 ,1,2,3, 张力岚1,2,3, 祁建民1,2, 张列梅1, 张立武,1,2,3,*
,1,2,3, 张力岚1,2,3, 祁建民1,2, 张列梅1, 张立武,1,2,3,*Genomics and genetic improvement in main bast fiber crops: advances and perspectives
XU Yi,1,2,3, ZHANG Li-Lan1,2,3, QI Jian-Min1,2, ZHANG Lie-Mei1, ZHANG Li-Wu,1,2,3,*通讯作者:
收稿日期:2020-05-1接受日期:2021-01-11网络出版日期:2021-06-12
| 基金资助: |
Received:2020-05-1Accepted:2021-01-11Online:2021-06-12
| Fund supported: |
作者简介 About authors
E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (706KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
徐益, 张力岚, 祁建民, 张列梅, 张立武. 主要麻类作物基因组学与遗传改良: 现状与展望[J]. 作物学报, 2021, 47(6): 997-1019. doi:10.3724/SP.J.1006.2021.04121
XU Yi, ZHANG Li-Lan, QI Jian-Min, ZHANG Lie-Mei, ZHANG Li-Wu.
黄麻(Corchorus spp.)、红麻(Hibiscus cannabinus)、苎麻(Boehmeria spp.)、亚麻(Linum usitatissimum)与工业大麻(Cannabis sativa L.)在种植历史上曾经超过300万公顷面积, 属于大面积栽培的麻类作物(以下简称主要麻类作物), 与粮、棉、油、菜并列为第五大作物群, 其种植历史可追溯到远古新石器至尧舜时代[1,2]。主要麻类作物是重要的天然韧皮部纤维作物, 其韧皮部纤维(麻皮)主要用于麻线、麻绳、麻袋、麻布等包装用纺织, 还用于家饰板材等多功能用途; 其天然活性成分在食品、日用、医药等领域价值逐步开发, 推动生命健康产业发展。主要麻类作物分布范围广, 在热带、亚热带、温带和寒带均有分布。我国的华南、长江流域、黄淮流域、淮河流域到东北(辽宁、吉林、黑龙江)、西北(新疆、宁夏、内蒙古)等均可以种植。主要麻类作物生物学产量高, 且耐盐碱、耐干旱、耐洪涝、适应性广, 不与粮、棉、油、菜争好地, 可用于盐碱地或重金属污染农田的植物修复[1], 发展主要麻类作物将带动我国逆境农业和麻类作物多功能利用的发展。
自1986年提出基因组学的概念, 其相关研究不断深入并被广泛接受, 目前基因组学已成为生命科学领域活跃且有影响的科学[3]。模式植物拟南芥[4]率先完成全基因组测序, 这是第一个完成的植物基因组序列, 随后水稻[5]也完成全基因组测序。随着测序技术的发展, 越来越多的植物进行了全基因组测序, 其中包括主要麻类作物(黄麻[6,7]、红麻[8]、苎麻[9,10]、亚麻[11,12]和工业大麻[13,14])。这些主要麻类作物的全基因组测序, 可以提供该作物全基因组的遗传信息, 尤其是对控制重要农艺性状的位点或基因的深入挖掘, 以及对调控复杂性状的分子网络的解析奠定了重要基础。
本文旨在总结主要麻类作物基因组学和遗传改良研究的最新进展, 对于引领加快麻类作物遗传育种研究具有重要意义。
1 主要麻类作物全基因组测序研究概况
以联合国粮食及农业组织的2007年至2018年数据来看, 在世界范围内, 黄麻和红麻在收获面积、产量和农业生产价值均占据麻类作物之首, 其次是亚麻、苎麻和工业大麻。其中我国黄麻和红麻的收获面积、产量和农业生产价值居世界第三(图1)。主要麻类作物基因组测序从2011年到2020年陆续公布, 分别涉及锦葵科、荨麻科、亚麻科和大麻科, 基因组最小的是圆果种黄麻336 Mb, 最大的是红麻1078 Mb (表1)。图1
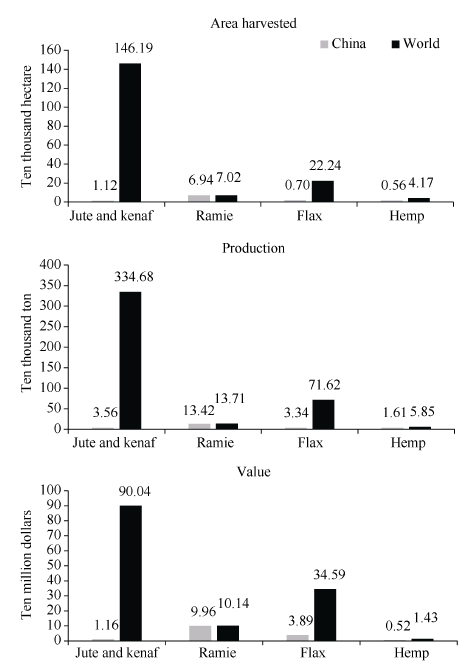 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1主要麻类作物在世界上的收获面积、产量和价值
收获面积和产量取自2009年至2018年数据平均值, 价值取自2007年至2016年数据平均值, 数据均来自联合国粮食及农业组织。
Fig. 1Harvesting area, production and value of main bast fiber crops in the world
The harvesting area and production are taken from the average values from 2009 to 2018, the values are from the average data from 2007 to 2016, and the data are all from the Food and Agriculture Organization of the United Nations.
Table 1
表1
表1主要麻类作物的基因组信息
Table 1
| 作物 Crop | 测序技术 Sequencing technology | 基因组大小 Genome size (Mb) | 科 Families | 染色体数目 Number of chromosomes | 繁殖方式 Reproduction mode | 文献 Reference | |
|---|---|---|---|---|---|---|---|
| 黄麻 Corchorus capsularis | 圆果种 C. capsularis | 二代测序 NGS | 338 | 锦葵科 Malvaceae | 2n=14 | 自花授粉 Self-pollination | [6] |
| 长果种 C. olitorius | 445 | 常异花授粉 Often cross-pollination | |||||
| 圆果种 C. capsularis | 三代测序 TGS | 336 | [7] | ||||
| 长果种 C. olitorius | 361 | ||||||
| 红麻 Hibiscus cannabinus | 二代+三代测序技术 NGS and TGS | 1078 | 锦葵科 Malvaceae | 2n=36 | 常异花授粉 Often cross-pollination | [8] | |
| 苎麻 Boehmeria nivea | 二代测序 NGS | 342 | 荨麻科 Urticaceae | 2n=28 | 无性繁殖 Asexual reproduction/异花授粉Cross-pollination | [9] | |
| 二代测序 NGS | 336 | [10] | |||||
| 亚麻 Linum usitatissimum | 二代测序 NGS | 373 | 亚麻科 Linaceae | 2n=30 | 自花授粉 Self-pollination | [11] | |
| 二代测序 NGS | 306,304,294 | [12] | |||||
| 工业大麻 Cannabis sativa | 二代测序 NGS | 534 | 大麻科 Cannabinaceae | 2n=20 | 异花授粉 Cross-pollination | [13] | |
| 二代测序 NGS | 808 | [14] | |||||
新窗口打开|下载CSV
1.1 黄麻基因组
黄麻基因组草图于2017年发表[6], 经组装发现, 长果种O-4和圆果种CVL-1基因组大小分别为445 Mb (13.04 Gb raw data, scaffold N50为3.3 Mb)和338 Mb (13.69 Gb raw data, scaffold N50为4.1 Mb), 分别有约8.4%和17.8%的数据无法覆盖, 表明该研究只是得到了黄麻基因组草图。为了获得高质量的黄麻参考基因组, 福建农林大学于2015年开展黄麻转录组[17]、基因组调研图和流式细胞仪分析。在此基础上, 以黄麻国家区域试验对照品种长果种宽叶长果和圆果种黄麻179为材料, 以三代测序技术 PacBio RS II为主体, Hi-C染色体构象捕获技术等技术辅助, 获得了长果种和圆果种黄麻染色体级别的参考基因组, 其基因组大小分别为361 Mb和336 Mb [7]。与已发表的黄麻长果种O-4和圆果种CVL-1基因组相比, 组装指标有了极大的提升。黄麻全基因组的系统发育树显示[6], 圆果种和长果种黄麻均与可可和雷蒙德氏棉聚为一类, 表明黄麻属于锦葵科(Malvaceae)。需要指出的是, 关于黄麻属于锦葵科还是椴树科(Tiliaceae)一直以来存在争议。为了验证这一结论, 考虑到叶绿体表现为母系遗传, 其基因组被广泛应用于植物的系统发育关系分析, 整理圆果种黄麻和长果种黄麻[15]以及红麻[16]的叶绿体基因组的系统发育关系结果发现, 黄麻圆果种和长果种在亲缘关系上与棉属更近(附图1)。这进一步佐证黄麻属于锦葵科, 而不是基于植物形态学分类的椴树科。
附图1
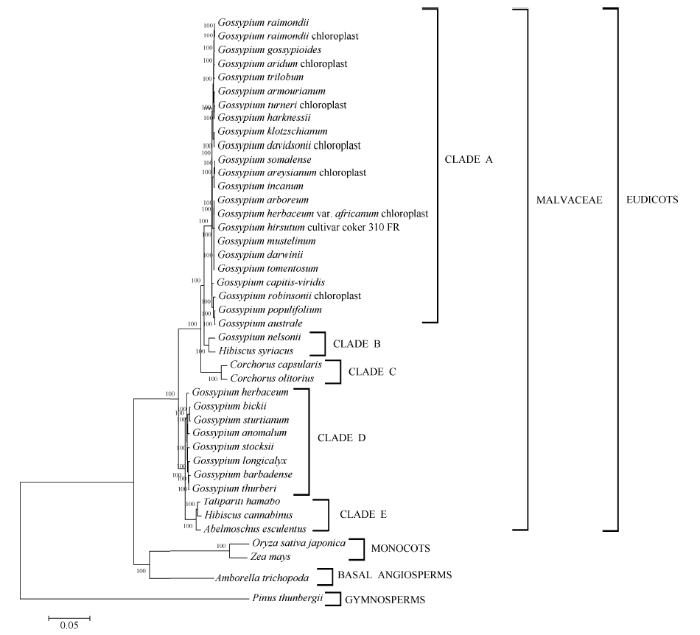 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT附图1基于叶绿体基因组序列的圆果种黄麻和长果种黄麻与锦葵科植物的系统发育关系
Fig. S1Phylogenetic relationship of Corchorus capsularis and Corchorus olitorius with species of the Malvaceae family based on the chloroplast genome sequences
1.2 红麻基因组
红麻(Hibiscus cannabinus, 2n=36)是锦葵科木槿属的一年生常异花授粉作物, 属于短日照喜温类型。2020年福建农林大学率先绘制高质量红麻参考基因组图谱[8], 该研究以红麻优良品种福红952为材料, 采用二代加三代的测序策略, 同时结合Hi-C染色体构象捕获技术和高密度遗传图谱, 完成红麻全基因组测序和组装工作, 其基因组大小约为1078 Mb, contig N50为2.73 Mb, 共鉴定到66,004个基因。红麻与拟南芥、水稻、亚麻和棉花的蛋白编码基因比较发现, 2195个基因是红麻所特有的, 包括67个NAC和47个MYB转录因子。比较基因组学分析发现, 红麻与雷蒙德氏棉在分化后存在一次独立的全基因组复制事件(whole genome duplications, WGD), 导致红麻绝大多数基因拷贝数加倍。利用核心种质重测序数据, 群体遗传学分析显示, 红麻起源于非洲, 沿着非洲南部、非洲西部的路线传播到亚洲, 并在传播过程中发生了2次驯化事件。在参考基因组基础上, 该研究确定了控制红麻叶形的基因为LATE MERISTEM IDENTITY 1 (LMI1)转录因子, 并用病毒诱导的基因沉默(virus-induced gene silencing, VIGS)验证了其基因功能。1.3 苎麻基因组
苎麻(Boehmeria nivea, 2n=28)为荨麻科苎麻属多年生宿根性草本植物, 单性花, 雌雄同株, 花序复穗状, 雄花花序生在茎的中下部, 雌花花序生于梢部。我国的苎麻产量占全世界苎麻产量的90%以上。2018年2个苎麻基因组草图陆续公布[9,10], 以Zhongzhu 1为测序品种, 得到一个341.9 Mb的基因组草图, contig N50和scaffold N50分别为22.62 kb和1226.36 kb, 鉴定出30,237个蛋白编码基因。苎麻作为荨麻科中第一个被测序的物种, 可对该物种的多样性和物种形成等研究提供参考。比较基因组学发现, 苎麻有1075个独有的基因家族, 包含4082个基因, 其中分别有5个和10个被注释为假定的CesA和WAT1相关蛋白。分析这15个基因所编码的蛋白结构域发现, 5个假定CesA蛋白和3个假定WAT1蛋白有相应的保守结构域, 表明这些基因可能分别是真实的CesA和WAT1相关基因, 且与苎麻纤维产量有关。与其他4个物种(川桑、大枣、西瓜和亚麻)的CesA和WAT1相关蛋白的系统发育分析进一步证实, 5个CesA和3个WAT1相关基因是苎麻特有的。进化树分析发现106个自然选择的基因,
其中22个与氮代谢有关, 这些基因可能与苎麻的粗蛋白含量和营养生长性状驯化相关。这些研究为苎麻遗传育种和分子研究提供了理论基础。
1.4 亚麻基因组
2012年以CDC Bethune为测序品种, 得到一个373 Mb的基因组草图, contig N50为0.02 Mb, scaffold N50为0.69 Mb, 鉴定出43,484个基因[11]。最近, 甘肃省农业科学院分别对油用亚麻品种、纤维用亚麻品种和白胡麻测序得到染色体水平参考基因组[12], 组装得到的基因组大小为别为306.0、303.7和293.5 Mb, 其中contig N50/scaffold N50分别为131 kb/ 1235 kb、156 kb/700 kb和59 kb/384 kb, 在每个基因组中大约预测到43,500个编码基因和2600~2800个非编码RNA; 系统发育分析表明, 栽培亚麻和白胡麻在大约232万年前发生了分化, 自古被子植物的六倍化事件以来, 亚麻发生了2次WGD事件。在最近一次WGD事件中, MYB46/MYB83基因拷贝数发生了扩张, 可能促成了亚麻独特的次生细胞壁生物合成, 对MYB46/MYB83基因的定向选择可能形成了当前的油用和纤维用亚麻的形态类型。对83份亚麻种质资源重测序, 在油用亚麻中, 油脂相关基因(LOX、FatA、LTP和PDH-E2)和种子大小相关基因(BIN2和GW5)受到选择; 在纤维用亚麻中, 次生细胞壁生物合成相关基因(MYB46-1、XTH和ROPGAP3)和植物株高相关基因(GA3ox、GA20ox和GID)受到选择。1.5 工业大麻基因组
2011年工业大麻基因组草图发表[13], 该研究以一种广泛应用于药用的大麻品种Purple Kush为测序品种, 组装得到一个534 Mb的基因组草图。2020年5月公安部物证鉴定中心发表了一个高质量染色体水平的野生大麻参考基因组[14], 其基因组大小为808 Mb, contig N50为513.57 kb, scaffold N50为83 Mb, 相对于2011年发表的大麻基因组草图(contig N50为2.8 kb; scaffold N50为16.2 kb)有很大的提升。统计其重复序列高达74.75%, 注释到了38,828个蛋白编码基因。然而目前为止籽用大麻基因组还未公布, 待籽用大麻基因组公布之后, 以期比较基因组学在3种不同用途的大麻之间挖掘更丰富的变异位点, 为不同用途的大麻育种提供更多信息。2 主要麻类作物重要应用价值基因挖掘
纤维发育与非生物胁迫相关基因的挖掘一直是主要麻类作物遗传改良的重要方向。随着主要麻类作物全基因组测序陆续被报道, 很有必要系统总结回顾NGS (next generation sequencing)技术对主要麻类作物纤维发育相关基因挖掘(附表1)、响应各种非生物胁迫的遗传机制(附表2)和麻类作物特异性状候选基因的最新报道(附表3)。为了表述方便, 不同麻类作物分开阐述。每个麻类作物分别从纤维发育、非生物胁迫和麻类作物特异性状3个方面展开。Table S1
附表1
附表1主要麻类作物纤维发育候选基因信息
Table S1
| 基因 Gene | 作物 Crop | 功能或表型 Function or phenotype | 文献 Reference | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4CL | 黄麻, 红麻 Jute, kenaf | 参与木质素生物合成 Involved in lignin biosynthesis | [6, 8, 20] | |||||||||||
| 4CL1 | 苎麻 Ramie | 参与木质素生物合成 Involved in lignin biosynthesis | [47] | |||||||||||
| 4CL3 | 苎麻 Ramie | 参与黄酮类化合物的合成 Involved in the synthesis of flavonoids | [47] | |||||||||||
| APL | 黄麻, 红麻 Jute, kenaf | 参与纤维素生物合成 Involved in cellulose biosynthesis | [6, 8] | |||||||||||
| bHLH | 亚麻 Flax | 参与茎底部区域次生细胞壁沉积 Involved in the deposition of secondary cell wall at the bottom of stem | [66] | |||||||||||
| 基因 Gene | 作物 Crop | 功能或表型 Function or phenotype | 文献 Reference | |||||||||||
| bZIP | 亚麻 Flax | 参与茎底部区域次生细胞壁沉积 Involved in the deposition of secondary cell wall at the bottom of stem | [66] | |||||||||||
