 ,1,2,**, 周蓉,1,2,**, 陈雪1,2, 陈蕾1,2, 李加纳1,2, 王瑞,1,2,*
,1,2,**, 周蓉,1,2,**, 陈雪1,2, 陈蕾1,2, 李加纳1,2, 王瑞,1,2,*Location and InDel markers for candidate interval of the orange petal gene in Brassica napus L. by next generation sequencing
GUO Qing-Qing,1,2,**, ZHOU Rong,1,2,**, CHEN Xue1,2, CHEN Lei1,2, LI Jia-Na1,2, WANG Rui,1,2,*通讯作者: * 王瑞, E-mail:Ruiwang71@163.com
第一联系人:
收稿日期:2020-10-31接受日期:2021-01-13网络出版日期:2021-02-18
| 基金资助: |
Corresponding authors: * E-mail:Ruiwang71@163.com
First author contact:
Received:2020-10-31Accepted:2021-01-13Published online:2021-02-18
| Fund supported: |
作者简介 About authors
郭青青,E-mail:1833266719@qq.com
周蓉,E-mail:1822701415@qq.com

摘要
甘蓝型油菜花色的选育和改良已成为种质资源鉴定和材料创制的重要研究方向。目前为止, 甘蓝型油菜桔红花显性基因定位的研究还未见报道。本研究以甘蓝型油菜双单倍体(doubled haploid, DH)纯系黄花Y05和桔红花R08杂交, 分析桔红花性状遗传模式。在F2群体中选取30株极端桔红花和30株极端纯黄花构建叶片DNA子代池和花瓣RNA子代池, 对亲本和DNA子代池进行30×重测序, 对RNA子代池进行6G测序。以法国甘蓝型油菜Darmor-bzh为参考序列, QTL-seq流程和MMAPPR流程相互结合鉴定桔红花基因候选区间。利用IGV软件可视化分析候选区间内插入缺失(InDel)变异位点, 根据候选区间信息设计InDel引物。结果表明, 桔红花性状受1对显性主效基因控制, 全基因组重测序定位桔红花性状基因候选区间结果与转录组测序定位结果高度一致, 均位于法国甘蓝型油菜Darmor-bzh A07染色体18~19 Mb。聚丙烯酰胺凝胶电泳筛选到3个与桔红花性状紧密连锁共分离的InDel标记。这些研究为精细定位桔红花显性候选基因以及分子标记辅助选育甘蓝型油菜桔红花新材料提供新思路。
关键词:
Abstract
The petal color has been one of the major goals of breeding and genetic research in Brassica napus L. To date, there have been no reports about interval location of dominant orange petal gene trait in B. napus L. In this study, we constructed an F2 mapping population with 458 individuals from the cross between DH Y05 (yellow petal) and DH R08 (orange petal). Whole-genome re-sequencing of DNAs and transcriptome sequencing of RNAs were from two populations each composed of 30 individuals showing extreme opposite trait for a given phenotype in a segregating progeny. Then we performed 30× and 6G of sequencing. Darmor-bzh as the reference genome was aligned to sequence data from the two bulks and parents. QTL-seq and Mutation Mapping Analysis Pipeline for Pooled RNA-seq (MMAPPR) workflow were applied to identify the candidate region of the orange petal gene. The insertion-deletion (InDel) sites can be visualized in candidate interval by Integrative Genomics Viewer (IGV). Based on these Indel variations, we used Vector and Blast to design InDel primers. The results indicated that the orange petal trait was controlled by a dominant major gene. A major candidate region was identified on chromosome A07 (18-19 Mb) of Darmor-bzh. Three InDel markers linked to the orange petal gene were screened by Polyacrylamide gel electrophoresis (PAGE). This study may provide a novel idea for fine mapping dominant orange petal gene as well as marker assisted selection.
