 ,2,*, 赵正武,1,*, 韩龙植,2,*
,2,*, 赵正武,1,*, 韩龙植,2,*Mapping QTLs for yield-related traits using chromosome segment substitution lines of Dongxiang common wild rice (Oryza rufipogon Griff.) and Nipponbare (Oryza sativa L.)
LUO Lan1,**, LEI Li-Xia1,**, LIU Jin2,3, ZHANG Rui-Hua4, JIN Gui-Xiu4, CUI Di2, LI Mao-Mao3, MA Xiao-Ding,2,*, ZHAO Zheng-Wu,1,*, HAN Long-Zhi,2,*通讯作者:
收稿日期:2020-08-13接受日期:2020-11-13网络出版日期:2020-12-29
| 基金资助: |
Received:2020-08-13Accepted:2020-11-13Online:2020-12-29
| Fund supported: |
作者简介 About authors
罗兰, E-mail:
雷丽霞, E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (3193KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
罗兰, 雷丽霞, 刘进, 张瑞华, 金桂秀, 崔迪, 黎毛毛, 马小定, 赵正武, 韩龙植. 利用东乡普通野生稻染色体片段置换系定位产量相关性状QTL[J]. 作物学报, 2020, 47(7): 1391-1401. doi:10.3724/SP.J.1006.2021.02054
LUO Lan, LEI Li-Xia, LIU Jin, ZHANG Rui-Hua, JIN Gui-Xiu, CUI Di, LI Mao-Mao, MA Xiao-Ding, ZHAO Zheng-Wu, HAN Long-Zhi.
水稻(Oryza sativa L.)是世界最重要的粮食作物之一, 全球一半以上的人口以稻米为主食[1]。品种改良对水稻增产具有非常重要的作用, 我国水稻品种改良经历了矮化育种、杂种优势利用和绿色超级稻培育3次飞跃, 产量从160千克增长到1000多千克[2], 但高产仍然是当前和今后相当一段时间内水稻育种的主要目标之一。水稻产量相关性状, 包括株高、有效分蘖数、每穗粒数和千粒重等。株高是株型的重要组成部分, 间接影响水稻产量; 分蘖数直接决定了每株穗数, 是产量的直接影响因子; 穗长、一二次枝梗数以及枝梗着粒数等与每穗粒数密切相关, 是影响产量的直接因素; 粒长、粒宽、粒厚以及籽粒灌浆程度又直接决定了千粒重[3]。
水稻株高、有效分蘖数、穗长、穗粒数和粒形等产量相关性状为多基因控制的数量性状, 易受种植环境影响。由于其重要性, 产量性状的遗传规律解析一直是研究的重点和热点, 特别是近年来分子生物学的发展, 部分性状的分子调控机制得到了很好的解析。例如矮化育种的株高性状由“绿色革命”基因SD1控制, SD1基因突变导致植株变矮[4,5], 从而解决了水稻的倒伏问题。分蘖数主要由MOC1调控, 该基因突变的植株只有1个主茎, 没有任何分蘖[6]。此外, D53[7,8]、D3[9]、HTD1[10]等基因也与水稻分蘖相关。DEP1是调控水稻穗粒数的关键基因, 功能获得性突变的DEP1基因能引起稻穗变短、直立、着粒密集[11]。此外, 与水稻每穗粒数相关的基因, 还包括GN1a[12]、GNP1[13]、GAD1[14]、NOG1[15]等。GS3是水稻粒长和粒重主效QTL, 通过负调控颖壳细胞数目控制水稻粒长[16]; GW2是粒宽和粒重主效QTL, 该基因突变能显著增加粒宽和粒重[17]; TGW6控制水稻千粒重, 来自Kasalath的近等基因系NIL (TGW6)能使日本晴增产15%[18]; 此外, 目前已克隆的粒形相关基因, 还包括GS5[19]、GW8[20]、GL7[21]、GS9[22]等。
综上所述, 虽然产量相关性状的遗传规律和分子调控机制研究取得了一些进展, 然而产量相关性状的QTL发掘仍然是重要的研究内容, 而且每隔一段时间依然有新发现。因此, 要想完全解析产量性状的遗传规律, 亟需利用新资源和新群体发掘更多新基因以完善水稻产量性状的调控网络。本研究利用东乡普通野生稻(O. rufipogon Griff.)为供体亲本, 日本晴为受体亲本构建的染色体片段置换系为研究材料, 多环境考察, 对株高、分蘖数等产量相关性状进行连锁分析, 以期发掘新位点, 为进一步解析产量相关性状的遗传基础、阐明产量性状的分子调控机制及遗传调控网络奠定基础。
1 材料与方法
1.1 试验材料
以江西东乡普通野生稻为供体亲本、日本晴为受体亲本, 构建的染色体片段置换系(chromosome segment substitution lines, CSSLs)为研究材料, 包括104个家系[23]。2019年将该套材料分别播种于北京、山东临沂和江西南昌3个地点进行多环境表型鉴定。北京于4月26日播种, 临沂于5月20播种, 南昌于5月14日播种, 成苗后按照当地大田种植密度单本插秧, 2行区, 每行15株; 其他按照水稻大田常规方法进行管理。由于东乡普通野生稻生育期长, 3个地点仅南昌可以抽穗。本文仅以日本晴为对照进行表型考察。1.2 产量相关性状考察
产量相关性状考察在水稻成熟期进行, 为消除边际效应, 每个置换系从第1行的第3株开始连续取5个植株, 考察株高和分蘖数, 从取样的5个植株上, 每株取4个穗,对穗长、一次枝梗数、一次枝梗粒数、二次枝梗数、二次枝梗粒数、每穗粒数、千粒重、粒长和粒宽进行考察, 参照崔国庆等[24]报道的方法测定。1.3 QTL定位
利用QTL IciMapping (V4.1)[25]软件的RSTEP-LRT- ADD方法进行加性QTL分析, 基因型来自本实验室前期发表的染色体片段置换系的基因型数据[23,26], LOD阈值设定为2.5, 其他参数设置为默认值。1.4 次级群体构建及BSA分析
QTL验证采用分离群体分组分析法(BSA)。CSSLs与日本晴杂交, 构建F2次级分离群体。2019年冬季种植于海南基地, 表型考察和统计分析后, 将分离群体中30~50个极端表型的单株叶片等量混合, 提取基因组DNA, 组成DNA混池, 然后进行Illumina PE150测序, 测序深度为50×。经过滤, 将有效测序数据通过BWA软件[27]比对到参考基因组(fttp://fttp.ensemblgenomes.org/pub/plants/release-28/fasta/oryza_sativa/dna/)。采用GATK3.8软件[28]进行多样本SNP检测, 以参考基因组为参考, 分别计算混池的SNP频率(SNP-index), 去掉SNP-index混池中都小于0.3且SNP深度都小于7的位点, 以提高定位效率。为了直观体现混池间差异区域, 计算2个混池SNP-index的差值: Δ(SNP-index) = SNP-index (混池1)-SNP-index (混池2), 选择1 Mb为窗口, 1 kb为步长, 计算每个窗口中Δ(SNP-index)的平均值来反应Δ(SNP-index)分布, 置换检验1000次[29], 最后画出曼哈顿图(Manhattan Plot)显示Δ(All-index)在染色体上的分布。2 结果与分析
2.1 CSSL和日本晴产量相关性状表型分析
CSSL群体和日本晴的产量相关性状, 在北京、南昌和临沂3个地点的表型分布如图1和表1所示。3个环境中, 产量相关性状在CSSL群体中均呈现连续分布, 并存在双向超亲分离现象, 说明产量相关性状属于受多基因控制的数量性状。在考察的11个性状中, 变异系数最大的是二次枝梗粒数, 北京、临沂和南昌分别为35.46%、41.80%和36.72%; 变异系数最小的是粒宽, 3个地点分别为3.09%、3.34%和4.64%。11个性状在3个地点表型差异明显, 而且不同地点的变异又各不相同, 综合分析临沂和南昌2个地点的变异要高于北京(表1), 说明产量相关性状的表现易受种植环境的影响。图1
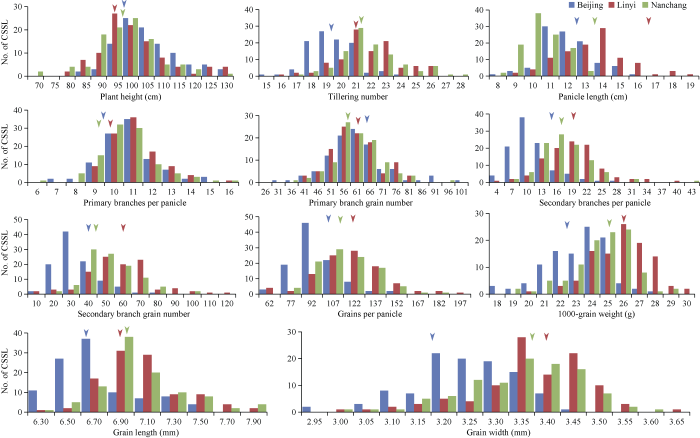 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1CSSL群体中11个产量相关性状的频率分布
纵坐标表示CSSL数量。蓝色、红色和绿色长柱分别代表CSSL中11个性状在北京、临沂和南昌的频率分布。蓝色、红色和绿色箭头表示日本晴的11个性状在北京、临沂和南昌的表现。
Fig. 1Frequency distribution of 11 yield-related traits in the CSSLs The vertical axis of each figure represents the number of CSSL individuals.
The blue, red and green rectangles represent the distribution of eleven yield-related traits in the CSSLs at the Beijing, Linyi and Nanchang locations, respectively. The blue, red, and green triangles represent the positions of the means of Nipponbare in Beijing, Linyi, and Nanchang, respectively.
