 ,1, 刘彻1, 宋皓1, 姚盼盼1, 苏沛霖1, 魏跃伟1, 杨永霞,1,*, 李青常,2,*
,1, 刘彻1, 宋皓1, 姚盼盼1, 苏沛霖1, 魏跃伟1, 杨永霞,1,*, 李青常,2,*Identification and analysis of non-specific lipid transfer protein family in tobacco
LI Peng,1, LIU Che1, SONG Hao1, YAO Pan-Pan1, SU Pei-Lin1, WEI Yao-Wei1, YANG Yong-Xia,1,*, LI Qing-Chang,2,*通讯作者: * 杨永霞, E-mail:yyx624@126.com;李青常, E-mail:ctsrc@126.com
收稿日期:2020-11-5接受日期:2021-04-26网络出版日期:2021-05-13
| 基金资助: |
Corresponding authors: * E-mail:yyx624@126.com;E-mail:ctsrc@126.com
Received:2020-11-5Accepted:2021-04-26Published online:2021-05-13
| Fund supported: |
作者简介 About authors
E-mail:lpeng1995@126.com

摘要
植物非特异性脂质转移蛋白(non-specific lipid transfer proteins, nsLTPs)可以在体外转移脂质, 调节植物的生长发育以及对环境非生物和生物胁迫作出反应等。本研究从烟草栽培品种K326 (Nicotiana tabacum L.)基因组中鉴定出74个nsLTPs基因, 对其系统发育关系、基因结构、保守基序、染色体定位、基因重复、启动子顺式作用元件、3D结构和激素与非生物胁迫处理下的表达模式进行了分析。结果表明, 根据8个半胱氨酸之间的间隔和序列相似性将其分为I、II、III、IV、V、VII、VIII和XIII八种类型, 相同类型的NtLTPs具有相似的内含子-外显子基因结构和保守基序模式, motif 2和motif 3是NtLTPs基因家族的特征基序。在进化过程中, 片段重复是NtLTPs基因家族扩展的主要原因, 干旱处理后的RNA-seq分析发现, 在进化过程中, 不同基因重复事件发生后功能分化模式存在差异。启动子分析表明, 它们含有多种光反应、激素和非生物胁迫响应顺式作用元件。进一步采用qRT-PCR分析发现, NtLTPs家族基因在烟草植株的不同组织和器官中具有不同的表达模式, 可以响应干旱、盐等非生物胁迫以及IAA、GA、SA等激素处理。研究结果为深入分析NtLTPs家族基因的功能提供了理论参考, 并为进一步的分子育种提供了理论基础。
关键词:
Abstract
Plant non-specific lipid transfer proteins (nsLTPs) can transfer lipids in vitro, regulate plant growth and development, and respond to environmental abiotic and biotic stresses. In this study, 74 nsLTPs genes were identified from the genome of Nicotiana tabacum variety K326, and we analyzed multiple characteristics of these genes, including phylogeny, gene structures, conserved motifs, protein domains, chromosome locations, cis-elements in the promoter sequences, 3D structure, and the expression patterns under different hormones and abiotic stresses. The results revealed that nsLTPs in tobacco could be divided into eight types, including type I, II, III, IV, V, VII, VIII, and XIII, according to the interval and sequence similarity between the eight cysteines. The same types of NtLTPs had similar intron-exon patterns and conserved motifs, motif 2 and motif 3 were the characteristic motifs of NtLTPs family. In the process of evolution, fragment duplication dominated the expansion of the NtLTPs family. RNA-seq analysis after drought treatment revealed that the functional differentiation patterns of repeat gene pairs were diverse during evolution period. Promoter analysis showed that they contained a variety of cis-acting elements in response to light response, hormones, and abiotic stress. Furthermore, qRT-PCR demonstrated that NtLTPs family genes had different expression patterns in different tissues and organs of tobacco plants, which could respond to abiotic stresses such as drought, salt, and hormone treatments (IAA, GA, and SA etc.). These results provide a theoretical reference for the in-depth analysis of the functions of NtLTPs family genes and molecular breeding.
Keywords:
PDF (3490KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
李鹏, 刘彻, 宋皓, 姚盼盼, 苏沛霖, 魏跃伟, 杨永霞, 李青常. 烟草非特异性脂质转移蛋白基因家族的鉴定与分析. 作物学报, 2021, 47(11): 2184-2198 DOI:10.3724/SP.J.1006.2021.04240
LI Peng, LIU Che, SONG Hao, YAO Pan-Pan, SU Pei-Lin, WEI Yao-Wei, YANG Yong-Xia, LI Qing-Chang.
