 ,, 王延周, 严理, 朱四元, 刘头明,*农业农村部麻类生物学与加工重点实验室/中国农业科学院麻类研究所, 湖南长沙 410205
,, 王延周, 严理, 朱四元, 刘头明,*农业农村部麻类生物学与加工重点实验室/中国农业科学院麻类研究所, 湖南长沙 410205Characterization of the expression profiling of circRNAs in the barks of stems in ramie
LI Fu,, WANG Yan-Zhou, YAN Li, ZHU Si-Yuan, LIU Tou-Ming,*Key Laboratory of Biological and Processing for Bast Fiber Crops, Ministry of Agriculture and Rural Affairs/Institute of Bast Fiber Crops, Chinese Academy of Agricultural Sciences, Changsha 410205, Hunan, China通讯作者:
收稿日期:2020-02-24接受日期:2020-07-2网络出版日期:2021-06-12
| 基金资助: |
Received:2020-02-24Accepted:2020-07-2Online:2021-06-12
| Fund supported: |
作者简介 About authors
E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (1873KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
李富, 王延周, 严理, 朱四元, 刘头明. 苎麻茎皮环状RNA表达谱分析[J]. 作物学报, 2021, 47(6): 1020-1030. doi:10.3724/SP.J.1006.2021.04042
LI Fu, WANG Yan-Zhou, YAN Li, ZHU Si-Yuan, LIU Tou-Ming.
苎麻[Boehmeria nivea (L.) Gaud.]是我国重要的天然纤维作物, 有着悠久的栽培和加工历史, 其纤维及纤维织品是我国重要的工业原料和传统的出口创汇产品。中国作为苎麻的主产国, 其种植面积占全世界90%以上, 因此在国际上苎麻又被称为“中国草”。苎麻纤维是一种韧皮纤维, 具有拉伸力强、纤维长等特点, 其单纤维最长可达55 cm [1], 主要成分为纤维素、半纤维素和木质素。
目前, 苎麻中已有大量可能与纤维发育相关的基因被鉴定, 这为深入解析其纤维形成机制奠定了基础。基于RACE技术, 多个苎麻纤维素合成酶基因(CesA)已被鉴定和克隆[2,3,4]。木质素作为纤维主要成分之一, 其多个生物合成酶基因, 如肉桂酸4-羟化酶(C4H)、香豆酸4-香豆酸:辅酶A连接酶(4CL)、肉桂酰辅酶a还原酶基因(CCR)也被鉴定和克隆[5,6,7], 并发现它们的表达水平与苎麻纤维木质素含量显著相关[8]。此外, 近10年来高通量测序技术快速发展, 也被广泛应用于苎麻纤维发育机制研究。Liu等[9]分析苎麻转录组共获得36个在韧皮部中高表达的苎麻CesA基因。Chen等[10]比较不同发育时期的苎麻韧皮组织转录组表达谱, 共鉴定到780个差异表达的基因, 其中含4个CesA基因、3个扩张蛋白基因、6个木聚糖水解酶基因, 它们很可能与苎麻的纤维发育相关。
非编码RNA是一类直接发挥催化和调控功能的转录本, 包含miRNA、长链非编码RNA (lncRNA)和环状RNA (circRNA)等[11]。Wang等[12]分析了苎麻纤维在4个不同发育时期的miRNA表达谱, 并在纤维伸长期和次生壁加厚期分别鉴定到150个和148个miRNA, 其中51个miRNA在纤维不同发育期呈现差异表达, 推测非编码RNA可能参与苎麻的纤维发育。circRNA是一类由mRNA前体经反向可变剪切而来的共价闭合且保守的环状转录本, 根据剪接位点可分为exonic circRNAs、intronic circRNAs、exonic-intronic circRNAs和intergenic circRNAs, 它们大多数具有较低的表达量, 且呈现组织特异性和细胞特异性表达。circRNA已知的功能包括隔离miRNA或蛋白质、转录调节和干扰可变剪接, 甚至翻译产生多肽[13]。植物circRNA主要集中于生长发育及生物和非生物逆境研究[14,15,16,17,18,19], 如Zhang等[14]在玉米和拟南芥中分别鉴定出2174个和1354个干旱相关的circRNA; Zuo等[15]鉴定出番茄163个冷胁迫响应的circRNA; Wang等[16]在野生型和有义/反义LeERF1转基因番茄果实中发现282个显著差异表达的cicrRNA; Wang等[18]在小麦中发现62个circRNA响应干旱逆境; Gao等[19]在葡萄中鉴定了475个响应寒冷的差异表达circRNA。尽管circRNA被发现广泛参与了植物的生长调控, 但有关它在植物纤维发育中的功能角色, 当前仍鲜有研究。本研究将开展苎麻纤维发育期的全基因组circRNA表达谱分析, 并筛选差异表达的circRNA, 旨在为理解circRNA在植物纤维发育中的调控角色奠定基础。
1 材料与方法
1.1 试验材料和取样
本试验以中苎1号为研究材料。2016年, 将中苎1号扦插苗栽种于中国农业科学院麻类研究所沅江田间试验基地。经切片显微观察, 茎中部韧皮纤维处于生长期, 次生壁正在加厚; 而茎顶部韧皮纤维尚未起始生长[10]。因此, 在2018年5月, 当具有两年龄的苎麻长到约1.5 m高时, 按照Chen等[10]方法, 分别取茎中部(the barks sample collected from the middle, MPS)和顶部(the barks sample collected from the top, TPS)的茎皮组织(图1)。对3株苎麻单独取样, 分别作为3个试验重复。组织样品经液氮速冻后置于-80℃低温保存, 以备后续试验使用。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1苎麻circRNA测序取样部位示意图
MPS和TPS分别指从茎中部和顶部收集的茎皮组织样品, 其中位于茎顶端的纤维细胞次生壁尚未加厚, 而处于茎中部的纤维细胞次生壁显著加厚(图片引自Chen等[10])。
Fig. 1A picture shows the sampling position for circRNA sequencing
MPS and TPS represent the barks sample collected from the middle and top stems, respectively. This picture displays that the secondary cellular walls (SCWs) of fiber cells from the top of the ramie stems do not initiate growth and those from middle part of the ramie stems are thickening. This figure is cited from the study of Chen et al.[10]
1.2 文库构建和高通量测序