2.2 产量相关性状QTL定位
3个环境下共检测到68个产量相关性状QTL, 分布于水稻的12条染色体上, LOD值在2.50~12.66之间, QTL贡献率为4.67%~27.79%, 其中15个QTL的贡献率大于15%, 8个QTL贡献率小于8%。6个QTL能在2个环境下同时被检测到, 分别是qPH2、qPL2-2、qTGW8-2、qGL2、qGL3-4和qGW8; 1个千粒重QTL-qTGW2在3个环境下被重复检测到(表2和图2)。具体分析如下:2.2.1 株高 共检测到4个株高QTL, LOD值在2.63~5.87之间, 贡献率变幅为8.99%~20.74%。qPH2位于2号染色体DX-C2-13标记附近, 在北京和南昌2个环境中都能被检测到, 贡献率分别为20.74%和10.90%; qPH5和qPH8分别位于5号染色体DX-C5-10和8号染色体DX-C8-12标记, 2个QTL仅在临沂环境中被检测到, 贡献率分别为11.80%和8.99%; qPH7位于7号染色体indel-c7-1标记处, 仅在北京环境中被检测到, 贡献率为9.86%。
2.2.2 穗长 共检测到穗长QTL 5个, LOD值在2.66~5.48之间, 贡献率变幅为8.06%~17.36%。qPL2-2位于2号染色体02-067标记处, 在临沂和南昌2个环境下均能被检测到, 贡献率分别为12.87%和11.88%; qPL1位于1号染色体01-009处, qPL2-1位于2号染色体DX-C2-13标记处, qPL10位于10号染色体DX-S10-1标记处, 3个QTL仅在北京环境下被检测到, 贡献率分别为9.17%、17.36%和8.06%; qPL5位于5号染色体05-028标记处, 仅在南昌环境中被检测到, 贡献率为10.42%。
2.2.3 分蘖数 检测到的有效分蘖数QTL 2个, LOD值在3.03~3.89之间。qTN1-1和qTN1-2分别位于1号染色体DX-C1-2和010-060标记处, 2个QTL均仅在临沂环境下被检测到, 贡献率分别为10.96%和13.32%。
2.2.4 一次枝梗数 共检测到7个一次枝梗数QTL, LOD值介于2.68~7.98之间, QTL贡献率在6.79%~18.54%范围内。qPBPP1-1和qPBPP1-2位于1号染色体DX-C1-6和DX-C1-10标记处, 仅在临沂环境下被检测到, 贡献率分别为9.25%和8.34%; qPBPP3、qPBPP5和qPBPP8-3分别位于3号染色体DX-C3-8、5号染色体DX-C5-3和8号染色体DX-S8-14标记处, 仅在北京环境下被检测到, 贡献率分别为9.86%、11.23%和18.54%; qPBPP8-1和qPBPP8-2分别位于8号染色体DX-C8-11和DX-C8-12标记处, 仅在南昌环境下被检测到, 贡献率分别为6.79%和13.99%。
2.2.5 一次枝梗粒数 共检测到8个一次枝梗粒数QTL, LOD值在2.61~9.27之间, 贡献率变幅为8.18%~17.32%。qPBGN1-1、qPBGN1-2、qPBGN3、qPBGN8-3和qPBGN11分别位于1号染色体DX-C1-3、DX-C1-10、3号染色体DX-C3-28、8号染色体DX-S8-14和11号染色体DX-S11-10标记处, 仅在北京环境下能检测到, 贡献率分别为9.85%、12.29%、8.18%、9.33%和9.86%; qPBGN1-3仅在临沂环境下能被检测到, 位于1号染色体01-046标记处, 贡献率为17.32%; 仅在南昌环境下被检测到的有qPBGN8-1和qPBGN8-2, 分别位于8号染色体DX-C8-11和DX-C8-12标记处, 贡献率分别为7.45%和16.35%。
2.2.6 二次枝梗数 共检测到8个二次枝梗数QTL, LOD值在3.09~9.46之间, 贡献率范围为6.71%~23.99%。qSBPP1、qSBPP2-1和qSBPP4-2分别位于1号染色体DX-C1-7、2号染色体DX-C2-1和4号染色体S4-12-2标记处, 仅在临沂环境下能检测到, 贡献率分别为8.86%、15.83%和15.75%; qSBPP2-2、qSBPP2-3、qSBPP4-1和qSBPP6-2分别位于2号染色体DX-C2-2、02-057、4号染色体DX-C4-12和6号染色体S6-9-1标记处, 仅在北京环境下能检测到, 贡献率分别为23.99%、12.01%、8.10%和6.71%; qSBPP6-1仅在南昌环境下能被检测到, 位于6号染色体DX-C6-2标记处, 贡献率为13.08%。
2.2.7 二次枝梗粒数 共检测到二次枝梗粒数QTL 10个, LOD值在2.68~12.66之间, 贡献率变幅为4.67%~27.79%。qSBGN2-1、qSBGN3-1、qSBGN4-1和qSBPP4-2分别位于2号染色体DX-C2-1、3号染色体DX-S3-16、4号染色体DX-C4-2和DX-C4-8标记处, 仅在临沂环境下能检测到, 贡献率分别为27.79%、6.99%、9.03%和4.67%; qSBGN2-2、qSBGN2-3、qSBGN4-3和qSBGN6-2分别位于2号染色体DX-C2-2、2号染色体02-057、4号染色体DX-C4-12和6号染色体S6-9-1标记处, 仅在北京环境下能检测到, 贡献率分别为21.39%、9.79%、10.55%和9.47%; qSBGN3-2和qSBGN6-1仅在南昌环境下能被检测到, 分别位于3号染色体DX-S3-17和6号染色体DX-C6-2标记处, 贡献率分别为15.34%和10.56%。
Table 1
表1
表1日本晴及CSSL群体产量相关性状表型统计分析
Table 1
| 性状 Trait | 北京Beijing | 临沂Linyi | 南昌Nanchang | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 日本晴 Nip | 平均值±标准差 Mean±SD | 变异系数 CV (%) | 幅度 Range | 日本晴 Nip | 平均值±标准差 Mean±SD | 变异系数 CV (%) | 幅度 Range | 日本晴 Nip | 平均值±标准差 Mean±SD | 变异系数 CV (%) | 幅度 Range | |
| 株高 PH (cm) | 96.30 | 108.02±10.07 | 9.33 | 87.75-133.75 | 94.64 | 103.34±10.95 | 10.59 | 80.30-134.82 | 95.00 | 100.31±9.76 | 9.73 | 70.20-130.00 |
| 穗长 PL (cm) | 19.68 | 19.97±1.53 | 7.68 | 15.07-24.94 | 21.59 | 22.29±2.03 | 9.10 | 16.64-26.96 | 21.74 | 22.32±2.01 | 9.01 | 17.78-28.03 |
| 分蘖数 TN | 13.00 | 14.30±2.14 | 14.96 | 8.80-20.00 | 17.21 | 16.24±2.89 | 17.78 | 9.00-24.17 | 13.60 | 11.83±1.58 | 13.38 | 7.80-15.40 |
| 一次枝梗数 PBPP | 10.23 | 10.72±1.42 | 13.27 | 7.17-15.17 | 10.69 | 10.99±1.56 | 14.18 | 8.42-19.67 | 9.63 | 10.52±1.50 | 14.27 | 6.38-15.75 |
| 一次枝梗粒数PBGN | 63.42 | 62.50±11.28 | 18.04 | 30.50-00.17 | 60.60 | 61.79±8.65 | 14.00 | 42.50-84.83 | 57.38 | 62.79±10.57 | 16.84 | 35.38-111.75 |
| 二次枝梗数 SBPP | 15.17 | 12.65±4.25 | 33.61 | 5.08-29.75 | 21.38 | 20.85±7.13 | 34.20 | 6.42-63.00 | 17.13 | 18.60±5.21 | 27.99 | 9.00-43.75 |
| 二次枝梗粒数 SBGN | 41.79 | 33.97±12.04 | 35.46 | 11.08-75.17 | 63.02 | 59.58±24.91 | 41.80 | 13.92-208.17 | 45.75 | 53.05±19.48 | 36.72 | 20.75-181.63 |
| 每穗粒数 GPP | 105.21 | 96.46±16.51 | 17.11 | 57.58-156.67 | 123.63 | 121.37±30.55 | 25.17 | 62.67-288.25 | 103.13 | 115.84±24.04 | 20.75 | 77.50-258.63 |
| 千粒重 TGW (g) | 23.81 | 23.21±1.92 | 8.28 | 17.95-28.30 | 26.58 | 25.81±1.76 | 6.82 | 19.71-30.01 | 25.68 | 24.51±2.03 | 8.26 | 16.39-28.87 |
| 粒长 GL (mm) | 6.60 | 6.74±0.29 | 4.36 | 6.21-7.57 | 6.94 | 7.04±0.29 | 4.17 | 6.34-7.89 | 6.93 | 7.07±0.40 | 5.66 | 6.27-8.85 |
| 粒宽 GW (mm) | 3.20 | 3.22±0.10 | 3.09 | 2.92-3.44 | 3.39 | 3.34±0.11 | 3.34 | 2.97-3.60 | 3.37 | 3.31±0.15 | 4.64 | 2.49-3.60 |
新窗口打开|下载CSV
2.2.8 每穗粒数 共检测到6个每穗粒数QTL, LOD值在3.08~5.14之间, 贡献率变幅为9.96%~17.43%。qGPP1、qGPP2和qGPP4分别位于1号染色体01-046、2号染色体DX-C2-1和4号染色体S4-12-2标记处, 仅在临沂环境下能检测到, 贡献率分别为13.92%、14.43%和12.58%; qGPP6-1位于6号染色体06-013标记处, 仅在南昌环境下能检测到, 贡献率分别为13.45%; qGPP6-2和qGPP11仅在北京环境下能被检测到, 分别位于6号染色体06-037和11号染色体DX-S11-10标记处, 贡献率分别为17.43%和9.96%。