Keywords:
PDF (13272KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
郭青青, 周蓉, 陈雪, 陈蕾, 李加纳, 王瑞. 甘蓝型油菜桔红花显性基因候选区域的NGS定位及InDel标记开发. 作物学报, 2021, 47(11): 2163-2172 DOI:10.3724/SP.J.1006.2021.04236
GUO Qing-Qing, ZHOU Rong, CHEN Xue, CHEN Lei, LI Jia-Na, WANG Rui.
甘蓝型油菜属于十字花科(Cruciferace)芸薹属(Brassica)植物, 是食用植物油、蛋白质饲料和能源的原料作物。近几年, 油菜花观光旅游已成为发展油菜产业和促进农民增收的重要内容[1]。因此, 加快选育创制不同遗传背景的油菜花色新品种具有重要的理论和现实意义。
油菜花色是指其成熟花瓣的颜色, 栽培油菜花色主要表现为黄色、白色及其中间衍生色。对油菜白花性状的研究已取得一系列成果[2,3,4], 由于不同****所用材料、方法不尽相同, 得出的花色遗传模式结论也有多种。部分****认为, 甘蓝型油菜白花性状由单个显性基因控制[5,6], 也有不少****发现, 白花性状由1对不完全显性基因控制, 表现为质量性状[4,7-10]。而董育红等[11]、黄萌等[12]、田露申等[13,14]和柳丽[15]均发现白花性状受1对不完全显性基因控制, 但表现出数量性状的遗传特点。而白菜型油菜白花性状由单个隐性基因控制[16], 芥菜型油菜白花性状由2对隐性基因控制[17]。与白花性状相比, 其他花色的遗传研究结果相对较少。李莓等[18]、淡亚彬[19]、Yao等[20]均发现甘蓝型油菜桔红花色由2对隐性核基因控制, Yao等[20]进一步将隐性基因确定为Bnpc1和Bnpc2。李莓等[18]还证实大白菜桔红色花由1对隐性基因控制。Zhang等[21]以黄花和桔红花大白菜为材料作遗传分析发现, 大白菜桔红花性状由单隐性基因BrOF控制。
至今已有多位****进行了花色相关基因染色体定位, 并开发出多个与花色紧密连锁的分子标记。刘雪平等[22]利用RAPD标记发现白花基因与高芥酸基因具有连锁关系, 并筛选到1个与黄花和低芥酸基因均连锁的标记S92-1400。Huang等[23]以甘蓝型白花和黄花油菜为材料, 获得了与白花基因连锁的3个AFLP和2个SSR标记, 其中位于白花基因两侧的标记EA06MG08和EA11MG12遗传距离分别为3.0 cM和3.2 cM。张豹等[6]结合SSR技术和QTL分析, 将甘蓝型油菜白花基因定位到了C03染色体上。随着二代测序技术的发展以及SNP标记的开发, 集团分离分析法(bulked segregant analysis, BSA)广泛应用于目标性状的基因定位。赵君伟[16]通过集团分离分析法以及SNP和InDel标记分析2330个单株, 将花色基因定位在白菜参考基因组A02染色体162 kb区间, 且SNP标记与白花基因共分离。陈雪等[24]利用二代测序技术和构建混池的原理将甘蓝型油菜白花性状基因定位在C03染色体52~55 Mb, 并在此区间760 kb内筛选出6个与该基因紧密连锁共分离的SSR标记。
甘蓝型油菜桔色花基因定位方面文献很少, 仅有Yao等[20]将甘蓝型油菜隐性桔红花基因Bnpc1定位在C09染色体1.000~5.464 Mb区间; 将Bnpc2基因锁定在1.86 cM区间, 同时筛选出1个共显性标记BnA09-22与Bnpc2紧密连锁。目前为止, 关于甘蓝型油菜桔红花显性基因定位的研究还未见报道。本研究基于BSA的原理, 利用甘蓝型油菜F2花色分离群体, 分别进行叶片重测序和花瓣转录组测序, 定位桔红花显性基因候选区间, 同时通过IGV软件可视化候选区间InDel变异, 开发InDel连锁标记, 以期为精细定位桔红花显性基因以及分子标记转育甘蓝型油菜桔红花新材料奠定基础。