非特异性脂质转移蛋白(nsLTPs)是一类小的、碱性蛋白质, 分子量约为6.5 kD至10.5 kD, 在高等植物中含量丰富, 占总可溶性蛋白的4%左右[1]。NsLTPs家族的特征是它们都有一个包含8个半胱氨酸的基序(Eight Cysteine Motif, ECM), 其一般形式为C-Xn-C-Xn-CC-Xn-CXC-Xn-C-Xn-C (“X”代表任何氨基酸, “n”表示氨基酸残基数), 三级结构可以形成带有4个或5个α-螺旋的疏水腔, 在体外可以结合或转移脂肪酸、脂肪酰基-CoA、磷脂、糖脂和角质单体等疏水分子[2,3]。
NsLTPs是由多个基因组成的复杂家族, 自发现至今, 其分类方法不断完善。目前nsLTPs的分类主要有3种, 一是根据nsLTPs的分子量大小, 将nsLTPs分为2种类型: nsLTPI (分子量大约为9 kD)和nsLTP II (分子量大约为7 kD)[4]。二是Boutrot等[5]在对水稻、小麦和拟南芥的全基因组分析中, 根据序列相似性以及8个半胱氨酸之间的氨基酸残基数量将nsLTPs分为9种类型(I、II、III、IV、V、VI、VII、VIII、IX型)。基于这个分类方法, 而后在其他物种中略有修改, Liu等[6]将135个茄科nsLTPs基因分为5种类型(I、II、IV、IX和X), 额外增加一个X型, I型被划分为了Ia、Ib、Ic、Id 4个亚型; D’Agostino等[7]将番茄中64个nsLTPs基因分为6个类型(I、II、III、IV、X和XI型), 额外增加了一个XI型。Li等[8]将马铃薯中83个nsLTPs基因分为I、II、IV、V、VII、VIII、XII和XIII 8种类型, 增加XII和XIII 2种类型。三是Edstam等[9]在对苔藓、蕨类等隐花植物的nsLTPs分类时, 根据nsLTPs内含子的位置、编码蛋白中潜在的糖基磷脂酰肌醇修饰位点和半胱氨酸之间的间隔, 将nsLTPs划分为10种类型(I、II、C、D、E、F、G、H、J和K)。与此类似的分类方法也被应用于许多物种中, 在对330个小麦、63个玉米和70个大麦nsLTPs基因的分类中, 根据此方法划分为I、II、C、D、G 5种类型[10,11,12]。
NsLTPs涉及参与植物许多生理生化过程, 具有多种生物学功能[13], 包括植物的有性生殖、信号传导、植物角质层蜡质的生物合成、花粉和纤维的伸长、细胞凋亡和果实成熟等过程[14,15,16,17,18,19,20]。NsLTPs参与生物与非生物胁迫应激反应。nsLTPs可以作为一种天然的抗菌肽物质, 参与SAR信号转导过程, 与植物系统获得性抗性有关, 并在细胞程序性死亡、植物抗逆和抗病过程中起着重要作用[21]。研究发现, 拟南芥AtLTP3参与对干旱胁迫的响应[22]。过表达拟南芥AtLTP4.4基因可使谷胱甘肽水平升高, 增强抗氧化防御能力, 提高拟南芥对单端孢菌毒素的抗性[23]。水稻中过量表达LTP1和LTP2基因可提高对真菌和细菌病害的抗性[24]。黄瓜CsLTP_2基因在对根结线虫感染的防御中起重要作用[25]。过表达小麦TaLTP5基因可以提高小麦对禾旋孢腔菌和禾谷镰孢菌的抵抗能力[26]。除此之外, nsLTPs还是植物性食物和花粉中的过敏原, 引起过敏反应[27,28]。
虽然在一些物种中对nsLTPs基因家族已有相关报道, 但烟草作为一种重要的模式和经济作物, 还没有相关报道, 仅对个别基因家族成员的功能进行研究。研究发现, 烟草NtLTP1在叶片表面长柄腺毛中大量表达, 过表达NtLTP1可使叶片脂质分泌增多, 显著提高对蚜虫的抗性[29]。过表达NtLTP4基因可以调控2个Na+转运体NHX1和HKT1, 改变烟草体内Na+的稳态, 进而减弱Na+对细胞的毒害作用, 增强烟草对盐胁迫的耐受性[30]。鉴于此, 本研究拟从烟草中鉴定nsLTPs基因家族, 并对其进行详细的生物信息学分析, 研究其在不同胁迫下的表达模式, 为进一步深入研究NtLTPs的功能提供理论基础。
1 材料与方法
1.1 烟草nsLTPs家族成员的搜索和鉴定
从PFAM数据库中下载nsLTPs蛋白保守的结构域PF00234和PF14368, 并以此为查询序列, 采用HMMER软件, 以阈值E<1e -5作为默认参数, 在茄科基因组网站(1.2 烟草nsLTPs的序列分析与系统发育树构建
使用在线工具SignaIP 5.0 (1.3 烟草nsLTPs的基因结构和motif分析
使用在线网站GSDS 2.0 (1.4 烟草nsLTPs的染色体定位与共线性分析
在线网站MapGene2 Chrome V2 (1.5 烟草nsLTPs蛋白的3D结构分析
使用SWISS-MODEL在线网站预测nsLTPs蛋白的3D结构, 然后用PyMoL软件进行可视化。1.6 烟草nsLTPs顺式作用元件分析
使用Plantcare (1.7 干旱处理下的RNA-seq分析
从NCBI的SRA数据库中下载六周左右的烟草品种K326幼苗在干旱处理下的RNA-seq数据(https://www.ncbi.nlm.nih.gov/bioproject/PRJNA691642), 并使用TBtools软件的FastQC和Trimmmonatic功能对测序数据进行质量控制, 去除接头污染和低质量碱基, 然后将其定位到烟草品种K326的参考基因组上, 最终获得过滤后的烟草品种K326在干旱处理下nsLTPs基因的表达量用于后续分析。基因表达量的计算使用FKPM法(每百万fragments中来自某一蛋白编码基因每千碱基长度的fragments数目), 使用TBtools软件的Different Gene Expression Analysis (DESeq2)插件作差异基因表达分析, 将FDR (adjusted P-value)小于0.05且表达差异大于2的基因视为差异表达基因, 最后使用TBtool软件的HeatMap功能对nsLTPs基因的FPKM值进行log2标准化后绘制差异基因表达量热图。1.8 植物材料与处理