图2
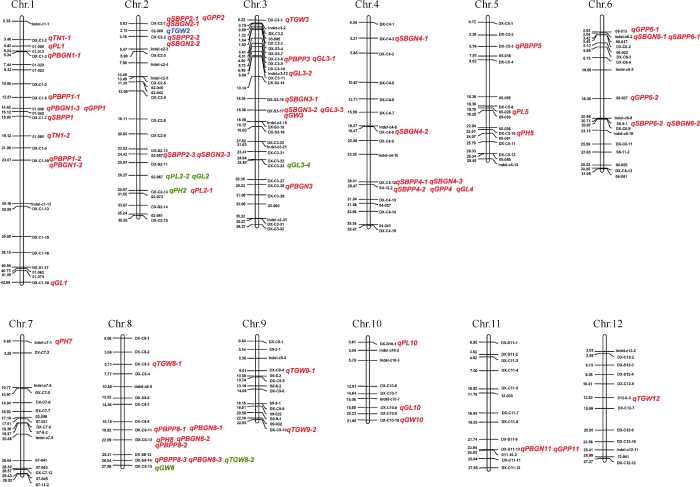 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2北京、临沂和南昌3个地点检测到的产量性状相关QTL在染色体上的分布
检测到的QTL具体信息如
Fig. 2Mapping locations of QTLs associated with yield-related traits detected in Beijing, Linyi, and Nanchang
The definitions of the abbreviations for the symbols representing the QTLs are listed in
2.2.9 千粒重 共检测到7个千粒重QTL, LOD值在2.90~5.28之间, 贡献率变幅为7.81%~14.36%。qTGW2位于2号染色体02-008标记处, 在北京、临沂和南昌3个环境中都能被检测到, 贡献率分别为14.36%、9.19%和10.96%; qTGW8-2位于8号染色体DX-S8-14标记处, 在临沂和南昌2个环境中能被同时检测到, 贡献率分别为8.54%和7.86%; qTGW3位于3号染色体DX-C3-1标记处, 仅在北京环境下检测到, 贡献率为10.64%; qTGW8-1和qTGW9-1分别位于8号染色体DX-C8-3和9号染色体DX-C9-4标记处, 仅在临沂环境中被检测到, 贡献率分别为11.68%和8.11%; qTGW9-2和qTGW12分别位于9号染色体DX-C9-10和12号染色体S12-6-3标记处, 仅在南昌环境中被检测到, 贡献率分别为7.81%和14.11%。
Table 2
表2
表2北京、临沂和南昌3地产量相关性状QTL分析
Table 2
| 性状 Trait | 位点 QTL | 染色体 Chr. | 标记名称 Marker | 北京Beijing | 临沂Linyi | 南昌Nanchang | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 阈值 LOD value | 贡献率 PVE (%) | 加性效应 Additive | 阈值 LOD value | 贡献率 PVE (%) | 加性效应 Additive | 阈值 LOD value | 贡献率 PVE (%) | 加性效应 Additive | |||||||||||||||||||||
| 株高PH | qPH2 | 2 | DX-C2-13 | 5.87 | 20.74 | 6.06 | 2.63 | 10.90 | 4.17 | ||||||||||||||||||||
| qPH5 | 5 | DX-C5-10 | 3.54 | 11.80 | 6.54 | ||||||||||||||||||||||||
| qPH7 | 7 | Indel-c7-1 | 2.93 | 9.86 | 5.91 | ||||||||||||||||||||||||
| qPH8 | 8 | DX-C8-12 | 2.80 | 8.99 | 5.52 | ||||||||||||||||||||||||
| 穗长PL | qPL1 | 1 | 01-009 | 3.06 | 9.17 | 0.61 | |||||||||||||||||||||||
| qPL2-1 | 2 | DX-C2-13 | 5.48 | 17.36 | 0.86 | ||||||||||||||||||||||||
| qPL2-2 | 2 | 02-067 | 3.26 | 12.87 | 1.06 | 3.37 | 11.88 | 1.07 | |||||||||||||||||||||
| qPL5 | 5 | 05-028 | 3.39 | 10.42 | 2.08 | ||||||||||||||||||||||||
| qPL10 | 10 | DX-S10-1 | 2.66 | 8.06 | 0.94 | ||||||||||||||||||||||||
| 分蘖数TN | qTN1-1 | 1 | DX-C1-2 | 3.03 | 10.96 | -1.31 | |||||||||||||||||||||||
| qTN1-2 | 1 | 010-060 | 3.89 | 13.32 | -1.70 | ||||||||||||||||||||||||
| 一次枝梗数PBPP | qPBPP1-1 | 1 | DX-C1-6 | 2.77 | 9.25 | 1.03 | |||||||||||||||||||||||
| qPBPP1-2 | 1 | DX-C1-10 | 2.68 | 8.34 | 2.33 | ||||||||||||||||||||||||
| qPBPP3 | 3 | DX-C3-8 | 3.08 | 9.86 | -0.69 | ||||||||||||||||||||||||
| qPBPP5 | 5 | DX-C5-3 | 3.48 | 11.23 | 1.05 | ||||||||||||||||||||||||
| qPBPP8-1 | 8 | DX-C8-11 | 4.23 | 6.79 | -1.41 | ||||||||||||||||||||||||
| qPBPP8-2 | 8 | DX-C8-12 | 7.98 | 13.99 | 1.36 | ||||||||||||||||||||||||
| 一次枝梗数PBPP | qPBPP8-3 | 8 | DX-S8-14 | 5.43 | 18.54 | 1.23 | |||||||||||||||||||||||
| 一次枝梗粒数PBGN | qPBGN1-1 | 1 | DX-C1-3 | 3.00 | 9.85 | 4.05 | |||||||||||||||||||||||
| qPBGN1-2 | 1 | DX-C1-10 | 3.74 | 12.29 | -17.18 | ||||||||||||||||||||||||
| qPBGN1-3 | 1 | 01-046 | 4.10 | 17.32 | 4.94 | ||||||||||||||||||||||||
| qPBGN3 | 3 | DX-C3-28 | 2.61 | 8.18 | -5.57 | ||||||||||||||||||||||||
| qPBGN8-1 | 8 | DX-C8-11 | 4.70 | 7.45 | -10.34 | ||||||||||||||||||||||||
| qPBGN8-2 | 8 | DX-C8-12 | 9.27 | 16.35 | 10.25 | ||||||||||||||||||||||||
| qPBGN8-3 | 8 | DX-S8-14 | 2.90 | 9.33 | 6.36 | ||||||||||||||||||||||||
| qPBGN11 | 11 | DX-S11-10 | 3.11 | 9.86 | 6.12 | ||||||||||||||||||||||||
| 二次枝梗数SBPP | qSBPP1 | 1 | DX-C1-7 | 3.26 | 8.86 | 3.50 | |||||||||||||||||||||||
| qSBPP2-1 | 2 | DX-C2-1 | 5.52 | 15.83 | 10.49 | ||||||||||||||||||||||||
| qSBPP2-2 | 2 | DX-C2-2 | 9.46 | 23.99 | 7.76 | ||||||||||||||||||||||||
| qSBPP2-3 | 2 | 02-057 | 5.26 | 12.01 | 2.71 | ||||||||||||||||||||||||
| qSBPP4-1 | 4 | DX-C4-12 | 3.68 | 8.10 | -1.51 | ||||||||||||||||||||||||
| qSBPP4-2 | 4 | S4-12-2 | 5.50 | 15.75 | -5.73 | ||||||||||||||||||||||||
| qSBPP6-1 | 6 | DX-C6-2 | 3.17 | 13.08 | 2.93 | ||||||||||||||||||||||||
| 二次枝梗数SBPP | qSBPP6-2 | 6 | S6-9-1 | 3.09 | 6.71 | 2.27 | |||||||||||||||||||||||
| 二次枝梗粒数SBGN | qSBGN2-1 | 2 | DX-C2-1 | 12.66 | 27.79 | 56.08 | |||||||||||||||||||||||
| qSBGN2-2 | 2 | DX-C2-2 | 8.56 | 21.39 | 20.58 | ||||||||||||||||||||||||
| qSBGN2-3 | 2 | 02-057 | 4.33 | 9.79 | 6.87 | ||||||||||||||||||||||||
| qSBGN3-1 | 3 | DX-S3-16 | 3.90 | 6.99 | 20.08 | ||||||||||||||||||||||||
| qSBGN3-2 | 3 | DX-S3-17 | 4.03 | 15.34 | 17.26 | ||||||||||||||||||||||||
| qSBGN4-1 | 4 | DX-C4-2 | 4.93 | 9.03 | -15.61 | ||||||||||||||||||||||||
| qSBGN4-2 | 4 | DX-C4-8 | 2.68 | 4.67 | -11.22 | ||||||||||||||||||||||||
| qSBGN4-3 | 4 | DX-C4-12 | 4.64 | 10.55 | -4.84 | ||||||||||||||||||||||||
| qSBGN6-1 | 6 | DX-C6-2 | 2.85 | 10.56 | 9.59 | ||||||||||||||||||||||||
| qSBGN6-2 | 6 | S6-9-1 | 4.21 | 9.47 | 7.58 | ||||||||||||||||||||||||