1 材料与方法
1.1 试验材料
甘蓝型油菜纯系黄花Y05和纯系桔红花R08是由西南大学李加纳油菜生物学团队通过小孢子加倍选育的双单倍体(DH)纯系材料。2018年将纯系黄花Y05与纯系桔红花R08杂交获得F1, 次年获得F2。观察F1和F2群体的花色分离。从F2群体中选取极端桔红花和极端黄花构建桔红花子代池和黄花子代池, 用于桔红花基因候选区间定位。1.2 田间试验和性状调查
2019年3月花期, F1单株自交获得F2。2019年9月对亲本和F2进行育苗播种, 10月将单株移栽到西南大学歇马油菜基地试验田, 行距为0.2 m, 株距为0.2 m。2020年1月苗期对F2群体458个单株插牌编号。2020年3月记录458个单株的盛花期花色。1.3 子代池构建
2020年1月苗期, 对亲本及F2群体每个单株插牌编号, 取幼嫩叶0.2 g。使用OMEGA HP Plant DNA试剂盒提取2个亲本以及F2代的30株极端黄花和30株极端桔红花单株幼嫩叶DNA。等量混合F2代单株幼嫩叶DNA, 构建桔红花叶片DNA子代池和黄花叶片DNA子代池。初花期时, 在F2群体中选取极端纯桔红花和极端纯黄花各30株, 取每株刚张开的花瓣0.15 g, 使用EZ-10 Total RNA Mini-Preps Kits试剂盒提取RNA。等量混合RNA, 构建桔红花花瓣RNA子代池和黄花花瓣RNA子代池。2个叶片DNA子代池和2个亲本叶片DNA建库类型为DNA-350 bp, 以illumina HiSeq PE150方法测序, 测序深度为30×。2个花瓣RNA子代池建库类型为DNA-300 bp, 以illumina HiSeq PE125方法测序, 测序数据量为6 G。1.4 数据处理
启动QTL-seq流程[25], 过滤低质量reads; 将过滤后的亲本reads与法国甘蓝型油菜参考组Darmor-bzh Brassica_napus.v4.1.fa (http://www.geno scope.cns.fr/brassicanapus/)比对并替换SNP, 构建亲本参考组, 再将亲本reads比对到新构建的亲本参考基因组上, 找出错配造成的SNP, 用于后续排除和过滤。将质控过滤后的2个DNA子代池reads分别与亲本参考基因组比对。使用Coval Refine过滤, Coval Call检测变异位点, 计算2个叶片DNA子代池SNP-index及2个子代池SNP-index的差值delta (SNP-index)。利用R包制作delta (SNP-index)滑窗分析图, 鉴定桔红花显性基因候选区间。利用Bwa软件将2个花瓣RNA子代池测序数据与法国甘蓝型油菜参考基因组Darmor-bzh比对, 得到sam文件后进行sort排序, 去除PCR重复, 建立索引文件。用GATK软件确定重新比对区域, 最大限度降低InDel附近比对错误率, 获得2个花瓣RNA子代池的bam文件(http://www.broadinstitute. org/gatk/)。启动MMAPPR分析流程[26], 计算SNP频率、欧氏距离Loess fit of ED4检测峰值和鉴定桔红花基因候选区间。
1.5 引物设计与电泳
