以分子标记技术为基础, 通过构建甘薯分子连锁图谱, 进而定位相关性状的数量性状位点(quantitative trait loci, QTL), 是目前甘薯分子育种的重要方向[5]。近年来, 甘薯QTL定位的研究主要集中在淀粉含量、干物质含量及β-胡萝卜素含量等品质性状。唐道彬等[6]在万薯5号遗传图谱上定位到3个QTL, 在商丘52-7遗传图谱上定位到14个QTL, 17个QTL共解释淀粉含量8.4%~ 40.5%的表型变异率; Zhao等[7]在徐薯18和徐781图谱上共定位到27个与干物质含量相关的QTL, 可解释表型变异的9.0%~45.1%; 李爱贤等[8]在漯徐薯8号和郑薯20图谱上定位到10个和7个与β-胡萝卜素含量相关的QTL, 可解释表型变异的33.1%~62.1%。鲜薯产量性状[9,10]以及抗性性状[11,12,13]的遗传图谱构建及QTL定位也有一些报道。但目前针对甘薯地上部地下部表型性状的图谱构建及QTL定位研究还很少。
因此, 本研究利用徐紫薯8号和美国红的F1分离群体为材料, 通过对分枝数、茎蔓直径、最长蔓长、叶柄长度、节间长度5个重要农艺性状进行QTL定位, 检测相关性状紧密连锁的分子标记, 以期为甘薯品种早期筛选及分子标记辅助选择育种提供技术支撑。
1 材料与方法
1.1 群体构建
本研究以紫肉甘薯品种徐紫薯8号为母本, 白肉甘薯品种美国红为父本配制杂交组合, 通过严格去雄授粉方式, 于2016—2018年杂交构建包含274个分离单株的基础作图群体。徐紫薯8号是江苏徐淮地区徐州农业科学研究所育成的紫甘薯品种, 美国红是从国外引进的优质高淀粉品种。两亲本遗传差异大, 除叶形、薯皮色、薯肉色等地上部地下部表型性状外, 在花青苷含量、淀粉含量、干物质含量、茎线虫病及黑斑病抗性等方面也有显著差异, 适于甘薯分子遗传图谱构建及相关性状的QTL定位。1.2 材料种植
分离群体各株系及双亲于2019年和2020年6月中旬种植在徐州市现代农业试验示范基地, 采用3行区、每行10株的大田种植方式, 垄距85 cm, 株距23 cm, 试验设3次重复; 10月中旬收获。1.3 重要农艺性状的调查
在生育期90 d, 参照《甘薯种质资源描述规范和数据标准》[14], 调查双亲及274份作图群体的分枝数、茎蔓直径、最长蔓长、叶柄长度、节间长度5个地上部表型性状。每个材料测量6次重复, 取平均值进行QTL分析。1.4 甘薯基因组DNA提取与SSR分析
取双亲及分离群体各株系的幼嫩展开叶, 采用改良CTAB法[15]提取基因组DNA。参考Meng等[16]及徐紫薯8号转录组测序结果, 筛选多态性好的SSR引物, 由上海生工生物工程股份有限公司合成。SSR标记的PCR反应体系共20 μL, 包含模板DNA (50 ng μL-1) 1 μL、上下游引物(10 μmol L-1)各0.5 μL、2×Taq Mix 8 μL、ddH2O 10 μL。PCR反应程序为95℃预变性5 min; 95℃变性30 s, 55℃复性30 s, 72℃延伸30 s, 35个循环; 72℃继续延伸5 min; 最后4℃降温保存10 min。PCR产物用6%的聚丙烯酰胺凝胶电泳分离。参考高闰飞[17]的方法银染显色。1.5 数据记载与标记命名
利用双亲及其杂交F1代分离群体中随机抽取的2个性状差异大的分离单株筛选多态性好的SSR引物, 然后利用筛选到的多态性引物对F1群体进行标记基因型检测。多态性标记按从上到下的顺序, 分子量越大的标号越小; 同一标记处有条带的记为1, 没有的记为0, 模糊或缺失的记为2; 属于单标记(Simplex)、双标记(Duplex)、三标记(Triplex)和共有标记(Double-simplex)的多态性标记, 其后缀分别用s、d、t和ds表示; 对每个多态性标记的分离比进行卡平方检验, 在α=0.05水平表现显著差异的标记以*表示, 在α=0.01水平表现极显著差异的标记以**表示[18]。例如SSR-78A-2s*表示引物78A的第2条多态性条带, 属于单标记类型, 在α=0.05水平表现显著差异。1.6 分子遗传图谱构建及相关QTL定位
1.6.1 双亲图谱构建 本研究利用JoinMap 4.0软件, 选择CP分析模型。首先利用Simplex和Double-simplex标记构建框架图, LOD值≥5, 然后将Duplex和Triplex标记插入到框架图谱的每一个LGs中, 得到最终的分子遗传图谱[18]。1.6.2 相关性状QTL定位 利用QTL分析软件MapQTL 5.0, 以复合区间作图法(composite interval mapping, CIM)[18]进行分枝数、茎蔓直径、最长蔓长、叶柄长度、节间长度的QTL定位。