| 每穗粒数GPP | qGPP1 | 1 | 01-046 | 4.39 | 13.92 | 14.35 | |||||||||||||||||||||||
| qGPP2 | 2 | DX-C2-1 | 4.02 | 14.43 | 37.54 | ||||||||||||||||||||||||
| qGPP4 | 4 | S4-12-2 | 3.55 | 12.58 | -19.21 | ||||||||||||||||||||||||
| qGPP6-1 | 6 | 06-013 | 3.26 | 13.45 | 20.53 | ||||||||||||||||||||||||
| qGPP6-2 | 6 | 06-037 | 5.14 | 17.43 | 14.15 | ||||||||||||||||||||||||
| qGPP11 | 11 | DX-S11-10 | 3.08 | 9.96 | 10.70 | ||||||||||||||||||||||||
| 千粒重TGW | qTGW2 | 2 | 02-008 | 4.39 | 14.36 | -2.79 | 3.26 | 9.19 | -1.98 | 4.21 | 10.96 | -2.56 | |||||||||||||||||
| qTGW3 | 3 | DX-C3-1 | 3.27 | 10.64 | 1.31 | ||||||||||||||||||||||||
| qTGW8-1 | 8 | DX-C8-3 | 4.07 | 11.68 | -3.14 | ||||||||||||||||||||||||
| qTGW8-2 | 8 | DX-S8-14 | 3.02 | 8.54 | -1.12 | 3.07 | 7.86 | -1.27 | |||||||||||||||||||||
| qTGW9-1 | 9 | DX-C9-4 | 2.90 | 8.12 | 1.33 | ||||||||||||||||||||||||
| qTGW9-2 | 9 | DX-C9-10 | 3.02 | 7.81 | -2.15 | ||||||||||||||||||||||||
| qTGW12 | 12 | S12-6-3 | 5.28 | 14.11 | -2.38 | ||||||||||||||||||||||||
| 粒长GL | qGL1 | 1 | DX-C1-18 | 5.29 | 16.65 | 0.42 | |||||||||||||||||||||||
| qGL2 | 2 | 02-067 | 3.13 | 10.71 | 0.13 | 2.86 | 7.12 | 0.15 | |||||||||||||||||||||
| qGL3-1 | 3 | DX-C3-8 | 3.89 | 13.44 | 0.17 | ||||||||||||||||||||||||
| qGL3-2 | 3 | indel-c3-12 | 3.08 | 9.22 | 0.12 | ||||||||||||||||||||||||
| qGL3-3 | 3 | DX-S3-17 | 5.75 | 15.14 | 0.37 | ||||||||||||||||||||||||
| qGL3-4 | 3 | DX-C3-23 | 4.78 | 14.90 | 0.17 | 5.69 | 14.95 | 0.25 | |||||||||||||||||||||
| qGL4 | 4 | S4-12-2 | 3.14 | 10.68 | 0.18 | ||||||||||||||||||||||||
| qGL10 | 10 | DX-C10-8 | 3.55 | 8.88 | 0.20 | ||||||||||||||||||||||||
| 粒宽GW | qGW3 | 3 | DX-S3-17 | 2.50 | 10.06 | -0.11 | |||||||||||||||||||||||
| qGW8 | 8 | DX-C8-15 | 4.59 | 17.86 | -0.07 | 3.53 | 14.49 | -0.07 | |||||||||||||||||||||
| qGW10 | 10 | DX-C10-10 | 2.56 | 10.33 | -0.08 | ||||||||||||||||||||||||
新窗口打开|下载CSV
2.2.10 粒长 共检测到8个粒长QTL, LOD值在2.86~5.75之间, 贡献率变幅为7.12%~16.65%。qGL2位于2号染色体02-067标记处, 在临沂和南昌2个环境中都能被检测到, 贡献率分别为10.71%和7.12%; qGL3-4位于3号染色体DX-C3-23标记处, 在北京和南昌2个环境中都能被检测到, 贡献率分别为14.90%和14.95%; qGL1和qGL3-2分别位于1号染色体DX-C1-18和3号染色体indel-c3-12标记处, 仅在北京环境中被检测到, 贡献率分别为16.65%和9.22%; qGL3-1和qGL4仅在临沂环境中被检测到, 分别位于3号染色体DX-C3-8和4号染色体S4-12-2标记处, 贡献率分别为13.44%和10.68%; 在南昌环境下能够被检测到QTL有qGL3-3和qGL10, 分别位于3号染色体DX-S3-17和10号染色体DX-C10-8标记处, 贡献率分别为15.14%和8.88%。
2.2.11 粒宽 共检测到3个粒宽QTL, LOD值在2.50~4.59之间, 贡献率介于10.06%~17.86%之间。qGW8位于8号染色体DX-C8-15标记处, 在北京和临沂2个环境中都能被检测到, 贡献率分别为17.86%和14.49%; qGW3和qGW10分别位于3号染色体DX-S3-17和10号染色体DX-C10-10标记处, 仅在南昌环境下能够被检测到, 贡献率为10.06%和10.33%
2.3 QTL验证
利用已构建的次级分离群体, 对株高QTL qPH7、一次枝梗数QTL qPBPP8-2和粒宽QTL qGW10三个位点进行了验证。将含有qPH7置换系L13、qPBPP8-2置换系L78和qGW10置换系L90分别与日本晴杂交, 构建F2次级分离群体。分别考察了相应群体530个单株的株高、402个单株的一次枝梗数和198个单株的粒宽, 发现3个性状在分离群体中均呈连续分布(图3-A, C, E)。通过高通量测序, 极端矮株混池获得188,787,238个Reads, 极端高株混池获得190,156,356个Reads, 检测到602,546个SNP, Δ(All-index)在染色体上的分布如图3-B; 在5号染色体的23,053,001~25,260,000区间、6号染色体的7,388,001~20,383,000区间和7号染色体的1,727,001~3,832,000区间有3个明显的QTL, qPH7处于7号染色体区间内。极端多一次枝梗混池获得217,405,862个Reads, 极端少一次枝梗混池获得230,822,574个Reads, 检测到221,953个SNP, Δ(All-index)在染色体上的分布如图3-D; 在7号染色体的1~1,552,000区间、8号染色体的201,001~2,764,000区间以及18,259,001~25,357,000区间有3个明显的QTL, qPBPP8-2在8号染色体18,259,001~25,357,000区间内。极端窄粒混池获得200,257,918个Reads, 极端宽粒混池获得205,590,678个Reads, 检测到166,301个SNP, Δ(All-index)在染色体上的分布如图3-F; 在1号染色体的4,069,001~4,803,000区间、5号染色体的2,584,001~5,802,000区间以及10号染色体17,095,001~23,206,000区间有3个明显的QTL, qGW10在10号染色体区间内。因此, qPH7、qPBPP8-2和qGW10三个QTL是真实存在的。
图3
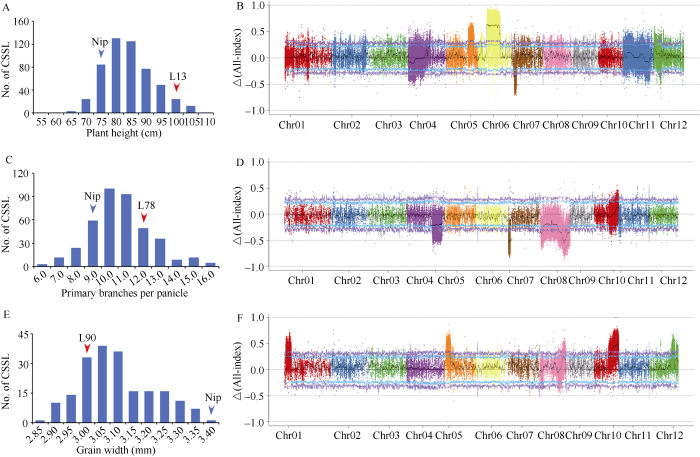 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3次级分离群体株高、一次枝梗数和粒宽性状的频率分布和?(SNP-index)曼哈顿图
A: qPH7位点次级分离群体中株高的频率分布; B: 株高极端混池?(SNP-index)曼哈顿图; C: qPBPP8-2位点次级分离群体中一次枝梗数的频率分布; D: 一次枝梗数极端混池?(SNP-index)曼哈顿图; E: qGW10位点次级分离群体中粒宽的频率分布; F: 粒宽极端混池?(SNP-index)曼哈顿图。蓝色和红色箭头分别表示日本晴和对应染色体片段置换系3个性状的表现。
Fig. 3Frequency distributions of plant height, primary branches per panicle and grain width in the secondary segregated populations and Manhattan plot of ?(SNP-index)
A: frequency distribution of plant height in secondary segregated population related of qPH7; B: ?(SNP-index) graph of plant height extreme mixed pools; C: frequency distribution of primary branches per panicle in secondary segregated population related of qPBPP8-2; D: ?(SNP-index) graph of primary branches per panicle extreme mixed pools; E: frequency distribution of grain width in secondary segregated population related of qGW10; F: ?(SNP-index) graph of grain width extreme mixed pools. The blue and red triangles represent the positions of the means of Nipponbare and its related CSSL, respectively.