烟草品种K326采用漂浮育苗, 并在恒定的生长条件(28℃, 16 h光照, 8 h黑暗, 600~800 μmol光子m-2 s-1, 相对湿度为60~70%, CO2浓度为400~450 μmol mol-1)下进行培养, 直到生长至4片真叶(生根期)时进行处理, 分别采用100 µmol L-1的脱落酸(abscisic acid, ABA)、茉莉酸甲酯(methyl jasmonate, MeJA)、赤霉素(gibberellins, GA)、生长素(auxin, IAA)和2 mmol L-1的水杨酸(salicylic acid, SA)喷洒幼苗, 将幼苗放放置于边长为40 cm的方形托盘中, 每种激素喷施60 mL。另外, 于37℃下处理幼苗48 h模拟高温逆境。干旱和盐胁迫处理是将幼苗分别在浓度为200 mmol L-1甘露醇和200 mmol L-1 NaCl的溶液中培养48 h。以上处理, 分别在0、1、3、6、9、12、24和48 h采集从上往下数第2片叶的叶片组织样本, 每个时刻3个重复。组织表达特异性分析是在开花期收集烟草植株不同组织和器官(根、茎、上部叶、中部叶、下部叶和花), 所有采集的样品均在液氮中迅速冷冻, -80℃保存, 用于分析NtLTPs基因的表达模式。每个样品进行3个生物学重复和3个技术重复。1.9 实时荧光定量PCR
使用植物总RNA分离试剂盒提取每个样品的总RNA。HiScript II Reverse Transcriptase (NO.R201- 01/02)试剂盒(南京诺唯赞生物科技股份有限公司)进行反转录。利用Primer 5.0软件设计实时荧光定量(Quantitative Real-time, qRT) PCR引物, 以L25作为内参基因(表1)。qRT-PCR反应体系包含1 μL cDNA、5 μL AceQ Universal SYBR qPCR Master Mix (Vazyme, 中国)、正/反向引物各0.5 μL和3 μL ddH2O。在实时荧光定量PCR仪(Eppendorf, 德国)上进行PCR反应, 程序为95℃预变性10 min; 95℃变性10 s, 60℃退火30 s, 35个循环。采用2-ΔΔCT算法计算基因的相对表达量。Table 1
表1
表1引物列表
Table 1
| 引物名称Primer name | 引物序列Primer sequence (5′-3′) |
|---|---|
| L25-F | CCCCTCACCACAGAGTCTGC |
| L25-R | AAGGGTGTTGTTGTCCTCAATCTT |
| Nitab4.5_0000125g0010-F | CGGATCGCCGGAGTGTTTGC |
| Nitab4.5_0000125g0010-R | GCCACATTTGCCAGGGAGGG |
| Nitab4.5_0004362g0040-F | AAGCCGATTTGCGTTGTATG |
| Nitab4.5_0004362g0040-R | CTTAGGCAGTTTCATAGCAG |
新窗口打开|下载CSV
2 结果与分析
2.1 烟草nsLTPs基因家族成员的鉴定
从烟草品种K326基因组中搜索得到122条NtLTPs序列。使用NCBI的Blastp和CD-Search功能对NtLTPs进行分析, 删除了47条不包含完整ECM基序、含有其他结构域及注释不完整的序列和1条富含脯氨酸的序列, 剩余74条NtLTPs序列用于以下分析。2.2 烟草nsLTPs基因家族成员的序列分析与分类
根据Boutrot等[5]的分类方法, 基于ECM的序列相似性以及8个半胱氨酸之间的间隔, 将烟草品种K326的nsLTPs序列划分为8种类型: I、II、III、IV、V、VII、VIII、XIII, 没有VI和IX型, 并依据同为茄科的马铃薯中的分类方法, 新增了一个XIII型[23]。对每种类型NtLTPs的8个半胱氨酸之间的间隔进行统计(表2)。I型中的NtLTPs数量最多, 有38条, 其次为IV型, 有10条, III型和V型的数量最少, 各有1条。VII型和VIII的主要区别在于第1个半胱氨酸和第2个半胱氨酸之间的数量, 而XIII型的不同之处在于第6个和第7个半胱氨酸之间的氨基酸残基数量。Table 2
表2
表2烟草nsLTPs八个半胱氨酸基序(ECM)的多样性
Table 2
| 类型 Type | 数量 Number of members | ECM | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I | 38 | C | X9,24 | C | X12-16 | CC | X19,20,22 | CXC | X21-23 | C | X10-15,25 | C |
| II | 6 | C | X7 | C | X13 | CC | X8 | CXC | X23 | C | X6,10 | C |
| III | 1 | C | X9 | C | X14 | CC | X9 | CXC | X12 | C | X6 | C |
| IV | 10 | C | X9,10 | C | X14-16 | CC | X9,12 | CXC | X24 | C | X7-10 | C |
| V | 1 | C | X14 | C | X14 | CC | X11 | CXC | X24 | C | X10 | C |
| VII | 5 | C | X9 | C | X14,16 | CC | X12 | CXC | X25,27 | C | X9 | C |