2 结果与分析
2.1 分子遗传图谱构建
利用筛选的110对多态性好的SSR引物对F1群体进行标记基因型检测, 分别获得220个和219个多态性标记用于徐紫薯8号和美国红图谱构建。其中母本徐紫薯8号图谱由24个LGs组成, 包括144个标记, 图谱总长1325.8 cM, 标记间平均距离9.2 cM (图1); 父本美国红图谱由21个LGs组成, 包括132个标记, 图谱总长1088.6 cM, 标记间平均距离8.2 cM (图2)。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1徐紫薯8号分子连锁图谱
Fig. 1Linkage maps of Xuzishu 8
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2美国红分子连锁图谱
Fig. 2Linkage maps of Meiguohong
2.2 重要农艺性状的QTL定位
分别于2019年和2020年测定了徐紫薯8号、美国红及其F1分离群体的分枝数、茎蔓直径、最长蔓长、叶柄长度、节间长度。利用2019年、2020年及2年平均群体数据进行QTL分析, 将至少在1年和2年平均数据中同时存在的QTL作为稳定的QTL。2.2.1 分枝数的表型分析与QTL初步定位 利用SAS 9.4软件对甘薯分枝数双亲值及其在F1群体中的分布进行统计分析和作图发现, 分枝数在F1群体中明显分离, 表现为连续分布, 且呈现明显的单峰曲线, 表明甘薯分枝数是由多基因控制的数量性状, 可以用于后续的QTL定位研究(表1和图3)。
Table 1
表1
表1甘薯分枝数双亲值及其在F1群体中的分布
Table 1
| 年份 Year | 母本(个) Maternal | 父本(个) Paternal | 最大值(个) Max. | 最小值(个) Min. | 平均值(个) Mean | 标准差 SD | 偏度 Skewness | 峰度 Kurtosis |
|---|---|---|---|---|---|---|---|---|
| 2019 | 14.83 | 6.67 | 75.00 | 7.00 | 23.00 | 10.47 | 1.37 | 3.29 |
| 2020 | 14.20 | 8.00 | 25.80 | 2.33 | 11.95 | 3.70 | 0.64 | 1.03 |
| 平均Average | 14.52 | 7.33 | 66.00 | 6.20 | 18.14 | 8.56 | 2.04 | 6.41 |
新窗口打开|下载CSV
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3分枝数在F1分离群体中的频率分布图
Fig. 3Frequency distribution of branch number in F1 population
通过复合区间作图法对甘薯分枝数进行QTL分析, 在2019年和2年平均数据中共同检测到1个控制甘薯分枝数的QTL, 命名为qBNm-1p, 位于父本美国红连锁图谱的第9 LGs的标记SSR-Z135-3d**和SSR-234-1s**之间, 表现为正向效应, 可解释表型变异的53.2% (图4)。
图4
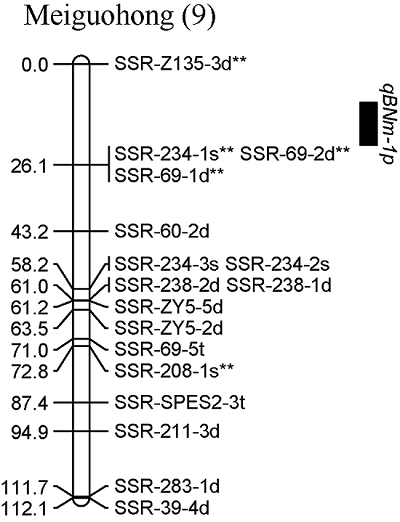 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4甘薯分枝数QTL在分子连锁图谱上的分布
Fig. 4Distribution of QTLs for branch number in genetic linkage map of sweetpotato
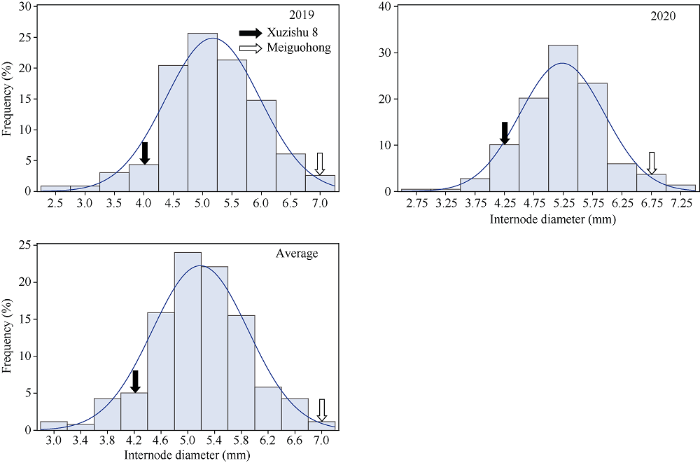
2.2.2 茎蔓直径的表型分析与QTL初步定位 茎蔓直径在F1群体中明显分离, 表现为连续分布, 且呈现明显的单峰曲线, 表明甘薯茎蔓直径是由多基因控制的数量性状, 可以用于后续的QTL定位研究(表2和图5)。
Table 2
表2
表2甘薯茎蔓直径双亲值及其在F1群体中的分布
Table 2