| C3H | 红麻 Kenaf | 参与木质素生物合成 Involved in lignin biosynthesis | [24] | |||||||||||
| C4H | 红麻, 苎麻 Kenaf, ramie | 参与木质素生物合成 Involved in lignin biosynthesis | [8, 47] | |||||||||||
| CAD | 黄麻, 红麻, 苎麻 Jute, kenaf, ramie | 参与纤维素生物合成 Involved in cellulose biosynthesis | [19, 24, 47] | |||||||||||
| CCR | 黄麻, 红麻 Jute, kenaf | 参与木质素生物合成 Involved in lignin biosynthesis | [6, 8, 20] | |||||||||||
| CCoAOMT | 黄麻,红麻,苎麻 Jute, kenaf, ramie | 参与木质素生物合成 Involved in lignin biosynthesis | [6, 24, 47] | |||||||||||
| CesA | 亚麻 Flax | 沉默后, 韧皮纤维的数量和结构受到了严重的影响 The quantity and structure of phloem fibers were seriously affected after silencing | [62-63] | |||||||||||
| CesA1 | 红麻, 苎麻 Kenaf, ramie | 参与初生细胞壁的纤维素沉积 Involved in cellulose deposition of primary cell wall | [8, 42] | |||||||||||
| CesA2 | 苎麻 Ramie | 参与木质素生物合成 Involved in lignin biosynthesis | [43] | |||||||||||
| CesA3 | 红麻, 苎麻 Kenaf, ramie | 参与初生细胞壁的纤维素沉积 Involved in cellulose deposition of primary cell wall | [8, 43] | |||||||||||
| CesA4 | 黄麻, 红麻, 苎麻, 工业大麻 Jute, kenaf, ramie, hemp | 参与次生细胞壁中纤维素沉积 Involved in cellulose deposition in secondary cell wall | [6, 8, 44, 80] | |||||||||||
| CesA6 | 红麻 Kenaf | 参与初生细胞壁的纤维素沉积 Involved in cellulose deposition of primary cell wall | [8] | |||||||||||
| CesA7 | 黄麻, 红麻, 工业大麻 Jute, kenaf, hemp | 协调木聚糖型S层的沉积 Coordinating the S-layers deposition in the xylan-type | [8, 19, 80] | |||||||||||
| CesA8 | 红麻, 工业大麻 Kenaf, hemp | 参与次生细胞壁中纤维素沉积 Involved in cellulose deposition in secondary cell wall | [8, 80] | |||||||||||
| CML15b | 亚麻 Flax | PLR1基因转录调控的关键因子 Key factors for transcription regulation of PLR1 gene | [68] | |||||||||||
| COB | 工业大麻 Hemp | 在次级生长的下胚轴中有较高表达量 Highly expressed in hypocotyls undergoing secondary growth | [80] | |||||||||||
| COBL4 | 工业大麻 Hemp | 在次级生长的下胚轴中有较高表达量 Highly expressed in hypocotyls undergoing secondary growth | [80] | |||||||||||
| COMT | 黄麻, 红麻, 亚麻 Jute, kenaf, flax | 参与木质素生物合成 Involved in lignin biosynthesis | [6, 8, 20, 24, 67] | |||||||||||
| CSL | 黄麻, 红麻, 亚麻 Jute, kenaf, flax | 参与纤维素生物合成 Involved in cellulose biosynthesis | [8, 17, 62] | |||||||||||
| F5H | 红麻, 苎麻 Kenaf, ramie | 参与木质素生物合成 Involved in lignin biosynthesis | [8, 24, 47] | |||||||||||
| FLA6 | 黄麻 Jute | 协调木聚糖型S层的沉积 Coordinating the S-layers deposition in the xylan-type | [19] | |||||||||||
| FLA11 | 工业大麻 Hemp | 影响植物细胞壁中纤维素、阿拉伯糖和半乳糖的含量 Impact cellulose, arabinose and galactose content in plant cell walls | [80, 82] | |||||||||||
| FLA12 | 工业大麻 Hemp | 影响植物细胞壁中纤维素、阿拉伯糖和半乳糖的含量 Impact cellulose, arabinose and galactose content in plant cell walls | [80, 82] | |||||||||||
| G2-like | 亚麻 Flax | 参与茎底部区域次生细胞壁沉积 Involved in the deposition of secondary cell wall at the bottom of stem | [66] | |||||||||||
| GRAS | 亚麻 Flax | 分支数候选基因Candidate gene for the number of branches | [112] | |||||||||||
| 基因 Gene | 作物 Crop | 功能或表型 Function or phenotype | 文献 Reference | |||||||||||
| HAT22 | 黄麻, 红麻 Jute, kenaf | 参与纤维素生物合成 Involved in cellulose biosynthesis | [6, 8] | |||||||||||
| HB8 | 红麻 Kenaf | 参与纤维形成 Involved in fiber formation | [8] | |||||||||||
| HCA2 | 红麻 Kenaf | 参与纤维形成 Involved in fiber formation | [8] | |||||||||||
| HCT | 红麻 Kenaf | 参与木质素生物合成 Involved in lignin biosynthesis | [8, 24] | |||||||||||
| KANADI | 红麻 Kenaf | 参与纤维形成 Involved in fiber formation | [8] | |||||||||||
| Kor | 黄麻, 红麻 Jute, kenaf | 参与纤维素生物合成 Involved in cellulose biosynthesis | [8, 17] | |||||||||||
| LBD1 | 红麻 Kenaf | 参与纤维形成 Involved in fiber formation | [8] | |||||||||||
| MP | 红麻 Kenaf | 参与纤维形成 Involved in fiber formation | [8] | |||||||||||
| MYB | 亚麻 Flax | 参与茎底部区域次生细胞壁沉积 Involved in the deposition of secondary cell wall at the bottom of stem | [66] | |||||||||||
| MYB46 | 红麻, 苎麻, 亚麻, 工业大麻 Kenaf, ramie, flax, hemp | 参与次生细胞壁生物发生和木聚糖生物合成 Involved in secondary cell wall biogenesis and xylan biosynthesis | [8, 12, 48, 80, 82] | |||||||||||
| MYB58 | 红麻 Kenaf | 参与韧皮纤维的次生细胞壁合成 Involved in the secondary cell wall synthesis of phloem fibers | [8] | |||||||||||
| MYB83 | 黄麻, 红麻, 亚麻 Jute, kenaf, flax | 参与纤维素生物合成 Involved in cellulose biosynthesis | [6, 8, 12] | |||||||||||
| MYB85 | 红麻 Kenaf | 参与韧皮纤维的次生细胞壁合成 Involved in the secondary cell wall synthesis of phloem fibers | [8] | |||||||||||
| MYB103 | 红麻 Kenaf | 参与韧皮纤维的次生细胞壁合成 Involved in the secondary cell wall synthesis of phloem fibers | [8] | |||||||||||
| NAC19 | 苎麻 Ramie | 上调MYB46的表达 Up regulation of MYB46 expression | [48] | |||||||||||
| NAC24 | 苎麻 Ramie | 上调MYB46的表达 Up regulation of MYB46 expression | [48] | |||||||||||
| NST1 | 工业大麻 Hemp | 纤维分化的主要调节因子 Major regulator of fibre differentiation | [80-81] | |||||||||||
| PAL | 红麻, 苎麻 Kenaf, ramie | 参与木质素生物合成 Involved in lignin biosynthesis | [8, 47] | |||||||||||
| NST1 | 红麻 Kenaf | 参与韧皮纤维的次生细胞壁合成 Involved in the secondary cell wall synthesis of phloem fibers | [8] | |||||||||||
| PHAC1 | 亚麻 Flax | 表达量与纤维拉力强度成正比 The expression is proportional to the tensile strength of the fiber | [68] | |||||||||||
| PIN1 | 红麻 Kenaf | 参与纤维形成 Involved in fiber formation | [8] | |||||||||||
| PL | 亚麻 Flax | 株高候选基因 Candidate gene for plant height | [112] | |||||||||||
| PLR1 | 亚麻 Flax | 参与木质素生物合成 Involved in lignin biosynthesis | [68] | |||||||||||
| ROPGAP3 | 亚麻 Flax | 参与韧皮纤维的次生细胞壁合成 Involved in the secondary cell wall synthesis of phloem fibers | [12] | |||||||||||
| SND1 | 红麻 Kenaf | 参与韧皮纤维的次生细胞壁合成 Involved in the secondary cell wall synthesis of phloem fibers | [8] | |||||||||||
| SND2 | 红麻 Kenaf | 参与韧皮纤维的次生细胞壁合成 Involved in the secondary cell wall synthesis of phloem fibers | [8] | |||||||||||
| SND3 | 红麻 Kenaf | 参与韧皮纤维的次生细胞壁合成 Involved in the secondary cell wall synthesis of phloem fibers | [8] | |||||||||||
| Susy | 黄麻, 红麻 Jute, kenaf | 参与纤维素生物合成 Involved in cellulose biosynthesis | [8, 17] | |||||||||||
| 基因 Gene | 作物 Crop | 功能或表型 Function or phenotype | 文献 Reference | |||||||||||
| TDIF | 红麻 Kenaf | 参与纤维形成 Involved in fiber formation | [8] | |||||||||||
| TOUCH4 | 工业大麻 Hemp | 将木葡聚糖靶向于次生细胞壁S1层的初生-次生细胞壁交界 Participate in the targeting of xyloglucan to the primary- secondary cell wall junction in the secondary cell wall S1 layer | [80] | |||||||||||
| UGPase | 黄麻 Jute | 参与纤维素生物合成 Involved in cellulose biosynthesis | [17] | |||||||||||
| UGT | 亚麻 Flax | 株高候选基因 Candidate gene for plant height | [112] | |||||||||||
| WAT1 | 苎麻 Ramie | 高纤维品种驯化中的受选择基因 Selection of highly domesticated fiber varieties | [45] | |||||||||||
| WLIM1 | 工业大麻 Hemp | 捆绑肌动蛋白丝来促进纤维的延伸, 与PAL-box结合来促进木质素生物合成基因的木质化 Fibre extension by bundling the actin filament but also fibre lignification by promoting the lignin/lignin-like biosynthetic genes via binding to the PAL-box | [80, 83] | |||||||||||
| WOX4 | 黄麻, 红麻 Jute, kenaf | 参与纤维素生物合成 Involved in cellulose biosynthesis | [6, 8] | |||||||||||
| WRKY | 黄麻 Jute | 纤维改良 Fiber improvement | [20] | |||||||||||
| WRKY36 | 亚麻 Flax | 通过与PLR1启动子中的W-box结合, 参与木质素生物合成 Involved in lignin biosynthesis by binding with W-box in PLR1 promoter | [69] | |||||||||||
| XTH | 亚麻 Flax | 分支数候选基因 Candidate gene for the number of branches | [112] | |||||||||||
| XTH5 | 工业大麻 Hemp | 韧皮纤维的伸长依赖于XTH的活性 Bast fibre extension depends on the activities of XTH | [80] | |||||||||||
| XTH8 | 工业大麻 Hemp | 韧皮纤维的伸长依赖于XTH的活性 Bast fibre extension depends on the activities of XTH | [80] | |||||||||||
| XTH15 | 工业大麻 Hemp | 在二次生长的较老大麻下胚轴中表达较多 More expressed in the elongating hemp hypocotyl | [80] | |||||||||||
| XTH22 | 工业大麻 Hemp | 在二次生长的较老大麻下胚轴中表达较多 More expressed in the elongating hemp hypocotyl | [80] | |||||||||||
新窗口打开|下载CSV
Table S2
附表2
附表2主要麻类作物响应非生物胁迫候选基因信息
Table S2