利用植物RNA提取试剂TRIzol (Invitrogen, CA, USA)提取6个样品的总RNA。之后对6个RNA样品单独构建circRNA测序文库, 具体流程如下: 使用试剂盒(Ribo-Zero Gold rRNA Removal Kit, Illumina)去除total RNA中的核糖体RNA, 并使用Agilent 2100Bioanalyzer (Agilent Technologies)验证纯化效果; RNaseR反应体系(TruSeq Stranded Total RNA with Ribo-Zero Plant, Illumina, USA)消化线性RNA, 将反应产物纯化, 并打断使其片段化; 以片段化环状RNA为模板, 使用反转录试剂盒(SuperScript III First-Strand Synthesis System for RT-PCR, Invitrogen)先后合成第一链cDNA和第二链cDNA, 并使用试剂盒(PureLink Quick Gel Extraction and PCR Purification Combo Kit, Invitrogen)纯化回收、黏性末端修复、cDNA的3°末端加上碱基“A”并连接接头, 最后进行PCR扩增; 构建好的文库用Agilent2100 Bioanalyzer (Agilent Technologies)和StepOnePlus Real-time PCR System (ABI)质检, 合格后的文库则通过Illumina测序平台(HiSeq 2500, Illumina, USA)进行高通量测序, 从而获得原始的序列reads。1.3 circRNA预测
对原始序列进行过滤, 主要包括去除接头污染的序列reads、未知碱基N含量大于5%的reads及低质量的序列reads, reads中超过20%的碱基为质量值低于30的碱基(即碱基错误率大于0.032), 从而获得高质量的clean reads。将clean reads比对到苎麻参考基因组上[20], 然后通过CIRI[21]、find_circ [22] 2个软件预测circRNA, 并且依据circRNA起始、终止位置来整合2个软件的预测结果。若2个circRNA的起始和终止位置均相近(10个碱基以内), 则将这2个circRNA合并为1个。1.4 circRNA表达定量及差异表达分析
本研究根据比对至circRNA两端的junction reads数来对circRNA表达定量[23]。因circRNA是由CIRI和find_circ 2个软件预测而来, 故就每个circRNA, 我们计算2个软件注释的junction reads数的平均值, 将其作为该circRNA最终的junction reads数。采用RPB (junction reads per billion mapped reads, 即比对上基因组的所有reads标准化到十亿后跨过backspliced位点的junction reads数目)对各样品作均一化处理。原始的clean reads序列及其表达定量已被提交到DDBJ/EMBL/GenBank数据库(序列号位GSE130587)。利用DEseq2软件[24]筛选差异表达的circRNA。利用DEseq2软件计算circRNA在茎顶部和茎中部的表达量差异倍数和P值, 通过Benjamini-Hochberg统计方法[25]对P值进行校正。对于每个circRNA, 若其校正的P值小于0.05, 表达差异大于2倍, 则认为该circRNA在2个组织中存在表达差异。
1.5 荧光定量PCR (qRT-PCR)分析
分别提取茎顶部和茎中部RNA (3个生物学重复, 共6个样品), 用DNase I (Fermentas, Canada)进行消化处理, 去除其中的DNA。之后, 利用M-MuLV反转录试剂盒(Fermentas)合成第1链cDNA。使用iTaq Universal SYBR Green super mix试剂盒(Bio-Rad, USA)配置qRT-PCR反应体系, 并在iQ5实时荧光PCR仪上进行PCR。PCR反应程序: 第一步95℃ 30 s; 第二步40个循环: 95℃ 15 s, 60 ℃ 30 s。就每个circRNA, 每个样品进行5个PCR反应重复。18S核糖体RNA在植物各器官中呈现组成性表达, 因此被用作内参基因[12]。circRNA和内参基因引物见表1。依据Livak的2-ΔΔCT方法[26]计算各circRNA在2个组织中的相对表达水平和标准误。Table 1
表1
表1用于qRT-PCR分析的苎麻差异表达circRNA和内参基因引物序列
Table 1
| circRNA名称 circRNA ID | 正向引物 Forward primer (5°-3°) | 反向引物 Reverse primer (5°-3°) |
|---|---|---|
| scaffold270:40371|40783 | TGGCATGAATCACCGAACCTG | TGTCATGACGCGCCCATTCCT |
| scaffold6:1851520|1851960 | ATGAAATCAGGGAATGCCAA | CTCCCAAACTTCACCGTC |
| scaffold360:24869|25462 | CTTTCCTTGCTTCGCCAAC | TTCCCTGCTACTCTATTGCT |
| scaffold360:11517|11838 | TCCACTAGCGTCATTCGAAG | CTAGGTCAGAAAAGCGAGAAC |
| scaffold36:1536277|1547559 | TTGATCTTGGCCATGTCCCGA | ACGGTTCAATAAGGCGAGA |
| scaffold110:48151|61235 | AGAATCCACAATACCGTCAGC | TTGTGTATTCATTTGGCCCAT |
| scaffold30:600099|602644 | ATAAAGAGTCGTTGGGCCTT | TGAACTTCCTTTCCGCCAGA |
| scaffold11:2168138|2169190 | GTCCACAGAGCGATCATATGTC | TTTTAGGCACTTACCGGGTT |
| scaffold360:15097|17687 | CGTTTGACTTGATCCCGACAG | CAGAGCAAATGAGGGTATAGCAA |
| scaffold128:291760|298584 | ACAAAAAGCAATGGCAATTACGG | AACAGACTCACCAATGGCATC |
| 18S | ATGATAACTCGACGGATCGC | CTTGGATGTGGTAGCCGTTT |
新窗口打开|下载CSV
2 结果与分析
2.1 circRNA的鉴定
经测序, 6个样品共产生大约6.46亿个原始序列reads, 经过滤后得到共计4.45亿个高质量的clean reads, 序列长度合计达到66.7 Gb (表2)。每个样品过滤后均有约74.0百万个clean reads, 且其Q30值均达到96%以上, 说明测序获得的数量和质量均足以开展下一步的circRNA分析。Table 2
表2
表2苎麻6个茎皮组织样本的circRNA测序数据统计
Table 2
| 样品 Sample | 原始read数 Raw read number (Million) | Clean read数 Clean read number (Million) | 总长度 Total length (Gb) | Q30值 Q30 value (%) | Clean reads产出率 Yielded ratio of clean reads (%) |
|---|---|---|---|---|---|
| MPS1 | 101.23 | 74.3 | 11.1 | 96.4 | 73.4 |
| MPS2 | 106.02 | 74.5 | 11.2 | 96.0 | 70.3 |
| MPS3 | 107.77 | 74.3 | 11.1 | 96.7 | 68.9 |
| TPS1 | 102.76 | 74.0 | 11.1 | 96.0 | 72.0 |
| TPS2 | 119.18 | 73.7 | 11.1 | 96.1 | 61.9 |
| TPS3 | 109.41 | 73.7 | 11.1 | 96.4 | 67.4 |
新窗口打开|下载CSV