使用GATK软件获得2个亲本叶片和2个子代池叶片bam文件, samtools截取4个bam文件候选区间, IGV软件查看2个亲本和2个子代池截取文件, 寻找桔红花基因候选区间InDel, 锁定起始和终止物理位置, 使用Vector和Blast设计InDel引物, 由上海生工生物工程技术服务有限公司合成引物序列。随机选取F2群体极端黄花和桔红花各11株幼嫩叶的DNA作为模板用于PCR扩增。PCR体系包含2.2 μL模板、0.25 μL dNTP、前后引物各0.36 μL、0.31 μL Taq酶(2.5 U μL)、1.9 μL 10×PCR buffer (含Mg2+), 最后补充ddH2O至8.90 μL。PCR程序为94℃ 5 min; 94℃ 30 s, 52~60℃ 30 s, 72℃ 30 s, 共35个循环; 72℃ 5 min; 4℃保存。8%聚丙烯酰胺凝胶电泳分离PCR扩增产物, 最后用银染法进行显影。2 结果与分析
2.1 黄花Y05和桔红花R08组合后代表型观察和遗传分离
甘蓝型油菜黄花纯系Y05与桔红花纯系R08正反交F1代均为桔红花, F2代纯桔红花和纯黄花分离明显(图1), 呈显著的质量性状或主效基因分布特点。并且桔红花的花药呈现桔红色, 与黄花的黄色花药有明显区别。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1甘蓝型油菜黄花Y05和桔红花R08花瓣形态比较
A1与A2: 黄花Y05; B1与B2: 桔红花R08。
Fig. 1Morphology comparision of yellow petal Y05 and orange petal R08 in Brassica napus L.
A1 and A2: yellow petal Y05; B1 and B2: orange petal R08.
2年田间试验数据显示, 桔红花单株和黄花单株的实际值分别为108:41和333:125, 接近3:1的分离比例, 按1对基因的控制模式进行χ2测验, χ2值分别为0.37和1.16, 符合理论预测, 即桔红花和黄花性状分离比符合3:1的分离规律(表1), 推测桔红花性状受1对显性基因或主效基因控制。
Table 1
表1
表1黄花Y05与桔红花R08杂交F2群体花色分离比例
Table 1
| 群体 Population | 总株数 Total plants | 桔红花 Orange petal | 黄花 Yellow petal | 期望比 Expected ratio | χ2 | P |
|---|---|---|---|---|---|---|
| 2019 | 149 | 108 | 41 | 3:1 | 0.37 | > 0.05 |
| 2020 | 458 | 333 | 125 | 3:1 | 1.16 | > 0.05 |
新窗口打开|下载CSV
2.2 重测序DNA子代池数据分析
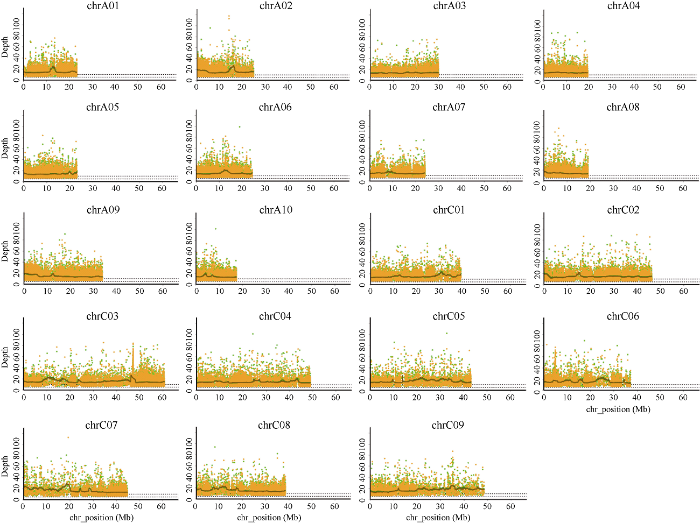
对亲本和子代池叶片重测序质控过滤后的数据进行测序深度和覆盖度分析(图2)发现, 19条染色体上测序深度平均为20×, 覆盖度和SNP检出率达到饱和。因此所有样本数据量足够, 测序质量合格, 测序数据与法国甘蓝型油菜参考组Darmor-bzh比对结果正常, 可用于变异分析和鉴定目标性状基因候选区间。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2甘蓝型油菜19条染色体上重测序平均深度和覆盖度
黑线: 利用滑动窗口计算得到的平均测序深度曲线。
Fig. 2Average depth of re-sequencing and coverage of bases on 19 chromosomes in Brassica napus L.
