 ,1, 杨多凤1, 丁林云1, 赵汀2, 张军2, 梅欢2, 黄楚珺2, 高阳1, 叶莉1, 高梦涛1, 严孙艺2, 张天真1,2, 胡艳,1,2,*
,1, 杨多凤1, 丁林云1, 赵汀2, 张军2, 梅欢2, 黄楚珺2, 高阳1, 叶莉1, 高梦涛1, 严孙艺2, 张天真1,2, 胡艳,1,2,*Identification of a cotton flower organ mutant 182-9 and cloning of candidate genes
YANG Qin-Li,1, YANG Duo-Feng1, DING Lin-Yun1, ZHANG Ting2, ZHANG Jun2, MEI Huan2, HUANG Chu-Jun2, GAO Yang1, YE Li1, GAO Meng-Tao1, YAN Sun-Yi2, ZHANG Tian-Zhen1,2, HU Yan,1,2,*通讯作者: *胡艳, E-mail:njauhuyan@njau.edu.cn
收稿日期:2020-09-10接受日期:2021-01-13网络出版日期:2021-03-11
| 基金资助: |
Corresponding authors: *E-mail:njauhuyan@njau.edu.cn
Received:2020-09-10Accepted:2021-01-13Published online:2021-03-11
| Fund supported: |
作者简介 About authors
E-mail:739768392@qq.com

摘要
棉花是世界性的重要经济作物, 是天然纤维的主要来源。棉花生殖生长过程现蕾、开花、结铃都直接影响棉花主要经济性状——棉纤维的产量和品质。本研究在转基因棉花材料中发现了1个花器官突变体, 命名为182-9, 其花器官呈现瓣化特征。PCR和Southern杂交证明突变体182-9中的外源基因已整合到基因组中, 且为单拷贝插入。通过基因组重测序进行序列比较发现, 突变体182-9基因组中外源T-DNA插入位点为染色体A11:59086840。PCR和Southern杂交对插入位点进行了进一步验证。根据棉花基因组注释结果, 在基因组插入位点附近有3个候选基因(GH_A11G2251、GH_A11G2252和GH_A11G2253)。其中GH_A11G2251为AP2类基因。已有研究证明, AP2类基因为花器官ABC模型中控制萼片和花瓣形成的A类功能基因。qRT-PCR结果显示, GH_A11G2251在转基因受体W0的花瓣、雌蕊和雄蕊3个组织中的表达与突变体182-9中存在显著性差异。本研究为进一步深入探究棉花花器官发育的分子机制研究提供了参考。
关键词:
Abstract
Cotton is an important economic crop and the main source of natural fiber in the world. The budding, flowering and bolling during cotton growth and development directly affect the yield and quality of cotton fiber that are the main economic traits of cotton. In this study, we found a flower organ mutant (named 182-9) in transgenic cottons, which displayed the floral organ petaloid feature. PCR and Southern blotting confirmed that the foreign T-DNA was integrated into the 182-9 genome with a single copy. Comparative analysis of the resequencing data revealed that the exogenic T-DNA was inserted in the 182-9 on chromosome A11: 59086840. The insertion site was further verified by PCR and southern blot. According to the gene annotation of cotton genome, there were three candidate genes of GH_A11G2251, GH_A11G2252, and GH_ A11G2253, near to the insertion site. GH_ A11G2251 encoded AP2 genes controlling the formation of sepals and petals in the ABC model of flower organs as previous report. qRT-PCR showed that there were significant differences in the expression level of GH_A11G2251 in petals, pistil and stamens of transgenic receptor W0 and mutant 182-9. Our study provided the basis for further study of molecular mechanism in cotton floral organ development.
Keywords:
PDF (3635KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
杨琴莉, 杨多凤, 丁林云, 赵汀, 张军, 梅欢, 黄楚珺, 高阳, 叶莉, 高梦涛, 严孙艺, 张天真, 胡艳. 棉花花器官突变体的鉴定及候选基因的克隆[J]. 作物学报, 2021, 47(10): 1854-1862 DOI:10.3724/SP.J.1006.2021.04208
YANG Qin-Li, YANG Duo-Feng, DING Lin-Yun, ZHANG Ting, ZHANG Jun, MEI Huan, HUANG Chu-Jun, GAO Yang, YE Li, GAO Meng-Tao, YAN Sun-Yi, ZHANG Tian-Zhen, HU Yan.