3 讨论
普通野生稻是亚洲栽培稻的近缘野生种, 在长期的进化过程中, 保留了栽培稻丢失的高产优质、抗病虫、耐逆和营养高效利用等有利基因和优异单倍型, 但一些不利性状往往和有利性状紧密连锁, 限制了野生稻的利用效率, 构建普通野生稻染色体片段置换系在一定程度上可以解决这个难题。近年来, 我国水稻、特别是北方粳稻产量一直徘徊不前, 利用普通野生稻丰富的遗传多样性, 拓宽育种亲本的遗传基础, 是改良现有品种、提高水稻产量的有效途径。本实验室前期针对粳稻育种材料, 利用东乡普通野生稻为供体亲本, 日本晴为受体亲本, 构建了一套CSSL[23]。本研究在此基础上, 2019年分别在北京、临沂和南昌种植该CSSL, 考察了11个与产量相关的性状。统计分析发现, 11个性状都存在超亲分离现象(图1和表1), 而且, 有些如落粒、长芒、高植株等野生稻不利性状与有利性状连锁已打破(数据没有提供), 可以直接选择一些家系供改良粳稻育种亲本利用。CSSL不仅可以为拓宽育种亲本的遗传基础提供基因资源, 而且还是定位与克隆重要数量性状基因的理想群体, 特别是对微效QTL的发掘具有良好的效果。本研究在3地对11个产量相关性状进行了表型鉴定, 结合CSSL分子标记数据, 共检测到68个QTL, QTL贡献率介于4.67%~27.79%, 贡献率最大的是二次枝梗粒数QTL qSBGN2-1, 贡献率为27.79%; 贡献率最小的是二次枝梗粒数QTL qSBGN4-2, 贡献率仅有4.69% (表2)。检测到的一二次枝梗数、枝梗粒数、每穗粒数及粒形等驯化相关性状QTL数量较多, 也说明普通野生稻在这些性状上具有更为丰富的遗传多样性, 可以为这些性状相关基因的克隆和性状改良提供多样的基因资源。
环境显著影响数量性状QTL定位的数量和质量, 特别是产量相关性状的表现受环境影响更大。本研究在北京、临沂和南昌3地种植CSSL群体和亲本日本晴, 各地表型差异较大, 即使是高遗传力的粒长和粒宽性状也差异显著(图1), 不可避免地影响到定位QTL的可重复性。检测到的68个QTL中, 在北京环境下检测到的有29个, 临沂26个, 南昌21个; 6个QTL能在2个环境下被重复检测到, 仅有1个QTL在3个环境下被重复检测到。利用前期已构建的F2次级分离群体采用BSA法分别对株高qPH7、一次枝梗数qPBPP8-2和粒宽qGW10三个位点进行了验证, 虽然结果显示相应的3个分离群体的株高、一次枝梗数和粒宽均呈连续分布, 受多个基因控制, 但是本文检测的3个QTL在BSA分析中都能重现。因此, 一些在单环境中检测到的QTL可能也是真实可靠的(图3)。68个QTL中, 有24个QTL与已报道的QTL/基因在染色体上的位置相同或邻近。株高QTL qPH2和穗长QTL qPL2-1与Ghd2邻近[30], qPH5与EUI1邻近[31,32]。一次枝梗粒数QTL qPBGN1-1与Gn1a邻近[12]; 二次枝梗数QTL qSBPP2-2和二次枝梗粒数QTL qSBGN2-2与LRK1邻近[33]; 每穗粒数QTL qGPP6-1与gpl6.1、 qGPP11和qPBGN11与gpp11.1区间重叠[34,35]。在粒形相关QTL中, 本研究中的相关QTL大多与已克隆的粒形基因位置重叠, 例如qTGW3与qGW-3[36]、qGL2与GS2/OsGRF4[37,38,39]、qGL3-1与OsLG3[40]、qGL3-3和qGW3与GS3[16]、qGL3-4与GL3.1[41]、qGL4与OsMKKK10[42]以及qGW8和qTGW8-2与GW8[20]。此外, 穗长QTL qPL2-2、一次枝梗数QTL qPBPP8-3和一次枝梗粒数QTL qPBGN8-3、二次枝梗数QTL qSBPP4-2和每穗粒数QTL qGPP4分别与粒形基因GS2/OsGRF4、GW8和OsMKKK10位置相同或者邻近。除了24个QTL与已报道的位点/基因位置相近外, 其余44个为本研究新发现位点。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s00122-011-1657-xURLPMID:21805338 [本文引用: 1]

Rice seed size is an important agronomic trait in determining the yield potential, and four seed size related genes (GS3, GW2, qSW5/GW5 and GIF1) have been cloned in rice so far. However, the relationship among these four genes is still unclear, which will impede the process of gene pyramiding breeding program to some extent. To shade light on the relationship of above four genes, gene expression analysis was performed with GS3-RNAi, GW2-RNAi lines and CSSL of qSW5 at the transcriptional level. The results clearly showed that qSW5 and GW2 positively regulate the expression of GS3. Meanwhile, qSW5 can be down-regulated by repression of GW2 transcription. Additionally, GIF1 expression was found to be positively regulated by qSW5 but negatively by GW2 and GS3. Moreover, the allelic effects of qSW5 and GS3 were detailedly characterized based on a natural population consisting of 180 rice cultivars. It was indicated that mutual interactions exist between the two genes, in which, qSW5 affecting seed length is masked by GS3 alleles, and GS3 affecting seed width is masked by qSW5 alleles. These findings provide more insights into the molecular mechanisms underlying seed size development in rice and are likely to be useful for improving rice grain yield.
DOI:10.1093/dnares/9.1.11URLPMID:11939564 [本文引用: 1]
A rice semidwarfing gene, sd-1, known as the
DOI:10.1073/pnas.132266399URLPMID:12077303 [本文引用: 1]
The introduction of semidwarf rice (Oryza sativa L.) led to record yield increases throughout Asia in the 1960s. The major semidwarfing allele, sd-1, is still extensively used in modern rice cultivars. The phenotype of sd-1 is consistent with dwarfism that results from a deficiency in gibberellin (GA) plant growth hormones. We propose that the semidwarf (sd-1) phenotype is the result of a deficiency of active GAs in the elongating stem arising from a defective 20-oxidase GA biosynthetic enzyme. Sequence data from the rice genome was combined with previous mapping studies to locate a putative GA 20-oxidase gene (Os20ox2) at the predicted map location of sd-1 on chromosome 1. Two independent sd-1 alleles contained alterations within Os20ox2: a deletion of 280 bp within the coding region of Os20ox2 was predicted to encode a nonfunctional protein in an indica type semidwarf (Doongara), whereas a substitution in an amino acid residue (Leu-266) that is highly conserved among dioxygenases could explain loss of function of Os20ox2 in a japonica semidwarf (Calrose76). The quantification of GAs in elongating stems by GC-MS showed that the initial substrate of GA 20-oxidase activity (GA53) accumulated, whereas the content of the major product (GA20) and of bioactive GA1 was lower in semidwarf compared with tall lines. We propose that the Os20ox2 gene corresponds to the sd-1 locus.
DOI:10.1038/nature01518URLPMID:12687001 [本文引用: 1]
Tillering in rice (Oryza sativa L.) is an important agronomic trait for grain production, and also a model system for the study of branching in monocotyledonous plants. Rice tiller is a specialized grain-bearing branch that is formed on the unelongated basal internode and grows independently of the mother stem (culm) by means of its own adventitious roots. Rice tillering occurs in a two-stage process: the formation of an axillary bud at each leaf axil and its subsequent outgrowth. Although the morphology and histology and some mutants of rice tillering have been well described, the molecular mechanism of rice tillering remains to be elucidated. Here we report the isolation and characterization of MONOCULM 1 (MOC1), a gene that is important in the control of rice tillering. The moc1 mutant plants have only a main culm without any tillers owing to a defect in the formation of tiller buds. MOC1 encodes a putative GRAS family nuclear protein that is expressed mainly in the axillary buds and functions to initiate axillary buds and to promote their outgrowth.
DOI:10.1038/nature12878URLPMID:24336215 [本文引用: 1]
Strigolactones (SLs), a newly discovered class of carotenoid-derived phytohormones, are essential for developmental processes that shape plant architecture and interactions with parasitic weeds and symbiotic arbuscular mycorrhizal fungi. Despite the rapid progress in elucidating the SL biosynthetic pathway, the perception and signalling mechanisms of SL remain poorly understood. Here we show that DWARF 53 (D53) acts as a repressor of SL signalling and that SLs induce its degradation. We find that the rice (Oryza sativa) d53 mutant, which produces an exaggerated number of tillers compared to wild-type plants, is caused by a gain-of-function mutation and is insensitive to exogenous SL treatment. The D53 gene product shares predicted features with the class I Clp ATPase proteins and can form a complex with the alpha/beta hydrolase protein DWARF 14 (D14) and the F-box protein DWARF 3 (D3), two previously identified signalling components potentially responsible for SL perception. We demonstrate that, in a D14- and D3-dependent manner, SLs induce D53 degradation by the proteasome and abrogate its activity in promoting axillary bud outgrowth. Our combined genetic and biochemical data reveal that D53 acts as a repressor of the SL signalling pathway, whose hormone-induced degradation represents a key molecular link between SL perception and responses.
DOI:10.1038/nature12870URLPMID:24336200 [本文引用: 1]
Strigolactones (SLs) are a group of newly identified plant hormones that control plant shoot branching. SL signalling requires the hormone-dependent interaction of DWARF 14 (D14), a probable candidate SL receptor, with DWARF 3 (D3), an F-box component of the Skp-Cullin-F-box (SCF) E3 ubiquitin ligase complex. Here we report the characterization of a dominant SL-insensitive rice (Oryza sativa) mutant dwarf 53 (d53) and the cloning of D53, which encodes a substrate of the SCF(D3) ubiquitination complex and functions as a repressor of SL signalling. Treatments with GR24, a synthetic SL analogue, cause D53 degradation via the proteasome in a manner that requires D14 and the SCF(D3) ubiquitin ligase, whereas the dominant form of D53 is resistant to SL-mediated degradation. Moreover, D53 can interact with transcriptional co-repressors known as TOPLESS-RELATED PROTEINS. Our results suggest a model of SL signalling that involves SL-dependent degradation of the D53 repressor mediated by the D14-D3 complex.