| 基因 Gene | 作物 Crop | 功能或表型 Function or phenotype | 文献 Reference | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| α-amylase | 苎麻 Ramie | 响应干旱和高盐逆境胁迫 Response to drought and high salt stress | [53] | |||||||
| A-ARR | 黄麻 Jute | 通过细胞分裂素途径响应盐胁迫 Response to salt stress through cytokinin pathway | [21] | |||||||
| ABF | 黄麻 Jute | 通过ABA信号通路响应盐胁迫 Response to salt stress through the ABA signaling pathway | [21] | |||||||
| ACO1 | 苎麻 Ramie | 响应干旱和高盐逆境胁迫 Response to drought and high salt stress | [51] | |||||||
| AHP | 黄麻 Jute | 通过细胞分裂素途径响应盐胁迫 Response to salt stress through cytokinin pathway | [21] | |||||||
| AP2/ERF | 红麻 Kenaf | 在盐胁迫转录组中检测到的差异表达因子 Differential expression factors detected in transcriptome under salt stress | [27] | |||||||
| AP2/EREBP | 红麻 Kenaf | 在盐胁迫转录组中检测到的差异表达因子 Differential expression factors detected in transcriptome under salt stress | [27] | |||||||
| ARF | 红麻 Kenaf | 在盐胁迫转录组中检测到的差异表达因子 Differential expression factors detected in transcriptome under salt stress | [27] | |||||||
| 基因 Gene | 作物 Crop | 功能或表型 Function or phenotype | 文献 Reference | |||||||
| AvrL2 | 亚麻 Flax | 抵御锈病感染 Resist rust infection | [76, 107] | |||||||
| AvrL567 | 亚麻 Flax | 抵御锈病感染 Resist rust infection | [76] | |||||||
| AvrM14 | 亚麻 Flax | 抵御锈病感染 Resist rust infection | [76, 107] | |||||||
| AvrP123 | 亚麻 Flax | 抵御锈病感染 Resist rust infection | [76] | |||||||
| AvrP4 | 亚麻 Flax | 抵御锈病感染 Resist rust infection | [76] | |||||||
| AvrM | 亚麻 Flax | 抵御锈病感染 Resist rust infection | [76] | |||||||
| B-ARR | 黄麻 Jute | 通过细胞分裂素途径响应盐胁迫 Response to salt stress through cytokinin pathway | [21] | |||||||
| bHLH | 红麻 Kenaf | 在盐胁迫转录组中检测到的差异表达因子 Differential expression factors detected in transcriptome under salt stress | [27] | |||||||
| bZIP | 红麻, 苎麻 Kenaf, ramie | 响应干旱和高盐逆境胁迫 Response to drought and high salt stress | [27, 52] | |||||||
| CAX3 | 亚麻 Flax | 基因产物可能通过Ca2+介导的胞内调节参与了亚麻对高酸性、高Al3+浓度的响应 Gene product may be involved in the response of flax to high acidity and high Al3+ concentration through Ca2+ mediated intracellular regulation | [73] | |||||||
| CCAAT | 黄麻 Jute | 在盐胁迫转录组中检测到的差异表达因子 Differential expression factors detected in transcriptome under salt stress | [21] | |||||||
| CRE1 | 黄麻 Jute | 通过细胞分裂素途径响应盐胁迫 Response to salt stress through cytokinin pathway | [21] | |||||||
| G6PDH1 | 苎麻 Ramie | 响应重金属镉胁迫 Response to cadmium stress | [57] | |||||||
| GS2 | 苎麻 Ramie | 转基因烟草能提高生物产量和氮利用效率 Transgenic tobacco can improve biomass and nitrogen utilization | [54] | |||||||
| HB | 黄麻 Jute | 在盐胁迫转录组中检测到的差异表达因子 Differential expression factors detected in transcriptome under salt stress | [21] | |||||||
| HDA2 | 红麻 Kenaf | 盐胁迫和干旱胁迫的响应基因 Response genes of salt and drought stress | [30] | |||||||
| HDA8 | 红麻 Kenaf | 盐胁迫和干旱胁迫的响应基因 Response genes of salt and drought stress | [30] | |||||||
| HDA9 | 红麻 Kenaf | 盐胁迫和干旱胁迫的响应基因 Response genes of salt and drought stress | [30] | |||||||
| HDA19 | 红麻 Kenaf | 盐胁迫和干旱胁迫的响应基因 Response genes of salt and drought stress | [30] | |||||||
| HHDA6 | 红麻 Kenaf | 盐胁迫和干旱胁迫的响应基因 Response genes of salt and drought stress | [30] | |||||||
| HSF | 黄麻 Jute | 在盐胁迫转录组中检测到的差异表达因子 Differential expression factors detected in transcriptome under salt stress | [21] | |||||||
| HSF | 亚麻 Flax | 响应高温胁迫 Response to high temperature stress | [70] | |||||||
| JAS | 亚麻 Flax | 响应土壤营养胁迫的响应 Response to soil nutrient stress | [71] | |||||||
| KCS | 黄麻 Jute | 转基因增强了植株的抗旱性 Transgene enhances plant drought resistance | [23] | |||||||
| MADS-box | 亚麻 Flax | 参与调节植物生长发育和参与耐铝性细胞壁修饰的酶 Enzymes involved in the regulation of plant growth and development and in the modification of aluminum resistant cell wall | [74] | |||||||
| MYB | 黄麻 Jute | 在盐胁迫转录组中检测到的差异表达因子 Differential expression factors detected in transcriptome under salt stress | [21] | |||||||
| MYB83 | 苎麻 Ramie | 响应重金属镉胁迫 Response to cadmium stress | [56] | |||||||
| NAC | 红麻 Kenaf | 在盐胁迫转录组中检测到的差异表达因子 Differential expression factors detected in transcriptome under salt stress | [27] | |||||||
| 基因 Gene | 作物 Crop | 功能或表型 Function or phenotype | 文献 Reference | |||||||
| NAC | 亚麻 Flax | 参与调节植物生长发育和参与耐铝性细胞壁修饰的酶 Enzymes involved in the regulation of plant growth and development and in the modification of aluminum resistant cell wall | [74] | |||||||
| NRAMP1 | 苎麻 Ramie | 响应重金属镉胁迫 Response to cadmium stress | [58] | |||||||
| PCS1 | 苎麻 Ramie | 响应重金属镉胁迫 Response to cadmium stress | [55] | |||||||
| PP2C | 黄麻 Jute | 通过ABA信号通路响应盐胁迫 Response to salt stress through the ABA signaling pathway | [21] | |||||||
| PYL | 黄麻 Jute | 通过ABA信号通路响应盐胁迫 Response to salt stress through the ABA signaling pathway | [21] | |||||||
| RLK | 黄麻 Jute | 抗旱机制的负调控因子 Negative regulatory factors of drought resistance mechanism | [22] | |||||||
| SnRK2 | 黄麻 Jute | 通过ABA信号通路响应盐胁迫 Response to salt stress through the ABA signaling pathway | [21] | |||||||
| SRT2 | 红麻 Kenaf | 盐胁迫和干旱胁迫的响应基因 Response genes of salt and drought stress | [30] | |||||||
| TCP | 红麻 Kenaf | 在盐胁迫转录组中检测到的差异表达因子 Differential expression factors detected in transcriptome under salt stress | [27] | |||||||
| WD40-1 | 红麻 Kenaf | ABA和MeJA信号转导途径、盐和干旱胁迫应答途径的关键枢纽基因 Key genes of ABA and MeJA signal transduction pathway, salt and drought stress response pathway | [28] | |||||||
| WRKY | 黄麻, 红麻, 苎麻 Jute, kenaf, ramie | 在盐胁迫和干旱胁迫转录组中检测到的差异表达因子 Differential expression factors detected in transcriptome under salt and drought stress | [21, 27, 29, 50] | |||||||
新窗口打开|下载CSV
Table S3
附表3
附表3主要麻类作物特异性状候选基因信息
Table S3
| 基因 Gene | 作物 Crop | 功能或表型 Function or phenotype | 文献 Reference | ||||
|---|---|---|---|---|---|---|---|
| LMI1 | 红麻 Kenaf | 叶形基因 Leaf shape gene | [8] | ||||
| GI | 红麻 Kenaf | 参与红麻光周期调控 Involved in the photoperiod regulation of kenaf | [26] | ||||
| CO | 红麻 Kenaf | 参与红麻光周期调控 Involved in the photoperiod regulation of kenaf | [26] | ||||
| MADS-box | 红麻 Kenaf | 可能参与了红麻细胞质雄性不育 May be involved in the cytoplasmic male sterility of kenaf | [35] | ||||
| TIR1 | 红麻 Kenaf | 编码一个富含亮氨酸重复的F-box蛋白 Encodes a leucine rich F-box protein | [38-39] | ||||
| atp9 | 红麻, 苎麻 Kenaf, ramie | 细胞质雄性不育 Cytoplasmic male sterility | [32, 61] | ||||
| atp6 | 红麻, 苎麻 Kenaf, ramie | 细胞质雄性不育 Cytoplasmic male sterility | [35-37, 61] | ||||
| ATPA | 苎麻 Ramie | 育性相关的基因 Fertility related genes | [60] | ||||
| PLA2 | 亚麻 Flax | 参与甘油磷脂代谢途径和醚酯代谢途径 Involved in glycerophosphatidic and ether ester metabolic pathways | [77] | ||||
| PLC | 亚麻 Flax | 参与甘油磷脂代谢和醚酯代谢和肌醇磷酸代谢 Involved in the metabolism of glycerophosphatide, ether ester and inositol phosphate | [77] | ||||
| PDAT1 | 亚麻 Flax | 参与TAG合成途径 Involved in TAG synthesis pathway | [78] | ||||
| DGAT1 | 亚麻 Flax | 参与TAG合成途径 Involved in TAG synthesis pathway | [78] | ||||
| DGAT2 | 亚麻 Flax | 参与TAG合成途径 Involved in TAG synthesis pathway | [78] | ||||
| FAD2a | 亚麻 Flax | 参与不饱和脂肪酸积累 Involved in the accumulation of unsaturated fatty acids | [79] | ||||
| FAD3a | 亚麻 Flax | 参与不饱和脂肪酸积累 Involved in the accumulation of unsaturated fatty acids | [79] | ||||
| 基因 Gene | 作物 Crop | 功能或表型 Function or phenotype | 文献 Reference | ||||
| FAD3b | 亚麻 Flax | 参与不饱和脂肪酸积累 Involved in the accumulation of unsaturated fatty acids | [79] | ||||
| PHO1 | 亚麻 Flax | 千粒重候选基因Candidate gene for the 1000-seed weight | [112] | ||||
| AEE1 | 工业大麻 Hemp | 参与调控大麻素合成途径 Involved in the regulation of cannabinoid synthesis pathway | [13] | ||||
| OLS | 工业大麻 Hemp | 参与调控大麻素合成途径 Involved in the regulation of cannabinoid synthesis pathway | [13, 86] | ||||
| THCAS-like1 | 工业大麻 Hemp | 编码大麻素原酸形成 Encodes cannabinoid acid formation | [13] | ||||
| THCAS-like2 | 工业大麻 Hemp | 编码大麻素原酸形成 Encodes cannabinoid acid formation | [13] | ||||
| THCAS-like3 | 工业大麻 Hemp | 编码大麻素原酸形成 Encodes cannabinoid acid formation | [13] | ||||