棉花是世界上重要的经济作物和油料作物, 在世界上超过100个国家和地区种植。棉花纤维是纺织工业天然纤维的重要来源[1]。纤维由棉花胚珠表皮细胞突起形成[2], 因此棉花花器官的分化和发育直接影响棉花的产量和纤维品质。在植株的生长发育过程中, 植物顶端分生组织由发育成叶原基转变为开始发育成花原基[3], 是营养生长到生殖生长的转变。花器官的分化和发育是被子植物生命周期的重要阶段[4]。典型的花器官分为4个部分, 从外到内依次是萼片、花瓣、雄蕊、心皮, 它们组成了花发育的4个轮回[5,6]。
1991年, Coen和Meyerowitz[7]首次系统地解释了花发育相关基因(主要是MADS盒基因)在不同花器官发育中的作用。已知MIKC型的MADS-box基因是植物特有的, 它们在开花植物(被子植物)生殖发育中的作用尤为重要[8]。绝大多数MIKC型基因属于花器官决定基因, 它建立了被子植物花发育的基本框架, 保障了“AE模型”[9]的保守性, 并参与调控开花时间、花器官和花分生组织识别、果实发育、营养器官发育[10,11]等过程。除了MADS-box基因家族外, 还有一类APETALA2 (AP2)基因在花发育过程中扮演了重要角色。AP2基因属于花发育模型中A类基因。最早发现拟南芥胚珠和雌配子体发育所需的AINTEGUMENTA基因与花型同源异形基因AP2有关。编码的蛋白质AINTEGUMENTA (ANT)属于AP2相关基因家族, 是胚珠发育所必需的[12,13]。已有研究表明, 拟南芥中ANT基因在其子房及雌配子体发育过程中发挥了关键作用[13,14,15,16]。同时为了更好地理解ANT基因在控制胚珠发育中的作用, 研究人员从拟南芥中分离出1个ANT雌性不育突变体。在该突变体的胚珠中, 胚珠不发育。当ANT突变体与花同源突变体AP2结合时, 花器官结构就会完全丧失, 表明这2个基因共同启动了花发育[12]。
有关棉花花器官发育相关基因已有报道。Wang等[17]从陆地棉中分别克隆到了1个SQUAMOSA类似基因和1个开花启动因子GhFPF1基因, 并在转基因拟南芥中进行了功能验证。Zhang等[18,19]对GhLFY和GhSPL基因家族进行了鉴定和分析, 证实GhSOC1与GhSPL3和GhLFY基因启动子结合以调节开花。而后研究确定了GhSOC1和GhMADS42的功能及其调控机制, 为早期成熟棉花品种发展提供了有用的信息[19]。研究表明GhMADS12基因属于PI亚家族, 为B类功能基因, 基因GhMADS13属于AGL6亚家族, 行使C类基因功能[20]。烟草植物中导入GhMADS12和GhMADS13基因, 证实了基因在相应花器官发育中的作用[20]。此外, AP2基因在植物的花发育网络调控中也扮演了重要角色。了解AP2基因与其他基因的相互作用关系, 有利于我们更好地理解花发育调控的基本框架[21]。已知AP1与AP2同属于A类功能基因, 它们共同调控花萼和花瓣的发育, 且2个基因被证明有一定的叠加效应, 它们共同影响了花原基的建成[16]。在花的发育过程中, AG基因与AP2基因作用最为明显。已有研究表明, 在ag突变体中, 很多AG负调控因子中AP2的作用最强, AG将AP2的表达限制在花器官形成的第1、2轮回中[22,23]。miR172对AP2的表达也有一定的调控作用。由于AP2结构中存在1个miR172的结合位点, 因此二者结合之后, miR172在翻译水平抑制了AP2蛋白质的形成[21]。总之, 在各类基因相互作用下形成的复杂调控网络, 共同调节了植物的花发育进程。
本研究在转基因棉花中发现了1个花器官突变体182-9。突变体表现为花器官异常, 萼片相对细长偏大, 花瓣和雄蕊结构异常。Southern杂交表明, T-DNA在基因组中为单拷贝插入。利用基因组重测序数据进行比较基因组分析, 鉴定出T-DNA在突变体182-9基因组的插入位点进行了准确定位。插入位点附近存在3个候选基因, 其中包括1个AP2类基因, 在突变花器官中表达量显著低于正常花器官。推测可能由于T-DNA的插入导致1个AP2类基因的表达发生差异变化, 从而引起花器官突变表型。
1 材料与方法
1.1 试验材料
花器官突变体182-9为转基因植株。植物材料种植于浙江大学农生环9号温室(培养条件: 温度22℃, 光照16 h/黑暗8 h, 相对湿度75%~80%)。试验中转基因载体为GoPGF-RNAi, 载体骨架序列为pBI121载体(图1)保存于本实验室。图1
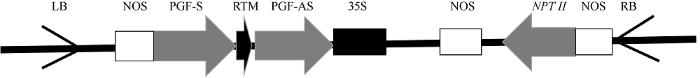 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1转基因载体T-DNA序列
Fig. 1T-DNA structure of the transgenic vector
1.2 DNA、RNA提取及cDNA合成
采用CTAB法提取转基因受体W0和花器官突变体182-9新鲜叶片DNA。按照EASY spin Plus植物RNA快速提取试剂盒(Adlab, 北京)指示说明书, 分别提取W0和182-9的萼片、花瓣、雄蕊和雌蕊4个组织的RNA。植物材料于液氮中速冻后保存于-80℃超低温冰箱中备用。使用反转录试剂盒将各组织RNA反转成cDNA。1.3 Southern印记杂交