Black lines: refer to the average sequencing depth curve using a sliding window.
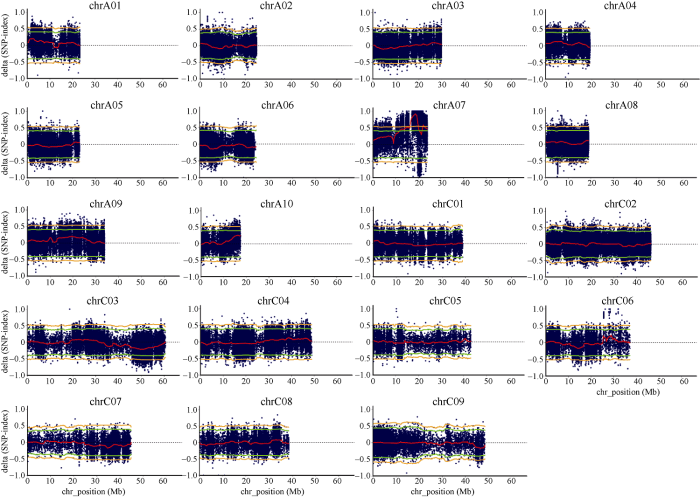
过滤掉桔红花和黄花2个子代池中SNP-index均小于0.3的位点, 分别计算每个子代池的SNP-index和2个子代池之间的delta (SNP-index)。以滑动窗口法(窗口大小为2 Mb, 滑动步长为50 kb)对delta (SNP-index)在19条染色体上作图。置信区间设置为95%和99%, 置信水平以上窗口作为候选区间(图3)。除chrA07染色体外, 其余18条染色体delta (SNP- index)红线都在置信区间范围内。Darmor-bzh A07染色体18~19 Mb区间delta (SNP-index)峰值达到极显著水平, 显示甘蓝型油菜桔红花显性基因在此候选区间之内。
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3甘蓝型油菜19条染色体上delta (SNP-index)分布
蓝点: delta (SNP-index)位点; 红线: 利用滑动窗口数计算的delta (SNP-index)的变化趋势; 绿线: 显著性为95%的阈值; 橘线: 显著性为99%的阈值。
Fig. 3Delta (SNP-index) on 19 chromosomes in Brassica napus L.
Blue dot: delta (SNP-index); Red line: sliding window average of delta (SNP-index); Green lines: sliding window average of 95%-confidence interval upper/lower side; Orange line: sliding window average of 99%-confidence interval upper/lower side.
2.3 转录组子代池数据分析
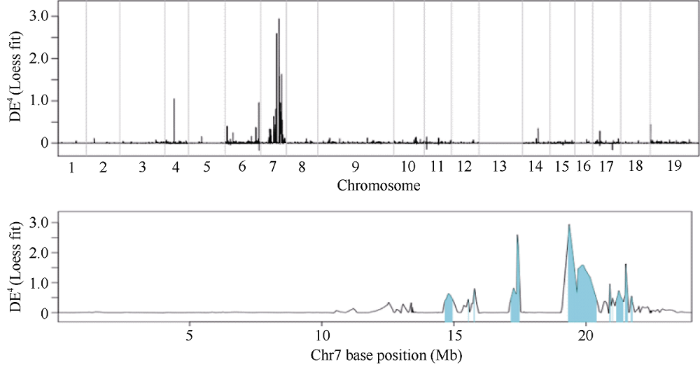
更改参考组Darmor-bzh基因序列染色体名称(chrA01~chrA10、chrC01~chrC09更改为chr1~chr19), 使用GATK软件处理黄花花瓣子代池和桔红花花瓣子代池RNA测序数据, 获得2个RNA子代池bam文件。启动MMAPPR流程, 利用2个子代池SNP频率计算ED4 (Loess fit), 以0.6为阈值, 桔红花显性基因定位在chr7(A07)染色体18~19 Mb (图4)。花瓣转录组测序定位桔红花显性基因染色体区域与叶片DNA重测序定位结果高度一致。图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4甘蓝型油菜19条染色体上ED4 (Loess fit)和A07染色体上ED4 (Loess fit)
Fig. 4ED4 (Loess fit) on 19 chromosomes and A07 chromosome in Brassica napus L.