| VIII | 7 | C | X6 | C | X12,14 | CC | X12 | CXC | X25,27 | C | X8 | C |
| XIII | 6 | C | X9 | C | X14 | CC | X12 | CXC | X26,30 | C | X8 | C |
新窗口打开|下载CSV
信号肽预测结果显示, 有67条NtLTPs序列含有N-末端信号肽, 占总序列的90.5%, 信号肽长度约为16~31个氨基酸。通过计算NtLTPs全长蛋白的分子量和等电点发现, 烟草nsLTPs蛋白的分子量相对较小, 大部分为10~15 kD, 而大多数nsLTPs蛋白的等电点在8~10间(图1)。
图1
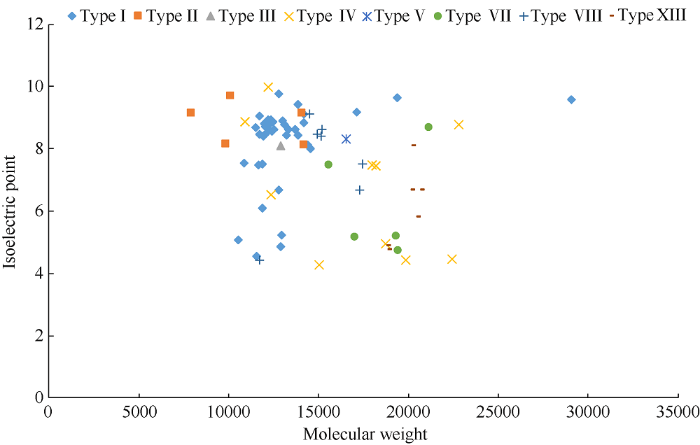 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1烟草nsLTPs蛋白的分子量和等电点的分布
Fig. 1Distributions of molecular weights and isoelectric points of tobacco nsLTPs
2.3 烟草nsLTPs的系统发育分析
使用MEGA 6.0软件, 采用邻接法构建了烟草nsLTPs的系统发育树(图2)。除IV型外, 其余7个类型的烟草nsLTPs分别聚在一起。图2
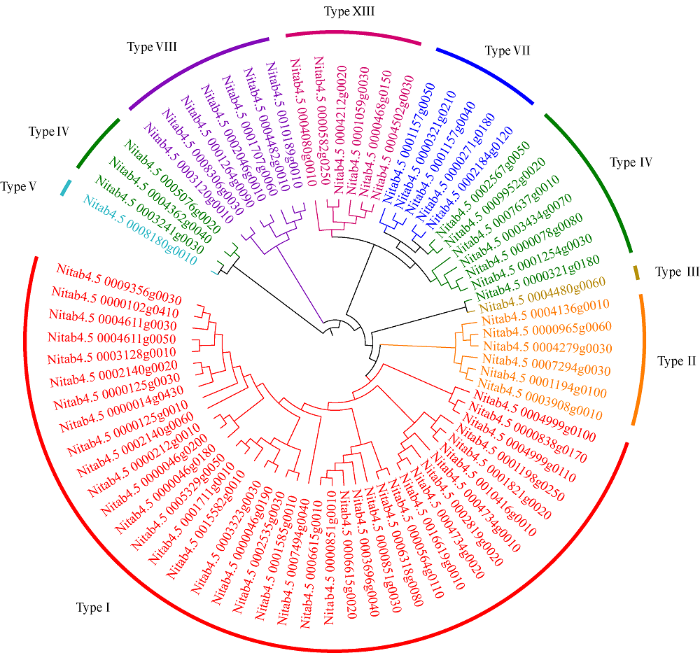 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2烟草中nsLTPs家族的系统发育关系
Fig. 2Phylogenetic relationship of nsLTPs family in tobacco
2.4 烟草nsLTPs的基因结构和motif分析
通过将烟草nsLTPs的基因组DNA与CDS进行比较, 分析了烟草nsLTPs的内含子-外显子结构(图3)。I型nsLTPs的大部分成员含有1~3个外显子, 其中14条基因没有内含子, 17条基因含有2个外显子和1个内含子, 6条基因含有3个外显子和2个内含子, 而Nitab4.5_0000102g0410含有5个外显子和4个内含子。II型中, 有3条基因没有内含子, 2条基因含有1个内含子, 1条基因含有2个内含子。IV型中, 有3条基因含有4个外显子, 3条基因含有3个外显子, 3条基因含有2个外显子, 1条基因不含内含子。VII型中大多数含有3个外显子, VIII型大多数含有2个外显子, 在XIII型中, 有3条基因含有3个外显子, 3条含有2个外显子的基因。图3
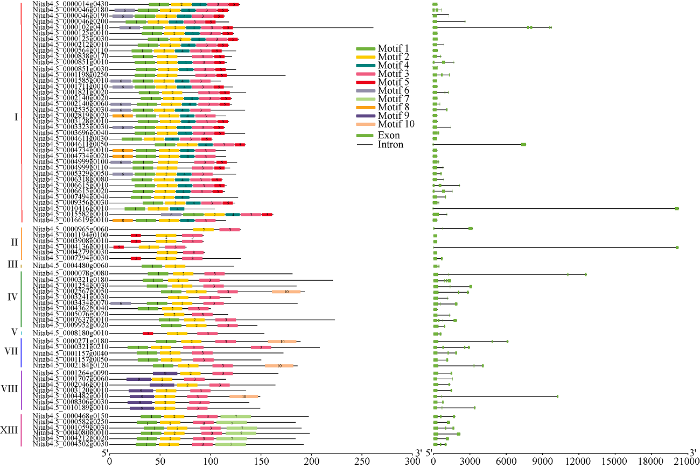 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3烟草nsLTPs蛋白的保守基序(中间部分)和nsLTPs基因的基因结构(右侧部分)
Fig. 3Conserved motifs of tobacco nsLTPs proteins (middle part) and gene structures of nsLTPs genes (right part) in tobacco
每种类型的NtLTPs都由相似的保守基序组成, motif 2和motif 3存在于所有类型的NtLTPs中。另外, 有些保守基序仅存在于少数类型中, 例如, motif 4仅在I型存在, motif 9仅在VIII型存在, motif 7仅在XIII型存在。
同时, 对每种类型的ECM基序进行seqlogo分析(图4)发现, 每种类型的nsLTPs蛋白都具有保守的ECM基序, 即保守的8个半胱氨酸残基。
图4
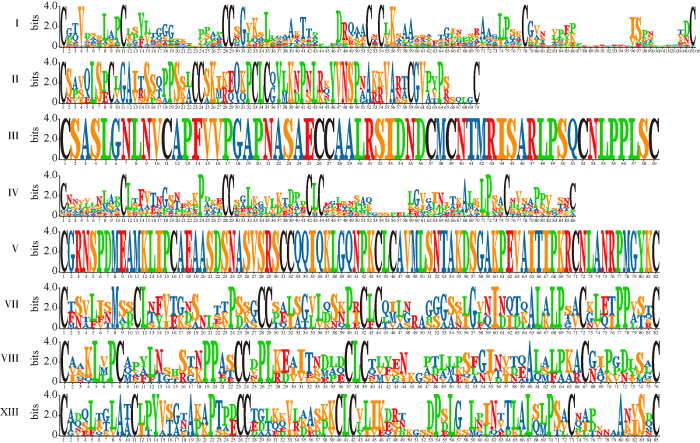 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4烟草nsLTPs的保守半胱氨酸结构域
Fig. 4Conserved cysteine domain of nsLTPs in tobacco
2.5 烟草nsLTPs的染色体定位与基因重复性分析
从烟草基因组中下载了烟草品种K326的染色体长度、NtLTPs基因在染色体上的位置等注释信息, 74条NtLTPs基因序列中有42条序列定位在染色体上, 以此, 绘制了42条基因在染色体上的分布图(图5)。结果发现, 17号染色体上的NtLTPs数量最多, 有7个; 2号和23号染色体上分别有5个。在3、5、6、12、15、18、21和24号染色体上, 分别只有1个基因, 其余6个染色体上不含NtLTPs家族基因。图5
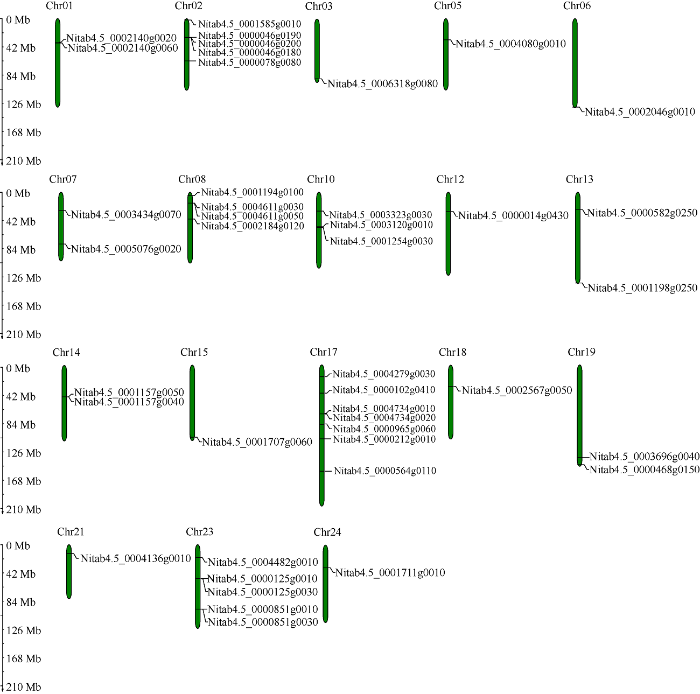 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5烟草nsLTPs基因在染色体上的分布
Fig. 5Distribution of nsLTPs genes on the chromosomes in tobacco
对42条可以定位到染色体上的NtLTPs序列进行了序列一致性分析, 将序列同一性≥70%视为1个基因对。共找到了7个基因对, 其中有6个基因对属于I型, 另外1对属于Ⅳ型。基因重复事件分为串联重复和片段重复, 基因重复事件导致了物种进化过程中基因家族的扩增[33]。按同一条染色体上的100 kb染色体片段中, 2个基因之间的间隔少于5个基因的标准, 判断是否为串联重复基因[34]。结果发现, 7个基因对中有1个串联重复基因对, 6个片段重复基因对, 对其进行了可视化分析(图6), 串联重复发生在17号染色体上。本研究中, 串联重复和片段重复事件均促进了烟草中nsLTPs家族的扩展, 而片段重复对于扩大烟草nsLTPs基因家族更为重要。