提取叶片DNA后, 取20 μg基因组DNA, 加入150 U的Hind III内切酶和20 μL限制性内切酶缓冲液, 加无菌重蒸水至总体积200 μL, 37℃保温过夜。Hind III内切酶(南京诺唯赞生物科技股份有限公司, 简称诺唯赞)酶切后的质粒作为阳性对照。以NPTII基因扩增产物(约196 bp)和T-DNA扩增产物做探针, 分别检测外源基因插入的拷贝数以及插入位点的验证。NPTII基因扩增引物序列为F: 5°-ACCTGTCCGGT GCCCTGAATGAACTGC-3°和R: 5°-GCCATGATGGAT ACTTTCTCGGCAGGAGC-3°; T-DNA插入片段扩增引物序列为F: 5°-TGGGTGATGGTTCACGTAGT-3°和R: 5°-CATAATCATGTTAAAATGCTTGG-3°。将酶切后的基因组DNA在0.8%琼脂糖凝胶中电泳过夜。通过纸吸印法转移到尼龙膜上, 参照DIG High Prime DNA Labeling and Detection Starter Kit I (Roche)试剂盒进行探针标记、预杂交、杂交及检测。1.4 T-DNA插入位点鉴定
为明确外源T-DNA插入基因组的准确位点, 本研究对花器官突变体182-9和受体W0进行高深度的Illumina PE150基因组重测序。对所获得数据进行过滤去除低质量reads。利用Blastn[24]软件将高质量reads比对pBI121载体序列。提取比对到pBI121载体序列的另一端数据, 使用软件hisat2[25]比对陆地棉TM-1参考基因组(V2.1) (cotton.zju.edu.cn), 估算插入位点。1.5 候选基因的克隆
从本实验室TM-1基因组(V2.1)数据库中获取候选基因GH_A11G2251、GH_A11G2252和GH_ A11 G2253三个基因的CDS序列, 设计引物对3个基因进行克隆, 引物序列分别为GH_A11G2251 (F: 5°- ATGAAGTCCATGAGCAATGATG-3°, R: 5°-CTAAG CATCTGTCCAGGCAGT-3°)、GH_A11G2252 (F: 5°- ATGCAAAAGGAGGCTGCTCTTTT-3°, R: 5°-CTATA GCTTACACATGAGC-3°)、GH_A11G2253 (F: 5°-ATG GAAGGAAGTATACCCTT-3°, R: 5°-CTAGACAAAT TCATGTAAACCACG-3°)。1.6 实时荧光定量PCR (RT-PCR)
以棉花Histone 3基因(Acc. No. AF024716, F: 5°- GGTGGTGTGAAGAAGCCTCAT-3°, R: 5°-AATTTC ACGAACAAGCCTCTGGAA-3°)为内参对表达数据进行标准化。使用诺唯赞SYBR Green I kit染料, 在荧光定量RT-PCR仪(ABI 7500)上进行分析, 以最小的样本阈值循环数(Ct值)和最高的荧光值为标准。反应体系与程序按照诺唯赞AceQ qPCR SYBR Green Master Mix试剂盒说明书进行。引物序列分别为: GH_A11G2251-QF: 5°-ATGGAGGTTAGCAATC AAGG-3°, GH_A11G2251-QR: 5°-AGATGAGATGGT GGAGATGG-3°; GH_A11G2252-QF: 5°-GTTTAGAC GAGTGGAAGCC-3°, GH_A11G2252-QR: 5°-AGTTT GGAGTGTCCTAATGC-3°; GH_A11G2253-QF: 5°-G GCCTATAAAGCGTGTACC-3°; GH_A11G2253-QR: 5°-TAATTCGGAGCTACAAGTGC-3°。采用2-ΔΔCt法计算相对表达量, 计算公式如下: 目的基因的相对表达量 = 2-ΔΔCt; 而ΔCt = Ct目的基因 - Ct内参。2 结果与分析
2.1 花器官突变体182-9表型鉴定
本研究以棉花野生型W0为受体, 在转基因材料中发现1个转基因克隆株系表现为花器官突变体, 命名为182-9。突变体花器官呈现明显的生理缺陷构造, 整个花器官显著变小, 外围苞叶相对卷曲窄小, 萼片相对细长偏大, 花瓣和雄蕊结构异常, 失去了花器官四轮结构, 均呈现类似细长叶片状态, 心皮中无胚珠(图2)。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2突变体182-9与转基因受体W0花器官表型比较
A: 转基因受体W0花器官整体结构与子房。B: 突变体182-9花器官结构。C: 转基因受体W0苞叶、萼片、花瓣、雌蕊雄蕊结构。D: 突变体182-9苞叶、萼片以及瓣化结构。标尺为1 cm。
Fig. 2Phenotype comparison of floral organ between the mutant 182-9 and transgenic receptor W0
A: the whole flower organ and ovary of transgenic receptor W0. B: floral organ of the mutant 182-9. C: bract, sepal, petal, pistil stamen of transgenic receptor W0. D: bracts, sepals, and petaloid structure of the mutant 182-9. Bar: 1 cm.