2.4 IGV可视化桔红花基因候选区间InDel变异
基于重测序和转录组定位桔红花基因候选区间结果, 使用samtools软件view参数截取亲本和两子代池bam文件中的A07染色体18~19 Mb区间。对截取的4个bam文件排序、建立索引后导入IGV软件, 可视化查看两亲本P1 (Y05)、P2 (R08)和桔红花子代池(R)、黄花子代池(Y)中A07染色体18~19 Mb区间的InDel变异(图5), 滑动IGV显示窗口, 在此区间找到3个遗传稳定的small InDel。亲本之间的InDel差异在2个子代池中稳定表现和遗传。图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5IGV可视化候选区间InDel变异
Fig. 5Visualization of InDel variation in candidate intervals by IGV
2.5 InDel引物设计和聚丙烯酰胺电泳
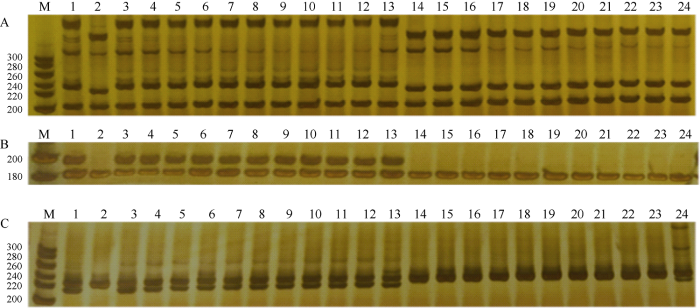
3个InDel均位于A07染色体18~19 Mb, IGV软件锁定其物理位置, 结合使用Vector和Blast设计InDel 引物(表2)。聚丙烯酰胺凝胶电泳结果表明, 3个InDel标记均能准确区分F2极端黄花和极端桔红花单株, 仅图6-C中24桔红花单株条带为交换株(图6), 推断桔红花性状候选基因与这3个small InDel标记紧密连锁共分离。Table 2
表2
表2基于候选区间设计的InDel引物序列
Table 2
| 引物名称 Primer name | InDel位点 InDel site | InDel | 正向引物序列 Positive Primer sequence (5′-3′) | 反向引物序列 Reverse primer sequence (5′-3′) |
|---|---|---|---|---|
| InDel-65 | 18,968,634-18,968,641 | D7 | GGGTCGAGATACGGTTACGG | CGTCGGTAAGTCTTCTTCACC |
| InDel-69 | 18,949,999-18,950,007 | D8 | TACCGAGCTCCAGGTCTCTC | TGCGCAACTGTAACCATTCT |
| InDel-56 | 18,789,060-18,789,066 | D6 | ACAAGCAAGGCCTATATTTTGC | TCGGGTGAAGCAAATGTGGA |
新窗口打开|下载CSV
图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6黄花和桔红花亲本及F2代单株电泳
M: 20 bp ladder; A: InDel-65; B: InDel-69; C: InDel-56; 1: 黄花亲本; 2: 桔红花亲本; 3~13: F2群体11个黄花单株; 14~24: F2群体11个桔红花单株。
Fig. 6Electrophoretic bands of parents and individuals of F2 population
1: parents with yellow petal; 2: parents with orange petal; 3-13: 11 yellow petal plants in F2 population; 14-24: 11 orange petal plants in F2 population.