图6
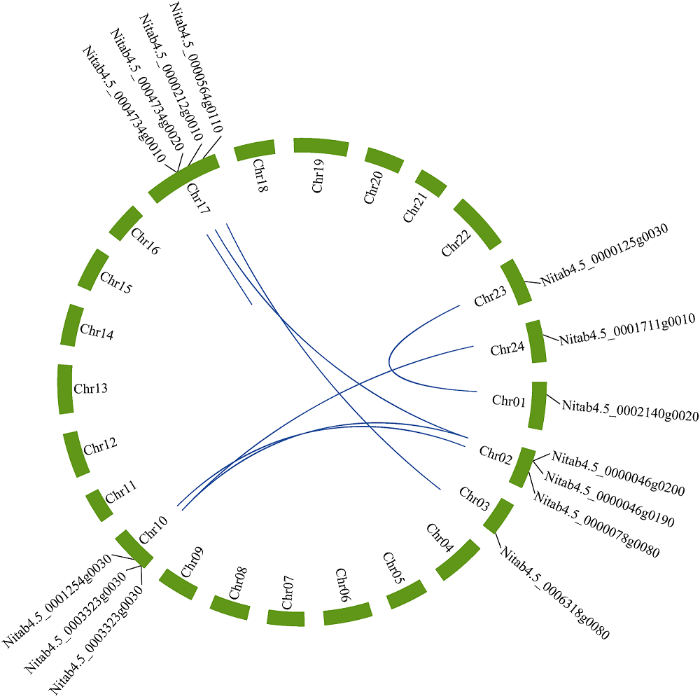 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6烟草中nsLTPs基因的基因重复
Fig. 6Gene duplications of nsLTPs genes in tobacco
此外, 计算了重复基因对之间的Ka/Ks值以评估烟草nsLTPs的分子进化速率, Ka/Ks的比值可用作选择压力的标志(图7)。Ka/Ks的比值大于1表示正选择, 小于1表示纯化选择, 等于1则表示中性选择[35]。结果发现, Ka值在0.01到0.20之间, Ka/Ks值均小于1, 这表明来自烟草品种K326的所有重复nsLTPs基因都是由重复事件后的纯化选择压力驱动的。
图7
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7重复的NtLTPs基因之间的Ka和Ka/Ks值的频率分布
Fig. 7Frequency distribution of Ka and Ka/Ks values of duplicated NtLTPs gene pairs
2.6 烟草nsLTPs的3D结构预测
选取8种类型中的9条nsLTPs蛋白序列为代表预测了其蛋白3D结构, 其中, I型NtLTPs的数量较多, 选择了2条。由图8可知, 烟草nsLTPs蛋白的3D结构具有nsLTPs蛋白的典型特征, 即4个α-螺旋以及在内部形成的一个疏水空腔。图8
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8烟草中9个nsLTPs蛋白的3D结构
Fig. 83D-structure of nine nsLTPs proteins in tobacco
2.7 烟草nsLTPs的顺式作用元件分析
启动子中的顺式作用元件可以作为转录因子的结合位点, 并参与基因表达的调控。顺式作用元件分布的规律可能揭示了功能和调控水平的差异。使用PlantCARE在线网站预测分析了74个烟草nsLTPs基因的转录起始位点2000 bp的上游区域。由图9可知, 一共筛选了21种顺式作用元件。这些元件可以分为3种类型。第1类与植物激素有关, 包括ABRE、AuxRR-core、CGTCA-motif、GARE-motif、P-box、TCA-element、TGACG-motif和TGA-element。第2种类型与非生物胁迫有关, 包括TC-rich repeats、TATC-box、ARE、GC-motif、LTR、MBS、X3.AF1. binding site和MBSI。第3类是光响应有关的元件, 包括ACE、G-box、MRE、Sp1和GT1-motif。不同烟草nsLTPs的启动子中相关元件的分布规律不同, 表明它们可能执行不同的功能。其中, 与光响应有关的元件如G-box元件、抗氧化反应元件ARE和ABA响应元件ABRE, 在大多数NtLTPs家族基因启动子中存在, 表明它们可能在调节NtLTPs的功能方面起到重要作用。图9
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图9烟草nsLTPs启动子的顺式作用元件分析
Fig. 9Cis-acting elements of nsLTPs promoter in tobacco
2.8 干旱处理下的转录组差异基因表达分析
对于从NCBI SRA数据库获得的烟草品种K326苗期干旱处理的转录组数据, 从中筛选nsLTPs基因家族数据, 使用FPKM算法计算了nsLTPs基因的表达量, 以FDR小于0.05且表达差异大于2为差异标准, 有30个nsLTPs基因在干旱处理后显现显著性基因表达差异, log2标准化后绘制差异基因表达量热图(图10), 结果发现, 在干旱处理下, 14个nsLTPs基因表达量显著性上调, 包含9个I型, 2个II型, IV、VIII和XIII型各1个; 16个nsLTPs基因表达量显著性下调, 包含4个I型, 7个IV型, 2个VII型、1个VIII型和2个XIII型; 表明不同的nsLTPs家族基因在干旱处理下显示出不同的表达模式, 虽然表达模式和类型之间没有特定的对应关系, 但I型和IV型在干旱处理后显著性上调和下调的基因数目最多。图10
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图10干旱处理下烟草品种K326的差异基因热图
使用每个样本的FPKM数值log2标准化后绘制差异基因表达量热图。CK表示未进行干旱处理, D表示干旱处理, 均包含3个重复。红色和蓝色分别表示表达量高和低。
Fig. 10Relative expression profile of variety K326 under drought treatment in tobacco
The relative expression levels of nsLTPs are measured by FPKM value in RNA-seq data and the relative expression heatmap are drew after FPKM normalized by log2. CK represents no drought treatment, and D represents drought treatment. Red and white color indicate high and low expression levels, respectively.