基于这些高质量的序列, 本研究在苎麻茎顶部和中部韧皮组织中共鉴定到5268个circRNA, 其中539个位于基因间(intergenic), 而4729个(89.8%)则位于基因内部(intragenic)。对这4729个基因进行GO (gene ontology)功能分类发现, 它们主要归类到结合(binding)、催化活性(catalytic activity)、细胞过程(cellular process)、代谢过程(metabolic process)、膜(membrane)等功能类别(Q < 0.01)(图2)。
图2
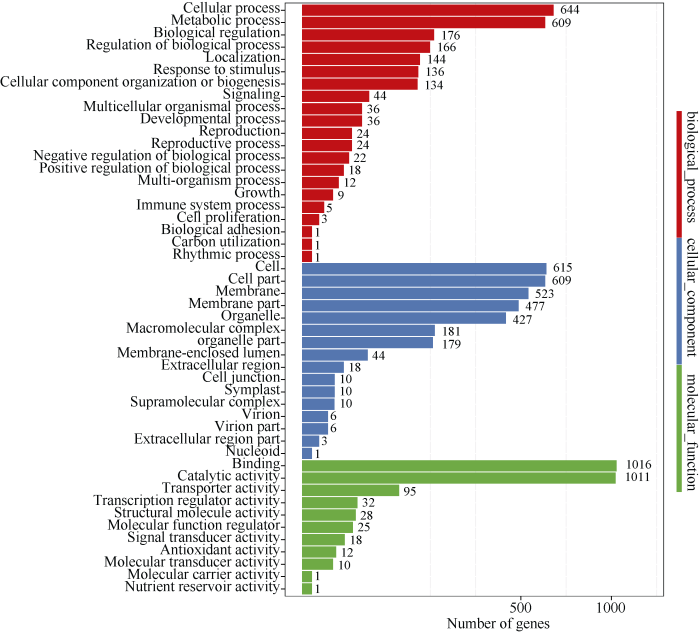 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2可转录circRNA的4729个苎麻基因GO功能分类
Fig. 2GO functional classification of 4729 transcribed circRNAs genes in ramie
2.2 circRNA表达谱分析
显微观察显示, 苎麻茎中部韧皮纤维处于生长期, 次生壁正在加厚; 而茎顶部韧皮纤维尚未起始生长(图1)。比较5268个苎麻circRNA在2个茎韧皮部位中的表达水平发现, 相对于TPS, 在MPS韧皮中有77个circRNA显著上调表达, 仅1个circRNA表达水平下调(图3)。因茎中部和顶部的纤维处于不同发育时期, 说明这78个circRNA在2个不同纤维发育时期存在表达差异。在这78个circRNA中, 11个circRNA的表达水平差异倍数达到30倍以上(表3)。其中scaffold13:3497301|3498287和scaffold7:2328081|2328970在MPS中的表达分别上调了42.7倍和42.0倍, 它们分别定位在苎麻基因whole_GLEAN_10021135和whole_GLEAN_10025341中; whole_GLEAN_10021135编码贝壳杉烯酸氧化酶, whole_GLEAN_10025341则为酪蛋白激酶编码基因。此外, 在这78个差异表达的circRNA中, 74个(94.9%)位于基因内部, 其中scaffold1:2974233| 2974487被定位在一个knotted-1-like Homeobox基因(whole_GLEAN_10029667)中, 其上调表达倍数达到30.6。图3
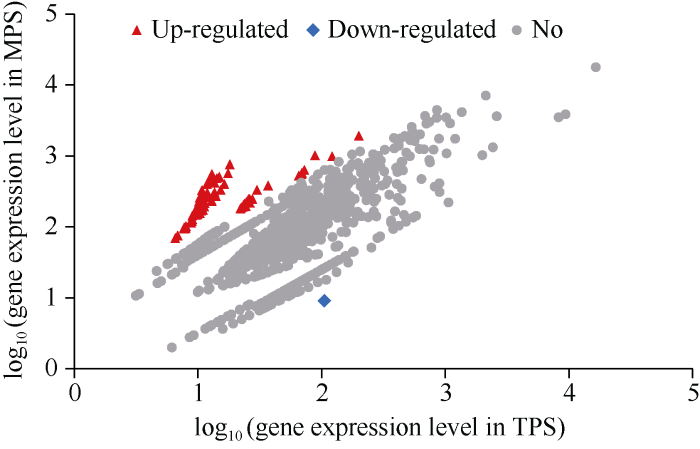 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3苎麻MPS和TPS茎皮中差异表达circRNA鉴定
图中红色、蓝色和灰色的数据点分别代表上调表达、下调表达及无表达差异的circRNA; 横坐标和纵坐标分别代表某circRNA在TPS和MPS组织中的read数的对数值。缩写同
Fig. 3Identification of differentially expressed circRNA by comparing gene expression level between TPS and MPS libraries
Red dots represent transcripts more prevalent in the MPS library, blue dots show those present at a lower frequency in the MPS, and gray dots indicate transcripts that did not change significantly; X and Y axes represent the logarithm of read number for each circRNA in TPS and MPS libraries, respectively. Abbreviations are the same as those given in
Table 3
表3
表3苎麻MPS和TPS茎皮中差异表达circRNA信息表
Table 3
| circRNA名称 circRNA ID | 类型 Type | MPS read数 MPS-reads number | TPS read数 TPS-reads number | 差异倍数 Differential fold (MPS/TPS) | 调控方向 Direction regulated | 校正P值 Adjusted P-value | 对应基因 Gene located by circRNA | 对应基因功能 Annotationof gene located by circRNA |
|---|---|---|---|---|---|---|---|---|
| scaffold13:3497301|3498287 | Introgenic | 553.7 | 13.0 | 42.7 | 上调 Up-regulated | 2.6E-04 | whole_GLEAN_10021135 | Ent-kaurenoic acid oxidase 1 |
| scaffold7:2328081|2328970 | Iintrogenic | 760.3 | 18.1 | 42.0 | 上调Up-regulated | 2.8E-04 | whole_GLEAN_10025341 | Casein kinase 1 |
| scaffold27:618745|619712 | Introgenic | 484.5 | 12.5 | 38.9 | 上调Up-regulated | 3.9E-04 | whole_GLEAN_10015712 | Cleavage and polyadenylation specificity factor subunit 3-I |
| scaffold24:109942|110887 | Introgenic | 461.0 | 12.8 | 35.9 | 上调Up-regulated | 5.4E-04 | whole_GLEAN_10016716 | Protein APEM9 |
| scaffold68:19731|20827 | Introgenic | 433.3 | 12.1 | 35.7 | 上调Up-regulated | 5.6E-04 | whole_GLEAN_10010106 | Protein CHROMATIN REMODELING 5 |
| scaffold777:22008|22923 | Introgenic | 485.5 | 14.2 | 34.2 | 上调Up-regulated | 6.7E-04 | whole_GLEAN_10001638 | Non-specific phospholipase C3 |
| scaffold1:1651705|1652476 | Introgenic | 507.7 | 14.9 | 34.1 | 上调Up-regulated | 6.7E-04 | whole_GLEAN_10029483 | Monogalactosyldiacylglycerol synthase |
| scaffold11:2168138|2169190 | Intergenic | 401.2 | 11.8 | 33.9 | 上调Up-regulated | 6.8E-04 | ||
| scaffold14:2019005|2020131 | Introgenic | 568.2 | 17.5 | 32.4 | 上调Up-regulated | 8.2E-04 | whole_GLEAN_10020529 | Peroxisome biogenesis protein 1 |
| scaffold1:2974233|2974487 | Introgenic | 418.1 | 13.7 | 30.6 | 上调Up-regulated | 1.0E-03 | whole_GLEAN_10029667 | Homeobox protein knotted-1-like 1 |
| scaffold4:2794969|2795547 | Introgenic | 328.8 | 10.8 | 30.5 | 上调Up-regulated | 1.0E-03 | whole_GLEAN_10027195 | Lissencephaly-1 homolog |