3 讨论
芸薹属中甘蓝型油菜桔红花研究报道文献较少, 仅有李莓等[18]、淡亚彬[19]、Yao等[20]发现甘蓝型油菜桔红花性状由2对隐性基因控制。而本研究所用甘蓝型油菜桔红花材料性状表现为1对显性基因或主效基因遗传特点, 表型观察发现, 除花瓣颜色为桔红色外, 花药颜色同时为桔红色, 与黄花的黄色花药明显不同, 且能稳定遗传。因而, 本研究材料的桔红花基因应与前人所研究的甘蓝型油菜桔红花基因在功能和位点上有所区别。对有参考序列物种进行全基因组重测序, 在全基因组水平扫描并检测碱基突变位点, 通过计算SNP变异和频率, 可以快速进行正向遗传学性状定位。近年来基于二代测序的BSA分析技术成为快速准确定位质量性状或主效基因的有力工具。因其不需构建复杂定位群体, 不依赖遗传图谱和大量分子标记, 且周期短、效率高, 目前已在多种作物中用于定位质量性状或主效基因, 快速筛选靶基因以获得紧密连锁分子标记[27,28,29,30,31,32]。在利用QTL-seq重测序定位甘蓝型油菜桔色花文献中, Yao等[20]将甘蓝型油菜桔红花隐性基因定位于C09染色体1.00~5.46 Mb区间。丁戈等[33]将控制桔黄花隐性基因定位在C09染色体区域4.64~8.28 Mb。QTL-seq原理是基于F2群体或重组自交系(RIL)群体开发的代码流程, 但这2篇文献中作者均以后代较为复杂的回交群体作为研究对象, 会在一定程度上使SNP频率偏分离, 导致定位区间显示较大, delta (SNP-index)信号不明显。
本研究对甘蓝型油菜桔红花显性基因F2花色分离群体进行叶片DNA重测序和花瓣RNA转录组测序, 在DNA水平和RNA转录本表达量水平分别使用QTL-seq和MMAPPR鉴定桔红花显性基因候选区间。MMAPPR实际上是欧式距离算法的BSR分析, 其优势是检测到的信号均在基因区域, 根据样品表达量可以初步验证候选位点或候选基因。但RNA-seq外显子拼接成转录本, 与参考组比对检测基因组变异位点时存在gap, 导致部分区域SNP鉴定不准确。另外, RNA水平上有时会出现RNA编辑现象, 造成非DNA水平上的SNP突变。因此, MMAPPR方法检测SNP的准确性没有QTL-seq高, 并且RNA转录本测序时, 由于每条基因表达量不均一, 会对后续SNP鉴定造成干扰。同时, 同源染色体等位基因表达量的不均衡还可能会出现低估杂合位点的情况。
传统分子标记的应用多集中在RAPD、AFLP标记等, 由于其稳定性、重演性差, 现已很少用于作物标记开发。SSR串联重复序列具有一定的物种稳定性, 可以通过NGS测序批量化设计某一物种的SSR引物数据库。此类重复序列一般分布在基因间区, 开发的SSR标记也位于基因间区。不过SSR位点易发生突变, 导致后续结果分析模棱两可。而InDel作为高通量分子标记, 具有各类遗传标记的优点, 且开发成本随测序技术的发展不断降低。InDel标记的开发完全基于序列差异, 开发过程中存在较少的无多型位点。相比于其他分子标记, 基因组同一位点仅有极小概率发生相同长度大小的InDel变异, 避免了特异性和复杂性带来的后续分析模糊[34]。
以往在水稻[35]、玉米[36]、棉花[37]、大麦[38]、油菜[39]等农作物中开发InDel标记时, 均以所用材料的测序数据与参考组比对, 鉴定InDel变异, 根据插入缺失序列, 在全基因组上开发InDel引物公共数据库。全基因组上的InDel引物数量丰富, 即使在染色体某一区域选择InDel引物筛选验证标记, 工作量也很大。而本研究通过对分离群体进行重测序, 结合使用QTL-seq和MMAPPR分析流程, 以法国甘蓝型油菜Darmor-bzh序列为参考组, 叶片DNA重测序和花瓣RNA转录组测序相互验证, 快速鉴定出甘蓝型油菜桔红花显性基因位于A07染色体18~19 Mb候选区间。同时利用IGV软件对亲本、子代池bam文件实现桔红花显性基因候选区间InDel变异的可视化, 直观地展示了InDel所处染色体物理区间以及来源于亲本之间的InDel差异在两子代池中的表现和遗传情况。在可视化直接查看两亲本和两子代池染色体特定区域InDel变异的基础上设计InDel引物, 通过聚丙烯酰胺凝胶电泳在F2花色分离群体高效开发桔红花显性候选基因连锁InDel分子标记, 可为桔红花显性基因精细定位奠定研究基础, 同时为开发农作物质量性状或主效基因连锁标记提供一种快速有效的解决思路。
4 结论
甘蓝型油菜桔红花性状由1对显性基因或主效基因控制。桔红花性状显性基因候选区间定位于A07染色体18~19 Mb区间, 在此区间开发和验证了3个与桔红花显性基因紧密连锁共分离的InDel标记。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 2]
[本文引用: 2]
DOIURL [本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 3]
[本文引用: 3]
[本文引用: 2]
[本文引用: 2]
DOIURL [本文引用: 5]