DOI:10.1093/pcp/pci022URLPMID:15659436 [本文引用: 1]
In this study, we analyzed five tillering dwarf mutants that exhibit reduction of plant stature and an increase in tiller numbers. We show that, in the mutants, axillary meristems are normally established but the suppression of tiller bud activity is weakened. The phenotypes of tillering dwarf mutants suggest that they play roles in the control of tiller bud dormancy to suppress bud activity. However, tillering dwarf mutants show the dependence of both node position and planting density on their growth, which implies that the functions of tillering dwarf genes are independent of the developmental and environmental control of bud activity. Map-based cloning of the D3 gene revealed that it encodes an F-box leucine-trich repeat (LRR) protein orthologous to Arabidopsis MAX2/ORE9. This indicates the conservation of mechanisms controlling axillary bud activity between monocots and eudicots. We suggest that tillering dwarf mutants are suitable for the study of bud activity control in rice and believe that future molecular and genetic studies using them may enable significant progress in understanding the control of tillering and shoot branching.
DOI:10.1007/s00425-005-0007-0URLPMID:16021500 [本文引用: 1]
A rice htd-1 mutant, related to tillering and dwarfing, was characterized. We show that the htd-1 mutant increases its tiller number by releasing axillary buds from dormant stage rather than by initiating more axillary buds. The dwarf is caused by averagely reducing each internode and panicle. Based on this dwarfing pattern, the htd-1 mutant could be grouped into dn-type dwarf defined by Takeda (Gamma Field Symp 16:1, 1977). In addition, the dwarfing of the htd-1 mutant was found independent of GA based on the analyses of two GA-mediated processes. Based on the quantitative determination of IAA and ABA and application of the two hormones exogenously to the seedlings, we inferred that the high tillering capacity of the htd-1 mutant should not be attributed to a defect in the synthesis of IAA or ABA. The genetic analysis of the htd-1 mutant indicated that the phenotypes of high tillering and dwarf were controlled by a recessive gene, termed htd1. By map-based cloning, the htd1 gene was fine mapped in a 30-kb DNA region on chromosome 4. Sequencing the target DNA region and comparing the counterpart DNA sequences between the htd-1 mutant and other rice varieties revealed a nucleotide substitution corresponding to an amino acid substitution from prolin to leucine in a predicted rice gene, OsCCD7, the rice orthologous gene of AtMAX3/CCD7. With the evidence of the association between the presence of one amino acid change in OsCCD7 and the abnormal phenotypes of the htd-1 mutant, OsCCD7 was identified as the candidate of the HTD1 gene.
DOI:10.1038/ng.352URLPMID:19305410 [本文引用: 1]
Grain yield is controlled by quantitative trait loci (QTLs) derived from natural variations in many crop plants. Here we report the molecular characterization of a major rice grain yield QTL that acts through the determination of panicle architecture. The dominant allele at the DEP1 locus is a gain-of-function mutation causing truncation of a phosphatidylethanolamine-binding protein-like domain protein. The effect of this allele is to enhance meristematic activity, resulting in a reduced length of the inflorescence internode, an increased number of grains per panicle and a consequent increase in grain yield. This allele is common to many Chinese high-yielding rice varieties and likely represents a relatively recent introduction into the cultivated rice gene pool. We also show that a functionally equivalent allele is present in the temperate cereals and seems to have arisen before the divergence of the wheat and barley lineages.
DOI:10.1126/science.1113373URLPMID:15976269 [本文引用: 2]
Most agriculturally important traits are regulated by genes known as quantitative trait loci (QTLs) derived from natural allelic variations. We here show that a QTL that increases grain productivity in rice, Gn1a, is a gene for cytokinin oxidase/dehydrogenase (OsCKX2), an enzyme that degrades the phytohormone cytokinin. Reduced expression of OsCKX2 causes cytokinin accumulation in inflorescence meristems and increases the number of reproductive organs, resulting in enhanced grain yield. QTL pyramiding to combine loci for grain number and plant height in the same genetic background generated lines exhibiting both beneficial traits. These results provide a strategy for tailormade crop improvement.
[本文引用: 1]
DOI:10.1105/tpc.16.00379URLPMID:27634315 [本文引用: 1]
Cultivated rice (Oryza sativa) was domesticated from wild rice (Oryza rufipogon), which typically displays fewer grains per panicle and longer grains than cultivated rice. In addition, wild rice has long awns, whereas cultivated rice has short awns or lacks them altogether. These changes represent critical events in rice domestication. Here, we identified a major gene, GRAIN NUMBER, GRAIN LENGTH AND AWN DEVELOPMENT1 (GAD1), that regulates those critical changes during rice domestication. GAD1 is located on chromosome 8 and is predicted to encode a small secretary signal peptide belonging to the EPIDERMAL PATTERNING FACTOR-LIKE family. A frame-shift insertion in gad1 destroyed the conserved cysteine residues of the peptide, resulting in a loss of function, and causing the increased number of grains per panicle, shorter grains, and awnless phenotype characteristic of cultivated rice. Our findings provide a useful paradigm for revealing functions of peptide signal molecules in plant development and helps elucidate the molecular basis of rice domestication.
DOI:10.1038/s41467-017-01501-8URLPMID:29133783 [本文引用: 1]
During rice domestication and improvement, increasing grain yield to meet human needs was the primary objective. Rice grain yield is a quantitative trait determined by multiple genes, but the molecular basis for increased grain yield is still unclear. Here, we show that NUMBER OF GRAINS 1 (NOG1), which encodes an enoyl-CoA hydratase/isomerase, increases the grain yield of rice by enhancing grain number per panicle without a negative effect on the number of panicles per plant or grain weight. NOG1 can significantly increase the grain yield of commercial high-yield varieties: introduction of NOG1 increases the grain yield by 25.8% in the NOG1-deficient rice cultivar Zhonghua 17, and overexpression of NOG1 can further increase the grain yield by 19.5% in the NOG1-containing variety Teqing. Interestingly, NOG1 plays a prominent role in increasing grain number, but does not change heading date or seed-setting rate. Our findings suggest that NOG1 could be used to increase rice production.
DOI:10.1007/s00122-008-0913-1URLPMID:19020856 [本文引用: 2]
Comparative sequencing of GS3, the most important grain length (GL) QTL, has shown that differentiation of rice GL might be principally due to a single nucleotide polymorphism (SNP) between C and A in the second exon. A total of 180 varieties representing a wide range of rice germplasm were used for association analysis between C-A mutation and GL in order to confirm the potential causal mutation. A cleaved amplified polymorphic sequence (CAPS) marker, SF28, was developed based on the C-A polymorphism in the GS3 gene. A total of 142 varieties carried allele C with GL from 6.4 to 8.8 mm, while the remaining 38 varieties carried allele A with GL from 8.8 to 10.7 mm. Twenty-four unlinked SSR markers were selected to genotype 180 varieties for population structure analysis. Population structure was observed when the population was classified to three subpopulations. Average GL of either genotype A or genotype C within japonica among the three subpopulations had no significant difference from that in indica, respectively, although indica rice had longer grains on average than japonica in the 180 varieties. However, genotype C always had longer grain length on average than genotype A among three subpopulations. The mutation could explain 79.1, 66.4 and 34.7% of GL variation in the three subpopulations, respectively. These results clearly confirmed the mutation between C and A was highly associated with GL. The SF28 could be a functional marker for improvement of rice grain length.
DOI:10.1038/ng2014URLPMID:17417637 [本文引用: 1]
Grain weight is one of the most important components of grain yield and is controlled by quantitative trait loci (QTLs) derived from natural variations in crops. However, the molecular roles of QTLs in the regulation of grain weight have not been fully elucidated. Here, we report the cloning and characterization of GW2, a new QTL that controls rice grain width and weight. Our data show that GW2 encodes a previously unknown RING-type protein with E3 ubiquitin ligase activity, which is known to function in the degradation by the ubiquitin-proteasome pathway. Loss of GW2 function increased cell numbers, resulting in a larger (wider) spikelet hull, and it accelerated the grain milk filling rate, resulting in enhanced grain width, weight and yield. Our results suggest that GW2 negatively regulates cell division by targeting its substrate(s) to proteasomes for regulated proteolysis. The functional characterization of GW2 provides insight into the mechanism of seed development and is a potential tool for improving grain yield in crops.
DOI:10.1038/ng.2612URLPMID:23583977 [本文引用: 1]
Increases in the yield of rice, a staple crop for more than half of the global population, are imperative to support rapid population growth. Grain weight is a major determining factor of yield. Here, we report the cloning and functional analysis of THOUSAND-GRAIN WEIGHT 6 (TGW6), a gene from the Indian landrace rice Kasalath. TGW6 encodes a novel protein with indole-3-acetic acid (IAA)-glucose hydrolase activity. In sink organs, the Nipponbare tgw6 allele affects the timing of the transition from the syncytial to the cellular phase by controlling IAA supply and limiting cell number and grain length. Most notably, loss of function of the Kasalath allele enhances grain weight through pleiotropic effects on source organs and leads to significant yield increases. Our findings suggest that TGW6 may be useful for further improvements in yield characteristics in most cultivars.
DOI:10.1038/ng.977URLPMID:22019783 [本文引用: 1]
Increasing crop yield is one of the most important goals of plant science research. Grain size is a major determinant of grain yield in cereals and is a target trait for both domestication and artificial breeding(1). We showed that the quantitative trait locus (QTL) GS5 in rice controls grain size by regulating grain width, filling and weight. GS5 encodes a putative serine carboxypeptidase and functions as a positive regulator of grain size, such that higher expression of GS5 is correlated with larger grain size. Sequencing of the promoter region in 51 rice accessions from a wide geographic range identified three haplotypes that seem to be associated with grain width. The results suggest that natural variation in GS5 contributes to grain size diversity in rice and may be useful in improving yield in rice and, potentially, other crops(2).