| scaffold13:1482426|1482614 | Introgenic | 288.1 | 10.8 | 26.7 | 上调Up-regulated | 1.7E-03 | whole_GLEAN_10020870 | Threonine-protein kinase PBL28 |
| scaffold42:394825|395928 | Introgenic | 303.4 | 11.9 | 25.5 | 上调Up-regulated | 2.0E-03 | whole_GLEAN_10012793 | U11/U12 small nuclear ribonucleoprotein 65 kDa protein |
| scaffold110:48151|61235 | Introgenic | 256.8 | 10.3 | 24.9 | 上调Up-regulated | 2.2E-03 | whole_GLEAN_10007543 | Protein TIC 40 |
| scaffold6:1851520|1851960 | Introgenic | 400.8 | 16.3 | 24.5 | 上调Up-regulated | 2.3E-03 | whole_GLEAN_10025870 | DEAD-box ATP-dependent RNA helicase 50 |
| scaffold6:319819|320593 | Introgenic | 239.6 | 10.5 | 22.9 | 上调Up-regulated | 2.9E-03 | whole_GLEAN_10025672 | GBF-interacting protein 1 |
| scaffold22:2080855|2081636 | Introgenic | 308.2 | 13.6 | 22.6 | 上调Up-regulated | 3.1E-03 | whole_GLEAN_10017734 | SOK1 kinase belonging to the STE20/SPS1/GC kinase family |
| scaffold38:1007107|1007440 | Introgenic | 229.5 | 10.2 | 22.5 | 上调Up-regulated | 3.1E-03 | whole_GLEAN_10013247 | Lipoxygenase 6 |
| scaffold4:984097|984791 | Introgenic | 225.4 | 10.3 | 21.8 | 上调Up-regulated | 3.5E-03 | whole_GLEAN_10026982 | V-type proton ATPase subunit a1 |
| scaffold36:1130226|1130557 | Introgenic | 332.4 | 15.3 | 21.8 | 上调Up-regulated | 3.5E-03 | whole_GLEAN_10013939 | Primary amine oxidase |
| scaffold9:3087645|3088126 | Introgenic | 261.8 | 12.2 | 21.5 | 上调Up-regulated | 3.7E-03 | whole_GLEAN_10024168 | Transcription factor bHLH144 |
| scaffold69:390306|390963 | Introgenic | 230.6 | 11.2 | 20.6 | 上调Up-regulated | 4.2E-03 | whole_GLEAN_10010042 | Formin-like protein 16 |
| scaffold244:78865|79334 | Introgenic | 216.4 | 10.9 | 19.9 | 上调Up-regulated | 4.7E-03 | whole_GLEAN_10004368 | Uncharacterized mitochondrial protein ymf40 |
| scaffold11:2226699|2233286 | Introgenic | 273.2 | 13.9 | 19.6 | 上调Up-regulated | 4.9E-03 | whole_GLEAN_10021948 | Cold-regulated 413 plasma membrane protein 2 |
| scaffold23:488065|489466 | Introgenic | 198.8 | 10.2 | 19.4 | 上调Up-regulated | 5.1E-03 | whole_GLEAN_10017916 | Phosphoinositide phosphatase SAC1 |
| scaffold59:336176|337027 | Introgenic | 192.9 | 10.1 | 19.1 | 上调Up-regulated | 5.4E-03 | whole_GLEAN_10011259 | Glycosylphosphatidylinositol anchor synthesis protein |
| scaffold270:63772|64261 | Introgenic | 215.3 | 11.3 | 19.0 | 上调Up-regulated | 5.5E-03 | whole_GLEAN_10004135 | ATP synthase protein YMF19 |
| scaffold21:626345|627257 | Introgenic | 179.7 | 9.7 | 18.6 | 上调Up-regulated | 5.9E-03 | whole_GLEAN_10019902 | Poly [ADP-ribose] polymerase 1 |
| scaffold41:981509|982441 | Introgenic | 232.5 | 12.9 | 18.0 | 上调Up-regulated | 6.5E-03 | whole_GLEAN_10013030 | WD40 repeat protein |
| scaffold25:1518597|1519148 | Introgenic | 183.3 | 10.3 | 17.7 | 上调Up-regulated | 6.8E-03 | whole_GLEAN_10016591 | Protein LSD1 |
| scaffold69:199217|199851 | Introgenic | 194.3 | 11.2 | 17.3 | 上调Up-regulated | 7.4E-03 | whole_GLEAN_10010022 | U-box domain-containing protein 14 |
| scaffold37:1378235|1379257 | Introgenic | 163.1 | 9.5 | 17.2 | 上调Up-regulated | 7.5E-03 | whole_GLEAN_10013509 | Uncharacterized protein At1g51745 |
| scaffold41:312471|314479 | Introgenic | 168.0 | 10.2 | 16.5 | 上调Up-regulated | 8.5E-03 | whole_GLEAN_10012948 | Nucleolar GTPase 2 |
| scaffold30:600099|602644 | Introgenic | 156.9 | 9.6 | 16.3 | 上调Up-regulated | 8.9E-03 | whole_GLEAN_10015039 | Protein CROWDED NUCLEI 3 |