| 年份 Year | 母本 Maternal (mm) | 父本 Paternal (mm) | 最大值 Max. (mm) | 最小值 Min. (mm) | 平均值 Mean (mm) | 标准差 SD | 偏度 Skewness | 峰度 Kurtosis |
|---|---|---|---|---|---|---|---|---|
| 2019 | 3.93 | 6.88 | 7.04 | 2.52 | 5.17 | 0.80 | -0.21 | 0.39 |
| 2020 | 4.19 | 6.90 | 7.26 | 2.80 | 5.23 | 0.72 | 0.06 | 0.75 |
| 平均Average | 4.06 | 6.89 | 7.19 | 2.96 | 5.19 | 0.72 | -0.13 | 0.54 |
新窗口打开|下载CSV
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5茎蔓直径在F1分离群体中的频率分布图
Fig. 5Frequency distribution of internode diameter in F1 population
对甘薯茎蔓直径进行QTL分析, 在2019年和2年平均数据中共同检测到1个控制甘薯茎蔓直径的QTL, 命名为qIDm-1n, 位于父本美国红连锁图谱的第9 LGs的标记SSR-234-1s**和SSR-60-2d之间, 表现为负向效应, 可解释表型变异的16.7% (图6)。
图6
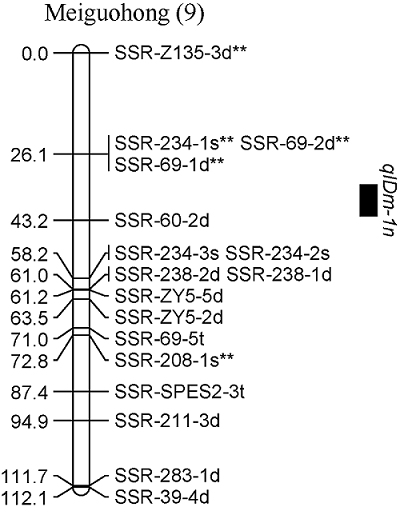 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6甘薯茎蔓直径QTL在分子连锁图谱上的分布
Fig. 6Distribution of QTLs for internode diameter in genetic linkage map of sweetpotato
2.2.3 最长蔓长的表型分析与QTL初步定位 最长蔓长在F1群体中明显分离, 表现为连续分布, 且呈现明显的单峰曲线, 表明甘薯最长蔓长是由多基因控制的数量性状, 可以用于后续的QTL定位研究(表3和图7)。
Table 3
表3
表3甘薯最长蔓长双亲值及其在F1群体中的分布
Table 3
| 年份 Year | 母本 Maternal (cm) | 父本 Paternal (cm) | 最大值 Max. (cm) | 最小值 Min. (cm) | 平均值 Mean (cm) | 标准差 SD | 偏度 Skewness | 峰度 Kurtosis |
|---|---|---|---|---|---|---|---|---|
| 2019 | 178.33 | 185.00 | 425.00 | 75.00 | 178.09 | 52.82 | 0.93 | 2.41 |
| 2020 | 182.00 | 195.00 | 361.00 | 77.00 | 172.74 | 47.83 | 0.50 | 0.60 |
| 平均Average | 180.17 | 190.00 | 393.00 | 75.00 | 174.68 | 43.86 | 0.63 | 2.20 |
新窗口打开|下载CSV
图7
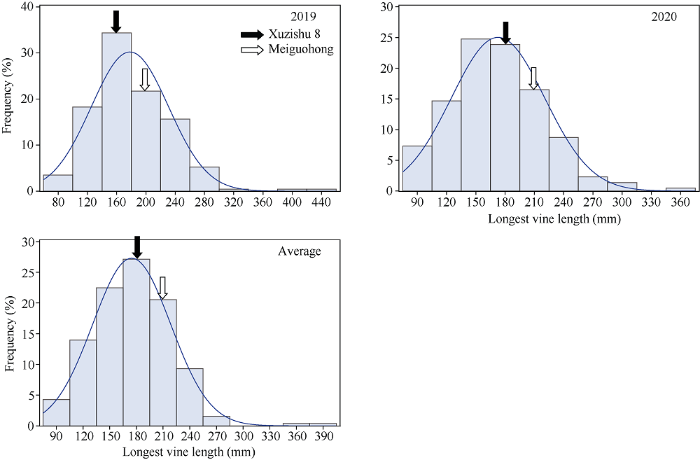 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7最长蔓长在F1分离群体中的频率分布图
Fig. 7Frequency distribution of the longest vine length in F1 population
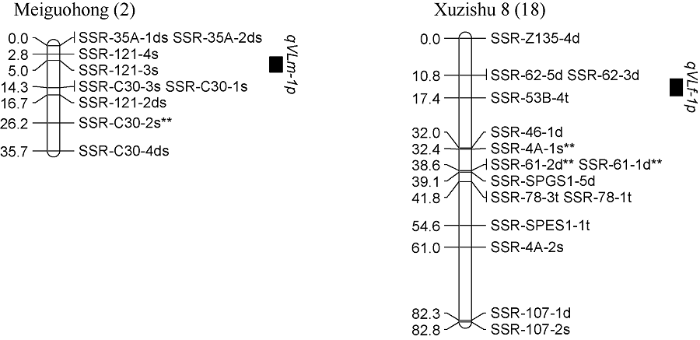
对甘薯最长蔓长进行QTL分析, 共检测到2个控制甘薯最长蔓长的QTL, 其中qVLm-1p位于父本美国红连锁图谱的第2 LGs的标记SSR-121-4s和SSR-C30-1s之间, 且与标记SSR-121-3s紧密连锁, 表现为正向效应, 可解释表型变异的13.7%; qVLf-1p位于母本徐紫薯8号连锁图谱的第18 LGs的标记SSR-62-3d和SSR-53B-4t之间, 表现为正向效应, 可解释表型变异的9.5% (表4和图8)。
Table 4
表4
表4甘薯最长蔓长的QTL分析
Table 4