| THCAS-like4 | 工业大麻 Hemp | 编码大麻素原酸形成 Encodes cannabinoid acid formation | [13] | ||||
| CMK | 工业大麻 Hemp | 表达量和大麻素含量呈现显著正相关 The expression level was positively correlated with cannabinoid content | [86] | ||||
| MDS | 工业大麻 Hemp | 表达量和大麻素含量呈现显著正相关 The expression level was positively correlated with cannabinoid content | [86] | ||||
| HDS | 工业大麻 Hemp | 表达量和大麻素含量呈现显著正相关 The expression level was positively correlated with cannabinoid content | [86] | ||||
| HDR | 工业大麻 Hemp | 表达量和大麻素含量呈现显著正相关 The expression level was positively correlated with cannabinoid content | [86] | ||||
| GPP(lsu) | 工业大麻 Hemp | 表达量和大麻素含量呈现显著正相关 The expression level was positively correlated with cannabinoid content | [86] | ||||
| PT | 工业大麻 Hemp | 在始果期苞片腺毛中对大麻素合成积累起着关键作用 Played key roles in the biosynthesis and accumulation of cannabinoids in the glandular hairs of bract at initial-fruiting stage | [86] | ||||
| THCAS | 工业大麻 Hemp | 在始果期苞片腺毛中对大麻素合成积累起着关键作用 Played key roles in the biosynthesis and accumulation of cannabinoids in the glandular hairs of bract at initial-fruiting stage | [86] | ||||
| TPS18VF | 工业大麻 Hemp | 编码橙花醇/芳樟醇合成酶 Nerolidol/linalool synthases | [87] | ||||
| TPS19BL | 工业大麻 Hemp | 编码橙花醇/芳樟醇合成酶 Nerolidol/linalool synthases | [87] | ||||
| TPS16CC | 工业大麻 Hemp | 编码大根香叶烯B合酶 Germacrene B synthase | [87] | ||||
| TPS20CT | 工业大麻 Hemp | 编码四甲基环癸二烯甲醇合酶 Hedycaryol synthase | [87] | ||||
新窗口打开|下载CSV
2.1 黄麻重要应用价值基因挖掘
2.1.1 黄麻纤维发育相关基因 纤维发育的分子机制解析属于麻类作物遗传改良研究的重要方向。为鉴定纤维发育相关的基因, 在黄麻纤维发育关键时期进行转录组测序[17], 共组装出48,914个unigenes, 其中有5个Susy、3个UGPase、9个CesA、18个CSL、2个Kor (Korrigan), 它们可能参与黄麻纤维素生物合成。长果种黄麻茎皮转录组[18]分析显示, 次生代谢产物的生物合成在代谢途径中显著富集, 其中26个基因持续上调, 包括4个参与葡聚糖代谢的基因, 调控茎皮韧皮部的发育。对一个纤维缺陷突变体进行转录组测序[19]发现, CAD7在突变体的韧皮部组织中的任何生长阶段都显著下调, 且CAD7在生长早期的表达下调是伴随着次生细胞壁特异性基因纤维素合成酶A7 (CesA7)和阿拉伯半乳聚糖糖蛋白6 (FLA6)的共同上调, 这可能与黄麻纤维中协调木聚糖S型的沉积有关。黄麻全基因组测序显示[6], 在纤维素合成途径中MYB83、WOX4、APL和HAT22的表达较高。抑制差减杂交[20]被成功应用于黄麻纤维发育过程中相关基因的鉴定, 以纤维素含量极低的突变体作为试验组, 纤维素含量正常的品种作为对照组, 共鉴定出377个功能已知的基因。其中, COMT、CCR和4CL在正常植株中的表达明显高于突变体, 表明这些基因可能参与黄麻木质素合成, 且4CL是起主要作用。2.1.2 黄麻非生物胁迫响应基因 解析麻类作物非生物胁迫响应的分子机制是其遗传改良的又一重要方向, 包括盐胁迫和干旱胁迫等。黄麻盐胁迫下转录组测序[21]显示, 在叶和根中分别检测到32个和196个差异表达转录因子, 其中有11个在2个组织中均被检测到; 在叶片中的差异表达转录因子家族包括HB和HSF, 而在根中的差异表达转录因子家族包括MYB、WRKY和CCAAT。脱落酸ABA信号通路是植物盐胁迫反应的核心, 在此途径上发现了20个差异表达基因, 包括编码PP2C、SnRK2、ABF (上调)和PYL (下调)的基因。在细胞分裂素途径上发现, 在高浓度盐胁迫下, 叶和根中的CRE1、B-ARR和A-ARR均下调, 而大多数AHP基因上调, 表明它们负调控细胞分裂素信号以提高植物的耐盐性。黄麻耐旱品种和敏感品种的比较转录组分析[22], 在干旱敏感型品种中, 鉴定出34个转录因子差异表达基因和23个蛋白激酶差异表达基因, 其中包括19个类受体激酶(receptor like kinases, RLKs)。这些RLKs在干旱胁迫下大部分下调, 说明它们是黄麻抗旱机制的负调控因子。此外, 3-ketoacyl-CoA合酶(3-ketoacyl-CoA synthase, KCS)的表达能显著提高黄麻抗旱性, 构建KCS基因的过表达和基因编辑载体, 并用于黄麻植株的转化, 发现KCS基因的过表达不仅增加了叶绿体的数量, 还增强了植株的抗旱性, 提高了木质素含量、生长速率、茎皮厚度和茎粗[23]。
2.1.3 黄麻特异性状相关基因 黄麻2个栽培种(或亚种)在表型上存在较大差异, 如长果种的果荚为长条形状, 种子颜色为墨绿色; 圆果种的果荚为球形, 种子颜色为棕色等。不仅如此, 长果种为常异花授粉作物, 圆果种为自花授粉作物, 然而造成2个栽培种间形态或重要性状差异的遗传机制目前还有待深入研究。
2.2 红麻重要应用价值基因挖掘
2.2.1 红麻纤维发育相关基因 与黄麻纤维不同的是红麻韧皮纤维中纤维素含量相对较低, 木质素含量较高。红麻基因组[8]不仅揭示了一些参与韧皮纤维次生壁合成和纤维素沉积的基因, 如: MYB、SND和CesA等, 还结合QTL定位鉴定出MYB83和MYB103这2个可能参与细胞壁形成的基因, 而MYB83也是红麻在驯化过程中的受选择基因。因此MYB83可能在红麻纤维发育过程中发挥重要作用。转录组分析揭示出317个淀粉和糖代谢途径的基因, 可能参与纤维素生物合成[24]。在此之前CAD、CCoAOMT、C3H、HCT、F5H和COMT被报道在木质素生物合成中起关键作用[25]。红麻在开花后纤维累积速度显著降低, 因此延后红麻开花期也是提高纤维产量的一个途径, 在红麻光周期调控途径中可能存在类似拟南芥GI-CO-FT的保守途径: 短日照条件下, GI表达量上调促进CO表达, 从而促进植物开花, GI和CO这2个基因在光敏感和光钝感种质中表达存在差异[26], 说明这2个基因可能参与红麻光周期调控。2.2.2 红麻非生物胁迫响应基因 红麻本身作为一种耐旱、耐盐碱、耐贫瘠、易栽培的作物[1], 研究红麻耐盐碱机制, 培育红麻耐盐碱种质可将红麻种植转向盐碱地, 减少用地压力。比较盐胁迫下红麻的转录谱[27]发现, 盐胁迫下转录因子NAC、BZIP、AP2/EREBP、ARF、AP2/ERF、bHLH、TCP均上调, 有8个NAC转录因子和2个WRKY转录因子表达下调, 这些转录因子可能通过多种不同途径参与了红麻的耐盐性。基于转录组, 不少****对红麻非生物胁迫产生响应的基因进入了深入挖掘, 认为WD40-1基因是红麻ABA和MeJA信号转导途径、盐和干旱胁迫应答途径的关键枢纽基因[28]; 外源镉和盐处理能够显著诱导WRKY20表达[29], 该基因可能参与调控红麻逆境胁迫反应, 但仍需要更多试验验证WRKY20在红麻逆境胁迫中的生物学功能。Wei等[30]分离鉴定红麻6个HDACs基因(HDA2、HHDA6、HDA8、HDA9、HDA19、SRT2), 亚细胞定位显示, HDA2和HDA8位于细胞核中, HDA6和HDA19位于细胞核和胞质中, 而HDA9位于细胞核和质膜中, 这6个基因均为红麻盐胁迫和干旱胁迫的响应基因。
2.2.3 红麻特异性状相关基因 作物细胞质雄性不育(cytoplasmic male sterile, CMS)是作物杂种优势利用的主要途径, 2004年选育出红麻CMS系K03A[31], 实现了三系配套, 自此红麻育性相关研究越来越多, 与红麻育性相关的基因也被陆续报道。赵艳红等[32]在红麻线粒体基因ATP9中发现一个与雄性不育细胞质相关的47 bp缺失, 这47 bp缺失片段分别与线粒体转运信号和GFP的ATP9融合, 形成2个嵌合基因MTS-HM184-GFP和MTS-HM184,将含嵌合基因的载体转入烟草发现, 部分转基因植株为雄性不育后半不育[33], 并基于ATP9基因开发的分子标签MM556可用于红麻不育细胞质的鉴定[34]。对不育系和在保持系花药进行比较转录组分析[35]发现, ATP6在三羧酸循环(tricarboxylic acid cycle, TCA cycle)中起直接作用, 在不育系中表达量下降了5.6倍。随后ATP6基因也被报道因为其CDS中存在33-bp的缺失和3-bp的插入, 导致红麻细胞质雄性不育[36]。ATP6与MYC2b蛋白之间存在互作, 导致红麻细胞质雄性不育, 而ATP6与MYC2a、MYC2b蛋白之间均存在互作时, 红麻表现为可育[37]。TIR1基因编码一个富含亮氨酸重复的F-box蛋白, 而F-box蛋白被报道出在花器官发育中有重要功能[38], 且在花药期不育系中TIR1基因表达显著高于保持系, 这可能导致花药败育[39]。目前认为雄性不育主要包括由线粒体基因和核基因共同控制, 然而又有报道通过RNA编辑技术发现叶绿体基因也可能与红麻细胞质雄性不育有关[40]。红麻细胞质雄性不育基因及其调控机制还有待深入研究。
2.3 苎麻重要应用价值基因挖掘
2.3.1 苎麻纤维发育相关基因 苎麻纤维中纤维素含量高于黄麻且木质素含量远低于黄麻, 纤维品质比黄麻好。转录组测序[41]鉴定出36个纤维素合酶基因, 其中有33个在茎皮中的表达远高于其他组织。随后CesA1[42]、CesA2、CesA3[43]和CesA4[44]也都被陆续报道在韧皮部或木质部表达参与次生细胞壁合成。对苎麻野生种和栽培种的转录组进行比较[45]发现, 高纤维素含量品种的驯化可能与WAT1基因的正向选择有关。WAT1相关的基因在苎麻基因组[10]中也被注释为与纤维产量的相关基因。对木质素含量的遗传基础进行分析发现[46], 3个位于木质素含量QTL区域的基因, 在纤维发育的2个不同阶段都有不同的表达, 且它们分别编码MYB蛋白、莽草酸羟基肉桂酰转移酶和漆酶。Tang等[47]发现, PAL、C4H、4CL1、F5H、CCAOMT和CAD的表达量均与木质素含量成正比, 但苎麻4CL3重组蛋白在催化过程中以肉桂酸为底物, 因此对木质素合成有负调控作用, 推测苎麻4CL3主要参与苎麻黄酮类化合物的合成, 4CL1主要参与木质素的合成。探索NAC与MYB对调控苎麻纤维次生细胞壁发育的作用发现[48], NAC19和NAC24过表达能够上调苎麻次生壁发育关键基因MYB46的表达量, 从而导致次生壁纤维的生物合成增加; NAC19有可能直接参与细胞程序性死亡调控次生细胞壁发育, 而MYB46可能在苎麻纤维成熟中发挥重要作用。对不同区段的茎皮进行转录组测序[49]发现, 乙烯和赤霉素可能参与的苎麻韧皮纤维组织发育, 这说明研究调控苎麻纤维发育的相关基因可以间接从乙烯和赤霉素合成酶入手。2.3.2 苎麻非生物胁迫响应基因 非生物胁迫严重影响苎麻的产量与品质。基于苎麻转录组, 筛选出8个WRKY转录因子, 与其他作物已报道的10个具有抗旱功能的WRKY转录因子对比, 推断这个8个WRKY转录因子也可能参与苎麻干旱胁迫响应[50]。ACO1[51]、bZIP1[52]和α-amylase[53]也被证实响应苎麻ABA、干旱和高盐逆境胁迫, 将苎麻GS2基因转入烟草发现, 其显著提高转基因植株的生物量和氮利用效率[54], 从而减轻外界非生物胁迫。PCS1[55]、MYB83[56]、G6PDH1[57]和NRAMP1[58]也都被陆续报道出能响应重金属镉胁迫。此外, Chen等[59]发现73个miRNA, 它们介导苎麻对重金属镉胁迫的响应。
2.3.3 苎麻特异性状相关基因 苎麻是一个杂种优势利用率较高的作物, 对苎麻育性相关基因的挖掘能为其雄性不育机理研究奠定基础。目前已经报道出的与苎麻育性相关的基因有ATPA[60]、ATP6和ATP9[61]。
2.4 亚麻重要应用价值基因挖掘
2.4.1 亚麻纤维发育相关基因 亚麻纤维中纤维素含量高于黄麻, 木质素含量远低于黄麻, 纤维品质优于黄麻。基于亚麻基因组信息, 鉴定出32个与亚麻纤维发育相关的候选基因, 其中16个编码纤维素合成酶(CesA), 16个编码纤维素合成酶类蛋白(Csl)[62], 利用VIGS沉默这些基因后, 韧皮纤维的数量和结构受到了严重的影响[63]。亚麻纤维素生物合成的调节涉及CesA的表达调控、胞质中纤维素合酶复合体的装配、纤维素的沉积等多个方面, 但对Csl蛋白的功能尚不清楚[64], 而CesA基因的转录水平主要依赖于产生的蛋白质的功能亚类和植物发育阶段[65]。对亚麻不同茎段的转录组分析显示[66], 在次生细胞壁形成的晚期韧皮纤维中(茎底部区域), 编码金属硫蛋白、脂质转移蛋白以及参与蛋白质合成/翻译和泛素介导降解的候选基因表达量增加; 与顶杆区相比, 分化后期的韧皮纤维中有156个基因上调, 其中有与细胞壁相关的基因; 与其他茎段相比, 参与次生细胞壁沉积的转录因子在韧皮纤维中的表达减少。除以上基因家族外, COMT基因主要参与S-木质素的生物合成, 转基因植物在COMT活性被抑制时, 木质素含量减少[67]; PHAC1基因表达量与亚麻纤维拉力强度成正比[68]; Ca2+信号通路通过对PLR1基因的转录调控, 在木质素生物合成的ABA调控中起着至关重要的作用, 而CML15b是该调控的关键调节因子[68]; WRKY36通过与PLR1启动子中的W-box结合, 在响应ABA和尖孢镰刀菌诱导的木质素生物合成中发挥调节作用[69]。2.4.2 亚麻非生物胁迫响应基因 亚麻基因组测序完成为全基因组水平分析非生物胁迫响应基因提供了前提条件。在亚麻全基因组中共找到137个NBS类抗病基因[70], 34个HSF基因响应高温胁迫[71]。转录组测序发现[72], WRKY和JAS基因家族在亚麻对土壤营养胁迫的响应中起着重要的作用, CAX3基因产物可能通过Ca2+介导的胞内调节参与了亚麻对高酸性、高Al3+浓度的响应[73], MADS-box和NAC转录因子参与调节植物生长发育和参与亚麻耐铝性细胞壁修饰的酶[74]。干旱胁迫对亚麻产量和油质量的影响主要表现在生育期, 选择性扫描发现了与脱落酸途径、生长素信号、Ca2+信号、光合作用调节和干旱应答转录因子有关的各种基因[75]。亚麻锈病感染转录组[76]揭示6个Avr基因(AvrM、AvrM14、AvrL2、AvrL567、AvrP123 (AvrP)和AvrP4)参与了亚麻抵御锈病感染, 在感染早期表达量达到一个峰值, 随着孢子形成逐渐下降。
2.4.3 亚麻特异性状相关基因 籽用亚麻种子含油量一般为30%~45%, 早在史前时代就被作为油料作物种植。对亚麻未成熟胚中磷脂酶基因家族的转录组学分析[77]发现, PLA2在亚麻中主要参与甘油磷脂代谢途径和醚酯代谢途径; PLC除参与甘油磷脂代谢和醚酯代谢之外, 还在肌醇磷酸代谢途径中发挥重要作用。在TAG合成途径中发现7个关键基因, 其中PDAT1、DGAT1和DGAT2基因的动态表达模式与含油量的动态积累模式显著正相关, PDAT1的动态表达模式与亚麻酸的动态积累模式显著正相关, 且在高油高亚麻酸材料的种子动态发育阶段中PDAT1基因的累积表达量也显著高于低油低亚麻酸材料, 因此PDAT1可能是影响籽用亚麻(胡麻)不同品种(系)中含油量和亚麻酸含量的关键基因[78]。全基因组关联分析和结合转录组测序[79], 筛选出10个候选基因, 其中6个基因参与了重要的脂肪酸代谢途径, 其中一些基因还具有上下游调节关系。对不同亚麻酸含量的亚麻品种进行不同时期的品质测定发现, FAD2a、FAD3a和FAD3b这3个基因参与不饱和脂肪酸积累过程。
2.5 工业大麻重要应用价值基因挖掘