| scaffold34:2347785|2349055 | Introgenic | 144.0 | 9.0 | 16.0 | 上调Up-regulated | 9.3E-03 | whole_GLEAN_10017394 | B3 domain-containing protein07g0563300 |
| scaffold360:24869|25462 | Introgenic | 174.8 | 11.0 | 15.9 | 上调Up-regulated | 9.4E-03 | whole_GLEAN_10003361 | Cytochrome c oxidase subunit 2 |
| scaffold1:5693805|5694897 | Introgenic | 169.3 | 10.6 | 15.9 | 上调Up-regulated | 9.5E-03 | whole_GLEAN_10030060 | Internal alternative NAD(P)H-ubiquinone oxidoreductase A1 |
| scaffold244:79500|79823 | Introgenic | 155.0 | 9.9 | 15.7 | 上调Up-regulated | 9.8E-03 | whole_GLEAN_10004368 | Uncharacterized mitochondrial protein ymf40 |
| scaffold270:63801|64263 | Introgenic | 155.3 | 10.1 | 15.4 | 上调Up-regulated | 1.0E-02 | whole_GLEAN_10004135 | ATP synthase protein YMF19 |
| scaffold112:248381|248869 | Introgenic | 148.3 | 9.6 | 15.4 | 上调Up-regulated | 1.0E-02 | whole_GLEAN_10007469 | Peptidyl-prolyl cis-trans isomerase CYP71 |
| scaffold36:1536277|1547559 | Introgenic | 150.7 | 9.9 | 15.2 | 上调Up-regulated | 1.1E-02 | whole_GLEAN_10013997 | Cytochrome P450 71A1 |
| scaffold360:12593|13058 | Intergenic | 155.4 | 10.4 | 15.0 | 上调Up-regulated | 1.1E-02 | ||
| scaffold1:142468|143089 | Introgenic | 128.5 | 9.0 | 14.3 | 上调Up-regulated | 1.3E-02 | whole_GLEAN_10029278 | Protein Mut11 |
| scaffold29:718451|719832 | Introgenic | 122.4 | 8.8 | 13.9 | 上调Up-regulated | 1.4E-02 | whole_GLEAN_10015276 | E4 SUMO-protein ligase PIAL2 |
| scaffold360:15097|17687 | Introgenic | 121.8 | 8.8 | 13.9 | 上调Up-regulated | 1.4E-02 | whole_GLEAN_10003361 | Cytochrome c oxidase subunit 2 |
| scaffold39:1205365|1205747 | Intergenic | 121.1 | 8.9 | 13.6 | 上调Up-regulated | 1.5E-02 | ||
| scaffold19:1034720|1049699 | Introgenic | 115.4 | 8.8 | 13.1 | 上调Up-regulated | 1.6E-02 | whole_GLEAN_10018479 | Short-chain dehydrogenase/reductase 2b |
| scaffold27:1733501|1744156 | Introgenic | 102.4 | 8.0 | 12.8 | 上调Up-regulated | 1.7E-02 | whole_GLEAN_10015833 | Large ribosomal RNA subunit accumulation protein YCED homolog 2 |
| scaffold270:115352|115747 | Introgenic | 98.1 | 7.7 | 12.7 | 上调Up-regulated | 1.8E-02 | whole_GLEAN_10004138 | Hypothetical protein |
| scaffold11:3427880|3429252 | Introgenic | 95.6 | 7.8 | 12.3 | 上调Up-regulated | 2.0E-02 | whole_GLEAN_10022104 | Probable ubiquitin-like-specific protease 2B |
| scaffold5:2904984|2905988 | Introgenic | 96.1 | 7.9 | 12.1 | 上调Up-regulated | 2.0E-02 | whole_GLEAN_10026531 | Transcription factor bHLH68 |
| scaffold22:2902269|2903224 | Introgenic | 1019.2 | 88.3 | 11.5 | 上调Up-regulated | 2.0E-02 | whole_GLEAN_10017861 | TATA-binding protein-associated factor BTAF1 |
| scaffold41:1051261|1051835 | Introgenic | 333.0 | 30.0 | 11.1 | 上调Up-regulated | 2.5E-02 | whole_GLEAN_10013037 | Probable E3 ubiquitin-protein ligase RHB1A |
| scaffold423:8362|8891 | Introgenic | 75.7 | 6.9 | 11.0 | 上调Up-regulated | 2.6E-02 | whole_GLEAN_10002921 | NADH-ubiquinone oxidoreductase chain 2 |
| scaffold50:971580|972301 | Introgenic | 75.7 | 6.9 | 11.0 | 上调Up-regulated | 2.6E-02 | whole_GLEAN_10022800 | Uncharacterized conserved protein |
| scaffold29:144492|145002 | Introgenic | 1922.2 | 200.1 | 9.6 | 上调Up-regulated | 2.6E-02 | whole_GLEAN_10015203 | A Plastidial ribosomal protein S1 |
| scaffold423:7458|7843 | Introgenic | 69.4 | 6.5 | 10.6 | 上调Up-regulated | 2.8E-02 | whole_GLEAN_10002921 | NADH-ubiquinone oxidoreductase chain 2 |
| scaffold41:1051261|1051640 | Introgenic | 383.7 | 36.9 | 10.4 | 上调Up-regulated | 3.0E-02 | whole_GLEAN_10013037 | Probable E3 ubiquitin-protein ligase RHB1A |