| QTL名称 QTL name | 连锁图谱 Linkage group | QTL位置 QTL location (cM) | 年份 Year | 遗传效应 Genetic effect | 贡献率 R2 (%) |
|---|---|---|---|---|---|
| qVLm-1p | Meiguohong (2) | 5.006 | 2020 | 正向Positive | 18.0 |
| 平均Average | 13.7 | ||||
| qVLf-1p | Xuzishu 8 (18) | 15.775 | 2019 | 正向Positive | 12.4 |
| 平均Average | 9.5 |
新窗口打开|下载CSV
图8
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8甘薯最长蔓长QTL在分子连锁图谱上的分布
Fig. 8Distribution of QTLs for the longest vine length in genetic linkage map of sweetpotato
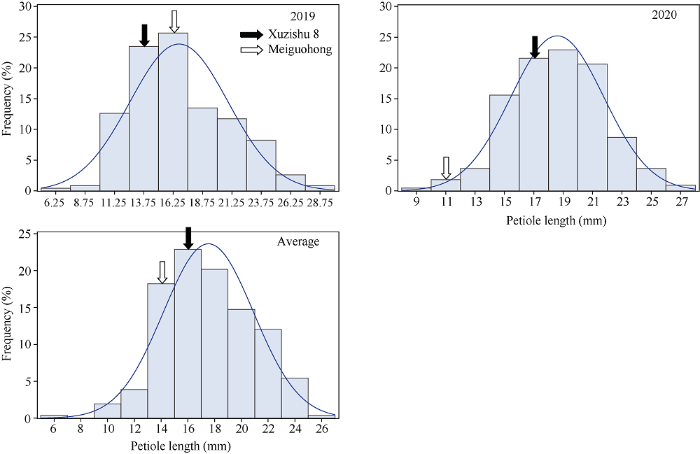
2.2.4 叶柄长度的表型分析与QTL初步定位 叶柄长度在F1群体中明显分离, 表现为连续分布, 且呈现明显的单峰曲线, 表明甘薯叶柄长度是由多基因控制的数量性状, 可以用于后续的QTL定位研究(表5和图9)。
Table 5
表5
表5甘薯叶柄长度双亲值及其在F1群体中的分布
Table 5
| 年份 Year | 母本 Maternal (cm) | 父本 Paternal (cm) | 最大值 Max. (cm) | 最小值 Min. (cm) | 平均值 Mean (cm) | 标准差 SD | 偏度 Skewness | 峰度 Kurtosis |
|---|---|---|---|---|---|---|---|---|
| 2019 | 14.17 | 15.48 | 27.98 | 5.80 | 16.74 | 4.18 | 0.47 | -0.24 |
| 2020 | 17.94 | 10.92 | 26.64 | 8.80 | 18.60 | 3.16 | -0.05 | 0.10 |
| 平均Average | 16.05 | 13.20 | 25.24 | 5.80 | 17.54 | 3.38 | 0.02 | -0.14 |
新窗口打开|下载CSV
图9
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图9叶柄长度在F1分离群体中的频率分布图
Fig. 9Frequency distribution of petiole length in F1 population
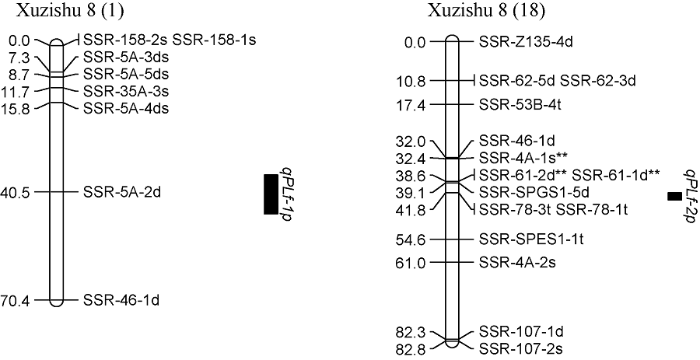
对甘薯叶柄长度进行QTL分析, 共检测到2个控制甘薯叶柄长度的QTL, 其中qPLf-1p位于母本徐紫薯8号连锁图谱的第1 LGs的标记SSR-5A-4ds 和SSR-46-1d之间, 且与记SSR-5A-2紧密连锁, 表现为正向效应, 可解释表型变异的11.3%; qPLf-2p位于母本徐紫薯8号连锁图谱的第18LGs的标记SSR-SPGS1-5d和SSR-SPES1-1t之间, 且与标记SSR-78-3t紧密连锁, 表现为正向效应, 可解释表型变异的8.8% (表6和图10)。
Table 6
表6
表6甘薯叶柄长度的QTL分析
Table 6
| QTL名称 QTL name | 连锁图谱 Linkage group | QTL位置 QTL location (cM) | 年份 Year | 遗传效应 Genetic effect | 贡献率 R2 (%) |
|---|---|---|---|---|---|
| qPLf-1p | Xuzishu 8 (1) | 40.483 | 2019 | 正向Positive | 6.5 |
| 平均Average | 11.3 | ||||
| qPLf-2p | Xuzishu 8 (18) | 41.837 | 2020 | 正向Positive | 6.5 |
| 平均Average | 8.8 |
新窗口打开|下载CSV
图10
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图10甘薯叶柄长度QTL在分子连锁图谱上的分布