2.2 突变体182-9的分子检测
对转基因突变体182-9进行NPT II基因PCR检测发现, 182-9为转基因阳性克隆, 外源T-DNA片段已经整合到基因组中(图3)。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3PCR检测转基因材料(182-9) NPTII基因
M: DNA分子量Marker; W0: 阴性对照; 1: 转基因株系182-9; 2: 转基因株系182-36; 3: 转基因株系182-91; 4: 转基因株系182-150; 5: 转基因株系182-172; 6: 转基因株系182-173; 7: 转基因株系182-187。
Fig. 3PCR detection of NPTII gene in transgenic mutant 182-9
M: DNA molecular-weight markers; W0: negative control; 1: transgenic line 182-9; 2: transgenic line 182-36; 3: transgenic line 182-91; 4: transgenic line 182-150; 5: transgenic line 182-172; 6: transgenic line 182-173; 7: transgenic line 182-187.
2.3 突变体182-9的Southern检测
为进一步确定T-DNA在突变体182-9中的插入拷贝数, 本研究根据外源基因NPT II设计探针, 与转基因获得的不同克隆的基因组DNA (包括突变体182-9)进行Southern杂交。同时, 以质粒DNA为阳性对照, 野生型W0为阴性对照。结果显示, 突变体182-9中T-DNA已稳定整合到基因组中且为单拷贝插入(图4)。图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4Southern杂交检测突变体182-9中外源基因NPTII的整合
M: DNA marker; 1: 转基因受体W0; C: 质粒; 2: 转基因株系182-187; 3: 转基因株系182-36; 4: 转基因株系182-173; 5: 转基因株系182-9 (箭头所示)。
Fig. 4Southern blotting of NPTII gene in mutant 182-9 genome
M: DNA marker; 1: transgenic receptor W0; C: plasmid; 2: transgenic line 182-187; 3: transgenic line 182-36; 4: transgenic line 182-173; 5: transgenic line 182-9 (indicated by the arrow).