| scaffold128:291760|298584 | Introgenic | 244.6 | 25.7 | 9.5 | 上调Up-regulated | 3.7E-02 | whole_GLEAN_10007591 | Histone-lysine N-methyltransferase ATXR6 |
| scaffold42:575382|577290 | Introgenic | 248.1 | 26.2 | 9.5 | 上调Up-regulated | 3.7E-02 | whole_GLEAN_10012809 | Uncharacterized conserved protein |
| scaffold4:4247567|4248126 | Introgenic | 238.8 | 25.3 | 9.4 | 上调Up-regulated | 3.8E-02 | whole_GLEAN_10027396 | ALBINO3-like protein 2 |
| scaffold26:228677|229118 | Introgenic | 233.3 | 25.6 | 9.1 | 上调Up-regulated | 4.1E-02 | whole_GLEAN_10016157 | Protein BTR1 |
| scaffold81:336505|338444 | Introgenic | 223.2 | 24.5 | 9.1 | 上调Up-regulated | 4.1E-02 | whole_GLEAN_10009183 | Protein FORGETTER 1 |
| scaffold8:474393|475508 | Introgenic | 633.7 | 72.5 | 8.7 | 上调Up-regulated | 4.1E-02 | whole_GLEAN_10024439 | Uncharacterized protein At3g06530 |
| scaffold3:3339987|3341286 | Introgenic | 246.4 | 27.5 | 8.9 | 上调Up-regulated | 4.2E-02 | whole_GLEAN_10028049 | Aspartate aminotransferase 3 |
| scaffold133:143853|144812 | Introgenic | 212.1 | 23.8 | 8.9 | 上调Up-regulated | 4.3E-02 | whole_GLEAN_10006440 | Protein RNA-directed DNA methylation 3 |
| scaffold16:2445618|2446250 | Introgenic | 203.6 | 23.3 | 8.7 | 上调Up-regulated | 4.5E-02 | whole_GLEAN_10019477 | Alternative NAD(P)H-ubiquinone oxidoreductase C1 |
| scaffold270:115352|115722 | Introgenic | 202.7 | 23.3 | 8.7 | 上调Up-regulated | 4.5E-02 | whole_GLEAN_10004138 | Hypothetical protein |
| scaffold8:2674752|2676656 | Introgenic | 580.2 | 69.2 | 8.4 | 上调Up-regulated | 4.6E-02 | whole_GLEAN_10024738 | COMPASS-like H3K4 histone methylase component WDR5A |
| scaffold360:11517|11838 | Intergenic | 560.1 | 68.9 | 8.1 | 上调Up-regulated | 4.9E-02 | ||
| scaffold12:2523138|2525034 | Introgenic | 529.5 | 65.4 | 8.1 | 上调Up-regulated | 4.9E-02 | whole_GLEAN_10021469 | Tetraspanin-18 |
| scaffold40:1450202|1450949 | Introgenic | 188.2 | 22.5 | 8.3 | 上调Up-regulated | 5.0E-02 | whole_GLEAN_10013727 | Proline-rich receptor-like protein kinase PERK8 |
| scaffold270:40371|40783 | Introgenic | 991.9 | 121.6 | 8.2 | 上调Up-regulated | 5.0E-02 | whole_GLEAN_10004133 | Hypothetical protein |
| scaffold7:3133443|3134407 | Introgenic | 218.9 | 26.3 | 8.3 | 上调Up-regulated | 5.0E-02 | whole_GLEAN_10025435 | DEAD-box ATP-dependent RNA helicase 13 |
| scaffold69:125839|129078 | Introgenic | 195.2 | 23.4 | 8.3 | 上调Up-regulated | 5.0E-02 | whole_GLEAN_10010014 | Exocyst complex component EXO70A1 |
| scaffold38:37152|37816 | Introgenic | 181.8 | 22.1 | 8.2 | 上调Up-regulated | 5.0E-02 | whole_GLEAN_10013105 | DNA replication licensing factor MCM2 |
| scaffold9:1148862|1149242 | Introgenic | 196.3 | 24.4 | 8.0 | 上调Up-regulated | 5.0E-02 | whole_GLEAN_10023894 | Pyrophosphate-energized vacuolar membrane proton pump |
| scaffold2:6347151|6348009 | Introgenic | 9.1 | 105.2 | 11.5 | 下调Down-regulated | 2.3E-02 | whole_GLEAN_10029246 | N-Acetylglucosamine kinase |
新窗口打开|下载CSV
为了验证这些circRNA的差异表达结果, 本研究随机选取了10个circRNA, 通过qRT-PCR进一步分析它们在2个组织中的表达水平。由图4可知, 10个circRNA均呈现出差异表达, 且差异表达倍数明显高于测序分析结果, scaffold6:1851520| 1851960和scaffold30:600099|602644的差异表达倍数甚至达到上千倍。可见, 与高通量测序技术相比, qRT-PCR检测circRNA具有更高的灵敏度。说明这78个circRNA的差异表达是真实可靠的。
图4
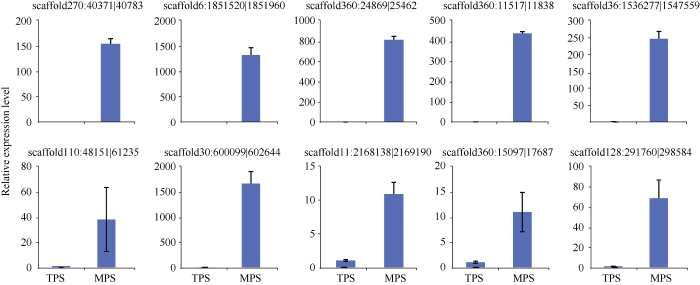 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4qRT-PCR分析10个苎麻circRNA在MPS和TPS茎皮中的相对表达水平
缩写同表2。
Fig. 4Relative expression level of ten circRNAs stem bark tissues of MPS and TPS by qRT-PCR in ramie
Abbreviations are the same as those given in Table 2.