Fig. 10Distribution of QTLs for petiole length in genetic linkage map of sweetpotato
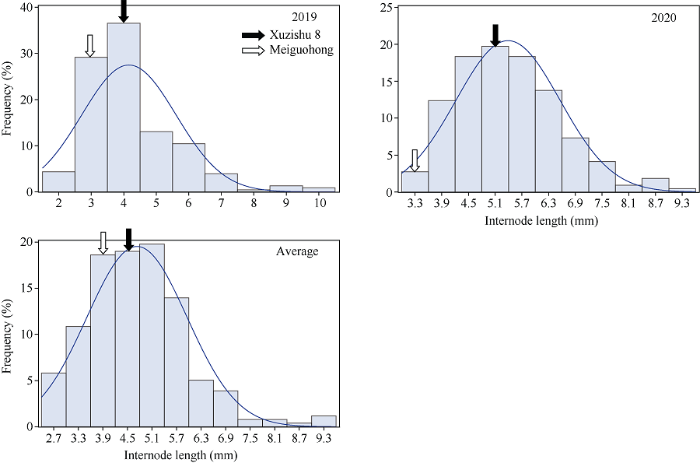
2.2.5 节间长度的表型分析与QTL初步定位 节间长度在F1群体中明显分离, 表现为连续分布, 且呈现明显的单峰曲线, 表明甘薯节间长度是由多基因控制的数量性状, 可以用于后续的QTL定位研究(表7和图11)。
Table 7
表7
表7甘薯节间长度双亲值及其在F1群体中的分布
Table 7
| 年份 Year | 母本 Maternal (cm) | 父本 Paternal (cm) | 最大值 Max. (cm) | 最小值 Min. (cm) | 平均值 Mean (cm) | 标准差 SD | 偏度 Skewness | 峰度 Kurtosis |
|---|---|---|---|---|---|---|---|---|
| 2019 | 4.08 | 3.95 | 10.35 | 2.00 | 4.16 | 1.45 | 1.46 | 2.78 |
| 2020 | 4.98 | 3.48 | 9.46 | 3.10 | 5.39 | 1.17 | 0.63 | 0.51 |
| 平均Average | 4.53 | 3.72 | 9.55 | 2.40 | 4.73 | 1.23 | 0.76 | 1.50 |
新窗口打开|下载CSV
图11
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图11节间长度在F1分离群体中的频率分布图
Fig. 11Frequency distribution of internode length in F1 population
对甘薯节间长度进行QTL分析, 共检测到5个控制甘薯节间长度的QTLs, 其中qILm-1p位于父本美国红连锁图谱的第2 LGs的标记SSR-121-4s和SSR-C30-1s之间, 且与标记SSR-121-3s紧密连锁, 表现为正向效应, 可解释表型变异的9.6%; 另外4个位于母本徐紫薯8号连锁图谱上, qILf-1p位于第1 LGs的标记SSR-5A-4ds和SSR-46-1d之间, 且与标记SSR-5A-2d紧密连锁, 表现为正向效应, 可解释表型变异的15.8%; qILf-2n位于第2 LGs的标记SSR-121-1s和SSR-C30-4ds之间, 且与标记SSR-226-2s紧密连锁, 表现为负向效应, 可解释表型变异的16.1%; qILf-3n位于第9 LGs的标记SSR-53B-4t和SSR-118-2s之间, 表现为负向效应, 可解释表型变异的28.1%; qILf-4p位于第18 LGs的标记SSR- 53B-4t和SSR-46-1d之间, 表现为正向效应, 可解释表型变异的20.6% (表8和图12)。
Table 8
表8
表8甘薯节间长度的QTL分析
Table 8
| QTL名称 QTL name | 连锁图谱 Linkage group | QTL位置 QTL location (cM) | 年份 Year | 遗传效应 Genetic effect | 贡献率 R2 (%) |
|---|---|---|---|---|---|
| qILm-1p | Meiguohong (2) | 5.006 | 2020 | 正向Positive | 9.8 |
| 平均Average | 9.6 | ||||
| qILf-1p | Xuzishu 8 (1) | 40.483 | 2019 | 正向Positive | 15.0 |
| 平均Average | 15.8 | ||||
| qILf-2n | Xuzishu 8 (2) | 13.470 | 2019 | 负向Negative | 13.1 |
| 平均Average | 16.1 | ||||
| qILf-3n | Xuzishu 8 (9) | 24.405 | 2019 | 负向Negative | 53.6 |
| 平均Average | 28.1 | ||||
| qILf-4p | Xuzishu 8 (18) | 21.380 | 2019 | 正向Positive | 43.5 |
| 平均Average | 20.6 |
新窗口打开|下载CSV
图12