2.9 烟草nsLTPs基因的表达模式分析
使用qRT-PCR分析了Nitab4.5_0000125g0010 (I型)和Nitab4.5_0004362g0040 (IV型)基因在根、茎、下部叶、中部叶、上部叶和花中的表达模式。由图11可知, I型Nitab4.5_0000125g0010基因在烟草品种K326中部叶表达水平最高, 而IV型Nitab4.5_0004362g0040基因在茎中表达水平最高, 表明NtLTPs基因的表达具有组织特异性, 不同基因组织间的表达模式也存在差异。图11
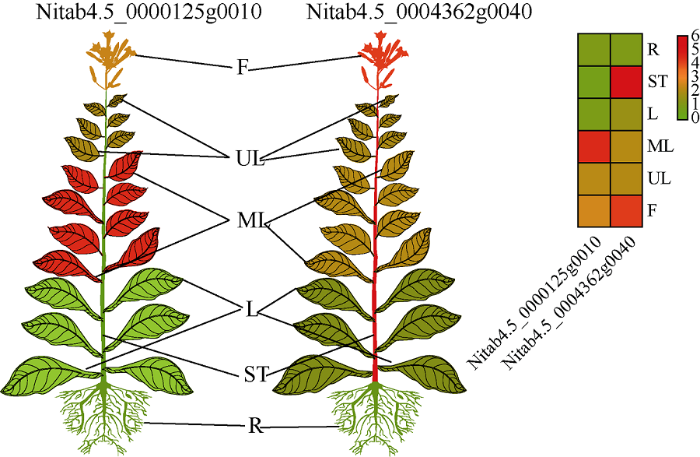 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图11Nitab4.5_0000125g0010 (I型)和Nitab4.5_0004362g0040基因(IV型)在烟草不同组织中的表达水平
以根中的相对表达量作为对照, 红色和绿色表示相对表达量高和低。R: 根; ST: 茎; L: 下部叶; ML: 中部叶; UL: 上部叶; F: 花。
Fig. 11Relative expression levels of Nitab4.5_0000125g0010 (type I) and Nitab4.5_0004362g0040 (type IV) in different tissues of tobacco
The relative expression levels in the roots were used as controls, with high and low relative expression levels in red and green, respectively. R: root; ST: stem; L: lugs; ML: middle leaf; UL: upper leaf; F: flower.
使用qRT-PCR分析了非生物胁迫(干旱、盐和高温)和激素胁迫(ABA、SA、MeJA、IAA和GA)处理下Nitab4.5_0000125g0010 (I型)和Nitab4.5_ 0004362g0040 (IV型)的表达模式(图12)。结果表明, NtLTPs家族基因可以响应多种激素和非生物胁迫, 不同类型的基因在相同的胁迫下表现出相似或不同的表达模式, 例如, 在37℃、NaCl和IAA处理下, 2个基因的表达模式相似; 而在甘露醇(Mannitol)和ABA处理后, Nitab4.5_0000125g0010基因的表达在6 h或9 h达到高峰, 随后降低, Nitab4.5_0004362g0040基因的表达只是略微升高, 随后下降。
图12
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图12Nitab4.5_0000125g0010 (黑色)和Nitab4.5_ 0004362g0040基因(灰色)在不同处理下的表达模式
用2-Δ∆CT方法计算相对基因表达量。将0 h的相对表达量设置为1。误差线表示3个生物学重复的标准偏差。37℃: 高温处理; NaCl: 盐处理; Mannitol: 甘露醇处理; GA: 赤霉素处理; IAA: 生长素处理; MeJA: 茉莉酸甲酯处理; ABA: 脱落酸处理; SA: 水杨酸处理。*表示在0.05水平上差异显著。
Fig. 12Relative expression patterns of Nitab4.5_0000125g0010 (black) and Nitab4.5_0004362g0040 (gray) under different treatments
The relative gene expression was calculated by 2-ΔΔCT method. The relative expression of 0 h was set to 1. Error bars represent the standard deviations of three biological replicates. 37°C: high temperature treatment; NaCl: salt treatment; Mannitol: mannitol treatment; GA: gibberellin treatment; IAA: auxin treatment; MeJA: methyl jasmonate treatment; ABA: abscisic acid treatment; SA: salicylic acid treatment. *: P < 0.05.