2.5.1 工业大麻纤维发育相关基因 工业大麻是一种多用途作物, 韧皮纤维主要用于纺织和生物复合工业。其下胚轴有一个主动伸长, 然后变厚形成初生和次生韧皮纤维的阶段, 因此工业大麻是研究二次生长过程的合适模型[80]。工业大麻下胚轴的二次生长与转录因子NST1、MYB46和WLIM1的上调有关[80,81,82,83]。韧皮纤维的伸长依赖于XTH的活性, 如XTH5和XTH8, 这些基因在伸长的大麻下胚轴中表达较多[80]。COBRA家族中参与纤维素生物合成的2个基因COB和COBL4, 以及次生细胞壁纤维素合成酶基因(CesA4、CesA7和CesA8)在二次生长的下胚轴中均有较高表达量[80]。FLA11和FLA12在工业大麻二次生长的下胚轴中表达量高, 此时初生和次生韧皮纤维都存在[80]。已知FLA基因家族会影响植物细胞壁中纤维素、阿拉伯糖和半乳糖的含量, 因此也可能有助于确定韧皮纤维细胞壁的组成[84]。RNA-Seq分析发现[80], 在下胚轴二次生长过程中涉及到葡萄糖醛酸苷生物合成的几个基因的上调, 特别是涉及到主干延伸(IRX10、IRX10L、IRX14和IRX15L)、主干乙酰化(ESK1、RWA3)和取代基甲基化(GXM3)有关的基因。2.5.2 工业大麻特异性状相关基因 工业大麻虽然是一种纤维作物, 但大麻素作为工业大麻所特有的一类次生代谢产物, 在抗肿瘤、神经系统保护、免疫调节和抗炎抗氧化等方面具有重要的药用价值[85], 因此工业大麻作为药用植物也有很大的前景。工业大麻全基因组测序[13]鉴定出2个参与调控大麻素合成途径的基因AEE1和OLS, 此外还鉴定出23个编码大麻素原酸(CBCA)形成的候选基因, 其中4个基因命名为THCAS-like1~THCAS-like4。在MEP和GPP途径后端的酶基因CMK、MDS、HDS、HDR和GPP (lsu)表达量和大麻素含量呈现显著正相关, 在大麻素合成途径中OLS、PT和THCAS 3个大麻中特有的酶基因主要在始果期苞片腺毛中对大麻素合成积累起着关键作用[86]。不同工业大麻品种的腺毛转录组分析, 鉴定出之前从未报道过的编码活性酶的基因, TPS18VF和TPS19BL编码橙花醇/芳樟醇合成酶, TPS16CC编码大根香叶烯B合酶和TPS20CT编码四甲基环癸二烯甲醇合酶[87]。大麻聚酮合酶(PKS)是催化大麻素生物合成的第一步, 有研究发现, 啤酒花衍生的转录因子和番茄浓密矮化病毒(TBSV)p19共同参与了PKS基因启动子的激活[88]。Laverty等[89]利用药用大麻和毒品大麻杂交群体构建遗传图谱发现, 大麻素的生物合成基因是不连锁的, 但产生THCA和CBDA合成酶底物的芳香族丙烯酰胺转移酶(AP)与已知的大麻素含量标记紧密连锁, 这为在大麻中如何产生大麻素提供了不同见解。
大麻作为雌雄异株植物, 性染色体进化是其又一特异性状。Prentout等[90]在大麻中发现了超过500个性连锁基因, 将这些性连锁基因定位到大麻基因组组合中, 其中有363个在1号染色体, 并且发现有1对染色体具有较大的非重组区, 进一步分析发现, 大麻具有一个强烈退化的Y染色体, 是迄今为止最古老的植物性染色体系统, 这有助于工业大麻性染色体鉴定和性别基因定位。
3 基于基因组学的麻类作物遗传改良
3.1 我国麻类作物品种改良历程
主要麻类作物在我国均拥有悠久的种植历史, 其品种改良大致经历了以下4个阶段: 第1阶段是地方种的搜集鉴定及引种利用。在此阶段不仅大量丰富了种质资源类型, 而且鉴定出一批具有优良性状的材料, 如黄麻品种D154和翠绿等。第2阶段是杂交育种或回交选育等常规育种方法选育优良种质。此阶段是利用第1阶段所得到的优异种质为基础材料进行育种工作, 在此阶段利用杂交与诱变育种相结合, 选育出一大批高纤维产量和高含油量的亚麻品种, 如黑亚2号和亚宁1号等。第3阶段为高产、优质、多抗育种阶段。在此之前由于长期使用少数几个性状优异的骨干材料作为亲本, 使麻类作物品种遗传基础狭窄, 导致作物平均单产和品质的降低以及病虫害的发生, 如: 亚麻枯萎病和红麻炭疽病, 因此育种家们在此阶段选育出一批高产、优质、多抗育种的新品种。第4阶段为优质高产专用新品种选育和现代生物技术辅助育种阶段。由于常规育种在进一步提高产量、品质和抗性等方面的局限性逐步显现, 且很难满足市场需求的优质专用种质培育, 与此同时现代生物技术快速发展, 利用生物技术辅助育种越来越受到重视, 因此麻类作物已经由单一的纤维用新品种选育向多用途、专用型新品种选育发展, 如亚麻轮选1号和天亚7号等, 红麻福红2号和福红992等。可见, 我国主要麻类作物的育种方法正在从常规育种转变为常规育种结合现代生物技术辅助育种方法, 而育种目标也在从高产转变为优质抗病高产并重, 逐步过渡到为了适用多用途而进行专用型品种的选育。其遗传改良已经从一个完全基于表型的过程, 在某种程度上转变为越来越依赖基于基因型的选择。这得益于基因组学革命极大地提高了我们对作物基因组的理解, 使得现代育种家可以更加关注一个新的时代, 即基于基因组测序的作物遗传改良。3.2 麻类作物育种技术的变迁
我国麻类作物育种工作开始于农家品种选育及外来优异种质引进结合常规育种手段, 如杂交育种、诱变育种等, 选育出性状优良种质。随着生物技术的高速发展, 常规育种手段结合现代生物技术的育种方法成了麻类作物的主流育种技术。较常见的3种基于生物技术的育种方法有, 一是分子标记辅助育种 (molecular marker assisted breeding, MAS)[91], 目前常用的分子标记有简单重复序列(simple sequence repeat, SSR)和单核苷酸多态性(single nucleotide polymorphism, SNP)等。这些分子标记在麻类作物中主要应用于物种演化及亲缘关系研究[92]、群体多样性研究[93]、标记辅助选择[94]和遗传图谱构建[95]等。另一种是利用全基因组选择(genomic selection, GS)[96,97], 即利用所有可用的标记进行育种值的预测。在GS中, 所有的位点、单倍型和标记效应都是在整个基因组水平上进行估计的, 根据已知的表型和基因型数据, 以计算代表性群体中每个个体的基因组估计育种值(genome estimation breeding values, GEBVs)。连锁不平衡(linkage disequilibrium, LD)在GS方法使用中是一个非常重要的参数[97], 对于LD衰减速率快的物种, 利用没有亲缘关系的自然群体, 可以使用数量较多的标记可以在不降低检测能力的前提下筛选出用于杂交育种的亲本选择。目前全基因选择育种已经广泛应用于模式作物育种, 玉米[98]和水稻[99]等粮食作物育种中有了较深入的研究, 但在麻类作物上还鲜有报道。全基因组选择育种研究在麻类作物遗传改良中正处于起步阶段, 随着基因型分析成本的降低和统计方法的发展, 基因组选择将会逐步完善, 加快我国麻类作物育种发展的步伐[100]。
还有一种是利用基因编辑育种。随着科学技术的进步, 被称为上帝之手的基因编辑应用于遗传改良, 已成为当今的作物遗传改良研究热点。序列特异性核酸酶, 包括锌指核酸酶(zinc-finger nuclease, ZFN)和转录激活物样效应核酸酶(transcription activator-like effector nuclease, TALEN)已被初步应用于真核生物中进行靶向基因组编辑[101]。另一种基于双链断裂的基因组编辑技术CRISPR/Cas9系统, 是在细菌和古菌群中利用规则间隔短回文重复序列(clustered regularly interspaced short palindromic repeats, CRISPR)自适应免疫系统的基础上发展起来的[101], 它与ZFN和TALEN相比, 操作更简单、精确度和效率也都更高。这种新技术目前已在麻类作物上有所应用, 如构建针对FAD2 基因的敲除载体[102], 为进一步实现在亚麻植株体内对基因高效率的定点编辑, 以及CRISPR/Cas9 系统在亚麻基因组编辑中的应用提供参考。
3.3 下一代测序(NGS)技术对麻类作物遗传改良带来新的策略提供可能
在下一代测序(next generation sequencing, NGS)技术之前, 一个标准的遗传连锁图谱是基于几百个分子标记来构建[103]。NGS技术的发展与许多其他新技术相结合, 使得DNA测序具有高通量和成本低的特点[104], 麻类作物基因组测序完成为基于自然变异的正向遗传学策略提供可能。目前SNP标记在主要麻类作物遗传育种研究中的应用鲜有报道, 首个黄麻SNP连锁图谱用于对RIL群体是否抗茎腐病进行基因分型, 准确率高达91% [94]。这表明, NGS技术对麻类作物高密度遗传图谱构建和重要性状定位以及标记辅助育种带来新的策略。随着NGS技术的发展, 我们可以对麻类作物的不同品种进行深度重测序和全基因组从头测序并进行等位基因变异分析, 通过序列比对找到了大量与重要性状相关的SNP位点, 为品质改良和新品种选育带来了新的科研方法, 加快了新品种的育种进程。在没有参考基因组之前, 基于酶切的简化基因组测序(restriction-site associated DNA sequencing, RAD-seq)[105]和基因分型测序(genotyping by sequencing, GBS)[106]被应用于植物中, 简化基因组测序技术具有不依赖于基因组序列, 而进行高通量的SNP标记开发的优点。为了解亚麻锈病致病机制, 基于RAD-seq技术检测到的SNP构建高密度遗传图谱, 检测到2个新的致病基因, 提高了对该病原体致病机制的认识[107]; 亚麻[108]和苎麻[109]也都利用GBS技术构建了高密度遗传图谱, 分别定位了株高和纤维相关QTL。
随着麻类作物参考基因组图谱绘制, 通过利用基因组所有染色体上的标记和性状之间的连锁不平衡(LD), 进行全基因组关联分析(genome-wide association study, GWAS), 能对自然群体或人工创造群体中的复杂表型进行基因鉴定[110]。112份苎麻核心种质GWAS发现6个与分枝数相关的候选基因[111], 为研究苎麻候选数量性状位点和控制苎麻分枝数的基因提供信息。亚麻GWAS报道较多[112,113,114,115], 鉴定出与纤维、亚麻疫病和亚麻种子油含量相关候选基因, 大量丰富了亚麻育种遗传资源。这些研究反映出, 下一代测序(NGS)技术对麻类作物自然群体的优良等位基因变异发掘以及基因组选择育种带来新的策略。
3.4 麻类作物品种改良的发展趋势
目前的麻类作物育种计划仍是依赖于将分子标记选择整合到常规育种方案中的表型选择, 即常规育种与生物技术相结合。过去, 麻类作物的育种目标大多是高产优质的纤维品种培育。但随着社会发展, 市场需求与产品多样化对麻类育种工作者提出新的挑战, 需要选育出高产高效、抗逆抗病、适宜轻简化机械化、优质专用的多用途麻类作物新品种。一是麻类纤维用途不能只是局限于纺织业。由于麻类纤维可自然降解, 因此以麻纤维为主要原料可制作环保型的包装带或可降解地膜等。麻类纤维具有纤维强力好的特点, 其地膜的抗拉强力大于普通地膜, 适宜机械铺膜, 不仅能减少塑料薄膜导致的白色污染, 而且能减轻劳动强度和生产成本。麻纤维还能用于建筑或家饰板材等用途。近年来关于麻纤维复合材料的研究越来越多, 这是由于与玻璃纤维复合材料相比麻纤维复合材料具有密度低、隔音效果好、价格低和人体亲和性好等优点。因此培育出更适合用于制作复合材料的新品种也是当下麻类作物育种的一大趋势。
二是为了适应现代机械化, 麻类作物育种需要满足轻简栽培需要。直播和机械化等轻简栽培技术能大大降低劳动成本, 目前已经成为水稻、玉米等作物的主流生产技术, 然而麻类作物想要适应轻简栽培还需要克服易倒伏和麻秆易断等问题。为满足麻类作物适应现代机械化, 育种家们需要培育出茎秆粗壮、耐密植、抗倒伏能力强和高产优质抗病的麻类作物新品种。对于短日照喜温类型麻类作物, 宜采取“南种北植”来延长其营养生长期, 提高纤维产量。
三是为了“不与粮争好地”, 培育适合逆境农业的品种。麻类作物在贫瘠土壤的种植, 能具有更强的生产潜能, 这是棉花所不能比拟的, 培育适合逆境农业的新品种不仅仅是要求麻类作物能在不良环境下保持正常生长, 更要稳定的达到高产和优产的特点。因此, 麻类作物育种工作要更注重耐盐碱、抗旱、重金属吸附和抗病虫等性能。
四是根据不同麻类作物特异性状, 培育不同用途的专用品种。如菜用黄麻、高大麻二酚(CBD)大麻品种、籽用大麻和籽用亚麻等。福建农林大学针对福建春夏蔬菜淡季的缺口, 以中晚熟、高产、食用品质优、抗炭疽病为选育目标, 着力开展菜用黄麻新品种的选育, 最终选育出福农系列菜用黄麻, 主食嫩茎和幼叶, 口感极佳。大麻素作为大麻独有的一类次生代谢物, 具有重要的医用价值, 培育高CBD大麻品种将一直是大麻遗传改良的重点方向, 这也是近年来大麻产业蓬勃发展的重要原因之一。亚麻籽油营养丰富, 不仅含α-亚麻酸这一人体所必须的脂肪酸, 而且亚油酸等不饱和脂肪酸含量高, 对皮肤具有优良的亲和力和渗透力, 能用于护肤产品原料, 为满足市场需求, 培育专用的籽用亚麻是亚麻育种工作必不可少的。
4 展望
从优良品种到地方品种和野生近缘种的种质资源, 将仍然是任何育种计划的基础。随着麻类作物参考基因组测序完成, 如何高效挖掘这些麻类种质资源的遗传变异和重要应用价值基因位点信息并应用到遗传改良, 值得探索。NGS革命的第一个效果是降低每个测试数据的标记成本[104], 在琼脂糖凝胶或毛细管测序仪上运行的SSRs或CAPS标记要比在高通量平台上运行的SNPs昂贵得多。然后, 开发一种全基因组选择的育种方法, 如GS[96], 正在成为可能; 并将有助于设计新的植物品种, 不仅应用于少数特定的农艺性状, 而且应用于基因组中几乎所有的基因位点; 以一种成本合理和相对快速的方式, 有望给下一代育种做出显著的贡献。目前基因组选择育种只是在水稻和玉米等主要农作物中有所报道, 若能将此育种方法应用于麻类作物, 必将加快麻类作物育种步伐。预计在不久的将来, NGS技术、生物信息学和表型自动化等方法在种质资源的遗传多样性中进一步应用。建立基于优异基因资源挖掘与种质创新的高通量基因型-表型数据库, 创制种质资源管理和共享平台。创新并集成分子标记辅助选择、GS、转基因等技术, 建立高效的快速育种技术体系, 将彻底改变麻类作物育种策略, 以实现更有效的遗传改良。麻类作物作为第五大作物群, 其遗传改良越来越受益于基因组学的发展, 通过基因组学能发现一批已知位点与功能的基因资源, 从而解决育种亲本贫乏和选择效率低的问题, 同时还能供转基因育种使用, 能大幅度推动主要麻类作物遗传改良。但应该指出的是, 这些重要基因以及位点的信息绝大数是基于或依赖于模式植物的同源基因信息。麻类作物纤维相对于模式植物而言, 有其独特之处。同样地, 麻类作物响应各种非生物胁迫和氮肥等高效利用等方面也有其特异之处。以红麻耐盐碱为例, 耐盐红麻品种在400 mmol L-1的NaCl浓度下可正常生长。完成麻类作物参考基因组图谱绘制, 采用正向和反向遗传学策略挖掘麻类作物的纤维和重要性状的作用机制值得关注, 可开展麻类作物种质资源形成和演化机制研究, 系统解析纤维产量、纤维品质、抗病耐逆等农艺性状形成的分子基础。主要麻类作物特异性状的相关基因有所报道, 但功能基因鉴定仍较困难, 如工业大麻性别相关的基因, 这也与功能标记的缺乏有关。同时, 在利用这些基因进行麻类作物遗传改良的过程中还面临着遗传转化体系及其基因编辑体系构建等挑战, 如在CRISPR-Cas9基因编辑系统中, 挖掘适用于特定麻类作物的内源U6启动子并建立相应的基因编辑体系, 仍需要摸索与优化。克服这些挑战将能显著提高主要麻类作物遗传改良效率。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 3]
[本文引用: 3]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:11935018 [本文引用: 1]
[本文引用: 4]
[本文引用: 2]
[本文引用: 2]
[本文引用: 3]
[本文引用: 2]
[本文引用: 3]
DOI:10.1111/j.1365-313X.2012.05093.xURLPMID:22757964 [本文引用: 2]