3 讨论
纤维广泛存在于植物各器官中, 具有支撑器官形态、植株直立生长, 及抵抗病原菌攻击等作用。此外, 植物纤维也是重要的工业原料, 广泛用于纺织、造纸等工业领域。可见, 不管是对植物本身, 还是对国民经济, 植物纤维具有非常重要的作用。目前, 有关植物纤维生物合成机制在模式植物拟南芥中研究的比较多, 发现其主要由一系列NAC (NAM, ATAF1/2和CUC2)和MYB (v-myb avian myloblastosis viral oncogene homolog)转录因子调控, 它们形成分层次的网络并逐级调控整个纤维发育过程[27]。蛋白翻译后修饰如磷酸化、泛素化等也参与植物纤维的发育调控[28,29,30]。此外, 非编码RNA也可参与植物的纤维发育调控, 如柑橘miR397a可以调控纤维发育控制基因LAC17和LAC4的表达[31], 而拟南芥miR319则靶向调控TCP4基因的表达, 进而抑制纤维发育[32]。circRNA是一类重要的非编码RNA, 广泛参与花发育、果实成熟、逆境响应等多个生命过程[33]。植物中存在大量的circRNA, 如PlantcircBase数据库[34]已收录了40,311个水稻(Oryza sativa) circRNA、38,938个拟南芥(Arabidopsis thaliana) circRNA、6302个玉米(Zea mays) circRNA、7806个大豆(Glycine max) circRNA和1976个番茄(Solanum lycopersicum) circRNA。Wang等[35]系统研究了244个玉米RNA-Seq样本和288个水稻RNA-Seq样本, 鉴定到38,785个玉米circRNA和63,048个水稻circRNA, 并发现在玉米中有11,206个circRNA响应干旱逆境, 6770个circRNA响应盐逆境, 而在水稻中有824、6313和5724个circRNA分别响应干旱、盐和冷逆境。Zhang等[36]首次发现可能与花生种子发育相关的347个差异表达circRNA, 并通过qRT-PCR验证了其中15个circRNA的表达水平。茉莉酸甲酯(methyl jasmonic acid, MeJA)在植物生长和防御中发挥关键作用, 拟南芥种子用MeJA处理后, 经高通量测序鉴定出8588个circRNA, 其中有385个circRNA响应MeJA处理[37]。已有研究表明, circRNA能够参与调控其宿主基因表达, 例如在水稻中过表达circRNA Os08circ16564降低其宿主基因AK064900在叶和花序中的表达[37]。Gao等[19]在低温胁迫下的葡萄叶片中鉴定了475个差异表达的circRNA, 通过异源过度表达一种源自甘油-3-P-酰基转移酶的circRNA (Vv-circATS1), 可以增强拟南芥(Arabidopsis thaliana)的耐寒性, 而源自同一序列的线性RNA则不能, 这为植物耐寒提供新见解。
当前对circRNA在植物纤维发育中的功能角色仍有待深入研究。本研究鉴定到78个苎麻circRNA在纤维发育不同时期呈现差异表达, 这将为研究circRNA在苎麻纤维发育中的功能奠定重要基础。在78个差异表达circRNA中, 有74个circRNA位于基因内部, 占比达94.9%, 这个比例高于苎麻总circRNA中该类型的比例(89.8%), 其表达是否影响相应基因的表达, 目前仍难以确定。例如knotted-1-like Homeobox家族是植物发育中重要的转录因子家族, 其多个成员, 如拟南芥KNAT7、棉花GhKNL1均被发现参与植物的纤维发育调控[38,39]。此外, 在纤维发育过程中, 胞外复合体酶EXO70A1主要负责转运纤维素合成酶[40], 而磷酸酶SAC1在纤维次生壁加厚过程中具有重要功能[41]。本研究发现, whole_GLEAN_10029667、whole_GLEAN_10017916和whole_GLEAN_10010014分别注释为拟南芥纤维发育基因KNAT7、SAC1和EXO70A1的同源基因,circRNAscaffold1:2974233|2974487、scaffold23:488065| 489466和scaffold69:125839|129078分别定位到这3个苎麻基因中。因circRNA可通过miRNA海绵功能、干扰可变剪切、结合蛋白等方式调控相应基因表达, 这3个circRNA的上调表达或许会影响到相应的whole_GLEAN_10029667、whole_GLEAN_10017916和whole_GLEAN_10010014表达, 进而可能在纤维发育中发挥功能。因此, 本研究将为我们深入研究这3个circRNA和相应基因的功能及它们的表达调控关系奠定基础。
4 结论
本研究首次在苎麻中鉴定到5268个circRNA, 并发现其中78个circRNA在纤维发育的2个不同时期呈现差异表达。这些circRNA的鉴定为研究苎麻生长发育, 尤其是纤维的生长发育调控功能提供重要基础。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1186/1471-2164-14-125URLPMID:23442184 [本文引用: 1]

BACKGROUND: Ramie fiber, extracted from vegetative organ stem bast, is one of the most important natural fibers. Understanding the molecular mechanisms of the vegetative growth of the ramie and the formation and development of bast fiber is essential for improving the yield and quality of the ramie fiber. However, only 418 expressed tag sequences (ESTs) of ramie deposited in public databases are far from sufficient to understand the molecular mechanisms. Thus, high-throughput transcriptome sequencing is essential to generate enormous ramie transcript sequences for the purpose of gene discovery, especially genes such as the cellulose synthase (CesA) gene. RESULTS: Using Illumina paired-end sequencing, about 53 million sequencing reads were generated. De novo assembly yielded 43,990 unigenes with an average length of 824 bp. By sequence similarity searching for known proteins, a total of 34,192 (77.7%) genes were annotated for their function. Out of these annotated unigenes, 16,050 and 13,042 unigenes were assigned to gene ontology and clusters of orthologous group, respectively. Searching against the Kyoto Encyclopedia of Genes and Genomes Pathway database (KEGG) indicated that 19,846 unigenes were mapped to 126 KEGG pathways, and 565 genes were assigned to http://starch and sucrose metabolic pathway which was related with cellulose biosynthesis. Additionally, 51 CesA genes involved in cellulose biosynthesis were identified. Analysis of tissue-specific expression pattern of the 51 CesA genes revealed that there were 36 genes with a relatively high expression levels in the stem bark, which suggests that they are most likely responsible for the biosynthesis of bast fiber. CONCLUSION: To the best of our knowledge, this study is the first to characterize the ramie transcriptome and the substantial amount of transcripts obtained will accelerate the understanding of the ramie vegetative growth and development mechanism. Moreover, discovery of the 36 CesA genes with relatively high expression levels in the stem bark will present an opportunity to understand the ramie bast fiber formation and development mechanisms.