2.4 插入位点在参考基因组中的定位
我们对转基因受体W0和突变体182-9进行基因组重测序, 分别产生了70.8 Gb和73.7 Gb的数据, 覆盖基因组约30倍(以棉花基因组2.5 Gb计算) (表1)。其中, Sample代表样品名, Raw Base (bp)表示原始数据产量, Clean Base是过滤之后的有效数据量, 即过滤后测序序列的个数乘以测序序列的长度, Q30即Phred 数值大于30的碱基占总体碱基的百分比。为鉴定T-DNA在受体W0基因组中的插入位点, 本研究将重测序reads分别比对到载体pBI121, 在转基因材料182-9和转基因受体W0重测序reads中, 分别有41条和0条序列和载体具有同源性。使用软件hisat 2 [25]将41条reads的另一端再比对到TM-1的参考基因组上发现, 共有15条reads能唯一匹配到陆地棉TM-1 A11染色体的59,086,746~59,086,947的区间, 推测位点为外源T-DNA的插入位点。Table 1
表1
表1重测序数据评估
Table 1
| 样品 Sample | 原始reads Raw reads | 过滤后reads Clean reads | 原始碱基数 Raw base (Gb) | 过滤后碱基数Clean base (Gb) | Q30 (%) | GC含量 GC content (%) |
|---|---|---|---|---|---|---|
| 182-9 | 245,670,776 | 245,536,479 | 73.70 | 73.66 | 91.81 | 35.43 |
| W0 | 236,081,132 | 235,926,310 | 70.82 | 70.78 | 89.16 | 35.51 |
新窗口打开|下载CSV
为进一步验证外源T-DNA的插入位点。根据重测序分析结果, 本研究利用SeqHunter1.0提取陆地棉TM-1基因组中该位点上下游2 kb序列, 设计特异性引物, 分别以突变体182-9和野生型W0的基因组DNA为模板, 扩增序列并测序。PCR扩增结果显示, 突变体182-9能扩增出约400 bp的特异性序列(图5-A)。测序结果证明, 突变体182-9基因组中T-DNA插入位点在染色体A11上的59,086,840位置, 红色部分为该位点附近的棉花基因组序列, 前半部分为载体序列(图5-B)。
图5
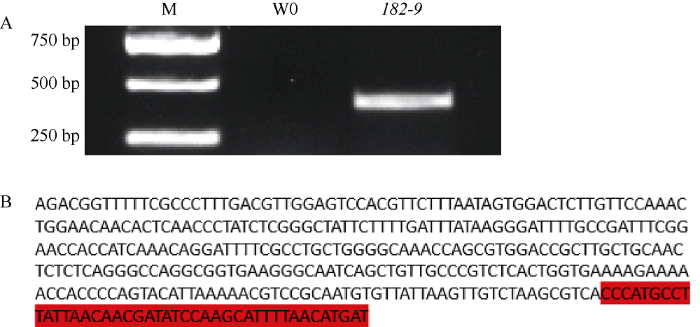 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5PCR检测外源T-DNA在基因组中插入位点
A: PCR扩增条带; B: 扩增序列的测序结果(红色部分为棉花基因组序列, 其他为T-DNA序列)。M: DNA marker; W0: 转基因受体; 182-9: 转基因株系。
Fig. 5PCR detection of the T-DNA insertion site in mutant 182-9
A: diagram of PCR detection; B: the sequencing results of the amplified sequence from 182-9 (the red part is the cotton genome sequence, and the others are T-DNA sequences). M: DNA marker; W0: transgenic receptor; 182-9: transgenic line.
根据PCR验证结果, 本研究以图5-B中的序列为探针, 与转基因突变体182-9基因组DNA进行Southern杂交。同时, 以探针序列为阳性对照, 转基因株系W0为阴性对照。结果进一步验证了182-9基因组中外源T-DNA以单拷贝插入染色体A11上的59,086,840位点(图6)。
图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6插入位点的Southern检测
M: DNA marker; C: 阳性质粒; 1: 转基因株系182-9; 2: 转基因受体W0。箭头所示为转基因182-9中特异性条带。
Fig. 6Southern blot detection of T-DNA insertion site
M: DNA marker; C: positive plasmid; 1: transgenic line 182-9; 2: transgenic receptor W0. The specific band of 182-9 is shown by an arrow.
2.5 候选基因的筛选和功能预测
根据外源T-DNA插入基因组中的定位结果, 本研究将外源插入T-DNA序列比对到陆地棉TM-1基因组, 根据陆地棉TM-1参考基因组注释结果(表2), 在插入位点400 kb范围内存在GH_A11G2251、GH_A11G2252和GH_A11G2253三个功能基因(图7)。根据拟南芥中同源基因的功能注释发现, GH_A11G2251为AP2类基因, 已有文献报道AP2类基因在花器官分化中起着重要作用, 为开花模型中A类基因。Table 2
表2
表2候选基因的注释信息
Table 2
| 基因ID Gene ID | 拟南芥中同源基因 Homologous genes in Arabidopsis | 功能描述 Function annotation |
|---|---|---|
| GH_A11G2251 | ANT | AP2类乙烯反应转录因子 AP2-like ethylene-responsive transcription factor ANT |
| GH_A11G2252 | At1g65750 | 假定核糖核酸酶H蛋白At1g65750 Putative ribonuclease H protein At1g65750 |
| GH_A11G2253 | NA | NA |
新窗口打开|下载CSV
图7
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7候选基因与插入位点的相对位置
Fig. 7Position of the candidate gene relative to the insertion site
为验证是否因为候选基因本身序列的突变而产生突变表型, 本研究对3个候选基因在转基因受体W0和花器官突变体182-9中的基因序列进行了比较发现, 3个候选基因CDS序列在182-9和W0之间并无差异(图8~图10), 表明T-DNA插入并未造成3个候选基因的碱基序列差异。
图8
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8GH_A11G2251在182-9和W0两个材料中的扩增序列比较
Fig. 8Comparison of the amplified sequences of GH_A11G2251 in mutant 182-9 and W0
图9
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图9GH_A11G2252在182-9和W0两个材料中的扩增序列比较
Fig. 9Comparison of the amplified sequences of GH_A11G2252 in mutant 182-9 and W0
图10
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图10GH_A11G2253在182-9和W0两个材料中的扩增序列比较
Fig. 10Comparison of the amplified sequences of GH_A11G2253 in mutant 182-9 and W0
选取W0和182-9典型花器官结构进行定量分析。q-PCR分析结果显示, GH_A11G2251在突变体182-9与转基因受体W0相应的花瓣(O2)、雄蕊(O3)和雌蕊(O4)中表现显著性差异, 在花瓣(O2)和雌蕊(O4)突变体表达量显著低于转基因受体。GH_A11 G2252在2个材料中表达无显著差异, GH_A11 G2253在转基因受体W0花器官和突变体182-9中几乎不表达(图11)。