Flax (Linum usitatissimum) is an ancient crop that is widely cultivated as a source of fiber, oil and medicinally relevant compounds. To accelerate crop improvement, we performed whole-genome shotgun sequencing of the nuclear genome of flax. Seven paired-end libraries ranging in size from 300 bp to 10 kb were sequenced using an Illumina genome analyzer. A de novo assembly, comprised exclusively of deep-coverage (approximately 94x raw, approximately 69x filtered) short-sequence reads (44-100 bp), produced a set of scaffolds with N(50) =694 kb, including contigs with N(50)=20.1 kb. The contig assembly contained 302 Mb of non-redundant sequence representing an estimated 81% genome coverage. Up to 96% of published flax ESTs aligned to the whole-genome shotgun scaffolds. However, comparisons with independently sequenced BACs and fosmids showed some mis-assembly of regions at the genome scale. A total of 43384 protein-coding genes were predicted in the whole-genome shotgun assembly, and up to 93% of published flax ESTs, and 86% of A. thaliana genes aligned to these predicted genes, indicating excellent coverage and accuracy at the gene level. Analysis of the synonymous substitution rates (K(s) ) observed within duplicate gene pairs was consistent with a recent (5-9 MYA) whole-genome duplication in flax. Within the predicted proteome, we observed enrichment of many conserved domains (Pfam-A) that may contribute to the unique properties of this crop, including agglutinin proteins. Together these results show that de novo assembly, based solely on whole-genome shotgun short-sequence reads, is an efficient means of obtaining nearly complete genome sequence information for some plant species.
[本文引用: 2]
[本文引用: 3]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
URLPMID:26666317 [本文引用: 2]
[本文引用: 1]
DOI:10.1007/s00438-015-1013-yURLPMID:25724692 [本文引用: 1]
We generated the bast transcriptomes of a deficient lignified phloem fibre mutant and its wild-type jute (Corchorus capsularis) using Illumina paired-end sequencing. A total of 34,163 wild-type and 29,463 mutant unigenes, with average lengths of 1442 and 1136 bp, respectively, were assembled de novo, ~77-79 % of which were functionally annotated. These annotated unigenes were assigned to COG (~37-40 %) and GO (~22-28 %) classifications and mapped to 189 KEGG pathways (~19-21 %). We discovered 38 and 43 isoforms of 16 and 10 genes of the upstream shikimate-aromatic amino acid and downstream monolignol biosynthetic pathways, respectively, rendered their sequence similarities, confirmed the identities of 22 of these candidate gene families by phylogenetic analyses and reconstructed the pathway leading to lignin biosynthesis in jute fibres. We also identified major genes and bast-related transcription factors involved in secondary cell wall (SCW) formation. The quantitative RT-PCRs revealed that phenylalanine ammonia-lyase 1 (CcPAL1) was co-down-regulated with several genes of the upstream shikimate pathway in mutant bast tissues at an early growth stage, although its expression relapsed to the normal level at the later growth stage. However, cinnamyl alcohol dehydrogenase 7 (CcCAD7) was strongly down-regulated in mutant bast tissues irrespective of growth stages. CcCAD7 disruption at an early growth stage was accompanied by co-up-regulation of SCW-specific genes cellulose synthase A7 (CcCesA7) and fasciclin-like arabinogalactan 6 (CcFLA6), which was predicted to be involved in coordinating the S-layers' deposition in the xylan-type jute fibres. Our results identified CAD as a promising target for developing low-lignin jute fibres using genomics-assisted molecular approaches.
[本文引用: 1]
[本文引用: 1]
URLPMID:29185475 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:27999968 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:30954076 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s11103-014-0214-9URLPMID:24934879 [本文引用: 1]
Ramie is an old fiber crop, cultivated for thousands of years in China. The cultivar ramie evolved from the wild species Qingyezhuma (QYZM, Boehmeria nivea var. tenacissima). However, the mechanism of domestication of this old fiber crop is poorly understood. In order to characterize the selective pattern in ramie domestication, orthologous genes between the transcriptomes of domesticated ramie variety Zhongzhu 1 (ZZ1) and wild QYZM were assessed using bidirectional best-hit method and ratio of non-synonymous (Ka) to synonymous (Ks) nucleotide substitutions was estimated. Sequence comparison of 56,932 and 59,246 unigenes from the wild QYZM and domesticated ZZ1, respectively, helped identify 10,745 orthologous unigene pairs with a total orthologous length of 10.18 Mb. Among these unigenes, 85 and 13 genes were found to undergo significant purifying and positive selection, respectively. Most of the selected genes were homologs of those involved in abiotic stress tolerance or disease resistance in other plants, suggesting that abiotic and biotic stresses were important selective pressures in ramie domestication. Two genes probably related to the fiber yield of ramie were subjected to positive selection, which may be caused by human manipulation. Thus, our results show the pervasive effects of artificial and natural selections on the accelerated domestication of ramie from its wild relative.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:31931705 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1186/s12870-018-1561-5URLPMID:30577815 [本文引用: 1]
BACKGROUND: MicroRNAs (miRNAs) regulate numerous crucial abiotic stress processes in plants. However, information is limited on their involvement in cadmium (Cd) stress response and tolerance mechanisms in plants, including ramie (Boehmeria nivea L.) that produces a number of economic valuable as an important natural fibre crop and an ideal crop for Cd pollution remediation. RESULTS: Four small RNA libraries of Cd-stressed and non-stressed leaves and roots of ramie were constructed. Using small RNA-sequencing, 73 novel miRNAs were identified. Genome-wide expression analysis revealed that a set of miRNAs was differentially regulated in response to Cd stress. In silico target prediction identified 426 potential miRNA targets that include several uptake or transport factors for heavy metal ions. The reliability of small RNA sequencing and the relationship between the expression levels of miRNAs and their target genes were confirmed by quantitative PCR (q-PCR). We showed that the expression patterns of miRNAs obtained by q-PCR were consistent with those obtained from small RNA sequencing. Moreover, we demonstrated that the expression of six randomly selected target genes was inversely related to that of their corresponding miRNAs, indicating that the miRNAs regulate Cd stress response in ramie. CONCLUSIONS: This study enriches the number of Cd-responsive miRNAs and lays a foundation for the elucidation of the miRNA-mediated regulatory mechanism in ramie during Cd stress.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s11103-016-0571-7URLPMID:27981388 [本文引用: 1]
Functional specialization of cells is among the most fundamental processes of higher organism ontogenesis. The major obstacle to studying this phenomenon in plants is the difficulty of isolating certain types of cells at defined stages of in planta development for in-depth analysis. A rare opportunity is given by the developed model system of flax (Linum usitatissimum L.) phloem fibres that can be purified from the surrounding tissues at the stage of the tertiary cell wall deposition. The performed comparison of the whole transcriptome profile in isolated fibres and other portions of the flax stem, together with fibre metabolism characterization, helped to elucidate the general picture of the advanced stage of plant cell specialization and to reveal novel participants potentially involved in fibre metabolism regulation and cell wall formation. Down-regulation of all genes encoding proteins involved in xylan and lignin synthesis and up-regulation of genes for the specific set of transcription factors transcribed during tertiary cell wall formation were revealed. The increased abundance of transcripts for several glycosyltransferases indicated the enzymes that may be involved in synthesis of fibre-specific version of rhamnogalacturonan I.