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图12甘薯节间长度QTL在分子连锁图谱上的分布
Fig. 12Distribution of QTL for internode length in genetic linkage map of sweetpotato
3 讨论
甘薯是高度杂合的六倍体块根类作物[19], 地下部块根的发育与地上部的生长存在复杂的相关性。赵大伟等[20]通过测定不同环境下文薯2号的产量和农艺性状认为, 分枝数与商品薯率呈显著正相关, 淀粉含量和干率与茎粗呈显著负相关; 崔翠等[21]的研究表明, 鲜薯产量与分枝数之间呈极显著负相关, 可通过适当减少蔓长、分枝数, 稳定藤叶重, 增加大中薯数等手段来达到高产育种的目的; 郑光武等[22]的研究则认为分枝数较多有利于提高甘薯产量; 后猛等[23]认为, 选择茎粗适中、蔓长适中、分枝数多、结薯数多、茎叶鲜重较大以及茎叶干率较低的株系可达到甘薯高产育种的目的; 余金龙[24]认为, 在甘薯育种中, 分枝数及主茎与分枝数的关系(最长蔓长/平均分枝长)可作为高产品种选育的重要参考。因此, 加大甘薯重要农艺性状的研究对于甘薯品种选育具有重要意义。甘薯很多重要的农艺性状都是多基因控制的数量性状, 数量性状的表达是许多QTL以不同大小、方向共同作用的结果[25]。通过研究不同QTL的遗传效应大小及方向, 找到控制数量性状的主效基因和微效基因, 采用标记辅助选择及转基因技术, 有可能将同一效应的不同基因聚合在一起, 从而培育出超亲遗传个体。由于甘薯遗传背景复杂, 甘薯遗传图谱构建及QTL定位研究相对较少, 远落后于水稻、小麦等其他主要作物[26]。直到20世纪90年代末, Ukoskit和Thompson才构建了第一张甘薯RAPD (Random Amplified Polymorphic DNA)分子遗传图谱[27]。此后, 越来越多的研究者根据Grattapaglia和Sederoff提出的“双假测交” (pseudo-testcross)策略[28]以及甘薯显性标记遗传预期分离比例[29]相继构建了针对不同性状的分子遗传连锁图谱, 并进行了相关性状的QTL定位。Kim等[30]2017年报道了甘薯重要农艺性状QTL定位的研究, 从15个主要农艺性状中只检测到4个性状的21个QTL位点, 其中3个与节间长度相关, 1个与薯皮厚度相关, 15个与薯皮主要颜色相关, 2个与薯皮次要颜色相关。其定位到的QTL主要是与薯皮、薯肉等相关的地下部性状, 对于地上部相关性状, 仅仅在节间长度中检测到了QTL位点。而本研究定位到的QTL是与甘薯产量密切相关的分枝数、茎蔓直径、最长蔓长、叶柄长度、节间长度等重要地上部农艺性状, 在育种上有较好的应用前景, 且以两年数据进行QTL分析, 增加了QTL定位的准确性。
本研究以地上部表型性状差异显著的徐紫薯8号和美国红为亲本构建甘薯分子遗传图谱, 从而开展相关性状QTL定位的研究。在构建图谱过程中, 存在着标记偏分离现象。出现这种现象主要是由于分离群体中的等位基因分离比例不符合预期的孟德尔分离比例造成的[31]。这种现象在自然界中是普遍存在的, 是生物进化的一种重要动力[32,33]。本研究用于构建母本图谱的144个SSR标记中, 有32个偏分离标记, 占22.2%; 用于构建父本图谱的132个SSR标记中, 有52个偏分离标记, 占39.4%。偏分离标记比例过高, 会在一定程度上影响图谱的准确性和可靠度, 因此, 后续可通过增加标记数量或剔除一些对图谱影响较大的标记来降低偏分离标记比例[34]。
双亲图谱由于标记数量较少, 无法覆盖90条染色体, 对于一些不在图谱上的QTL无法检测到。同时, 本研究QTL分析的数据是在2年的同一环境中得到的, 没有多点鉴定。因此, 下一步的工作除了构建更加精密的遗传图谱, 还要对相关性状进行不同环境的分析, 从而增加QTL定位的准确性。
4 结论
本研究初步构建了紫肉甘薯品种徐紫薯8号和白肉甘薯品种美国红的SSR分子连锁图谱, 可以提高已有遗传图谱的分子标记密度。定位出的与甘薯分枝数、茎蔓直径、最长蔓长、叶柄长度、节间长度等重要农艺性状相关的QTL, 可以开发与其连锁的分子标记, 辅助室内早代苗期筛选具有理想农艺性状的株系, 从而提高田间选择效率。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIPMID [本文引用: 1]