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
DOIPMID [本文引用: 1]

Forward genetic screens in model organisms are vital for identifying novel genes essential for developmental or disease processes. One drawback of these screens is the labor-intensive and sometimes inconclusive process of mapping the causative mutation. To leverage high-throughput techniques to improve this mapping process, we have developed a Mutation Mapping Analysis Pipeline for Pooled RNA-seq (MMAPPR) that works without parental strain information or requiring a preexisting SNP map of the organism, and adapts to differential recombination frequencies across the genome. MMAPPR accommodates the considerable amount of noise in RNA-seq data sets, calculates allelic frequency by Euclidean distance followed by Loess regression analysis, identifies the region where the mutation lies, and generates a list of putative coding region mutations in the linked genomic segment. MMAPPR can exploit RNA-seq data sets from isolated tissues or whole organisms that are used for gene expression and transcriptome analysis in novel mutants. We tested MMAPPR on two known mutant lines in zebrafish, nkx2.5 and tbx1, and used it to map two novel ENU-induced cardiovascular mutants, with mutations found in the ctr9 and cds2 genes. MMAPPR can be directly applied to other model organisms, such as Drosophila and Caenorhabditis elegans, that are amenable to both forward genetic screens and pooled RNA-seq experiments. Thus, MMAPPR is a rapid, cost-efficient, and highly automated pipeline, available to perform mutant mapping in any organism with a well-assembled genome.
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI [本文引用: 1]
InDel是指在近缘种或同一物种不同个体之间基因组同一位点的序列发生不同大小核苷酸片段的插入或缺失(insertion-deletion), 是同源序列比对产生空位(gap)的现象。InDel在基因组中分布广泛、密度大、数目众多。InDel多态性分子标记是基于插入/缺失位点两侧的序列设计特异引物进行PCR扩增的标记, 其本质仍属于长度多态性标记, 可利用便捷的电泳平台进行分型。InDel标记准确性高、稳定性好, 避免了由于特异性和复杂性导致的后续分析模糊。此外, InDel标记能扩增混合DNA样品和高度降解的微量DNA样品, 并进行有效分型。InDel标记目前已开始应用于动植物群体遗传分析、分子辅助育种以及人类法医遗传学、医学诊断等领域。随着位于功能基因上InDel标记的开发, 结合染色体步移和基因精细定位, 可将这些标记应用于相关物种经济性状的功能基因的筛选, 有利于优良基因的进一步开发和利用。
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}