[本文引用: 1]
[本文引用: 1]
URLPMID:30484148 [本文引用: 2]
DOI:10.1007/s00425-019-03172-9URLPMID:31037486 [本文引用: 1]
MAIN CONCLUSION: The involvement of a WRKY transcription factor in the regulation of lignan biosynthesis in flax using a hairy root system is described. Secoisolariciresinol is the main flax lignan synthesized by action of LuPLR1 (pinoresinol-lariciresinol reductase 1). LuPLR1 gene promoter deletion experiments have revealed a promoter region containing W boxes potentially responsible for the response to Fusarium oxysporum. W boxes are bound by WRKY transcription factors that play a role in the response to stress. A candidate WRKY transcription factor, LuWRKY36, was isolated from both abscisic acid and Fusarium elicitor-treated flax cell cDNA libraries. This transcription factors contains two WRKY DNA-binding domains and is a homolog of AtWRKY33. Different approaches confirmed LuWRKY36 binding to a W box located in the LuPLR1 promoter occurring through a unique direct interaction mediated by its N-terminal WRKY domain. Our results propose that the positive regulator action of LuWRKY36 on the LuPLR1 gene regulation and lignan biosynthesis in response to biotic stress is positively mediated by abscisic acid and inhibited by ethylene. Additionally, we demonstrate a differential Fusarium elicitor response in susceptible and resistant flax cultivars, seen as a faster and stronger LuPLR1 gene expression response accompanied with higher secoisolariciresinol accumulation in HR of the resistant cultivar.
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/s41598-019-41936-1URLPMID:30944362 [本文引用: 1]
Flax (Linum usitatissimum) is a cool season crop commercially cultivated for seed oil and stem fibre production. A comprehensive characterization of the heat shock factor (HSF) candidate genes in flax can accelerate genetic improvement and adaptive breeding for high temperature stress tolerance. We report the genome-wide identification of 34 putative HSF genes from the flax genome, which we mapped on 14 of the 15 chromosomes. Through comparative homology analysis, we classified these genes into three broad groups, and sub-groups. The arrangement of HSF-specific protein motifs, DNA-binding domain (DBD) and hydrophobic heptad repeat (HR-A/B), and exon-intron boundaries substantiated the phylogenetic separation of these genes. Orthologous relationships and evolutionary analysis revealed that the co-evolution of the LusHSF genes was due to recent genome duplication events. Digital and RT-qPCR analyses provided significant evidence of the differential expression of the LusHSF genes in various tissues, at various developmental stages, and in response to high-temperature stress. The co-localization of diverse cis-acting elements in the promoters of the LusHSF genes further emphasized their regulatory roles in the abiotic stress response. We further confirmed DNA-binding sites on the LusHSF proteins and designed guide RNA sequences for gene editing with minimal off-target effects. These results will hasten functional investigations of LusHSFs or assist in devising genome engineering strategies to develop high-temperature stress tolerant flax cultivars.
URLPMID:28105944 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s10681-019-2449-7URLPMID:33364632 [本文引用: 1]
Various insect pests attack wheat (Triticum aestivum L.) that can cause significant grain yield losses to the crop. Farmers usually depend on pesticides, however, smallholder farmers often have limited and ill-timed access to control methods, including insecticides. Host plant resistance is an alternative to protect grain yield and reduce costs to farmers. Three of the most serious pests of wheat are Diuraphis noxia (Kurdjumov), Mayetiola destructor (Say), and Cephus pygmeaus L. These pests occur in most of the wheat growing areas. However, they are of high importance in North Africa and West Asia. The aim of this study was to evaluate a set of wheat-alien translocations for resistance against D. noxia, M. destructor and C. pygmeaus. Genotypes of this germplasm set have already been reported to carry resistance against certain wheat aphid species. Genotypes 1RSam.1AL and MA1S.1RLe(1B), displayed high levels of resistance against D. noxia and C. pygmeaus, respectively. While three genotypes showed resistance reaction against M. destructor: 1Re(1D), 7A.7S-L5, and 7A.7S-Gb5. Except for the resistant genotype to C. pygmeaus, the other four genotypes were previously reported to carry resistance against Sitobion avenae Fabricius, Rhopalosiphum padi L. and Schizaphis graminum (Rondani). These five wheat-alien translocations are currently being used in the bread-wheat breeding programs at CIMMYT and ICARDA to transfer the multiple pest resistance in elite germplasm.
DOI:10.1371/journal.pone.0226106URLPMID:31830116 [本文引用: 1]
Secreted effectors of fungal pathogens are essential elements for disease development. However, lack of sequence conservation among identified effectors has long been a problem for predicting effector complements in fungi. Here we have explored the expression characteristics of avirulence (Avr) genes and candidate effectors of the flax rust fungus, Melampsora lini. We performed transcriptome sequencing and real-time quantitative PCR (qPCR) on RNA extracted from ungerminated spores, germinated spores, isolated haustoria and flax seedlings inoculated with M. lini isolate CH5 during plant infection. Genes encoding two categories of M. lini proteins, namely Avr proteins and plant cell wall degrading enzymes (CWDEs), were investigated in detail. Analysis of the expression profiles of 623 genes encoding predicted secreted proteins in the M. lini transcriptome shows that the six known Avr genes (i.e. AvrM (avrM), AvrM14, AvrL2, AvrL567, AvrP123 (AvrP) and AvrP4) fall within a group of 64 similarly expressed genes that are induced in planta and show a peak of expression early in infection with a subsequent decline towards sporulation. Other genes within this group include two paralogues of AvrL2, an AvrL567 virulence allele, and a number of genes encoding putative effector proteins. By contrast, M. lini genes encoding CWDEs fall into different expression clusters with their distribution often unrelated to their catalytic activity or substrate targets. These results suggest that synthesis of M. lini Avr proteins may be regulated in a coordinated fashion and that the expression profiling-based analysis has significant predictive power for the identification of candidate Avr genes.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.plantsci.2019.06.004URLPMID:31300147 [本文引用: 1]
Flax seeds have a high oil content and are rich in unsaturated fatty acids, which have advantageous effects in preventing chronic diseases, such as cardiovascular diseases. At present, flax seeds are mainly developed for oil. Therefore, it is of practical significance to identify the candidate genes of fatty acid metabolism in flax seeds for breeding flax seeds with high oil content. In the present study, a natural population of flax containing 224 samples planted in 3 different environments was studied. The genome-wide association analysis (GWAS) of seed fatty acid content was conducted based on specific length amplified fragment sequencing (SLAF-seq) data. Transcriptome sequencing (RNA-seq) of samples from 3 different periods (14 d, 21 d and 28 d after anthesis) during seed development of the low oil variety Shuangya 4 and the high oil variety NEW was performed. The candidate genes for seed fatty acid metabolism were identified by combined analysis of these 2 methods. GWAS detected 16 SNP loci significantly associated with seed fatty acid content, and RNA-seq analysis identified 11,802 differentially expressed genes between high and low oil samples. Pathway enrichment analysis revealed that some differentially expressed genes were classified into fatty acid-related pathways. After comparison of these differentially expressed genes with the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, 20 genes homologous to other species were obtained. After analysis, 10 candidate genes were screened by GWAS and RNA-seq screening. Of these 10 genes, qRT-PCR assays using flax seeds in 5 different developmental stages showed that the expression levels of 6 candidate genes were significantly correlated with 5 fatty acid contents in seeds of the high oil variety NEW. Through metabolic pathway analysis found that 6 genes were involved in important fatty acid metabolic pathways, and some of them also have upstream and downstream regulation relations. The present study combined GWAS and RNA-seq methods to identify candidate genes for fatty acid metabolism in flax seeds, which provided reference for screening of candidate genes with complex traits.
DOI:10.3389/fpls.2016.01733URLPMID:27917184 [本文引用: 6]
Cannabis sativa L. is an annual herbaceous crop grown for the production of long extraxylary fibers, the bast fibers, rich in cellulose and used both in the textile and biocomposite sectors. Despite being herbaceous, hemp undergoes secondary growth and this is well exemplified by the hypocotyl. The hypocotyl was already shown to be a suitable model to study secondary growth in other herbaceous species, namely Arabidopsis thaliana and it shows an important practical advantage, i.e., elongation and radial thickening are temporally separated. This study focuses on the mechanisms marking the transition from primary to secondary growth in the hemp hypocotyl by analysing the suite of events accompanying vascular tissue and bast fiber development. Transcriptomics, imaging and quantification of phytohormones were carried out on four representative developmental stages (i.e., 6-9-15-20 days after sowing) to provide a comprehensive overview of the events associated with primary and secondary growth in hemp. This multidisciplinary approach provides cell wall-related snapshots of the growing hemp hypocotyl and identifies marker genes associated with the young (expansins, beta-galactosidases, and transcription factors involved in light-related processes) and the older hypocotyl (secondary cell wall biosynthetic genes and transcription factors).
URLPMID:25294860 [本文引用: 1]
[本文引用: 1]
DOI:10.1105/tpc.113.116970URLPMID:24220634 [本文引用: 1]
LIN-11, Isl1 and MEC-3 (LIM)-domain proteins play pivotal roles in a variety of cellular processes in animals, but plant LIM functions remain largely unexplored. Here, we demonstrate dual roles of the WLIM1a gene in fiber development in upland cotton (Gossypium hirsutum). WLIM1a is preferentially expressed during the elongation and secondary wall synthesis stages in developing fibers. Overexpression of WLIM1a in cotton led to significant changes in fiber length and secondary wall structure. Compared with the wild type, fibers of WLIM1a-overexpressing plants grew longer and formed a thinner and more compact secondary cell wall, which contributed to improved fiber strength and fineness. Functional studies demonstrated that (1) WLIM1a acts as an actin bundler to facilitate elongation of fiber cells and (2) WLIM1a also functions as a transcription factor to activate expression of Phe ammonia lyase-box genes involved in phenylpropanoid biosynthesis to build up the secondary cell wall. WLIM1a localizes in the cytosol and nucleus and moves into the nucleus in response to hydrogen peroxide. Taken together, these results demonstrate that WLIM1a has dual roles in cotton fiber development, elongation, and secondary wall formation. Moreover, our study shows that lignin/lignin-like phenolics may substantially affect cotton fiber quality; this finding may guide cotton breeding for improved fiber traits.
DOI:10.1111/nph.13320URLPMID:25676073 [本文引用: 1]
Fasciclin-like arabinogalactan protein (FLA) families have been identified and characterised in key plant species, with some members exhibiting functional specialization. Here we identify the FLA family of Eucalyptus grandis, and investigate the roles of three single-FAS domain FLAs, with particular focus on secondary cell-wall formation and wood properties. We use various in-silico approaches to identify and characterise E. grandis genome FLAs, and perform phylogenetic comparisons with other species. For three key FLAs, we perform functional testing including promoter-reporter and overexpression transgenic approaches using eucalypts, poplar and tobacco. Of the 18 eucalypt FLAs identified, several were specifically and highly expressed in stems. The specificity to stem xylem vessel and fibre development was demonstrated with EniFLA1promoter:GUS studies in several species. Testing of select eucalypt FLAs resulted in altered wood development and properties, for example 35S:EgrFLA2 led to a 3 degree reduction in cellulose microfibril angle in eucalypt xylem fibres, and 35S:EgrFLA3 to a reduction in tobacco stem flexural strength. These results indicate that the eucalypt FLA family contains diverse members, and particular members with single FAS domains that are functionally specialized for secondary cell wall growth and properties.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:31138625 [本文引用: 1]
[本文引用: 1]
DOI:10.1101/gr.242594.118URLPMID:30409771 [本文引用: 1]
Cannabis sativa is widely cultivated for medicinal, food, industrial, and recreational use, but much remains unknown regarding its genetics, including the molecular determinants of cannabinoid content. Here, we describe a combined physical and genetic map derived from a cross between the drug-type strain Purple Kush and the hemp variety
DOI:10.1101/gr.251207.119URLPMID:32033943 [本文引用: 1]
Cannabis sativa-derived tetrahydrocannabinol (THC) production is increasing very fast worldwide. C. sativa is a dioecious plant with XY Chromosomes, and only females (XX) are useful for THC production. Identifying the sex chromosome sequence would improve early sexing and better management of this crop; however, the C. sativa genome projects have failed to do so. Moreover, as dioecy in the Cannabaceae family is ancestral, C. sativa sex chromosomes are potentially old and thus very interesting to study, as little is known about old plant sex chromosomes. Here, we RNA-sequenced a C. sativa family (two parents and 10 male and female offspring, 576 million reads) and performed a segregation analysis for all C. sativa genes using the probabilistic method SEX-DETector. We identified >500 sex-linked genes. Mapping of these sex-linked genes to a C. sativa genome assembly identified the largest chromosome pair being the sex chromosomes. We found that the X-specific region (not recombining between X and Y) is large compared to other plant systems. Further analysis of the sex-linked genes revealed that C. sativa has a strongly degenerated Y Chromosome and may represent the oldest plant sex chromosome system documented so far. Our study revealed that old plant sex chromosomes can have large, highly divergent nonrecombining regions, yet still be roughly homomorphic.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/s41598-017-15816-5URLPMID:29142201 [本文引用: 1]
Cannabis sativa has a complex history reflected in both selection on naturally occurring compounds and historical trade routes among humans. Iran is a rich resource of natural populationswhich hold the promise to characterize historical patterns of population structure and genetic diversity within Cannabis. Recent advances in high-throughput DNA sequencing technologies have dramatically increased our ability to produce information to the point that it is now feasible to inexpensively obtain population level genotype information at a large scale. In the present investigation, we have explored the use of Genotyping-By-Sequencing (GBS) in Iranian cannabis. We genotyped 98 cannabis samples 36 from Iranian locations and 26 accessions from two germplasm collections. In total, 24,710 high-quality Single Nucleotide Polymorphisms (SNP) were identified. Clustering analysis by Principal Component Analysis (PCA) identified two genetic clusters among Iranian populations and fineSTRUCTURE analysis identified 19 populations with some geographic partitioning. We defined Iranian cannabis in two main groups using the results of the PCA and discovered some strong signal to define some locations as population according to fineSTRUCTURE analyses. However, single nucleotide variant analysis uncovered a relatively moderate level of variation among Iranian cannabis.