DOI:10.1038/ng.2327URLPMID:22729225 [本文引用: 2]
Grain size and shape are important components of grain yield and quality and have been under selection since cereals were first domesticated. Here, we show that a quantitative trait locus GW8 is synonymous with OsSPL16, which encodes a protein that is a positive regulator of cell proliferation. Higher expression of this gene promotes cell division and grain filling, with positive consequences for grain width and yield in rice. Conversely, a loss-of-function mutation in Basmati rice is associated with the formation of a more slender grain and better quality of appearance. The correlation between grain size and allelic variation at the GW8 locus suggests that mutations within the promoter region were likely selected in rice breeding programs. We also show that a marker-assisted strategy targeted at elite alleles of GS3 and OsSPL16 underlying grain size and shape can be effectively used to simultaneously improve grain quality and yield.
DOI:10.1038/ng.3346URLPMID:26147619 [本文引用: 1]
Copy number variants (CNVs) are associated with changes in gene expression levels and contribute to various adaptive traits. Here we show that a CNV at the Grain Length on Chromosome 7 (GL7) locus contributes to grain size diversity in rice (Oryza sativa L.). GL7 encodes a protein homologous to Arabidopsis thaliana LONGIFOLIA proteins, which regulate longitudinal cell elongation. Tandem duplication of a 17.1-kb segment at the GL7 locus leads to upregulation of GL7 and downregulation of its nearby negative regulator, resulting in an increase in grain length and improvement of grain appearance quality. Sequence analysis indicates that allelic variants of GL7 and its negative regulator are associated with grain size diversity and that the CNV at the GL7 locus was selected for and used in breeding. Our work suggests that pyramiding beneficial alleles of GL7 and other yield- and quality-related genes may improve the breeding of elite rice varieties.
DOI:10.1038/s41467-018-03616-yURLPMID:29588443 [本文引用: 1]
Identification of grain shape determining genes can facilitate breeding of rice cultivars with optimal grain shape and appearance quality. Here, we identify GS9 (Grain Shape Gene on Chromosome 9) gene by map-based cloning. The gs9 null mutant has slender grains, while overexpression GS9 results in round grains. GS9 encodes a protein without known conserved functional domain. It regulates grain shape by altering cell division. The interaction of GS9 and ovate family proteins OsOFP14 and OsOFP8 is modulated by OsGSK2 kinase, a key regulator of the brassinosteroids signaling pathway. Genetic interaction analysis reveals that GS9 functions independently from other previously identified grain size genes. Introducing the gs9 allele into elite rice cultivars significantly improves grain shape and appearance quality. It suggests potential application of gs9, alone or in combination with other grain size determining genes, in breeding of rice varieties with optimized grain shape.
DOI:10.1016/j.plaphy.2019.09.041URLPMID:31593900 [本文引用: 3]
Dongxiang common wild rice (Oryza rufipogon Griff.) is believed to have the northernmost distribution of any wild rice species. Many favorable genes were lost during rice domestication, and Dongxiang common wild rice is a potential source of many genes related to biological and abiotic stress resistance and high grain yield. Despite its importance, japonica rice has not yet been used as a background material for the construction of introgression lines carrying Dongxiang common wild rice chromosome segments. In this study, we used Dongxiang common wild rice as the donor parent and Nipponbare (Oryza sativa L), a reference-sequence japonica cultivar, as the recurrent parent to develop a set of 104 chromosome segment substitution lines (CSSLs) through crossing and backcrossing with marker-assisted selection based on 203 polymorphic molecular markers evenly distributed across 12 chromosomes. The 104 CSSLs covered 87.94% of the genome of Dongxiang common wild rice. Each CSSL contained an average of four introgressed segments, and the average segment length was 3.3Mb. Quantitative trait locus (QTL) analysis of the panicle-related traits of the CSSLs at two sites (Beijing and Nanchang, Jiangxi Province) revealed 18 QTLs for eight traits. Among these 18 QTLs were two known grain length- and width-related genes and four novel QTLs. In addition, two QTLs were verified, and two novel QTLs were identified, for panicle neck length, a domestication-related trait. The developed CSSLs potentially represent a valuable population for detecting favorable genes in common wild rice and creating germplasm resources for japonica rice breeding.
[本文引用: 1]
[本文引用: 1]
DOI:10.1017/S0016672306008408URLPMID:17125584 [本文引用: 1]
Chromosome segment substitution (CSS) lines have the potential for use in QTL fine mapping and map-based cloning. The standard t-test used in the idealized case that each CSS line has a single segment from the donor parent is not suitable for non-idealized CSS lines carrying several substituted segments from the donor parent. In this study, we present a likelihood ratio test based on stepwise regression (RSTEP-LRT) that can be used for QTL mapping in a population consisting of non-idealized CSS lines. Stepwise regression is used to select the most important segments for the trait of interest, and the likelihood ratio test is used to calculate the LOD score of each chromosome segment. This method is statistically equivalent to the standard t-test with idealized CSS lines. To further improve the power of QTL mapping, a method is proposed to decrease multicollinearity among markers (or chromosome segments). QTL mapping with an example CSS population in rice consisting of 65 non-idealized CSS lines and 82 chromosome segments indicated that a total of 18 segments on eight of the 12 rice chromosomes harboured QTLs affecting grain length under the LOD threshold of 2.5. Three major stable QTLs were detected in all eight environments. Some minor QTLs were not detected in all environments, but they could increase or decrease the grain length constantly. These minor genes are also useful in marker-assisted gene pyramiding.
[本文引用: 1]
[本文引用: 1]
DOI:10.1093/bioinformatics/btp324URLPMID:19451168 [本文引用: 1]
MOTIVATION: The enormous amount of short reads generated by the new DNA sequencing technologies call for the development of fast and accurate read alignment programs. A first generation of hash table-based methods has been developed, including MAQ, which is accurate, feature rich and fast enough to align short reads from a single individual. However, MAQ does not support gapped alignment for single-end reads, which makes it unsuitable for alignment of longer reads where indels may occur frequently. The speed of MAQ is also a concern when the alignment is scaled up to the resequencing of hundreds of individuals. RESULTS: We implemented Burrows-Wheeler Alignment tool (BWA), a new read alignment package that is based on backward search with Burrows-Wheeler Transform (BWT), to efficiently align short sequencing reads against a large reference sequence such as the human genome, allowing mismatches and gaps. BWA supports both base space reads, e.g. from Illumina sequencing machines, and color space reads from AB SOLiD machines. Evaluations on both simulated and real data suggest that BWA is approximately 10-20x faster than MAQ, while achieving similar accuracy. In addition, BWA outputs alignment in the new standard SAM (Sequence Alignment/Map) format. Variant calling and other downstream analyses after the alignment can be achieved with the open source SAMtools software package. AVAILABILITY: http://maq.sourceforge.net.
[本文引用: 1]
DOI:10.1111/tpj.12105URLPMID:23289725 [本文引用: 1]
The majority of agronomically important crop traits are quantitative, meaning that they are controlled by multiple genes each with a small effect (quantitative trait loci, QTLs). Mapping and isolation of QTLs is important for efficient crop breeding by marker-assisted selection (MAS) and for a better understanding of the molecular mechanisms underlying the traits. However, since it requires the development and selection of DNA markers for linkage analysis, QTL analysis has been time-consuming and labor-intensive. Here we report the rapid identification of plant QTLs by whole-genome resequencing of DNAs from two populations each composed of 20-50 individuals showing extreme opposite trait values for a given phenotype in a segregating progeny. We propose to name this approach QTL-seq as applied to plant species. We applied QTL-seq to rice recombinant inbred lines and F2 populations and successfully identified QTLs for important agronomic traits, such as partial resistance to the fungal rice blast disease and seedling vigor. Simulation study showed that QTL-seq is able to detect QTLs over wide ranges of experimental variables, and the method can be generally applied in population genomics studies to rapidly identify genomic regions that underwent artificial or natural selective sweeps.
DOI:10.1093/jxb/erw344URLPMID:27638689 [本文引用: 1]
CONSTANS (CO)-like genes have been intensively investigated for their roles in the regulation of photoperiodic flowering, but very limited information has been reported on their functions in other biological processes. Here, we found that a CO-like gene, Ghd2 (Grain number, plant height, and heading date2), which can increase the yield potential under normal growth condition just like its homologue Ghd7, is involved in the regulation of leaf senescence and drought resistance. Ghd2 is expressed mainly in the rice (Oryza sativa) leaf with the highest level detected at the grain-filling stage, and it is down-regulated by drought stress conditions. Overexpression of Ghd2 resulted in significantly reduced drought resistance, while its knockout mutant showed the opposite phenotype. The earlier senescence symptoms and the transcript up-regulation of many senescence-associated genes (SAGs) in Ghd2-overexpressing transgenic rice plants under drought stress conditions indicate that Ghd2 plays essential roles in accelerating drought-induced leaf senescence in rice. Moreover, developmental and dark-induced leaf senescence was accelerated in the Ghd2-overexpressing rice and delayed in the ghd2 mutant. Several SAGs were confirmed to be regulated by Ghd2 using a transient expression system in rice protoplasts. Ghd2 interacted with several regulatory proteins, including OsARID3, OsPURalpha, and three 14-3-3 proteins. OsARID3 and OsPURalpha showed expression patterns similar to Ghd2 in rice leaves, with the highest levels at the grain-filling stage, whereas OsARID3 and the 14-3-3 genes responded differently to drought stress conditions. These results indicate that Ghd2 functions as a regulator by integrating environmental signals with the senescence process into a developmental programme through interaction with different proteins.