图11
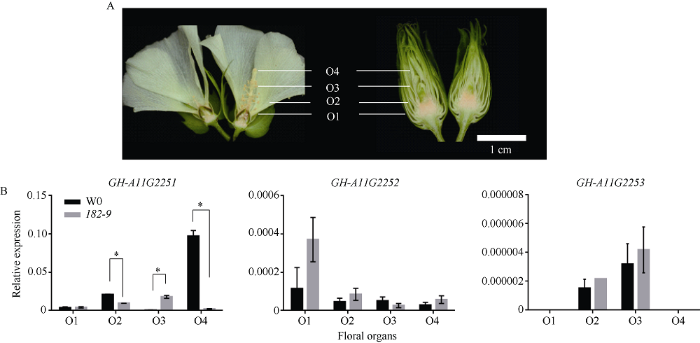 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图11候选基因的在花器官中表达分析
A: W0和182-9组织示意图; B: 候选基因在突变体182-9及其野生型W0中的表达水平。其中, *表示在0.05水平差异显著。
Fig. 11Relative expression levels of candidate genes in floral organs
A: the schematic diagram of floral organs of W0 and 182-9; B: the expression levels of the candidate genes in mutant 182-9 and wild-type W0. * means significant difference at the 0.05 probability level.
3 讨论
棉纤维为棉花胚珠表皮突起形成, 因此棉花的主要经济性状——棉纤维的品质和产量与棉花的生殖生长密切相关。但是, 目前关于棉花中花器官形成和发育的分子机制的研究报道并不多。本研究通过转基因得到花器官突变体182-9, 是研究花器官发育的良好材料。本研究中, 我们鉴定出了准确的外源T-DNA插入位点, 并在插入位点附近发现了1个AP2类基因家族基因。近年来, 关于AP2基因直接或间接参与花发育的某一具体调控仍存在许多问题, 对于其基因家族调节植物生长发育的方式及基因互作也不是十分清楚[21]。本研究中, 虽然T-DNA的插入位置与GH_A11G2251位置相差243 kb, 但是在DNA的三维空间上, 他们可能是靠近的。这种现象随着三维基因组研究[26]的发展, 已经有更多的证据证明了这种现象的存在。调控元件对基因的远程调控也已有报道。比如, 在玉米中, 已有报道证明一个控制光周期的ZmCCT9基因转录起始位点上游57 kb的Harbinger转座元件CTOR的插入调控了该基因的表达[27]。这种远程调控的作用机制为我们关于候选基因与突变表型之间的可能关系提供了一些理论依据。因此, 我们推测在突变体182-9中, T-DNA的插入可能引起了候选基因区域空间构象上的改变, 从而引起了基因的表达差异。突变体182-9花突变产生与GH_A11G2251的关系还需要更进一步的试验去证明。4 结论
本研究通过花器官突变体182-9与其野生型W0的表型差异, 为阐明调控棉花花发育的基因及突变机制提供了信息。鉴定出了T-DNA在转基因突变体中的准确插入位点A11:59,086,840, 并通过PCR以及Southern杂交进一步验证了位点鉴定的准确性。并在插入位点附近发现了一个可能性的功能基因, 在突变体182-9与转基因受体W0的花器官中存在显著差异表达。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
PMID [本文引用: 1]

In the present study, a haploid population from the cross of the two cultivated allotetraploid cottons, Gossypium hirsutum L. and Gossypium barbadense L., was developed by means of Vsg, a virescently marked semigamous line of Sea island cotton, and some target haploids were successfully doubled with colchicine. A molecular linkage map was constructed with 58 doubled and haploid plants. Among the total of 624 marker loci (510 SSRs and 114 RAPDs), 489 loci were assembled into 43 linkage groups and covered 3,314.5 centi-Morgans (cM). Using the monosomic and telodisomic genetic stocks, the linkage groups of the present map were associated with chromosomes of the allotetraploid genome, and some of the unassociated groups were connected to corresponding A or D subgenomes. Through the analysis of the assignment of the duplicated SSR loci in the chromosomes or the linkage groups, ten pairs of possible homoeologous chromosome (or linkage group) regions were identified. Among them, the pairs of Chrs. 1 and 15, Chrs. 4 and 22, and Chrs. 10 and 20 had already been determined as homoeologous by classical genetic and cytogenetic research, and the pair of Chrs. 9 and 23 had also been identified by the ISH method of molecular cytogenetics. But, from present research, it was assumed that Chrs. 5 and 18 might be a new pair of homoeologous chromosomes of the allotetraploid cotton genome detected by molecular mapping of the cotton genome.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
PMID [本文引用: 1]
Despite the differences in flower form, the underlying mechanism in determining the identity of floral organs is largely conserved among different angiosperms, but the details of how the functions of A, B, and C are specified varies greatly among plant species. Here, we report functional analysis of a Gerbera MADS box gene, GRCD1, which is orthologous to AGL2-like MADS box genes. Members of this group of genes are being reported in various species in growing numbers, but their functions remained largely unsettled. GRCD1 expression is detected in all four whorls, but the strongest signal is seen in the developing stamen and carpel. Downregulating GRCD1 expression by antisense transformation revealed