[本文引用: 2]
DOI:10.1186/s12870-019-2205-0URLPMID:31878891 [本文引用: 1]
BACKGROUND: Pueraria candollei var. mirifica, a Thai medicinal plant used traditionally as a rejuvenating herb, is known as a rich source of phytoestrogens, including isoflavonoids and the highly estrogenic miroestrol and deoxymiroestrol. Although these active constituents in P. candollei var. mirifica have been known for some time, actual knowledge regarding their biosynthetic genes remains unknown. RESULTS: Miroestrol biosynthesis was reconsidered and the most plausible mechanism starting from the isoflavonoid daidzein was proposed. A de novo transcriptome analysis was conducted using combined P. candollei var. mirifica tissues of young leaves, mature leaves, tuberous cortices, and cortex-excised tubers. A total of 166,923 contigs was assembled for functional annotation using protein databases and as a library for identification of genes that are potentially involved in the biosynthesis of isoflavonoids and miroestrol. Twenty-one differentially expressed genes from four separate libraries were identified as candidates involved in these biosynthetic pathways, and their respective expressions were validated by quantitative real-time reverse transcription polymerase chain reaction. Notably, isoflavonoid and miroestrol profiling generated by LC-MS/MS was positively correlated with expression levels of isoflavonoid biosynthetic genes across the four types of tissues. Moreover, we identified R2R3 MYB transcription factors that may be involved in the regulation of isoflavonoid biosynthesis in P. candollei var. mirifica. To confirm the function of a key-isoflavone biosynthetic gene, P. candollei var. mirifica isoflavone synthase identified in our library was transiently co-expressed with an Arabidopsis MYB12 transcription factor (AtMYB12) in Nicotiana benthamiana leaves. Remarkably, the combined expression of these proteins led to the production of the isoflavone genistein. CONCLUSIONS: Our results provide compelling evidence regarding the integration of transcriptome and metabolome as a powerful tool for identifying biosynthetic genes and transcription factors possibly involved in the isoflavonoid and miroestrol biosyntheses in P. candollei var. mirifica.
URLPMID:11290733 [本文引用: 2]
Recent advances in molecular genetic techniques will make dense marker maps available and genotyping many individuals for these markers feasible. Here we attempted to estimate the effects of approximately 50,000 marker haplotypes simultaneously from a limited number of phenotypic records. A genome of 1000 cM was simulated with a marker spacing of 1 cM. The markers surrounding every 1-cM region were combined into marker haplotypes. Due to finite population size N(e) = 100, the marker haplotypes were in linkage disequilibrium with the QTL located between the markers. Using least squares, all haplotype effects could not be estimated simultaneously. When only the biggest effects were included, they were overestimated and the accuracy of predicting genetic values of the offspring of the recorded animals was only 0.32. Best linear unbiased prediction of haplotype effects assumed equal variances associated to each 1-cM chromosomal segment, which yielded an accuracy of 0.73, although this assumption was far from true. Bayesian methods that assumed a prior distribution of the variance associated with each chromosome segment increased this accuracy to 0.85, even when the prior was not correct. It was concluded that selection on genetic values predicted from markers could substantially increase the rate of genetic gain in animals and plants, especially if combined with reproductive techniques to shorten the generation interval.
DOI:10.1038/nrg3097URLPMID:22207165 [本文引用: 2]
The completion of reference genome sequences for many important crops and the ability to perform high-throughput resequencing are providing opportunities for improving our understanding of the history of plant domestication and to accelerate crop improvement. Crop plant comparative genomics is being transformed by these data and a new generation of experimental and computational approaches. The future of crop improvement will be centred on comparisons of individual plant genomes, and some of the best opportunities may lie in using combinations of new genetic mapping strategies and evolutionary analyses to direct and optimize the discovery and use of genetic variation. Here we review such strategies and insights that are emerging.
DOI:10.1038/ng.1033URLPMID:22246502 [本文引用: 1]
Maize is both an exciting model organism in plant genetics and also the most important crop worldwide for food, animal feed and bioenergy production. Recent genome-wide association and metabolic profiling studies aimed to resolve quantitative traits to their causal genetic loci and key metabolic regulators. Here we present a complementary approach that exploits large-scale genomic and metabolic information to predict complex, highly polygenic traits in hybrid testcrosses. We crossed 285 diverse Dent inbred lines from worldwide sources with two testers and predicted their combining abilities for seven biomass- and bioenergy-related traits using 56,110 SNPs and 130 metabolites. Whole-genome and metabolic prediction models were built by fitting effects for all SNPs or metabolites. Prediction accuracies ranged from 0.72 to 0.81 for SNPs and from 0.60 to 0.80 for metabolites, allowing a reliable screening of large collections of diverse inbred lines for their potential to create superior hybrids.
URLPMID:27311694 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1186/s12864-016-3011-9URLPMID:27550217 [本文引用: 1]
BACKGROUND: Rust fungi are an important group of plant pathogens that cause devastating losses in agricultural, silvicultural and natural ecosystems. Plants can be protected from rust disease by resistance genes encoding receptors that trigger a highly effective defence response upon recognition of specific pathogen avirulence proteins. Identifying avirulence genes is crucial for understanding how virulence evolves in the field. RESULTS: To facilitate avirulence gene cloning in the flax rust fungus, Melampsora lini, we constructed a high-density genetic linkage map using single nucleotide polymorphisms detected in restriction site-associated DNA sequencing (RADseq) data. The map comprises 13,412 RADseq markers in 27 linkage groups that together span 5860 cM and contain 2756 recombination bins. The marker sequences were used to anchor 68.9 % of the M. lini genome assembly onto the genetic map. The map and anchored assembly were then used to: 1) show that M. lini has a high overall meiotic recombination rate, but recombination distribution is uneven and large coldspots exist; 2) show that substantial genome rearrangements have occurred in spontaneous loss-of-avirulence mutants; and 3) identify the AvrL2 and AvrM14 avirulence genes by map-based cloning. AvrM14 is a dual-specificity avirulence gene that encodes a predicted nudix hydrolase. AvrL2 is located in the region of the M. lini genome with the lowest recombination rate and encodes a small, highly-charged proline-rich protein. CONCLUSIONS: The M. lini high-density linkage map has greatly advanced our understanding of virulence mechanisms in this pathogen by providing novel insights into genome variability and enabling identification of two new avirulence genes.
DOI:10.1186/s12870-018-1366-6URLPMID:30086718 [本文引用: 1]
BACKGROUND: Flax is an important field crop that can be used for either oilseed or fiber production. Plant height and technical length are important characters for flax. For linseed flax, plants usually have a short technical length and plant height than those for fiber flax. As an important agronomical character for fiber and linseed flax, plant height is usually a selection target for breeding. However, because of limited technologies and methods available, there has been little research focused on discovering the molecular mechanism controlling plant height. RESULTS: In this study, two related recombinant inbred line (RIL) populations developed from crosses of linseed and fiber parents were developed and phenotyped for plant height and technical length in four environments. A consensus linkage map based on two RIL populations was constructed using SNP markers generated by genotyping by sequencing (GBS) technology. A total of 4497 single nucleotide polymorphism (SNP) markers were included on 15 linkage groups with an average marker density of one marker every 2.71 cM. Quantitative trait locus (QTL) mapping analysis was performed for plant height and technical length using the two populations. A total of 19 QTLs were identified for plant height and technical length. For the MH population, eight plant height QTLs and seven technical length QTLs were identified, five of which were common QTLs for both traits. For the PH population, six plant height and three technical length QTLs were identified. By comparing the QTLs and candidate gene information in the two population, two common QTLs and three candidate genes were discovered. CONCLUSIONS: This study provides a foundation for map-based cloning of QTLs and marker-assisted selection for plant height-related traits in linseed and fiber flax.
DOI:10.1038/s41598-017-13762-wURLPMID:29044147 [本文引用: 1]
Ramie fiber extracted from ramie stem bark (RSB) is a highly important natural fiber, and therefore, RSB is an economically important plant organ. The genetic basis of RSB traits is poorly understood. In the present study, fiber yield and three RSB traits (bark thickness, bark weight, and fiber output ratio) were subject to quantitative trait locus (QTL) analysis using an F2 agamous line population derived from two ramie varieties (Qingdaye and Zhongzhu 1). A total of 4338 high-quality single nucleotide polymorphisms were identified using the genotyping-by-sequencing technique and were subsequently used to construct a high-density genetic map spanning 1942.9 cM. Thereafter, QTL analysis identified five, two, four, and four QTLs for bark thickness, bark weight, fiber output ratio, and fiber yield, respectively. A 5.1 cM region that corresponded to a QTL for bark thickness (qBT4a) contained 106 candidate genes, and the Zhongzhu 1 allele of one of the genes, a putative MYB gene (evm. MODEL: scaffold7373.133_D1), included a 760-bp insertion that caused premature termination, thereby producing a protein that lacked part of the MYB domain. Because MYB transcription factors play central roles in regulating the development of secondary cellular walls and fiber biosynthesis, we propose evm. MODEL: scaffold7373.133_D1 as a likely candidate gene for qBT4a.
[本文引用: 1]
[本文引用: 1]
DOI:10.1186/s12870-018-1573-1URLPMID:30541445 [本文引用: 1]
DOI:10.3389/fpls.2017.02232URLPMID:29375606 [本文引用: 1]
Flax (Linum usitatissimum L.) is an important cash crop, and its agronomic traits directly affect yield and quality. Molecular studies on flax remain inadequate because relatively few flax genes have been associated with agronomic traits or have been identified as having potential applications. To identify markers and candidate genes that can potentially be used for genetic improvement of crucial agronomic traits, we examined 224 specimens of core flax germplasm; specifically, phenotypic data for key traits, including plant height, technical length, number of branches, number of fruits, and 1000-grain weight were investigated under three environmental conditions before specific-locus amplified fragment sequencing (SLAF-seq) was employed to perform a genome-wide association study (GWAS) for these five agronomic traits. Subsequently, the results were used to screen single nucleotide polymorphism (SNP) loci and candidate genes that exhibited a significant correlation with the important agronomic traits. Our analyses identified a total of 42 SNP loci that showed significant correlations with the five important agronomic flax traits. Next, candidate genes were screened in the 10 kb zone of each of the 42 SNP loci. These SNP loci were then analyzed by a more stringent screening via co-identification using both a general linear model (GLM) and a mixed linear model (MLM) as well as co-occurrences in at least two of the three environments, whereby 15 final candidate genes were obtained. Based on these results, we determined that UGT and PL are candidate genes for plant height, GRAS and XTH are candidate genes for the number of branches, Contig1437 and LU0019C12 are candidate genes for the number of fruits, and PHO1 is a candidate gene for the 1000-seed weight. We propose that the identified SNP loci and corresponding candidate genes might serve as a biological basis for improving crucial agronomic flax traits.
[本文引用: 1]
[本文引用: 1]
DOI:10.3389/fpls.2018.01982URLPMID:30693010 [本文引用: 1]
Pasmo is one of the most widespread diseases threatening flax production. To identify genetic regions associated with pasmo resistance (PR), a genome-wide association study was performed on 370 accessions from the flax core collection. Evaluation of pasmo severity was performed in the field from 2012 to 2016 in Morden, MB, Canada. Genotyping-by-sequencing has identified 258,873 single nucleotide polymorphisms (SNPs) distributed on all 15 flax chromosomes. Marker-trait associations were identified using ten different statistical models. A total of 692 unique quantitative trait nucleotides (QTNs) associated with 500 putative quantitative trait loci (QTL) were detected from six phenotypic PR datasets (five individual years and average across years). Different QTNs were identified with various statistical models and from individual PR datasets, indicative of the complementation between analytical methods and/or genotype x environment interactions of the QTL effects. The single-locus models tended to identify large-effect QTNs while the multi-loci models were able to detect QTNs with smaller effects. Among the putative QTL, 67 had large effects (3-23%), were stable across all datasets and explained 32-64% of the total variation for PR in the various datasets. Forty-five of these QTL spanned 85 resistance gene analogs including a large toll interleukin receptor, nucleotide-binding site, leucine-rich repeat (TNL) type gene cluster on chromosome 8. The number of QTL with positive-effect or favorite alleles (NPQTL) in accessions was significantly correlated with PR (R (2) = 0.55), suggesting that these QTL effects are mainly additive. NPQTL was also significantly associated with morphotype (R (2) = 0.52) and major QTL with positive effect alleles were present in the fiber type accessions. The 67 large effect QTL are suited for marker-assisted selection and the 500 QTL for effective genomic prediction in PR molecular breeding.
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}