[本文引用: 5]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
DOI:10.3389/fpls.2016.02024URLPMID:28105043 [本文引用: 2]
Circular RNAs (circRNAs) are a type of newly identified non-coding RNAs through high-throughput deep sequencing, which play important roles in miRNA function and transcriptional controlling in human, animals, and plants. To date, there is no report in wheat seedlings regarding the circRNAs identification and roles in the dehydration stress response. In present study, the total RNA was extracted from leaves of wheat seedlings under dehydration-stressed and well-watered conditions, respectively. Then, the circRNAs enriched library based deep sequencing was performed and the circRNAs were identified using bioinformatics tools. Around 88 circRNAs candidates were isolated in wheat seedlings leaves while 62 were differentially expressed in dehydration-stressed seedlings compared to well-watered control. Among the dehydration responsive circRNAs, six were found to act as 26 corresponding miRNAs sponges in wheat. Sixteen circRNAs including the 6 miRNAs sponges and other 10 randomly selected ones were further validated to be circular by real-time PCR assay, and 14 displayed consistent regulation patterns with the transcriptome sequencing results. After Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of the targeted mRNAs functions, the circRNAs were predicted to be involved in dehydration responsive process, such as photosynthesis, porphyrin, and chlorophyll metabolism, oxidative phosphorylation, amino acid biosynthesis, and metabolism, as well as plant hormone signal transduction, involving auxin, brassinosteroid, and salicylic acid. Herein, we revealed a possible connection between the regulations of circRNAs with the expressions of functional genes in wheat leaves associated with dehydration resistance.
DOI:10.1104/pp.18.01331URLPMID:30962290 [本文引用: 3]
Circular RNAs (circRNAs) are widely distributed and play essential roles in a series of developmental processes, although none have been identified or characterized in grapevines (Vitis vinifera). In this study, we characterized the function of grape circRNA and uncovered thousands of putative back-splicing sites by global transcriptome analysis. Our results indicated that several reported circRNA prediction algorithms should be used simultaneously to obtain comprehensive and reliable circRNA predictions in plants. Furthermore, the length of introns flanking grape circRNAs was closely related to exon circularization. Although the longer introns flanking grape circRNAs appeared to circularize more efficiently, a 20- to 50-nt region seemed large enough to drive grape circRNA biogenesis. In addition, the endogenous introns flanking circularized exon(s) in conjunction with reverse complementary sequences could support the accurate and efficient circularization of various exons in grape, which constitutes a new tool for exploring the functional consequences caused by circRNA expression. Finally, we identified 475 differentially expressed circRNAs in grape leaves under cold stress. Overexpression of Vv-circATS1, a circRNA derived from glycerol-3-P acyltransferase, improved cold tolerance in Arabidopsis (Arabidopsis thaliana), while the linear RNA derived from the same sequence cannot. These results indicate the functional difference between circRNA and linear RNA, and provide new insight into plant abiotic stress resistance.
URLPMID:29423997 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:28086788 [本文引用: 1]
URLPMID:25516281 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:31145998 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.molp.2017.03.003URLPMID:28315753 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
DOI:10.1111/j.1469-8137.2011.04016.xURLPMID:22236040 [本文引用: 1]
* The formation of secondary cell walls in cell types such as tracheary elements and fibers is a defining characteristic of vascular plants. The Arabidopsis transcription factor KNAT7 is a component of a transcription network that regulates secondary cell wall biosynthesis, but its function has remained unclear. * We conducted anatomical, biochemical and molecular phenotypic analyses of Arabidopsis knat7 loss-of-function alleles, KNAT7 over-expression lines and knat7 lines expressing poplar KNAT7. * KNAT7 was strongly expressed in concert with secondary wall formation in Arabidopsis and poplar. Arabidopsis knat7 loss-of-function alleles exhibited irregular xylem phenotypes, but also showed increased secondary cell wall thickness in fibers. Increased commitment to secondary cell wall biosynthesis was accompanied by increased lignin content and elevated expression of secondary cell wall biosynthetic genes. KNAT7 over-expression resulted in thinner interfascicular fiber cell walls. * Taken together with data demonstrating that KNAT7 is a transcriptional repressor, we hypothesize that KNAT7 is a negative regulator of secondary wall biosynthesis, and functions in a negative feedback loop that represses metabolically inappropriate commitment to secondary wall formation, thereby maintaining metabolic homeostasis. The conservation of the KNAT7 regulatory module in poplar suggests new ways to manipulate secondary cell wall deposition for improvement of bioenergy traits in this tree.
URLPMID:24831118 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}