that lack of GRCD1 caused homeotic changes in one whorl only: sterile staminodes, which normally develop in whorl 3 of marginal female florets, were changed into petals. This indicates that the GRCD1 gene product is active in determining stamen identity. Transgenic downregulation of GRCD1 causes a homeotic change similar to that in the downregulation of the Gerbera C function genes GAGA1 and GAGA2, but one that is limited to whorl 3. Downregulation of GRCD1 expression does not reduce expression of GAGA1 or GAGA2, or vice versa; and in yeast two-hybrid analysis, GRCD1 is able to interact with GAGA1 and GAGA2. We propose that a heterodimer between the GRCD1 and GAGA1/2 gene products is needed to fulfill the C function in whorl 3 in Gerbera.
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
PMID [本文引用: 1]
MIKC-type proteins represent a class of MADS-domain transcription factors and are defined by a unique domain structure: in addition to the highly conserved DNA-binding MADS-domain, they have three other domains ('I', 'K' and 'C'), with the keratin-like K-domain being the most highly conserved and characteristic one. The number and functional diversity of MIKC-type proteins increased considerably during land plant evolution, culminating in higher flowering plants, where they dominate the control of reproductive development from early to late stages. We wonder how one special class of proteins became important in the control of essentially all stages of a morphogenetic process. All MADS-domain proteins appear to bind to DNA as homo- or heterodimers and may function as part of ternary transcription factor complexes involving non-MADS-domain proteins. Only MIKC-type proteins, however, generate complex intrafamily interaction networks. These are based on the special potential of MIKC-type proteins to form complexes involving more than two homologous proteins constituting transcriptional regulators. We speculate that the potential to form heteromultimers of homologous proteins was achieved by the acquisition of the K-domain during evolution. There is emerging evidence that organismal complexity arises from progressively more elaborate regulation of gene expression. We hypothesize that combinatorial multimer formation of MIKC-type MADS-domain proteins facilitated an unusually efficient and rapid functional diversification based on gene duplication, sequence divergence and fixation. This 'networking' may have enabled a more sophisticated transcriptional control of target genes which was recruited for controlling increasingly complex and diverse developmental pathways during the rapid origin and diversification of plant reproductive structures. Therefore, MIKC-type proteins may owe their evolutionary 'success' and present-day developmental importance in part to their modular domain structure. Investigating the evolution of MIKC-type genes may thus help to better understand origin and diversification of gene regulatory networks.
URL [本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
PMID [本文引用: 1]
The function of MADS-box genes in flower and fruit development has been uncovered at a rapid pace over the past decade. Evolutionary biologists can now analyse the expression pattern of MADS-box genes during the development of different plant species, and study the homology of body parts and the evolution of body plans. These studies have shown that floral development is conserved among divergent species, and indicate that the basic mechanism of floral patterning might have evolved in an ancient flowering plant.