3 讨论
NsLTPs在植物界中广泛存在, 8个Cys通过4个二硫键连接形成4个α-螺旋并在内部形成一个疏水腔[36], 是结合和转移脂质的植物蛋白中功能最重要的一类。本研究鉴定的nsLTPs序列中, 我们删除了含有不完整的ECM基序的序列。以往的研究中, 由Boutrot等[5]和Edstam等[9]鉴定的nsLTPs序列包含不完整的ECM基序, Wang等[25]在黄瓜中鉴定出的39个nsLTPs都只含有5个Cys, Cys1、Cys2和Cys8缺失, 这与典型的nsLTPs不同。Wei等[11]排除了包含不完整ECM基序的序列, 认为包含不完整的8CM结构不能被认为是nsLTPs。据报道, 保守的8个半胱氨酸基序中2个二硫键丢失可能导致水稻在长日和低温条件下严重矮化[37]。目前, 还没有确定用于划分nsLTPs的统一标准, 不同的筛选方法可能导致结果的不同, 而且, Cys缺失的序列是否与包含完整8CM结构的nsLTPs序列具有相似的功能, 是否会发生功能的改变, 还需要进一步的研究。在本研究中, 我们从烟草品种K326基因组中鉴定出74个nsLTPs基因, 基于Boutrot等[5]的分类方法, 我们无法对所有的烟草nsLTPs进行分类。因此, 在Boutrot等分类方法的基础上, 结合系统发育关系, 基因结构以及保守motif的比较, 最终将其划分为8种类型(I、II、III、IV、V、VII、VIII和XIII), 没有VI型和IX型。在Boutrot等的分类方法中, 所有的VI型在Cys7之前的第4位为Val, Cys7之前的第10位为Met, 并以此来和IV型区分, 但是在我们鉴定的烟草nsLTPs中没有发现满足上述条件的序列。此外, 在I型中CC和CXC之间氨基酸残基的数量也有不同, 在本研究的分类中除19之外还有20和22, 这种现象也出现在棉花中(除19之外还含有20)[38]。XIII型为一个新型, 首次在同为茄科的马铃薯中出现, 我们的序列中有7条序列符合此分类特征, 在它们第6个和第7个Cys之间有26或30个氨基酸残基, 以此, 本研究把它们归为XIII型[8]。
内含子-外显子结构模式携带有基因家族进化的烙印[34]。植物在进化过程中倾向于保留较少内含子或短内含子的基因[40]。本研究中, 大多数NtLTPs基因含有1~3个内含子或没有内含子, 这与先前的研究结果一致[7,41]。物种在进化过程中, 已知大多数被子植物会经历整个基因组的一次或多次重复。水稻、小麦和拟南芥的多种nsLTPs亚型的系统进化分析表明, 基因和染色体片段的重复目前仍在继续。进化中, 重复的nsLTPs基因突变可能导致基因假基因化, 亚功能化(保留祖先基因的某些功能)或新功能化(即该基因获得全新功能), 从而会出现具有不同生物活性及结构和功能上与nsLTPs已有类型成员明显不同的蛋白[42]。本研究中, 片段重复对烟草NtLTPs基因家族的扩张更为重要。
干旱处理后的RNA-seq数据分析显示, 30个nsLTPs基因在干旱处理后显著上调或者下调表达, 说明nsLTPs家族在干旱调控方面具有重要作用。其中I型和Ⅳ型在干旱处理后显著性上调和下调的基因数目最多, 分别为9个和4个。对NtLTPs家族的基因重复分析结果也显示该家族的重复事件主要发生在I型和Ⅳ型间。但不同的重复基因对在干旱处理后呈现不同的表达模式, 比如重复基因对中的Nitab4.5_0003323g0030 (I型)干旱处理后表达量显著下调, 但Nitab4.5_0000046g0190 (I型)却检测不到表达量; 重复基因对Nitab4.5_ 0002140g0020 (I型)和Nitab4.5_0000125g0030 (I型)以及重复基因对Nitab4.5_0003323g0030 (I型)和Nitab4.5_0001711g0010 (I型)在干旱处理后则分别都呈现下调和上调的完全相反的趋势; 重复基因对Nitab4.5_0000212g0010 (I型)和Nitab4.5_ 0000046g0200 (I型)在干旱处理后均上调, 重复基因对Nitab4.5_0001254g0030 (IV型)和Nitab4.5_ 0000078g0080 (IV型)在干旱处理后均下调, 这表明不同基因在进化过程中重复事件发生后功能分化模式存在差异。
基因的表达受到启动子中顺式作用元件的调控, 启动子区域中顺式作用元件的数量和类型对于各种胁迫下的基因表达非常重要。NtLTPs基因家族的启动子中含有多个响应光照、高温、低温、干旱以及激素信号如脱落酸、茉莉酸甲酯、生长素、赤霉素等的顺式作用元件, 但不同基因含有的数量及类型存在差异, 说明不同类型的NtLTPs家族基因功能上可能存在分化。采用qRT-PCR分析NtLTPs在非生物胁迫和激素胁迫下的表达模式发现, NtLTPs家族基因可以响应多种激素和非生物胁迫, 不同类型的基因在不同胁迫下具有相似或者不同的表达模式, 暗示了功能上可能存在差异。其中, Nitab4.5_ 0000125g0010和Nitab4.5_0004362g0040基因在干旱处理下的RNA-seq结果都显著下调, 与甘露醇处理之后2个基因的表达模式基本一致(图11), 虽然在甘露醇处理之后有短暂的上升, 但随后呈现出下降的趋势。总之, 本研究对NtLTPs基因家族的分析对于将来进一步针对特定的基因研究其具体的功能提供了一定的依据。
4 结论
本研究首次从烟草品种K326中鉴定出74个nsLTPs家族基因, 根据8个半胱氨酸之间的间隔和序列相似性将其划分为8种类型: I、II、III、IV、V、VII、VIII和XIII型, 并对其进行了系统发育分析、内含子/外显子基因结构、保守motif和染色体定位分析, 相同类型的nsLTPs基因的内含子/外显子结构、保守motif和3D结构相似。基因重复和进化分析显示, 片段重复在nsLTPs家族的扩张中更为重要。对干旱处理下的RNA-seq数据进行分析表明, 不同的nsLTPs家族基因在干旱处理下具有不同的表达模式, I型和Ⅳ型基因显著性上调和下调的基因数目最多, 重复基因对的功能分化模式多样。NsLTPs基因启动子含有多种与光响应、激素和非生物胁迫相关的顺式作用元件, 采用qRT-PCR验证了不同激素(ABA、SA、MeJA、IAA和GA)和非生物胁迫(干旱、盐和高温)处理下Nitab4.5_ 0000125g0010和Nitab4.5_0004362g0040基因的表达模式, 结果表明, 他们对不同激素和非生物胁迫均有响应, 但在相同的胁迫下表现出相似或不同的表达模式。本研究对NtLTPs家族基因进行了较为全面的系统性分析, 研究结果为深入分析NtLTPs家族基因的功能提供了理论参考。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
DOIPMID [本文引用: 4]

Plant non-specific lipid transfer proteins (nsLTPs) are encoded by multigene families and possess physiological functions that remain unclear. Our objective was to characterize the complete nsLtp gene family in rice and arabidopsis and to perform wheat EST database mining for nsLtp gene discovery.In this study, we carried out a genome-wide analysis of nsLtp gene families in Oryza sativa and Arabidopsis thaliana and identified 52 rice nsLtp genes and 49 arabidopsis nsLtp genes. Here we present a complete overview of the genes and deduced protein features. Tandem duplication repeats, which represent 26 out of the 52 rice nsLtp genes and 18 out of the 49 arabidopsis nsLtp genes identified, support the complexity of the nsLtp gene families in these species. Phylogenetic analysis revealed that rice and arabidopsis nsLTPs are clustered in nine different clades. In addition, we performed comparative analysis of rice nsLtp genes and wheat (Triticum aestivum) EST sequences indexed in the UniGene database. We identified 156 putative wheat nsLtp genes, among which 91 were found in the 'Chinese Spring' cultivar. The 122 wheat non-redundant nsLTPs were organized in eight types and 33 subfamilies. Based on the observation that seven of these clades were present in arabidopsis, rice and wheat, we conclude that the major functional diversification within the nsLTP family predated the monocot/dicot divergence. In contrast, there is no type VII nsLTPs in arabidopsis and type IX nsLTPs were only identified in arabidopsis. The reason for the larger number of nsLtp genes in wheat may simply be due to the hexaploid state of wheat but may also reflect extensive duplication of gene clusters as observed on rice chromosomes 11 and 12 and arabidopsis chromosome 5.Our current study provides fundamental information on the organization of the rice, arabidopsis and wheat nsLtp gene families. The multiplicity of nsLTP types provide new insights on arabidopsis, rice and wheat nsLtp gene families and will strongly support further transcript profiling or functional analyses of nsLtp genes. Until such time as specific physiological functions are defined, it seems relevant to categorize plant nsLTPs on the basis of sequence similarity and/or phylogenetic clustering.