The southern root-knot nematode, Meloidogyne incognita, is a pest that decreases yield and the quality of sweetpotato [Ipomoea batatas (L.) Lam.]. There is a demand to produce resistant cultivars and develop DNA markers to select this trait. However, sweetpotato is hexaploid, highly heterozygous, and has an enormous genome (~3?Gb), which makes genetic linkage analysis difficult. In this study, a high-density linkage map was constructed based on retrotransposon insertion polymorphism, simple sequence repeat, and single nucleotide polymorphism markers. The markers were developed using F1 progeny between J-Red, which exhibits resistance to multiple races of M. incognita, and Choshu, which is susceptible to multiple races of such pest. Quantitative trait locus (QTL) analysis and a genome-wide association study detected highly effective QTLs for resistance against three races, namely, SP1, SP4, and SP6-1, in the Ib01-6 J-Red linkage group. A polymerase chain reaction marker that can identify genotypes based on single nucleotide polymorphisms located in this QTL region can discriminate resistance from susceptibility in the F1 progeny at a rate of 70%. Thus, this marker could be helpful in selecting sweetpotato cultivars that are resistant to multiple races of M. incognita.© The Author(s) 2019. Published by Oxford University Press on behalf of Kazusa DNA Research Institute.
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 3]
[本文引用: 3]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
PMID [本文引用: 1]
We have used a "two-way pseudo-testcross" mapping strategy in combination with the random amplified polymorphic DNA (RAPD) assay to construct two moderate density genetic linkage maps for species of Eucalyptus. In the cross between two heterozygous individuals many single-dose RAPD markers will be heterozygous in one parent, null in the other and therefore segregate 1:1 in their F1 progeny following a testcross configuration. Meiosis