PMID [本文引用: 2]
To understand better the role of genes in controlling ovule development, a female-sterile mutant, aintegumenta (ant), was isolated from Arabidopsis. In ovules of this mutant, integuments do not develop and megasporogenesis is blocked at the tetrad stage. As a pleiotropic effect, narrower floral organs arise in reduced numbers. More complete loss of floral organs occurs when the ant mutant is combined with the floral homeotic mutant apetala2, suggesting that the two genes share functions in initiating floral organ development. The ANT gene was cloned by transposon tagging, and sequence analysis showed that it is a member of the APETALA2-like family of transcription factor genes. The expression pattern of ANT in floral and vegetative tissues indicates that it is involved not only in the initiation of integuments but also in the initiation and early growth of all primorida except roots.
PMID [本文引用: 2]
Ovules play a central role in plant reproduction, generating the female gametophyte within sporophytic integuments. When fertilized, the integuments differentiate into the seed coat and support the development of the embryo and endosperm. Mutations in the AINTEGUMENTA (ANT) locus of Arabidopsis have a profound effect on ovule development. Strong ant mutants have ovules that fail to form integuments or a female gametophyte. Flower development is also altered, with a random reduction of organs in the outer three whorls. In addition, organs present in the outer three floral whorls often have abnormal morphology. Ovules from a weak ant mutant contain both inner and outer integuments but generally fail to produce a functional female gametophyte. We isolated the ANT gene by using a mutation derived by T-DNA insertional mutagenesis. ANT is a member of a gene family that includes the floral homeotic gene APETALA2 (AP2). Like AP2, ANT contains two AP2 domains homologous with the DNA binding domain of ethylene response element binding proteins. ANT is expressed most highly in developing flowers but is also expressed in vegetative tissue. Taken together, these results suggest that ANT is a transcription factor that plays a critical role in regulating ovule and female gametophyte development.
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 2]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 2]
[本文引用: 2]
[本文引用: 2]
URL [本文引用: 3]
URL [本文引用: 3]
DOIURL [本文引用: 1]
PMID [本文引用: 1]
The floral homeotic gene AGAMOUS specifies stamen and carpel fate in the central whorls of Arabidopsis flowers. Transcription of AGAMOUS RNA is restricted to the center of developing flowers by several, partially redundant negative regulators, one of which is the homeotic gene APETALA2. We have identified regulatory elements that mediate transcriptional repression of AGAMOUS by APETALA2 and found that several redundant elements respond independently to loss of APETALA2 activity. Thus, redundancy at the level of cis-regulatory sequences is independent of redundancy at the level of trans-regulators. We have also found that only the early, but not the late, effects of APETALA2 on AGAMOUS require the meristem-identity protein LEAFY, a positive regulator of AGAMOUS.Copyright 1999 Academic Press.
PMID [本文引用: 1]
The BLAST programs are widely used tools for searching protein and DNA databases for sequence similarities. For protein comparisons, a variety of definitional, algorithmic and statistical refinements described here permits the execution time of the BLAST programs to be decreased substantially while enhancing their sensitivity to weak similarities. A new criterion for triggering the extension of word hits, combined with a new heuristic for generating gapped alignments, yields a gapped BLAST program that runs at approximately three times the speed of the original. In addition, a method is introduced for automatically combining statistically significant alignments produced by BLAST into a position-specific score matrix, and searching the database using this matrix. The resulting Position-Specific Iterated BLAST (PSI-BLAST) program runs at approximately the same speed per iteration as gapped BLAST, but in many cases is much more sensitive to weak but biologically relevant sequence similarities. PSI-BLAST is used to uncover several new and interesting members of the BRCT superfamily.
DOIURL [本文引用: 2]
DOIPMID [本文引用: 1]
Chromosome conformation capture (3C) technologies can be used to investigate 3D genomic structures. However, high background noise, high costs, and a lack of straightforward noise evaluation in current methods impede the advancement of 3D genomic research. Here we developed a simple digestion-ligation-only Hi-C (DLO Hi-C) technology to explore the 3D landscape of the genome. This method requires only two rounds of digestion and ligation, without the need for biotin labeling and pulldown. Non-ligated DNA was efficiently removed in a cost-effective step by purifying specific linker-ligated DNA fragments. Notably, random ligation could be quickly evaluated in an early quality-control step before sequencing. Moreover, an in situ version of DLO Hi-C using a four-cutter restriction enzyme has been developed. We applied DLO Hi-C to delineate the genomic architecture of THP-1 and K562 cells and uncovered chromosomal translocations. This technology may facilitate investigation of genomic organization, gene regulation, and (meta)genome assembly.
DOIURL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}