DOIURL [本文引用: 1]
DOIURL [本文引用: 2]
DOIURL [本文引用: 2]
DOIURL [本文引用: 2]
[本文引用: 1]
DOIURL [本文引用: 2]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
DOIPMID [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 2]
DOIURL [本文引用: 1]
DOIURL [本文引用: 2]
DOIURL [本文引用: 1]
[本文引用: 1]
DOIPMID [本文引用: 1]
Baker's asthma is an important occupational allergic disease. Wheat lipid transfer protein (LTP) Tri a 14 is a major allergen associated with wheat allergy. No panel of wheat recombinant allergens for component-resolved diagnosis of baker's asthma is currently available.To evaluate the potential role of recombinant Tri a 14 as a novel tool for the diagnosis of baker's asthma, and to test the heat and proteolytic resistance of the wheat LTP allergen.A cDNA encoding Tri a 14 was isolated and sequenced, the recombinant allergen produced in Pichia pastoris and purified by chromatographic methods. Physicochemical and immunological comparison of the natural and recombinant forms of Tri a 14 was carried out by N-terminal amino acid sequencing, matrix-assisted laser desorption/ionization mass spectrometry, circular dichroism (CD) analysis, IgE immunodetection, and specific IgE determination and ELISA-inhibition assays using a pool or individual sera from 26 patients with baker's asthma. Thermal denaturation and simulated gastrointestinal digestion of both Tri a 14 forms were checked by spectroscopic and electrophoretic methods, respectively, and biological activity by basophil activation test (BAT).Natural and recombinant Tri a 14 were similarly folded, as indicated by their nearly identical CD spectra and heat denaturation profiles. A high interclass correlation coefficient (0.882) was found between specific IgE levels to both Tri a 14 proteins in individual sera from baker's asthma patients, but a slightly lower IgE-binding potency of rTri a 14 was detected by ELISA-inhibition assays. Natural and recombinant Tri a 14 elicited positive BAT in two and one out of three patients, respectively. Heat denaturation profiles and simulated gastrointestinal digestion assays indicated that Tri a 14 displayed a high heat and digestive proteolytic resistance, comparable to those of peach Pru p 3, the model food allergen of the LTP family.Recombinant Tri a 14 is a potential tool for baker's asthma diagnosis, based on its physicochemical and immunological similarity with its natural counterpart. Wheat Tri a 14 shows a high thermal stability and resistance to gastrointestinal digestion.
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 2]
PMID [本文引用: 1]
The past few years have seen the development of powerful statistical methods for detecting adaptive molecular evolution. These methods compare synonymous and nonsynonymous substitution rates in protein-coding genes, and regard a nonsynonymous rate elevated above the synonymous rate as evidence for darwinian selection. Numerous cases of molecular adaptation are being identified in various systems from viruses to humans. Although previous analyses averaging rates over sites and time have little power, recent methods designed to detect positive selection at individual sites and lineages have been successful. Here, we summarize recent statistical methods for detecting molecular adaptation, and discuss their limitations and possible improvements.
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL
PMID [本文引用: 1]
Eukaryotic phenotypic diversity arises from multitasking of a core proteome of limited size. Multitasking is routine in computers, as well as in other sophisticated information systems, and requires multiple inputs and outputs to control and integrate network activity. Higher eukaryotes have a mosaic gene structure with a dual output, mRNA (protein-coding) sequences and introns, which are released from the pre-mRNA by posttranscriptional processing. Introns have been enormously successful as a class of sequences and comprise up to 95% of the primary transcripts of protein-coding genes in mammals. In addition, many other transcripts (perhaps more than half) do not encode proteins at all, but appear both to be developmentally regulated and to have genetic function. We suggest that these RNAs (eRNAs) have evolved to function as endogenous network control molecules which enable direct gene-gene communication and multitasking of eukaryotic genomes. Analysis of a range of complex genetic phenomena in which RNA is involved or implicated, including co-suppression, transgene silencing, RNA interference, imprinting, methylation, and transvection, suggests that a higher-order regulatory system based on RNA signals operates in the higher eukaryotes and involves chromatin remodeling as well as other RNA-DNA, RNA-RNA, and RNA-protein interactions. The evolution of densely connected gene networks would be expected to result in a relatively stable core proteome due to the multiple reuse of components, implying that cellular differentiation and phenotypic variation in the higher eukaryotes results primarily from variation in the control architecture. Thus, network integration and multitasking using trans-acting RNA molecules produced in parallel with protein-coding sequences may underpin both the evolution of developmentally sophisticated multicellular organisms and the rapid expansion of phenotypic complexity into uncontested environments such as those initiated in the Cambrian radiation and those seen after major extinction events.
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}