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/cr.2008.28URLPMID:18268540 [本文引用: 1]
The rice Eui (ELONGATED UPPERMOST INTERNODE) gene encodes a cytochrome P450 monooxygenase that deactivates bioactive gibberellins (GAs). In this study, we investigated controlled expression of the Eui gene and its role in plant development. We found that Eui was differentially induced by exogenous GAs and that the Eui promoter had the highest activity in the vascular bundles. The eui mutant was defective in starch granule development in root caps and Eui overexpression enhanced starch granule generation and gravity responses, revealing a role for GA in root starch granule development and gravity responses. Experiments using embryoless half-seeds revealed that RAmy1A and GAmyb were highly upregulated in eui aleurone cells in the absence of exogenous GA. In addition, the GA biosynthesis genes GA3ox1 and GA20ox2 were downregulated and GA2ox1 was upregulated in eui seedlings. These results indicate that EUI is involved in GA homeostasis, not only in the internodes at the heading stage, but also in the seedling stage, roots and seeds. Disturbing GA homeostasis affected the expression of the GA signaling genes GID1 (GIBBERELLIN INSENSITIVE DWARF 1), GID2 and SLR1. Transgenic RNA interference of the Eui gene effectively increased plant height and improved heading performance. By contrast, the ectopic expression of Eui under the promoters of the rice GA biosynthesis genes GA3ox2 and GA20ox2 significantly reduced plant height. These results demonstrate that a slight increase in Eui expression could dramatically change rice morphology, indicating the practical application of the Eui gene in rice molecular breeding for a high yield potential.
DOI:10.1111/j.1467-7652.2009.00428.xURLPMID:19619185 [本文引用: 1]
In rice (Oryza sativa L.), the number of panicles, spikelets per panicle and grain weight are important components of grain yield. These characteristics are controlled by quantitative trait loci (QTLs) and are derived from variation inherent in crops. As a result of the complex genetic basis of these traits, only a few genes involved in their control have been cloned and characterized. We have previously map-cloned a gene cluster including eight leucine-rich repeat receptor-like kinase (LRK) genes in Dongxiang wild rice (Oryza rufipogon Griff.), which increased the grain yield by 16%. In the present study, we characterized the LRK1 gene, which was contained in the donor parent (Dongxiang wild rice) genome and absent from the recurrent parent genome (Guichao2, Oryza sativa L. ssp. indica). Our data showed that rice LRK1 is a plasma membrane protein expressed constitutively in leaves, young panicles, roots and culms. The over-expression of rice LRK1 results in increased panicles, spikelets per panicle, weight per grain and enhanced cellular proliferation, leading to a 27.09% increase in total grain yield per plant. The increased number of panicles and spikelets per panicle are associated with increased branch number. Our data suggest that rice LRK1 regulates rice branch number by enhancing cellular proliferation. The functional characterization of rice LRK1 facilitates an understanding of the mechanisms involved in cereal crop yield, and may have utility in improving grain yield in cereal crops.
[本文引用: 1]
DOI:10.1007/s00122-003-1270-8URLPMID:12736777 [本文引用: 1]
3.6 or a composite interval mapping LOD>3.9) were identified. For the traits measured in multiple environments, 47% of the QTLs were detected in at least two environments. The O. rufipogon allele was favorable for 53% of the yield and yield component QTLs, including loci for yield, grains per panicle, panicle length, and grain weight. Morphological traits related to the domestication process and/or weedy characteristics, including plant height, shattering, tiller type and awns, were found clustered on chromosomes 1 and 4. Comparisons to previous studies involving wild x cultivated crosses revealed O. rufipogon alleles with stable effects in multiple genetic backgrounds and environments, several of which have not been detected in studies between Oryza sativa cultivars, indicating potentially novel alleles from O. rufipogon. Some O. rufipogon-derived QTLs, however, were in similar regions as previously reported QTLs from Oryza sativa cultivars, providing evidence for conservation of these QTLs across the Oryza genus. In addition, several QTLs for grain weight, plant height, and flowering time were localized to putative homeologous regions in maize where QTLs for these traits have been previously reported, supporting the hypothesis of functional conservation of QTLs across the grasses.]]>
[本文引用: 1]
DOI:10.1038/nplants.2015.195URLPMID:27250747 [本文引用: 1]
Given the continuously growing population and decreasing arable land, food shortage is becoming one of the most serious global problems in this century(1). Grain size is one of the determining factors for grain yield and thus is a prime target for genetic breeding(2,3). Although a number of quantitative trait loci (QTLs) associated with rice grain size have been identified in the past decade, mechanisms underlying their functions remain largely unknown(4,5). Here we show that a grain-length-associated QTL, GL2, has the potential to improve grain weight and grain yield up to 27.1% and 16.6%, respectively. We also show that GL2 is allelic to OsGRF4 and that it contains mutations in the miR396 targeting sequence. Because of the mutation, GL2 has a moderately increased expression level, which consequently activates brassinosteroid responses by upregulating a large number of brassinosteroid-induced genes to promote grain development. Furthermore, we found that GSK2, the central negative regulator of rice brassinosteroid signalling, directly interacts with OsGRF4 and inhibits its transcription activation activity to mediate the specific regulation of grain length by the hormone. Thus, this work demonstrates the feasibility of modulating specific brassinosteroid responses to improve plant productivity.
DOI:10.1038/nplants.2015.203URLPMID:27250749 [本文引用: 1]
An increase in grain yield is crucial for modern agriculture(1). Grain size is one of the key components of grain yield in rice and is regulated by quantitative trait loci (QTLs)(2,3). Exploring new QTLs for grain size will help breeders develop elite rice varieties with higher yields(3,4). Here, we report a new semi-dominant QTL for grain size and weight (GS2) in rice, which encodes the transcription factor OsGRF4 (GROWTH-REGULATING FACTOR 4) and is regulated by OsmiR396. We demonstrate that a 2 bp substitution mutation in GS2 perturbs OsmiR396-directed regulation of GS2, resulting in large and heavy grains and increased grain yield. Further results reveal that GS2 interacts with the transcription coactivitors OsGIF1/2/3, and overexpression of OsGIF1 increases grain size and weight. Thus, our findings define the regulatory mechanism of GS2, OsGIFs and OsmiR396 in grain size and weight control, suggesting this pathway could be used to increase yields in crops.
DOI:10.1111/jipb.12473URL [本文引用: 1]
PANICLE TRAITS 2 (PT2), a novel gene from the Indica rice Chuandali (CDL), is reported. PT2 is synonymous with Growth-Regulating Factor 4 (OsGRF4), which encodes a growth-regulating factor that positively regulates grain shape and panicle length and negatively regulates seed shattering. Higher expression of OsGRF4 is correlated with larger grain, longer panicle and lower seed shattering. A unique OsGRF4 mutation, which occurs at the OsmiRNA396 target site of OsGRF4, seems to be associated with high levels of OsGRF4 expression, and results in phenotypic difference. Further research showed that OsGRF4 regulated two cytokinin dehydrogenase precursor genes (CKX5 and CKX1) resulting in increased cytokinin levels, which might affect the panicle traits. High storage capacity and moderate seed shattering of OsGRF4 may be useful in high-yield breeding and mechanized harvesting of rice. Our findings provide additional insight into the molecular basis of panicle growth.]]>
DOI:10.1186/s12915-017-0365-7URLPMID:28385155 [本文引用: 1]
BACKGROUND: Most agronomic traits in rice are complex and polygenic. The identification of quantitative trait loci (QTL) for grain length is an important objective of rice genetic research and breeding programs. RESULTS: Herein, we identified 99 QTL for grain length by GWAS based on approximately 10 million single nucleotide polymorphisms from 504 cultivated rice accessions (Oryza sativa L.), 13 of which were validated by four linkage populations and 92 were new loci for grain length. We scanned the Ho (observed heterozygosity per locus) index of coupled-parents of crosses mapping the same QTL, based on linkage and association mapping, and identified two new genes for grain length. We named this approach as Ho-LAMap. A simulation study of six known genes showed that Ho-LAMap could mine genes rapidly across a wide range of experimental variables using deep-sequencing data. We used Ho-LAMap to clone a new gene, OsLG3, as a positive regulator of grain length, which could improve rice yield without influencing grain quality. Sequencing of the promoter region in 283 rice accessions from a wide geographic range identified four haplotypes that seem to be associated with grain length. Further analysis showed that OsLG3 alleles in the indica and japonica evolved independently from distinct ancestors and low nucleotide diversity of OsLG3 in indica indicated artificial selection. Phylogenetic analysis showed that OsLG3 might have much potential value for improvement of grain length in japonica breeding. CONCLUSIONS: The results demonstrated that Ho-LAMap is a potential approach for gene discovery and OsLG3 is a promising gene to be utilized in genomic assisted breeding for rice cultivar improvement.
DOI:10.1073/pnas.1219776110URLPMID:23236132 [本文引用: 1]
Grain size and shape are important components determining rice grain yield, and they are controlled by quantitative trait loci (QTLs). Here, we report the cloning and functional characterization of a major grain length QTL, qGL3, which encodes a putative protein phosphatase with Kelch-like repeat domain (OsPPKL1). We found a rare allele qgl3 that leads to a long grain phenotype by an aspartate-to-glutamate transition in a conserved AVLDT motif of the second Kelch domain in OsPPKL1. The rice genome has other two OsPPKL1 homologs, OsPPKL2 and OsPPKL3. Transgenic studies showed that OsPPKL1 and OsPPKL3 function as negative regulators of grain length, whereas OsPPKL2 as a positive regulator. The Kelch domains are essential for the OsPPKL1 biological function. Field trials showed that the application of the qgl3 allele could significantly increase grain yield in both inbred and hybrid rice varieties, due to its favorable effect on grain length, filling, and weight.
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}