and gametic segregation in each individual can be directly and efficiently analyzed using RAPD markers. We screened 305 primers of arbitrary sequence, and selected 151 to amplify a total of 558 markers. These markers were grouped at LOD 5.0, theta = 0.25, resulting in the maternal Eucalyptus grandis map having a total of 240 markers into 14 linkage groups (1552 cM) and the paternal Eucalyptus urophylla map with 251 markers in 11 linkage groups (1101 cM) (n = 11 in Eucalyptus). Framework maps ordered with a likelihood support > or = 1000:1 were assembled covering 95% of the estimated genome size in both individuals. Characterization of genome complexity of a sample of 48 mapped random amplified polymorphic DNA (RAPD) markers indicate that 53% amplify from low copy regions. These are the first reported high coverage linkage maps for any species of Eucalyptus and among the first for any hardwood tree species. We propose the combined use of RAPD markers and the pseudo-testcross configuration as a general strategy for the construction of single individual genetic linkage maps in outbred forest trees as well as in any highly heterozygous sexually reproducing living organisms. A survey of the occurrence of RAPD markers in different individuals suggests that the pseudo-testcross/RAPD mapping strategy should also be efficient at the intraspecific level and increasingly so with crosses of genetically divergent individuals. The ability to quickly construct single-tree genetic linkage maps in any forest species opens the way for a shift from the paradigm of a species index map to the heterodox proposal of constructing several maps for individual trees of a population, therefore mitigating the problem of linkage equilibrium between marker and trait loci for the application of marker assisted strategies in tree breeding.
[本文引用: 1]
[本文引用: 1]
PMID [本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}